

Abstract
Background
The primary cause of morbidity and mortality in cystic fibrosis (CF) patients is lung infection by Pseudomonas aeruginosa. Therefore much work has been done to understand the adaptation and evolution of P. aeruginosa in the CF lung. However, many of these studies have focused on longitudinally collected single isolates, and only few have included cross-sectional analyses of entire P. aeruginosa populations in sputum samples. To date only few studies have used the approach of metagenomic analysis for the purpose of investigating P. aeruginosa populations in CF airways.
Results
We analysed five metagenomes together with longitudinally collected single isolates from four recently chronically infected CF patients. With this approach we were able to link the clone type and the majority of SNP profiles of the single isolates to that of the metagenome(s) for each individual patient.
Conclusion
Based on our analysis we find that when having access to comprehensive collections of longitudinal single isolates it is possible to rediscover the genotypes of the single isolates in the metagenomic samples. This suggests that information gained from genome sequencing of comprehensive collections of single isolates is satisfactory for many investigations of adaptation and evolution of P. aeruginosa to the CF airways.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2873-1) contains supplementary material, which is available to authorized users.
Keywords: Metagenomics, Longitudinal isolates, Pseudomonas aeruginosa, Cystic fibrosis
Background
Cystic fibrosis (CF) is a hereditary disease that causes malfunction of a chloride channel affecting the viscosity of the mucus on all muco-epithelial surfaces. Among other things, this results in impaired clearance of bacteria and other microorganisms from the airways with an associated increased risk of lung infections [1]. CF is the most common life-limiting genetic disorder in Caucasians, and lung infection with Pseudomonas aeruginosa is the primary cause of morbidity and mortality in CF patients [2, 3]. In the clinic, antibiotic treatment of these infections is usually based on the assumption that the bacterial populations in CF airways are homogeneous. In accordance with this assumption, several studies of the adaptation of P. aeruginosa to the CF airway environment with regard to e.g. resistance development [4, 5], metabolism [6], escape from the immune system [7], and transmission between niches in the airways of a patient [8] and between patients [9, 10], have primarily been carried out based on investigations of single longitudinally stored bacterial isolates [11–13].
However, it was recently shown that long-term bacterial infections of CF airways cannot solely be described as a “dominant lineage” model, where the infecting clone type adapts in a linear fashion, and new variants with increased fitness quickly outcompete their less fit ancestors [14]. Because of the heterogeneous environment of the CF airways, it is more likely a “diverse community” model that best describes the bacterial populations of the CF airways. This is a consequence of adaptive radiation and the development of different subpopulations with a high degree of polymorphic mutations [11, 14–16].
Thus, the question is whether genomic information from single isolates collected longitudinally from the same patient is sufficient for the characterization of adaptive and evolutionary processes in P. aeruginosa populations in CF airways. To answer this question, we have compared the sequences from longitudinally collected single isolates with single metagenomes from four CF patients. Therefore, rediscovery in the metagenomes of the genome sequences derived from the single isolates document that they constitute a substantial sub-population and thus are representative for the infecting population of the patient.
Methods
We included four CF patients followed at the CF clinic at Rigshospitalet, Copenhagen, Denmark. The age of the patients ranged from 15 to 31 years and they were all recently diagnosed as chronically infected with P. aeruginosa (Copenhagen criteria [17]).
Longitudinally collected single isolates
Genome sequenced longitudinally collected single isolates from the patients are described in details in Marvig et al. [13]. The single isolates included in this study cover P. aeruginosa sampled from: endolaryngeal suction, sputum samples, sinus samples taken at endoscopic sinus surgery, swabs from the sinuses, and bronchoalveolar lavage (BAL). Isolation and identification of P. aeruginosa from CF sputum samples was carried out as previously described [13].
Metagenomic samples
Sputum samples were collected at the CF clinic at Rigshospitalet and samples were processed a median of 2 days after expectoration (range: 1–3 days, Additional file 1: Table S1). During the lag-time between expectoration and processing the samples were stored at 4 °C.
Processing of metagenomic samples
The samples were treated with ca. 1:1 (v/v) 10× diluted Sputasol (Oxoid, c/o Thermo Fisher Scientific, UK) with continuous vigorous shaking for 30 min. for homogenisation.
The samples were divided into two fractions, one was plated on Pseudomonas isolation agar (PIA) plates and incubated in 24–72 h at 37 °C depending on when colonies appeared and before single colonies could no longer be picked. The single colonies were then grown in 96 well microtitter plates with 150 μl Luria Broth (LB) for 24–48 h. One hundred μl 50 % glycerol was added and the isolates were stored at −80°. The other fraction was directly subjected to DNA extraction (200–600 μl Sputasol treated sample), or stored at −20 °C until batch DNA purification could take place.
DNA extraction was carried out as in Lim et al. [18], with slight modifications: β-mercapoethanol was replaced by Sputasol treatment, centrifugation times were extended to 20 min at 3800×g, the volumes were adjusted to: 1.5 ml autoclaved milliQ, 100–200 μl DNase buffer and 3–6 μl DNase (depending on pellet size), and 1.5 SE buffer, with the Powersoil® DNA isolation kit (MO BIO Laboratories, USA) used according to the manufacturer, for DNA purification.
Sequencing and analysis of metagenomic reads
Libraries were prepared in triplicates with Nextera XT Sample Preparation kit (Illumina Inc., USA) and pooled prior to sequencing on an Illumina MiSeq® bench-top sequencer with MiSeq reagent kit V2, 300 cycles (Illumina Inc., USA), resulting in 150 bp paired end reads. Initial analysis of the reads (also used for species identification) was carried out using Novoalign V2.07.18 (Novocraft Technologies [19]) for alignment to a library of human (GRCh37, ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens/Assembled_chromosomes/seq/hs_ref_GRCh37.p13_chr*mfa.gz), bacterial, archaeal, viral, and fungal (The NCBI database, downloaded: November 11th 2013) sequences. All sequences aligning to the human genome were discarded.
The analysis of the P. aeruginosa population was performed according to Marvig et al. [13]. This implies: Alignment to the P. aeruginosa PAO1 reference genome (GenBank accession NC_002516.2; genome size of 6.4 Mb) with Bowtie 2 V2.0.2 [20] and The Genome Analysis Toolkit (GATK) V1.0.5083 [21] for realignment around indels. This simultaneously removed all non-P. aeruginosa reads from the metagenomic reads. The pileups of read alignments were performed with SAM tools V0.1.7 (r50) [22]. SNP calling from the metagenomic reads was carried out by manually identifying positions where mutations had previously been identified in the single isolates of the clone types from the same patients as the analysed metagenomes. The mutations of the single isolates have previously been discussed and presented in Marvig et al. [13]. Raw de novo assemblies of the metagenomes were carried out using Velvet [23] (version 1.2.10) with a k-mer length of 33 and the options set as follows: ‘-scaffolding no –ins_length 500 –cov_cutoff 3 –min_contig_lgth 500’. De novo-assembled genomes were aligned against each other using MUMmer3 [24] (version 3.23).
Maximum likelihood phylogenetic analysis was carried out with PAUP* [25] version 4.0b10 without root, using the alleles identified by the single isolate sequencing. The metagenomes were placed in the tree depending on their major alleles at positions where polymorphisms occurred. Maximum parsimonious phylogenetic analyses were also carried out with PAUP* version 4.0b10 using alleles of reference strain PAO1 as a root.
The rediscovery of SNPs from the single isolates in the metagenomes were done based on the assumption that if a SNP previously identified in the single isolates was present in more than 10 % of the reads, this was a rediscovered SNP. When looking at polymorphisms, only positions with a phred score >30 and with ≥4 read coverage were considered. This was then compared to the overall coverage of PAO1, to make the ratio presented in Fig. 6.
Fig. 6.
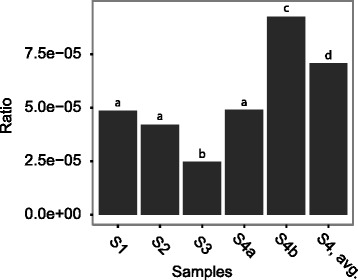
Polymorphic positions in the metagenomes. Because of the differing coverage of the PAO1 reference genome, the number of polymorphisms is shown as a ratio of polymorphisms and the coverage of each metagenome to PAO1. S4, avg. is the average of S4a and S4b. p <0.05 Fisher’s exact test with Holm correction
Diversity measurement by phylogenetic analysis of single isolates
Diversity is shown as the mean distance to the Line of Decent (LOD) [12]. LOD is the immediate line from the root, here based on PAO1 as out-group, to the latest sampled isolate (the red line in the phylogenetic trees in Fig. 5), and the mean distance to LOD is the mean number of SNPs from this line to the remaining isolates.
Fig. 5.
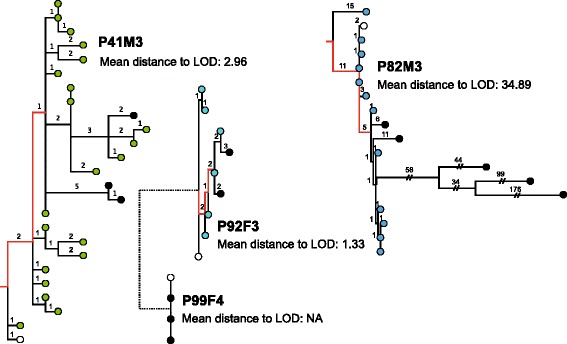
Mean distance to Line of Decent (LOD). Maximum parsimonious phylogenetic trees for all clone types identified in the metagenomes as being the latest. The red line in the trees indicates the LOD wherefrom SNPs (numbers on branches) have been counted. The LOD is set from the root (created for P41M3 and P82M3 using PAO1 as out-group) to the divergence of latest sampled isolates. White circles indicate the earliest sampled isolates and black circles indicates the latest sampled isolates from each patient, the coloured circles are comparable to the colours of Fig. 1. For each patient a mean distance to LOD is indicated below the patient name to the right of the corresponding tree. The mean distance to LOD of P82M3 is significantly different from the other patients, p <0.01, Fisher’s Exact test with Holm correction
Diversity measurement of metagenomic samples
Polymorphic positions were identified as described above. Because of the varying coverage of the different samples, the polymorphisms found were compared to the overall coverage of the PAO1 genome, and diversity was calculated as a ratio of polymorphisms and coverage. The reason, for using polymorphisms as a diversity measurement, was based on the assumption that the more positions a population diverge in, the more diverse is it likely to be.
As example: if we have e.g. two or three subpopulations they will differ from each other in a number of positions creating more ambiguous base calls and thus a higher ratio of polymorphisms, than a single homogeneous population. However, it is not possible to differentiate between two, three, or more different subpopulations based on this method. This is because of the possibility of a deep phylogenetic branching of two subpopulations and a shallow branching of three or more subpopulations.
Statistics
For comparisons of the rediscovery ratio of SNPs from single isolates in metagenomes and comparisons of diversity measurements of single isolates and metagenomes we used Fisher’s Exact Test with Holm correction for multiple testing, in R [26].
Results
Patient information and P. aeruginosa infection patterns
Four CF patients were enrolled in this study, median age 24 years; range 15–31, at the time of metagenome sampling. From each of the patients we have previously genome sequenced 9 to 27 longitudinally collected P. aeruginosa isolates covering 1–7 years of infection [13]. From the four patients we collected either one (n = 3) or two (n = 1) sputum samples for metagenomic analysis (Fig. 1a). Accordingly, sputum samples S1, S2, and S3 were sampled from patients P41M3, P99F4, and P92F3, respectively, and sputum samples S4a and S4b, separated by two weeks, were sampled from patient P82M3. The sputum samples used for the metagenome sequencing were collected approximately 1 year after the most recently genome sequenced single isolate. The time period between the most recently genome sequenced isolate and the metagenome is not critical, since the main question addressed here is whether or not the genotypes of the single isolates can be rediscovered in the metagenomic samples.
Fig. 1.
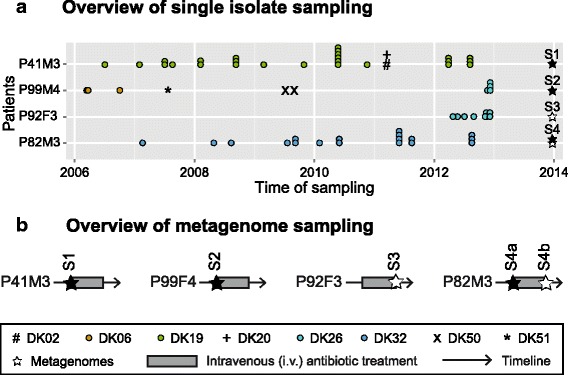
Overview. a Overview of single isolate sampling from four CF patients, clone types that are considered to be transient (found in 1–2 time points) are marked with #, +, x, or *, whereas clones considered to be persistent in the patient is marked by coloured circles. Metagenomes are marked with stars, black if sampled before i.v. antibiotic treatment, and white if sampled after i.v. antibiotic treatment. b Overview of metagenome sampling from patients, in correlation with 2-week i.v. antibiotic treatment
Three of the four patients (P41M3, P92F3, and P82M3) have infection patterns that are characteristic for the majority of the P. aeruginosa infected CF patients at the Copenhagen CF Center at Rigshospitalet [27], with a single primary clone type in the entire collection period. One patient (P99F4) has a change in clone type, where one clone type was outcompeted by another (Fig. 1a). All four patients in this study were recently diagnosed as chronically infected with P. aeruginosa according to the Copenhagen definitions at the time of metagenome sampling [17].
Processing of sputum sample reads
The metagenome sequences were aligned to a database containing all bacterial, fungal, and viral genome sequences deposited at NCBI (see Methods). With a median of 96 % of all bacterial reads (Additional file 2: Table S2), P. aeruginosa was the dominating microbial species in the patients, corresponding to their clinical diagnosis as chronically infected with P. aeruginosa. We further aligned reads from the sputum metagenomes to the P. aeruginosa PAO1 reference genome, as we have previously done for the single isolates [13]. In all cases, the metagenomes had an average coverage of 5.99 Mbp (range: 5.90–6.04 Mbp) of the 6.3 Mbp PAO1 reference genome, by >3 reads and a phred score >30 (Additional file 3: Table S3). This high genomic coverage ensured that the presence or absence of polymorphisms in the metagenomes could be determined at the majority of genomic positions. On average, sequenced positions were covered by 10 to 31 reads giving us the opportunity to identify subpopulations that are present in more than 10 % of the population at the positions with the lowest coverage (Additional file 3: Table S3).
In order to compare the P. aeruginosa population structure and diversity as displayed by the single isolates and the compliance with the metagenomic read assemblies, we conducted a three step analysis: 1) Identification of the dominant clone type(s) in the sputum samples, 2) investigation if mutations in the genomes of the single isolates were also identified in the metagenomes, i.e. rediscovery of SNPs in the metagenomes, and 3) comparison of diversity measurements of the populations represented by the single isolates and the metagenomes.
Identification of the dominant clone type(s)
To identify the P. aeruginosa clone types represented in the metagenomes, de novo assemblies of single isolates and metagenomes were compared. For each patient the clone types represented by the single isolates were compared with the metagenome(s) from the same patient.
For all four patients, the clone type of the most recently sampled single isolate corresponded to the clone type identified from the metagenome with less than 528 SNP of differences (median 131 SNPs, range 91–527 SNPs). In contrast, when comparing the metagenomes with single isolates of other clone types they differed by more than 16,268 SNPs (median 17,844 SNPs, range 16,269–30,918 SNPs) (Additional file 4: Table S4A and Table S4B).
This shows that for each patient the most recent clone type identified by the genome of the single isolate matches the dominating clone type in the P. aeruginosa population identified in the sputum sample metagenome.
Rediscovery of SNPs in the metagenomes
Previous investigations of genome evolution in the clonal lineages of P. aeruginosa strains from each of the four patients [13] identified SNPs accumulating in the clonal populations. If these SNPs are indeed present in actual propagating lineages of the P. aeruginosa population of these patients, they should also be present in the metagenome(s). When looking at all the SNPs identified in all the single isolates, it is expected that the ratio of rediscovery of SNPs between single isolates and metagenomes from the same patients should exceed the ratio determined between single isolates and metagenomes of different patients. Further, this ratio should reach a value of one if all mutations found in the single isolates are also present in the metagenome.
With the exception of patient P99F4 and P92F3, who are infected with the same clone type (DK26), the rediscovery of SNPs from the single isolates in the metagenome(s) of the same patient was found to be significantly higher than between patients (Fig. 2, p <0.05, Fisher’s exact test with Holm correction). This supports the specific linkage between single isolates and the P. aeruginosa population as a whole, as hypothesised above.
Fig. 2.
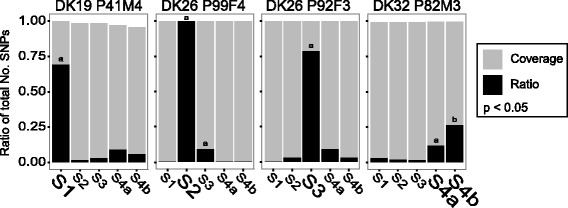
SNP rediscovery in metagenomes. Above each subfigure it is indicated which single isolates’ SNPs that have been sought rediscovered in the metagenomes (clone type and patient). The grey bars indicate the ratio of SNP positions that were sequenced in the metagenomes and the black bars indicate the ratio of the rediscovered reads to the sequenced positions. The metagenome(s) belonging to the same patient as the single isolates they are compared to is indicated with a larger font. NOTE, S2 and S3 are from different patients but the same clone type. P <0.05, Fisher’s exact test with Holm correction, significant differences are indicated by “a” and/or “b”
In one case (S2 from P99F4), the ratio of the rediscovery of SNPs reached one, suggesting that all SNPs identified in the single isolates are present in more than 10 % of the whole population. In all other cases the ratio was below one, which could be due to 1) not all mutations being fixed in the population, i.e. they were lost during the time of sampling of the single isolates (harbouring the mutations) until sampling of the metagenome, or 2) some of the mutations being present in only a small fraction (<10 %) of the population and therefore not sampled by the metagenomic reads. In the case of P92F3 the SNPs that were not rediscovered were only present in 11–22 % (Additional file 5: Table S5) of the single isolates, and thus could be explained by mutations not being fixed in the population.
The metagenomes S4a and S4b from patient P82M3 illustrate both explanations above: Firstly, the much lower ratio of rediscovery of SNPs in patient P82M3 compared to the other patients, may be explained by the presence of hypermutators in the P. aeruginosa population of P82M3. Hypermutators are known to accumulate many unfavourable mutations [28], which are not expected to remain in the population, thus leading to a low ratio of rediscovery (assuming that the mutations are not hitch-hiking with more favourable mutations). Secondly, the low coverage of the metagenomic samples (Additional file 3: Table S3) resulted in a higher percentage of the SNPs being rediscovered in the later metagenome (S4b) than in the early metagenome (S4a). The rediscovery of SNPs in the two metagenomes correspond to 26 % (122 of 461) and 12 % (54 of 461), respectively (Additional file 5: Table S5). This is contradictory since the mutations were previously identified in the single isolates and therefore must be present to some degree in S4a in order to be identified in S4b. This suggests that the subpopulation represented by the S4b metagenome is present below the limit of detection in the S4a metagenome sequences and is therefore not identified.
For patients P99F4 and P92F3 the similar rediscovery ratios of SNPs between the metagenomes and the single isolates can be explained by a co-infection of the same clone type, DK26. This relationship was noted previously and seems to be the consequence of a patient-to-patient transmission event of the DK26 clone from P92F3 to P99F4 [13], explaining the lack of differentiation between the two P. aeruginosa populations. However, despite this close relationship between the populations, Fig. 3 shows that it is possible to distinguish between the SNPs of the single isolates and the respective metagenomes.
Fig. 3.
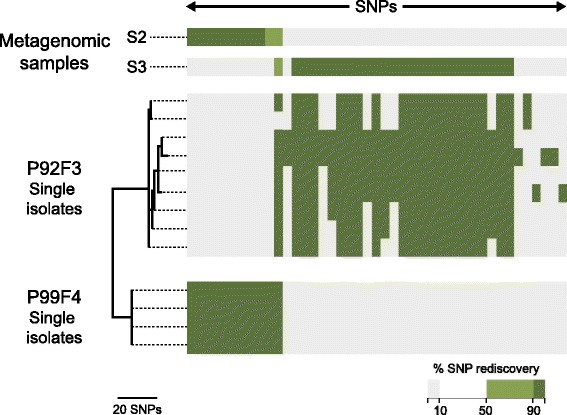
Patient specific correlation of rediscovered SNPs within a single clone type. A comparison of the SNPs found in the single isolates of the patients P99F4 and P92F3 as well as their respective metagenomes, S2 and S3. For the single isolates a dark green colour indicates the presence of SNPs and white the absence. For the metagenomes the percentage of reads covering the position of a SNP is indicated by dark green (>90 %), light green (51-90 %), or white (11-50 %) all considered to confirm the presence of the SNP in question. If the SNP is only found in <=10 % of the reads it is indicated by grey and not considered to be present in the metagenome(s)
We have identified SNPs in genome sequences of longitudinal single isolates, which seem to be characteristic and representative for the patient community, including cases of infections caused by patient-to-patient transmitted clones. This patient specific relationship between metagenomes and single isolates is further documented by the phylogenetic analysis of the single isolates and metagenomes of the hypermutator population of patient P82M3 (Fig. 4), which shows that despite the highly increased mutation rate, the metagenomes are placed within the phylogeny of the single isolates from the patient (Fig. 4). This phylogeny also shows that the single isolates are not clustered depending on their origin of sampling, indicating that the population is mixed between the upper and lower airways and that the different subpopulations are not limited to a specific spatial position in the airways.
Fig. 4.
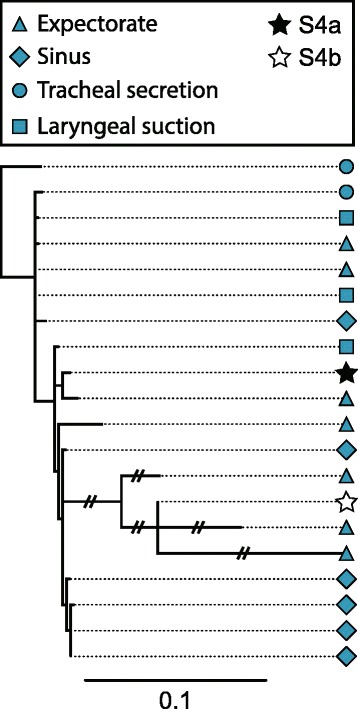
DK32 P82M3 maximum likelihood phylogeny including metagenomes. Blue shapes indicate single isolates sampled with different methods and from different locations (see legend) and stars indicate metagenomes. The scale bar indicates 0.1 likelihood of mutation
Diversity of the P. aeruginosa populations
In the single isolates, the diversity of the P. aeruginosa populations was determined from the phylogenies as the mean distance to the Line of Decent (LOD) (Fig. 5). For the metagenome-estimated diversity (Fig. 6) we used the number of polymorphisms normalised to the number of positions covered in the PAO1 genome in order to correct for differences in coverage between the different metagenomes (see Methods for details). Because S4a and S4b (patient P82M3) are representative of the same population we chose to merge the samples to carry out the inter-patient comparison of diversity (Fig. 6: “S4, avg.”). In both the LOD calculations and the number of polymorphisms we find, that the hypermutator population of patient P82M3 had the highest diversity and that the patient with the shortest period of infection (P92F3), as expected, harboured the least diverse population, to some degree validating our method of diversity calculations. We calculated 34.89 and 1.33 mean distances to LOD for the two single isolate populations, and diversity ratios of 7.08E-05, and 4.20E-05 for the metagenome populations from the two patients P82M3 and P92F3, respectively. Thus, in both cases of diversity measurements both single isolates and metagenomes we saw a significant difference between the diversity of the P. aeruginosa populations of patient P82M3 and P92F3 (p <0.05, Fisher’s Exact test with Holm correction) (Figs. 5 and 6).
When analysing further the single population of patient P82M3, the diversity calculations for the samples S4a and S4b illustrate that exhaustive sampling is essential, not only when using single isolates but also for metagenomic samples, in order to get the true picture of the population diversity. Because these two metagenomes represent a non-mutator and a hyper-mutator subpopulation, respectively, they have significantly different diversity ratios (4.90E-05 and 9.25E-05, respectively, p <0.05 Fisher’s Exact test with Holm correction).
Discussion
Airway infection in CF patients has attracted considerable interest as a model system for bacterial evolution and long-term human infections [29]. There is a number of reasons for this interest: 1) The infections are often mono-clonal lasting for decades, which can correspond to more than 100,000 bacterial generations [30], 2) sampling from the patients is relatively simple (sputum, BAL, suction), 3) the environmental conditions in the patient airways are very similar, and 4) isolate collections are found in many CF clinics covering long periods of sampling time. Several investigations of long-term CF airway infections based on the analysis of longitudinally collected single isolates of P. aeruginosa have been published in recent years [5, 6, 12, 13, 30–32], and some have also included cross-sectional analyses of the population diversity at the genomic level [11, 16, 33, 34]. One reason to question the validity of using single isolates to infer evolutionary dynamics of the entire population is the apparent heterogeneity of the P. aeruginosa population in the CF patients [11, 15, 16, 33–36].
In this study we have compared five meta-genomes obtained from four CF patient sputum samples with corresponding single, longitudinally collected, P. aeruginosa isolates, and a high degree of correlation was found within populations. The metagenomes were sampled from patients with the most common P. aeruginosa infection pattern, continuous culture of the same clone type, at the Copenhagen CF Centre [27], and they are therefore assumed to be representative for most of the CF patients and their lung infection. The collection of P. aeruginosa isolates from CF patients associated with the Copenhagen CF Clinic is comprehensive and characterized by frequent longitudinal sampling from the patients and frequent replicate isolates from individual patient samples. These features make the collection unique and useful for an assessment of the validity of single isolate analysis in relation to both biological and medical aspects. In general, single isolates and metagenome analyses depend on exhaustive sampling. Due to the possibility of temporal dominance by different subpopulations, as seen by the hyper-mutator population of P82M3, the metagenomic approach will also require multiple samples to reveal the profile and dynamics of P. aerugonosa populations. The results of this study, taken together with similar results from other studies [12], suggest that using comprehensive collections of longitudinally collected single isolates in the research of adaptation and evolution of P. aeruginosa to the CF airways will yield conclusive results.
One limitation of our study compared to e.g. that of Lieberman et al. [14] is the sequencing depth. This is especially true for the highly diverse population of P82M3, in which we were unable to identify subpopulations if present in less than 10 % of the population (the lowest coverage is 9.97). However, despite the lower sequencing depth, we were able to document a high degree of diversification of the populations in analogy with findings from Lieberman [14] and others [15, 16, 33]. In addition, we were also able to determine that the different subpopulations comprising this diversity differ in frequency over time. Especially in the hyper-mutator population, we noticed that the bacterial population is dominated by different subpopulations at different time points (Table 1 and Fig. 4).
Table 1.
SNPs rediscovered in S4a and S4b
| Total no. of reads on position | % of reads with the mutation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Position | Ref | Qry | Mutation (aa) | Mutation (bp) | PA no. | Gene name | S4a | S4b | S4a | S4b |
| 5671 | C | T | S466F | C1397T | PA0004 | gyrB | 15 | 13 | 7 | 100 |
| 2453983 | G | A | G106D | G317A | PA2231 | pslA | 17 | 17 | 24 | 0 |
| 2640133 | A | G | F43L | T127C | PA2386 | pvdA | 15 | 12 | 0 | 100 |
| 2926243 | C | G | E64Q | G190C | PA2586 | gacA | 5 | 11 | 0 | 100 |
| 3970113 | G | A | G172A | G58S | PA3545 | algG | 15 | 11 | 87 | 0 |
| 3971271 | G | A | G1330A | G444S | PA3545 | algG | 19 | 13 | 0 | 100 |
| 5551035 | A | C | V216G | T647G | PA4946 | mutL | 11 | 8 | 9 | 100 |
| 5677066 | T | C | T265A | A793G | PA5040 | pilQ | 12 | 17 | 17 | 100 |
| 6028514 | G | A | E325K | G973A | PA5361 | phoR | 5 | 12 | 100 | 0 |
Including examples of mutations not found in S4a but found in S4b (Ref reference base, Qry query base, PA no. PAO1 gene number, aa amino acid change, bp specific base pair substitution). NOTE: all mutation types are missense mutations
Population diversity was not the primary target of the investigations reported here due to the relatively short time frame of sampling and the resulting low number of mutations in the respective isolate genomes. However, the two cases of hyper-mutator isolates suggest that diversity is prevalent, resulting in significant population heterogeneity. This heterogeneity may be the result of spatial compartmentalisation of the CF airways and the confinement of different subpopulations to different niches [11, 35]. One obvious example of compartmentalization of the CF airways is illustrated by bacterial infections in both lungs and sinuses. Spatial isolation and adaptive radiation of different subpopulations in these niches has been suggested by Markussen [11] and Hansen [35]. In contrast, our current findings from longitudinally collected P. aeruginosa from upper and lower airways in younger CF patients [13] do not confirm this; instead, in accordance with Ciofu [37] and Johansen [38] we find that bacterial migration in both directions and consequential population mixing occurs between the upper and lower airways after a certain period of chronic infection (Fig. 4). Mixing of bacterial populations colonizing different airway compartments is also supported by other studies showing both genotypic and phenotypic overlap between samples from the upper and lower CF airways [37, 39].
Population mixing in CF airways is supported by frequent observations of clone type displacement; both in investigations of older CF patients with chronic P. aeruginosa lung infections [10] and in young patients with early colonization by P. aeruginosa. These findings are difficult to reconcile with spatial isolation and adaptive radiation, whether distribution of sub-populations is associated with the lung/sinus compartments or different sectors of the lungs as reported recently [40]. It is possible that the infections in fact switch between periods of adaptive radiation and spatial isolation of sub-populations resulting in diversity generation and periods of mixing caused by lung tissue changes. Such changes in population dynamics could explain the conflicting observations as well as the slow replacement of clone types, which sometimes take months or even years.
Conclusions
We find a consistency between the genomic changes identified in the single isolates and in the metagenomes, which can only be explained by the propagation of mutations identified by the analysis of single isolates within the specific patient’s P. aeruginosa population. These findings underline the relevance of comprehensive longitudinal sampling of single isolates of P. aeruginosa for investigations of adaptation and evolution. We also find it equally important for the metagnomic approach to have comprehensive sampling, in order to provide valuable information about the P. aeruginosa population dynamics of CF patient airway infections. It is, however important to emphasise that the conclusions to be drawn from this type of investigation will not provide a complete picture of the population diversity in the respective samples.
Abbreviations
BAL, bronchoalveolar lavage; CF, cystic fibrosis; DNA, deoxyribonucleic acid; LOD, line of decent; NCBI, national center for biotechnology information; P. aeruginosa, Pseudomonas aeruginosa; PIA, pseudomonas isolation agar; SNP, single nucleotide polymorphism
Acknowledgements
We thank laboratory technicians Pia Poss, and Helle Nordbjerg Andersen at Department of Clinical Microbiology at Rigshospitalet for their dedication to the project.
Funding
AL was funded by a Marie Curie Stipend, HKJ was funded by a clinical research stipend from The Novo Nordisk Foundation and Rigshospitalet Rammebevilling 2015–17 and Lundbeckfonden Grant R167-2013-15229.
Availability of data and materials
The.bam files have been deposited at the European Nucleotide Archive (ENA) http://www.ebi.ac.uk/ena/data/view/PRJEB14440 under accession PRJEB14440. See Additional file 1: Table S1 for the accession codes of individual samples (ERS1205965 - ERS1205969).
Authors’ contributions
LMS, RLM, AL, SM, and HKJ designed the experiments. LMS, AL and AK carried out experiments in the laboratory. LMS and RLM conducted the metagenome analysis. LMS, RLM, SM, and HKJ analyzed and interpreted the results. LMS, SM and HKJ wrote the manuscript. All authors commented on the manuscript and approved the final version.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The local ethics committee at the Capital Region of Denmark Region Hovedstaden (“De Videnskabsetiske Komiteer for Region Hovedstaden”) approved the use of the samples: registration number H-4-2015-FSP. All patients have given verbal informed consent. For patients below 18 years of age, informed consent was obtained from their parents.
Additional files
Sampling information of metagenomes. (XLSX 36 kb)
Species ratios in metagenomes. All species presented have been detected in >0.5 % of the bacterial reads of at least one metagenomic sample, if a species is detected with <0.01 % it is shown here as 0.00 %. (XLSX 47 kb)
Coverage of PAO1 by the metagenomes, coverage with >3 reads per position and phred score >30. (XLSX 37 kb)
Isolate and metagenome information of clone type (for the metagenomes this is the clone type that is supposed to be dominating, assumed from the single isolate information of the given patient). Table S4B: Genome comparisons of single isolates and metagenomes. (XLSX 42 kb)
Detailed overview of SNPs found in single isolates and the rediscovery in the metagenomic reads. For each isolate it is indicated whether the mutation has been discovered or not, indicated by 1 and 0 respectively. For each metagenome the percentage of reads (%) representing the SNP found in the single isolates are shown together with the actual number of reads that have been called for each base, C, G, A, and T. For the metagenome(s) it is indicated at the bottom of the column the number of SNPs rediscovered in >10, 50 and 90 % of the metagenomic reads, as well as the total number of SNPs covered by the metagenomic reads. (XLSX 239 kb)
Contributor Information
Lea M. Sommer, Email: lemad@bio.dtu.dk
Rasmus L. Marvig, Email: rasmus.lykke.marvig@regionh.dk
Adela Luján, Email: adem.lujan@gmail.com.
Anna Koza, Email: Akoza@biosustain.dtu.dk.
Tacjana Pressler, Email: tacjana.pressler@regionh.dk.
Søren Molin, Email: sm@bio.dtu.dk.
Helle K. Johansen, Phone: +45 3122 8406, Email: hkj@biosustain.dtu.dk
References
- 1.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. J Respir Crit Care Med. 2003;168(8):918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 2.Lyczak J, Cannon C, Pier G. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15(2). doi:10.1128/CMR.15.2.194. [DOI] [PMC free article] [PubMed]
- 3.O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373(9678):1891–904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 4.Yonezawa M, Takahata M, Matsubara N, et al. DNA gyrase gyrA mutations in quinolone-resistant clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1995;39(9). doi:10.1128/AAC.39.9.1970.Updated. [DOI] [PMC free article] [PubMed]
- 5.Marvig RL, Søndergaard MSR, Damkiær S, et al. Mutations in 23S rRNA confer resistance against azithromycin in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2012;56(8):4519–21. doi: 10.1128/AAC.00630-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marvig R, Damkiær S, Khademi S, et al. Within-host evolution of Pseudomonas aeruginosa reveals adaptation toward iron acquisition from hemoglobin. MBio. 2014 doi: 10.1128/mBio.00966-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldman M, Bryan R, Rajan S, et al. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect Immun. 1998;66(1):43–51. doi: 10.1128/iai.66.1.43-51.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahenthiralingam E, Campbell ME, Speert DP. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun. 1994;62(2):596–605. doi: 10.1128/iai.62.2.596-605.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCallum SJ, Corkill J, Gallagher M, et al. Superinfection with a transmissible strain of Pseudomonas aeruginosa in adults with cystic fibrosis chronically colonised by P. aeruginosa. Lancet. 2001;358(9281):558–60. doi: 10.1016/S0140-6736(01)05715-4. [DOI] [PubMed] [Google Scholar]
- 10.Jelsbak L, Johansen HK, Frost A-L, et al. Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect Immun. 2007;75(5):2214–24. doi: 10.1128/IAI.01282-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Markussen T, Marvig L, Gómez-Lozano M, et al. Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. MBio. 2014;5(5):1–10. doi: 10.1128/mBio.01592-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marvig RL, Johansen HK, Molin S, et al. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 2013;9(9). doi:10.1371/journal.pgen.1003741. [DOI] [PMC free article] [PubMed]
- 13.Marvig RL, Sommer LM, Molin S, et al. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat Genet. 2015;47(1):57–64. doi: 10.1038/ng.3148. [DOI] [PubMed] [Google Scholar]
- 14.Lieberman TD, Flett KB, Yelin I, et al. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat Genet. 2014;46(1):82–7. doi: 10.1038/ng.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mowat E, Paterson S, Fothergill JL, et al. Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am J Respir Crit Care Med. 2011;183(12):1674–9. doi: 10.1164/rccm.201009-1430OC. [DOI] [PubMed] [Google Scholar]
- 16.Feliziani S, Marvig RL, Luján AM, et al. Coexistence and within-host evolution of diversified lineages of hypermutable Pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet. 2014;10(10):e1004651. doi: 10.1371/journal.pgen.1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johansen HK, Nørregaard L, Gøtzsche PC, et al. Antibody response to Pseudomonas aeruginosa in cystic fibrosis patients: a marker of therapeutic success?--A 30-year cohort study of survival in Danish CF patients after onset of chronic P. aeruginosa lung infection. Pediatr Pulmonol. 2004;37(5):427–32. doi: 10.1002/ppul.10457. [DOI] [PubMed] [Google Scholar]
- 18.Lim YW, Schmieder R, Haynes M, et al. Metagenomics and metatranscriptomics: Windows on CF-associated viral and microbial communities. J Cyst Fibros. 2012;12(2):154–164. doi: 10.1016/j.jcf.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krawitz P, Rödelsperger C, Jäger M, et al. Microindel detection in short-read sequence data. Bioinformatics. 2010;26(6):722–9. doi: 10.1093/bioinformatics/btq027. [DOI] [PubMed] [Google Scholar]
- 20.Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Handsaker B, Wysoker A, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swofford DL. PAUP* phylogenetic analysis using parsimony (*and other methods). Version 4.0b10. Sinauer Assoc. 2002. doi:10.1159/000170955.
- 26.R Core Team. R: A language and environment for statistical computing. 2013. Available at: http://www.r-project.org/.
- 27.Johansen HK, Madsen LM, Marvig RL, et al. Rethinking Pseudomonas aeruginosa (PA) lung infection: using molecular microbiology rather than culture and antibodies. J Cyst Fibros. 2014;13(Suppl. 2):S33. doi: 10.1016/S1569-1993(14)60102-8. [DOI] [Google Scholar]
- 28.Arjan JA, Visser M, Zeyl CW, et al. Diminishing returns from mutation supply rate in asexual populations. Science. 1999;283(5400):404–406. doi: 10.1126/science.283.5400.404. [DOI] [PubMed] [Google Scholar]
- 29.Snitkin ES, Segre JA. Pseudomonas aeruginosa adaptation to human hosts. Nat Genet. 2015;47(1):2–3. doi: 10.1038/ng.3172. [DOI] [PubMed] [Google Scholar]
- 30.Yang L, Jelsbak L, Marvig RL, et al. Evolutionary dynamics of bacteria in a human host environment. Proc Natl Acad Sci U S A. 2011;108(18):7481–6. doi: 10.1073/pnas.1018249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bragonzi A, Paroni M, Nonis A, et al. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am J Respir Crit Care Med. 2009;180(2):138–145. doi: 10.1164/rccm.200812-1943OC. [DOI] [PubMed] [Google Scholar]
- 32.Marvig RL, Dolce D, Sommer LM, et al. Within-host microevolution of Pseudomonas aeruginosa in Italian cystic fibrosis patients. BMC Microbiol. 2015;15(1):218. doi: 10.1186/s12866-015-0563-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Workentine ML, Sibley CD, Glezerson B, et al. Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS One. 2013;8(4):e60225. doi: 10.1371/journal.pone.0060225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darch SE, Mcnally A, Harrison F, et al. Recombination is a key driver of genomic and phenotypic diversity in a Pseudomonas aeruginosa population during cystic fibrosis infection. Sci Rep. 2015;5(7649):1–12. doi: 10.1038/srep07649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansen SK, Rau MH, Johansen HK, et al. Evolution and diversification of Pseudomonas aeruginosa in the paranasal sinuses of cystic fibrosis children have implications for chronic lung infection. ISME J. 2012;6(1):31–45. doi: 10.1038/ismej.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams D, Evans B, Haldenby S, et al. Divergent, coexisting Pseudomonas aerugnosa lineages in chronic cystic fibrosis lung infections. Am J Respir Crit Care Med. 2015;191(7):775–785. doi: 10.1164/rccm.201409-1646OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciofu O, Johansen HK, Aanæs K, et al. P. aeruginosa in the paranasal sinuses and transplanted lungs have similar adaptive mutations as isolates from chronically infected CF lungs. J Cyst Fibros. 2013;12(6):729–736. doi: 10.1016/j.jcf.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Johansen HK, Aanaes K, Pressler T, et al. Colonisation and infection of the paranasal sinuses in cystic fibrosis patients is accompanied by a reduced PMN response. J Cyst Fibros. 2012;11(6):525–531. doi: 10.1016/j.jcf.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 39.Mainz JG, Naehrlich L, Schien M, et al. Concordant genotype of upper and lower airways P. aeruginosa and S. aureus isolates in cystic fibrosis. Thorax. 2009;64(6):535–40. doi: 10.1136/thx.2008.104711. [DOI] [PubMed] [Google Scholar]
- 40.Jorth P, Staudinger BJ, Wu X, et al. Regional isolation drives bacterial diversification within cystic fibrosis lungs. Cell Host Microbe. 2015;18(3):307–319. doi: 10.1016/j.chom.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The.bam files have been deposited at the European Nucleotide Archive (ENA) http://www.ebi.ac.uk/ena/data/view/PRJEB14440 under accession PRJEB14440. See Additional file 1: Table S1 for the accession codes of individual samples (ERS1205965 - ERS1205969).