

Abstract
Background
Meiotic recombination is a fundamental evolutionary process. It not only generates diversity, but influences the efficacy of natural selection and genome evolution. There can be significant heterogeneity in recombination rates within and between species, however this variation is not well understood outside of a few model taxa, particularly in forest trees. Eucalypts are forest trees of global economic importance, and dominate many Australian ecosystems. We studied recombination rate in Eucalyptus globulus using genetic linkage maps constructed in 10 unrelated individuals, and markers anchored to the Eucalyptus reference genome. This experimental design provided the replication to study whether recombination rate varied between individuals and chromosomes, and allowed us to study the genomic attributes and population genetic parameters correlated with this variation.
Results
Recombination rate varied significantly between individuals (range = 2.71 to 3.51 centimorgans/megabase [cM/Mb]), but was not significantly influenced by sex or cross type (F1 vs. F2). Significant differences in recombination rate between chromosomes were also evident (range = 1.98 to 3.81 cM/Mb), beyond those which were due to variation in chromosome size. Variation in chromosomal recombination rate was significantly correlated with gene density (r = 0.94), GC content (r = 0.90), and the number of tandem duplicated genes (r = −0.72) per chromosome. Notably, chromosome level recombination rate was also negatively correlated with the average genetic diversity across six species from an independent set of samples (r = −0.75).
Conclusions
The correlations with genomic attributes are consistent with findings in other taxa, however, the direction of the correlation between diversity and recombination rate is opposite to that commonly observed. We argue this is likely to reflect the interaction of selection and specific genome architecture of Eucalyptus. Interestingly, the differences amongst chromosomes in recombination rates appear stable across Eucalyptus species. Together with the strong correlations between recombination rate and features of the Eucalyptus reference genome, we maintain these findings provide further evidence for a broad conservation of genome architecture across the globally significant lineages of Eucalyptus.
Keywords: Eucalyptus globulus, Meiotic recombination, Crossover, Genomic features, Genetic diversity, Gene density
Background
Meiotic recombination is a fundamental evolutionary process, which is believed to have been central to the evolutionary success of eukaryotes. When crossover recombination occurs, DNA strands are exchanged between homologous chromosomes, resulting in allele shuffling. This generates novel genetic variation [1], by breaking the associations that occur both within and between linked genes [2]. As this genetic variation is sieved by natural selection, it shapes the adaptive evolution of living organisms and thus, indirectly, genome evolution.
Aside from allele shuffling, recent studies have identified four other processes by which recombination directly influences genome evolution [1, 3]. These are: GC-biased gene conversion, whereby recombination promotes GC enrichment at repair sites; Hotspot drive, through which a higher transmission of non-recombinant alleles likely contributes to rapid shifts in the location of recombination hotspots [4, 5]; through the potential Mutagenic effect of recombination itself, which causes structural mutations; and Indel drive, a neutral mechanism that may drive genome contraction by a higher transmission of chromosomal deletions compared to insertions [6, 7]. In combination, these processes enable recombination to substantially influence genome evolution at a variety of genomic scales.
Historically, recombination was studied through observation of chiasmata using microscopy [8], or by genetic linkage mapping using phenotypic markers. More recently, recombination has been studied through variation in recombination rate (RR); estimated from genetic distance (i.e., number of recombination events) divided by physical distance (the size in base pairs of DNA over which the genetic distance is measured) using three main approaches, linkage disequilibrium mapping, sperm typing and linkage mapping using molecular markers. The major differences between these approaches are whether they measure historical or current recombination and their applicability to populations, a single, or few individuals [9]. Linkage disequilibrium mapping estimates historical recombination at the population level, and is thus useful for understanding population evolution, but is affected by factors such as genetic drift, demography, mutation rate and natural selection. In contrast, both sperm typing and linkage mapping are little affected by these factors and provide direct estimates of current recombination across a single generation [9]. Linkage mapping is a classic technique widely used to study recombination in selfed or bi-parental families. Direct estimates of RR can be obtained from the genetic distance between pairs of linked markers compared to their physical distance. The resolution of linkage mapping studies is limited by sample size and the density of molecular markers. However, the use of high-throughput marker systems and whole genome sequences potentially allows fine scale study of RR, as well as comparison between sexes, populations and individuals [10].
Understanding variation in RR will contribute to a better understanding of genome evolution and is fundamental to many aspects of genetics. For example, knowing the genomic distribution of RR is important for predicting the potential of populations to respond to environmental change, as well as for breeding using both traditional and genomic approaches. Specifically, characterising RR will help predict the degree of linkage disequilibrium to guide marker densities needed for genomic selection, a technique which is showing promise for both animal and plant breeding [11]. It also has practical applications when trying to isolate the specific genes underlying quantitative trait loci (QTL) that have been detected in linkage mapping or association genetic studies, and for attempts to introduce favourable alleles into breeding lines [12, 13].
Most detailed studies of recombination and linkage disequilibrium have been accomplished in human, yeast, Drosophila or Arabidopsis, which have the required genomic resources (such as good reference genomes and high throughput marker systems). Although many of the factors which influence RR are conserved across eukaryotic organisms, empirical studies consistently report variation in RR between taxa, individuals and across genomes [9, 10, 14]. In view of this variation, it is important to obtain a broad understanding of RR across diverse taxa. This is particularly true for long-lived organisms such as forest trees which often have many genetic features, including low rates of selfing, high heterozygosity and high population genetic diversity [15, 16] which distinguish them from most model species. Trees often define and structure forest ecosystems; play vital ecosystem services, including climate-change mitigation; provide invaluable products; and are widely used for human recreation. Given their environmental and economic significance, understanding the genetic mechanisms which shape the evolution and diversity of trees is of major importance.
The genomic resources required to study RR have only recently become available in forest trees [17–19] thus there is little knowledge of the genomic distribution of RR in these organisms. Nonetheless, insights into the patterns of recombination in tree species have been gained from studies of linkage disequilibrium (e.g., Slavov et al. [20], reviewed by Thavamanikumar et al. [21]) and comparative mapping [22]. Assembled reference genomes are available for hardwood forest trees, such as Populus [17], Eucalyptus [19], Quercus [23] and Castanea [24] providing the opportunity to directly estimate genome-wide RR using linkage maps. Recent studies in Eucalyptus have compared genetic to physical map distance and reported RR averaged over the whole genome [25] or for each chromosome [26, 27]. Silva-Junior and Grattapaglia [27] also calculated high resolution estimates of linkage disequilibrium and ‘population scaled recombination’ rate (an estimate of the history of recombination in different genomic segments), and its genomic correlates. However, for direct estimates of RR there is little information regarding variation between individuals, factors influencing this variation, nor its correlation with genomic features in forest trees.
Eucalyptus is a diverse genus containing over 700 species across thirteen subgenera [28]. Most species belong to subgenus Symphyomyrtus including the majority of economically important plantation species [29], and the two species used in this study; E. grandis (section Latoangulatae) and E. globulus (section Maidenaria) [28]. Eucalypts generally have a mixed-mating system (i.e., they can reproduce both by selfing and outcrossing), and are mostly animal pollinated. Their outcrossing rate is variable, but generally high and most species are highly polymorphic [29, 30]. For example, recent findings indicate genome-wide diversity in E. grandis [27] is at the high end for plants, in line with other angiosperm trees such as Populus trichocarpa and an order of magnitude higher than most conifers, corroborating previous findings in eucalypt [19, 29]. All eucalypt species studied to date possess the same chromosome number (n = 11), although their genomes vary in size from 360 Mb (Corymbia variegata) to 640 Mb (Eucalyptus grandis) [29]. Despite this variation in genome size, there is a high degree of karyotype stability between species [29], which includes broad conservation of chromosome structure such as the location of centromeres and heterochromatin content within the subgenus Symphyomyrtus [31], and this is supported by comparative mapping studies [32, 33]. Specifically, there was no evidence for gross chromosomal rearrangements between (the 530 Mb genome of) E. globulus and an E. grandis/urophylla consensus map based on nearly 400 shared markers [33]. Re-sequencing of the E. globulus and E. grandis genomes suggested that the differences in genome size between these species are largely attributable to many small changes distributed throughout their genomes [19]. Further, the genomic distribution of diversity within and divergence between eucalypt species in subgenus Symphyomyrtus, appears stable and correlated with genomic architecture [34], providing further evidence for genome stability. In view of this stability, the numbering of chromosomes in most recent molecular studies in eucalypt has followed the consensus linkage map of Brondani et al. [35], and this order was used for the 11 major scaffolds in the E. grandis genome [19].
We here focus on Eucalyptus globulus, and exploit the recently published Eucalyptus grandis reference genome in conjunction with 10 linkage maps constructed with sequenced anchored markers. This experimental design allowed testing of the effects of various factors reported in other species to be associated with chromosomal variation in RR, including chromosome, sex, and cross-type; genomic features such as GC content, density of genes and transposable elements; and the population genetic parameters, genetic diversity and divergence (calculated from six species from subgenus Symphyomyrtus, [34]; see methods for detail).
Results
Variation between individuals
The difference in RR amongst parents (Fig. 1a) was highly significant (F9,90 = 6.1, P < 0.001) and due mainly to the high RR of two F1 parents (F1.4-F and F1.5-M) and low RR in the male and female parents of one of the F2 families (F2.C). The RR of the F1 families (3.1 ± 0.09 cM/Mb) was greater than that of the F2 families (2.8 ± 0.11 cM/Mb), but not significantly so (F1,8 = 3.9, P = 0.085). The F2 families had higher levels of heterozygosity than the F1 (2-tailed t8 = 7.04, P < 0.001). However while negative, the correlation between parental heterozygosity and RR was not significant. There was no significant differences in RR between sexes (F1,8 = 0.39, P = 0.549). The genome-wide RR ranged between 2.71 and 3.51 cM/Mb between the 10 E. globulus parents, and averaged 2.98 cM/Mb (Table 1). These values were calculated across linkage maps ranging in size (i.e., genetic distance, estimated by Haldane’s mapping function) from 1442 to 1787 cM (Table 2), averaging 1545 cM.
Fig. 1.
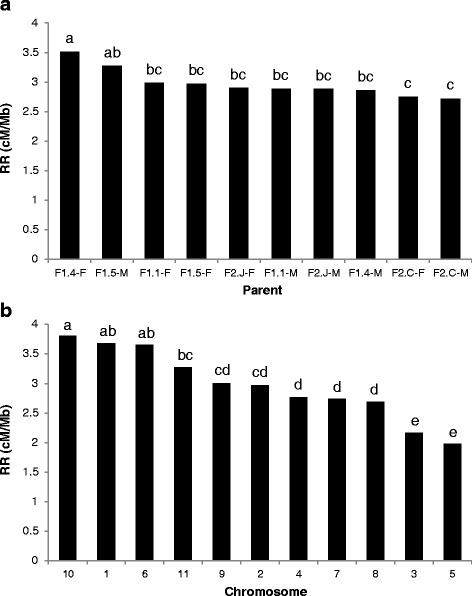
Variation in recombination rate between individuals and chromosomes in Eucalyptus globulus. a Mean genome-wide RR for each of the 10 Eucalyptus globulus individuals. b Mean RR in each chromosome across the 10 individuals. Means with different letters were significantly different in pairwise comparisons following the Tukey-Kramer (honestly significant difference, HSD) adjustment
Table 1.
Chromosome and genome scale descriptive statisticsa of the data used to study recombination in Eucalyptus
| Level and variable | Min | Max | Mean | CVe |
|---|---|---|---|---|
| Chromosome averages (n = 11) | ||||
| Physical size (Mb) | 39.02 | 80.10 | 55.10 | 0.29 |
| Diversity (global HHw)b | 0.196 | 0.229 | 0.196 | 0.04 |
| Divergence (global Fst)c | 0.385 | 0.405 | 0.354 | 0.03 |
| GC content (%) | 35.60 | 38.29 | 37.23 | 0.02 |
| RR (cM/Mb) | 1.98 | 3.81 | 2.98 | 0.20 |
| Individual genome-wide averages (n = 10) | ||||
| Genetic distance (cM) | 1442 | 1787 | 1545 | 0.08 |
| Physical size (Mb) | 508.3 | 569.5 | 543.1 | 0.03 |
| Genome coverage (%) | 84.13 | 93.55 | 89.37 | 0.03 |
| Heterozygosityd | 630 | 815 | 708 | 0.10 |
| RR (cM/Mb) | 2.71 | 3.51 | 2.98 | 0.08 |
aDiversity and divergence were estimated by Hudson et al. [34]. The remaining variables were estimated from our E. globulus data and the E. grandis genome [19]
bThe diversity within 6 Eucalyptus species, including E. globulus
cDivergence between the 6 Eucalyptus species
dEstimated from the total number of polymorphic markers derived from the DArT assay (see Methods)
eCoefficient of variation
Table 2.
Detailed summary of the linkage maps used in this study
| Scaffold ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cross | Pa | Scaffold Size (Mb)b | 40.3 | 64.2 | 80.1 | 42.0 | 74.7 | 53.9 | 52.4 | 74.3 | 39.0 | 39.3 | 45.5 |
| F1.1 | F | Interval Number | 8 | 8 | 15 | 6 | 17 | 10 | 12 | 11 | 10 | 5 | 8 |
| Total length (cM) | 122 | 170 | 174 | 99 | 125 | 199 | 147 | 206 | 132 | 91 | 122 | ||
| Phys. Size (Mb) | 36.1 | 58.0 | 67.6 | 40.6 | 71.0 | 52.6 | 51.7 | 64.7 | 37.2 | 26.5 | 41.5 | ||
| Mean RR (cM/Mb) | 3.38 | 2.93 | 2.57 | 2.44 | 1.76 | 3.78 | 2.84 | 3.19 | 3.55 | 3.44 | 2.94 | ||
| M | Interval Number | 7 | 10 | 16 | 8 | 11 | 10 | 9 | 14 | 8 | 4 | 9 | |
| Total length (cM) | 150 | 176 | 159 | 82 | 129 | 173 | 105 | 128 | 99 | 113 | 139 | ||
| Phys. Size (Mb) | 38.1 | 54.8 | 73.0 | 34.1 | 72.3 | 49.2 | 41.8 | 56.4 | 35.1 | 29.1 | 42.3 | ||
| Mean RR (cM/Mb) | 3.93 | 3.21 | 2.18 | 2.40 | 1.78 | 3.52 | 2.51 | 2.27 | 2.82 | 3.88 | 3.29 | ||
| F1.4 | F | Interval Number | 8 | 9 | 19 | 6 | 16 | 9 | 13 | 13 | 8 | 3 | 12 |
| Total length (cM) | 176 | 199 | 184 | 98 | 145 | 183 | 180 | 224 | 118 | 117 | 163 | ||
| Phys. Size (Mb) | 37.4 | 55.3 | 75.8 | 36.0 | 72.3 | 45.3 | 49.9 | 59.3 | 34.3 | 25.4 | 43.9 | ||
| Mean RR | 4.71 | 3.60 | 2.43 | 2.72 | 2.01 | 4.04 | 3.61 | 3.77 | 3.44 | 4.61 | 3.72 | ||
| M | Interval Number | 9 | 10 | 17 | 4 | 12 | 8 | 12 | 16 | 6 | 7 | 16 | |
| Total length (cM) | 153 | 144 | 142 | 110 | 143 | 154 | 115 | 177 | 82 | 99 | 137 | ||
| Phys. Size (Mb) | 39.3 | 51.8 | 74.5 | 34.9 | 66.3 | 38.9 | 51.2 | 71.9 | 33.5 | 31.0 | 41.5 | ||
| Mean RR (cM/Mb) | 3.90 | 2.78 | 1.91 | 3.15 | 2.16 | 3.96 | 2.25 | 2.46 | 2.45 | 3.19 | 3.30 | ||
| F1.5 | F | Interval Number | 7 | 7 | 17 | 7 | 10 | 9 | 11 | 14 | 9 | 9 | 11 |
| Total length (cM) | 115 | 142 | 136 | 101 | 109 | 165 | 152 | 170 | 104 | 104 | 144 | ||
| Phys. Size (Mb) | 34.4 | 47.9 | 67.9 | 35.6 | 59.4 | 40.2 | 51.9 | 65.3 | 36.1 | 27.6 | 42.1 | ||
| Mean RR (cM/Mb) | 3.35 | 2.96 | 2.00 | 2.84 | 1.84 | 4.11 | 2.93 | 2.60 | 2.88 | 3.77 | 3.42 | ||
| M | Interval Number | 6 | 14 | 12 | 7 | 12 | 11 | 8 | 10 | 7 | 11 | 11 | |
| Total length (cM) | 139 | 157 | 184 | 116 | 166 | 181 | 173 | 206 | 109 | 136 | 151 | ||
| Phys. Size (Mb) | 31.6 | 56.8 | 71.6 | 35.7 | 71.0 | 52.6 | 51.1 | 66.4 | 36.7 | 32.1 | 42.3 | ||
| Mean RR (cM/Mb) | 4.40 | 2.76 | 2.57 | 3.25 | 2.34 | 3.44 | 3.38 | 3.10 | 2.97 | 4.24 | 3.57 | ||
| F2.J | F | Interval Number | 6 | 13 | 17 | 8 | 14 | 11 | 10 | 15 | 9 | 10 | 10 |
| Total length (cM) | 107 | 167 | 139 | 101 | 129 | 163 | 101 | 158 | 105 | 162 | 168 | ||
| Phys. Size (Mb) | 36.1 | 56.0 | 76.1 | 36.7 | 69.7 | 43.3 | 47.2 | 66.4 | 34.8 | 36.7 | 43.4 | ||
| Mean RR (cM/Mb) | 2.96 | 2.98 | 1.83 | 2.75 | 1.85 | 3.76 | 2.14 | 2.38 | 3.02 | 4.42 | 3.87 | ||
| M | Interval Number | 10 | 9 | 17 | 12 | 13 | 14 | 14 | 15 | 8 | 14 | 16 | |
| Total length (cM) | 129 | 165 | 153 | 92 | 141 | 164 | 142 | 181 | 121 | 130 | 124 | ||
| Phys. Size (Mb) | 36.1 | 55.0 | 75.4 | 32.3 | 72.9 | 53.1 | 52.3 | 71.7 | 37.5 | 33.2 | 42.7 | ||
| Mean RR (cM/Mb) | 3.57 | 3.00 | 2.03 | 2.85 | 1.93 | 3.09 | 2.71 | 2.52 | 3.23 | 3.92 | 2.90 | ||
| F2.C | F | Interval Number | 5 | 13 | 16 | 9 | 14 | 15 | 10 | 14 | 11 | 9 | 8 |
| Total length (cM) | 105 | 164 | 160 | 110 | 144 | 168 | 133 | 137 | 117 | 120 | 118 | ||
| Phys. Size (Mb) | 35.0 | 61.1 | 69.7 | 38.0 | 67.0 | 52.2 | 51.1 | 64.7 | 38.0 | 35.0 | 42.3 | ||
| Mean RR (cM/Mb) | 3.00 | 2.68 | 2.30 | 2.90 | 2.15 | 3.22 | 2.60 | 2.12 | 3.08 | 3.43 | 2.79 | ||
| M | Interval Number | 6 | 11 | 18 | 11 | 12 | 12 | 14 | 17 | 8 | 8 | 12 | |
| Total length (cM) | 119 | 172 | 139 | 92 | 136 | 189 | 121 | 178 | 102 | 113 | 124 | ||
| Phys. Size (Mb) | 32.6 | 61.5 | 76.2 | 39.1 | 69.4 | 52.0 | 50.3 | 72.3 | 38.3 | 35.6 | 42.3 | ||
| Mean RR (cM/Mb) | 3.65 | 2.80 | 1.82 | 2.35 | 1.96 | 3.64 | 2.41 | 2.46 | 2.66 | 3.18 | 2.93 |
aP = Parent, F = Female, M = Male bPhys. Size = Physical Size
Variation between chromosomes
The 11 chromosomes of E. globulus differed markedly in RR. Averaged across parents, chromosome-wide RR ranged from 1.98 to 3.81 cM/Mb (Table 1). The difference between chromosome averages was highly significant when tested using parents as replicates (F10,90 = 34.0, P < 0.001), and was also significant (F10,89 = 92.7, P < 0.001) after removing the correlated effect of chromosome size (see discussion below). The variation between chromosomes was mainly due to the high RR in chromosomes 10, 1 and 6 and low RR in chromosomes 3 and 5 (Fig. 1b). The difference between chromosomes was moderately to highly positively correlated with previously published estimates for E. grandis x urophylla consensus maps ([26] n = 11, Pearson r = 0.90, P < 0.001; [25] n = 11, Pearson r = 0.92, P < 0.001) as well as parental maps [27] of E. grandis (n = 11, Pearson r = 0.72, P = 0.012) and E. urophylla (n = 11, Pearson r = 0.55) although the latter was not significant (P = 0.081). In no case was the interaction of chromosome with either sex (F10,80 = 1.17, P = 0.327) or cross type (F10,80 = 1.03, P = 0.426) significant, indicating that the chromosomal differences in RR were stable across sex and cross-type in this study.
Several genomic features of the E. grandis reference genome were correlated with the chromosomal variation in RR (Table 3). Chromosomes with high RR were smaller, had higher GC content, higher gene density, lower frequencies of several categories of transposable elements and fewer tandemly duplicated genes (Table 4). While the association with transposable elements as a whole was not significant (r = −0.59, P = 0.056), significant negative correlations were observed for specific classes of transposable elements (DNA transposable elements r = −0.76, P = 0.007 and uncategorised elements, i.e., those that could not be assigned to any specific transposable element class, r = −0.97, P < 0.001), and superfamilies within both the retrotransposons (long interspersed nuclear elements/short interspersed nuclear elements [LINE/SINE] r = −0.78, P = 0.005) and DNA transposable elements (helitrons r = −0.69, P = 0.019). Chromosomes with high RR also tended to have a lower density of tandem gene duplications (r = −0.72, P = 0.012). However, most of these genomic features were inter-correlated (Table 3) and when the effect of gene density (the most highly correlated feature in Table 3) was removed by calculation of partial correlation coefficients, correlations between RR and all other factors listed in Table 3 became insignificant. Partialling out the effect of chromosome size had little effect on the correlation between RR and other genome features, and the correlations involving gene density (partial r = 0.87, P < 0.001) and %GC (partial r = 0.76, P = 0.010) remained significant.
Table 3.
Pearson correlations among chromosome attributes (n = 11)
| RR | Chromosome length | GC content | Gene density | Transposable element density | Number of tandem duplicated genes | Diversity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chromosome length (bp) | −0.74 | ** | ||||||||||||
| GC content (%) | 0.90 | *** | −0.76 | ** | ||||||||||
| Gene density | 0.94 | *** | −0.70 | * | 0.91 | *** | ||||||||
| Transposable element density | −0.59 | ns | 0.32 | ns | −0.29 | ns | −0.62 | * | ||||||
| Number of tandem duplicated genes | −0.72 | * | 0.53 | ns | −0.70 | * | −0.75 | ** | 0.46 | ns | ||||
| Diversity (Global HHW)a | −0.75 | ** | 0.61 | * | −0.82 | ** | −0.85 | *** | 0.41 | ns | 0.78 | ** | ||
| Divergence (Global Fst)b | 0.90 | *** | −0.84 | ** | 0.95 | *** | 0.92 | *** | −0.40 | ns | −0.79 | ** | −0.91 | *** |
aThe diversity within 6 Eucalyptus species, including E. globulus
bDivergence between the 6 Eucalyptus species
Significance levels are: *** P < 0.001, ** P < 0.01, * P < 0.05, ns P ≥ 0.05
Table 4.
Chromosome mean values for the main attributes used in correlation analysesa
| Chromosome | Recombination rate (cM/Mb) | Chromosome length (Mbp)b | GC content (%) | Gene densityc | Transposable element densityd | Number of tandem duplicated genese | Diversity (Global HHW) | Divergence (Global Fst) |
|---|---|---|---|---|---|---|---|---|
| 1 | 3.68 | 40.30 | 0.380 | 63.55 | 401.86 | 0.088 | 0.181 | 0.400 |
| 2 | 2.97 | 64.24 | 0.364 | 53.05 | 392.64 | 0.092 | 0.206 | 0.376 |
| 3 | 2.16 | 80.09 | 0.357 | 42.84 | 416.24 | 0.103 | 0.229 | 0.354 |
| 4 | 2.77 | 41.98 | 0.372 | 53.03 | 430.05 | 0.085 | 0.188 | 0.395 |
| 5 | 1.98 | 74.73 | 0.353 | 44.35 | 418.50 | 0.100 | 0.215 | 0.359 |
| 6 | 3.66 | 53.89 | 0.377 | 71.49 | 361.55 | 0.077 | 0.175 | 0.400 |
| 7 | 2.74 | 52.45 | 0.368 | 52.70 | 412.18 | 0.092 | 0.219 | 0.373 |
| 8 | 2.69 | 74.33 | 0.375 | 56.09 | 439.17 | 0.090 | 0.189 | 0.383 |
| 9 | 3.01 | 39.02 | 0.375 | 61.00 | 398.21 | 0.096 | 0.194 | 0.391 |
| 10 | 3.81 | 39.36 | 0.383 | 69.77 | 406.39 | 0.090 | 0.188 | 0.405 |
| 11 | 3.27 | 45.51 | 0.377 | 67.35 | 389.91 | 0.088 | 0.165 | 0.403 |
aAll chromosome attributes are from the Eucalyptus grandis reference genome [19], except diversity and divergence which were estimated by Hudson et al. [34]
bEquivalent to 'Scaffold Size' in Table 2
c Number of genes per Mbp
d Number of transposable elements per Mbp
e The proportion of annotated genes which are tandem duplicates
Chromosomal variation in RR was also associated with differences in marker diversity (assuming Hard-Weinberg equilibrium [HHw]) and species divergence (measured as the proportion of the genetic variance in a sub population relative to the total genetic variance [Fst]). It was highly negatively correlated with the pooled genetic diversity (global HHw) within the six Eucalyptus species studied by Hudson et al. [34] (n = 11, r = −0.75, P = 0.008). The correlation between chromosomal RR and HHw of each individual species, including E. globulus, were similar in size and all also negative (r = −0.60 to −0.74). The trends in global diversity were also evident regardless of whether markers were intra- or intergenic. However, for global HHw the correlation was only significant for intragenic markers (r = −0.65, P = 0.032). The global Fst amongst the six species studied by Hudson et al. [34] was positively correlated with RR at the chromosome level (r = 0.90, P < 0.001). This relationship with Fst was also found for both intragenic (r = 0.93, P < 0.001) and intergenic (r = 0.75 P = 0.008) markers.
Discussion
This is the first study in forest trees to present direct, genome-wide, estimates of RR from multiple mapping pedigrees. We show RR varies between individuals as well as between chromosomes. The relative differences amongst chromosomes appear stable across eucalypt species, and are correlated with genomic features commonly related to RR in diverse taxa; including GC content, gene density and the density of specific transposable elements. Significantly, we show that the differences in RR are negatively correlated with species-wide genetic diversity at the chromosome level. The direction of this correlation is the opposite to that commonly observed (see reviews by Smukowski and Noor [9], and Cutter and Payseur [36]) and, we argue, is likely to reflect an interaction of selection and genome architecture.
Variation between individuals
The variation we found in genome-wide RR between individuals is consistent with reports in other studies [9, 37, 38] and may be due to environmental or genetic factors. RR has been shown to be affected by the environment in both plants and animals [39–41]. However, the role of the environment cannot be clarified in the present study as our parents come from different environments as well as being genetically distinct. Genetic factors which may influence RR, include sex and cross type. Sex specific differences have been reported in some taxa, with elevated RR often occurring in the gender subject to gametic selection [42]. However, no difference in RR between sexes was evident in E. globulus. There was a signal in our data, albeit not significant, that cross type could explain some of the variation between individuals, with lower average RR in the F2’s compared with the F1’s. However further study will be required to validate this. Irrespective of whether the genome-wide variation in RR we observed between individuals is due to genetic or environmental factors, it is clear that representative maps of the RR landscape in Eucalyptus species will need to integrate results from multiple crosses and individuals.
Our genome-wide average RR for E. globulus (2.98 cM/Mb) was higher than estimates from E. grandis (2.17 cM/Mb), E. urophylla (2.24 cM/Mb) [27], and E. grandis/urophylla consensus maps (1.58 cM/Mb and 1.95 cM/Mb; [25] and [26], respectively). However, differences in the scale over which RR was estimated is likely to have influenced the different estimates. Differences in genome coverage also no doubt influenced the estimates of RR between studies, particularly in the case of Kullan et al. [25], which estimated RR based on 152 windows of approximately 1 cM across the genome. Direct comparison of the estimates from different studies is also problematic because different mapping techniques produce different genetic distances and thus different estimates of genetic to physical distance. Our estimates of genetic distance (cM) were derived from Joinmap software, using the maximum likelihood algorithm, which uses Haldane’s mapping function. The estimates of genetic distance used by Petroli et al. [26] were derived from RECORD software [43]. RECORD produces distances very similar to the Joinmap maximum likelihood algorithm, if Haldane’s mapping function is used [44]. However, Silva-junior and Grattapaglia [27] used Kosambi’s mapping function, which produces smaller estimates of genetic distance relative to Haldane’s function. Similarly, both Kullan et al. [25] and Petroli et al. [26] used the regression algorithm and Kosambi’s mapping function in Joinmap, which produce considerably smaller genetic distances than Joinmap’s maximum likelihood algorithm. Therefore, the greater estimate of genome-wide RR in our study may simply reflect greater estimates of genetic distance due to methodological issues. Clearly, further study is required to draw robust conclusions regarding genome-wide differences in RR within and between eucalypt species, ideally using linkage maps constructed from similar techniques in multiple representatives of each species.
Variation between chromosomes
There was substantial variation in RR between chromosomes in our study, indeed chromosome was the most significant explanatory variable in our analysis of variance. Few studies in plants have examined genome-wide RR using multiple crosses, which are needed to provide the replication to test for significant differences between chromosomes. Nonetheless, chromosomal variation in RR has been demonstrated in a range of taxa [45, 46], including plants such as maize in which intraspecific variation was of a similar magnitude to our findings [14]. Chromosomal variation in RR is likely to at least in part reflect variation in chromosome size, due to the requirement for at least one crossover per chromosome per meiosis [45]. However, in our study the variation in RR between chromosomes remained significant after removing the correlated effect of chromosome size, arguing that other aspects of genomic architecture also contribute to this variation.
Despite the significant differences in genome-wide RR between individuals, three lines of evidence suggest that the relative RR in each chromosome is highly stable; not only within E. globulus, but between Eucalyptus species, and across evolutionary time. First, the differences between chromosomes in E. globulus were evident even when the significant effect of variation amongst the individuals was removed; and there was no significant interaction between chromosome and either sex or cross type within E. globulus. Indeed, the difference in RR amongst chromosomes was nearly two fold, whereas that amongst individuals was only 1.3 fold. Second, the high positive correlation between our chromosomal estimates of RR from E. globulus (section Maidenaria) and those in E. grandis/urophylla (section Latoangulatae) [25, 26] suggests the relative chromosomal differences in RR are stable across these Eucalyptus sections, despite an estimated 10 to15 Ma since their divergence [47]. Third, a positive correlation between GC content and RR is expected based on studies in other taxa (see below). The high positive chromosome-level correlation between GC content (from the E. grandis genome) and RR (measured from E. globulus map distance against the E. grandis genome), also points to stability of RR between E. grandis and E. globulus. Further, because our estimate of RR is from a single generation of recombination, while GC content in E. grandis reflects historical RR, the high positive correlation also points to a long term stability of RR at the chromosome level.
Association with genomic features and population genetic parameters
The chromosome level correlation between GC content and RR we observe is consistent with the theory of ‘GC-biased gene conversion’, whereby DNA repair during recombination promotes enrichment in GC bases, accounting for the positive correlation between RR and GC content seen in most eukaryotes studied to date [9, 48]. The particularly strong positive correlation observed between chromosome level RR and GC in E. globulus may reflect the unusually high GC content seen in the family Myrtales, compared with other eudicots [48]. In contrast, Arabidopsis is relatively GC poor and GC content and RR are uncorrelated, in line with a general pattern of greater GC enrichment in more derived plant genomes [48].
Our results also suggest a strong positive correlation between chromosomal estimates of RR and gene density. This finding is consistent with the positive correlation between RR and gene density observed at the whole-genome level in a range of taxa [9]. In plants such as maize, rice, wheat, and A. thaliana much of this correlation is attributable to the low density of genes in recombination-poor heterochromatin [49]. While in vertebrate genomes, Nam and Ellegren [7] hypothesise neutral processes associated with recombination, including a bias in deletions over insertions, contribute to a more compact genome structure and greater gene density in regions of elevated RR. However, the correlation between gene density and RR varies in strength and direction amongst taxa [9, 36, 50]. The positive correlation we observe at the chromosome level is particularly strong relative to other reports. Consistent with our findings, gene density was positively correlated with historical recombination rate in E. grandis (i.e., population scaled recombination rate, measured in 100 kb windows across the genome; [27]), suggesting the relationship between gene density and RR may be widespread in Eucalyptus, at least across the subgenus Symphyomyrtus.
A negative correlation between RR and some categories of transposable elements was also evident in our study. Transposable elements comprise over 50 % of most eukaryotes genomes and have a significant impact on genome evolution [51, 52]. In particular, it is well accepted that the accumulation of transposable elements is a major factor driving genome expansion [53]. The relative density of transposable elements across the genome reflects a balance between transposable element insertion and deletion. Transposable element deletion is believed to be primarily driven by recombinational processes, consistent with the observed transposable element accumulation in areas of low recombination in many taxa [49, 54] and in line with the correlation we observe. The mechanisms contributing to transposable elements accumulation in regions of low RR are the subject of some debate. Leading hypothesis involve relaxation of various forms of selection in these regions (see Dolgin and Charlesworth [55]); biased insertion of transposable elements in areas of low RR [56]; and a general deletion bias associated with recombination [7]. Regardless of the causative factors, in our study it is possible that in chromosomes with relatively low RR, a greater density of transposable elements has contributed to a reduced gene density, and thus contributed to the positive correlation we observe between RR and gene density at the chromosome level.
A notable finding to emerge from this study is our strong negative correlation between RR at the chromosome-level and species-wide genetic diversity. The common trend in animals is for a positive association between RR and genetic diversity, whereas the association in plants is more variable, but generally zero or positive [36]. The relationship between RR and genetic diversity may be affected by the scale and type of measures of RR (e.g., chromosome averages versus smaller genomic regions) and genetic diversity (i.e., within individuals, populations or species) both of which differ widely between studies [9]. Nevertheless, only in Oryza species (wild and domesticated rice) have negative correlations between RR and genetic diversity been previously reported [36]. In O. sativa, this association involved 100 kb intervals across three chromosomes (r = −0.183 and −0.314; for O. sativa ssp. indica and ssp. japonica, respectively) [57].
The more commonly observed positive correlation between RR and genetic diversity is often attributed to the impact of linked selection [9, 36]. Linked selection predicts a positive relationship between RR and genetic diversity due to the homogenising effects of selection being spread further in regions of low recombination [58]. However, as noted above there is disparity in this relationship between species [36] suggesting other factors also influence how selection impacts the genomic distribution of diversity, such as gene density [59]. Gene dense regions will be on average subject to greater selection, reducing diversity. Therefore, a more general prediction for the effect of selection across genomes is that the distribution of genetic diversity will be related to the density of elements under selection, i.e., gene density (assuming genes are the most likely targets of selection) [59]. This was the case in a recent study of E. grandis, where nucleotide diversity (at the population level) was negatively correlated with gene density in 100 kb windows across the E. grandis genome; and amongst the genomic features analysed, gene density yielded the highest correlation with nucleotide diversity, far exceeding the strength of the correlation between historical recombination rate and nucleotide diversity [27].
Thus when gene density and RR are strongly positively correlated, as in E. globulus, this may counter the influence of RR on linked selection, disrupting the relationship between RR and genetic diversity [36]. Indeed, there is growing evidence that the effects of linked selection are not confined to areas of low recombination but are often pervasive throughout genomes [36, 60], including in the forest tree poplar [61]. In support of this hypothesis, many plants exhibit a positive correlation between RR and gene density and a weak to no correlation between RR and genetic diversity [36, 60, 62]. In the case of Oryza sativa [57] and E. globulus the strong negative correlation between RR and diversity is likely to reflect, at least in part, the strong correlation between gene density and RR. However, in the case of eucalypts the contribution of other factors, such as their high genetic diversity [19, 27, 29] (see introduction) other facets of genome architecture, and the history of population size within each species, requires further investigation. Further, we cannot completely dismiss the possibly that other mechanisms are involved. For example, diversity per se could be affecting RR. It is well known that sequence divergence between individuals can inhibit recombination in adjacent regions of the genome, associated with the DNA mis-match repair system [63, 64] and this process could also contribute to the negative correlation between RR and diversity we observe.
Conclusions
Comparison of our results with previously published estimates provides evidence for stability in chromosomal level recombination rates across eucalypt species. Together with the previously reported stability in karyotypes, genome synteny/colinearity and the genomic distribution of population diversity and divergence across the globally significant lineages of Eucalyptus, our finding bode well for the transfer of genomic information between species within these lineages. We also demonstrate that the differences in RR between chromosomes are negatively correlated with species-wide genetic diversity at the chromosome level, which is opposite to the correlation observed in most other taxa. While our findings may in part reflect the scale of investigation, these insights into the recombination landscape of Eucalyptus provide testable hypotheses for future research in forest trees. Future research will investigate whether chromosomal differences in recombination rate are conserved between more distantly related eucalypt species, as well as investigating correlations between RR and genomic attributes at finer genomic scales.
Methods
Linkage map construction and comparison with physical map
Recombination rate (RR) was estimated in 10 unrelated genotypes, which were the parents of 5 full-sib pedigrees. These were all inter-provenance crosses and included three F1 (each with 183 to 184 individuals) and two ‘outbred F2’ (172 and 503 individuals) pedigrees. Together, these pedigrees sampled a diverse section of the natural distribution of E. globulus (see Hudson et al. [33], Freeman et al. [65]). All of the pedigrees had been previously used for linkage map construction [33, 65] and the progenies genotyped with diversity array technology ([DArT], DArT P/L Ltd Canberra, Australia) [66, 67] and microsatellite markers as described in Hudson et al. [33] and Freeman et al. [65]. To estimate RR in this study, we firstly rebuilt each of the 10 parental linkage maps using a subset of the markers used in the original studies. This was done to maximise the accuracy of RR estimates from the genome-anchored markers. The subset of markers were selected based on having a known and unique physical location on the BRASUZ1 reference genome of E. grandis [19] based on basic local alignment search tool (BLAST) searches, and linkage mapping criteria as described below.
For BLAST searches, adaptors were removed from the DArT sequences using Cutadapt V1.2.1 [68], then BlastN searches were performed in the Blastall V2.2.26 suite (alignment length > = 50 bp; e-value = 10−50). Two BLAST analyses were conducted. Firstly, an auto-blast amongst all marker sequences was performed to identify redundant markers. Secondly, the physical location of non-redundant markers was determined through a BLAST search against the reference genome. For the latter, BLAST results were classified according to the criteria developed by Salse et al. [69]: the cumulative percentage of sequence identity (CIP) and the cumulative alignment percentage (CALP), based on the length of high-scoring segment pairs (HSP) and the sum of all HSP lengths (AL). For each marker, the most plausible position on the BRASUZ1 reference genome was identified based on the CIP value with a maximum CALP (markers with CALP > 200 % were removed) which minimised RR.
Of the non-redundant markers anchored to unique genome positions, four main mapping criteria were used to select the markers used to estimate RR. Ordered by priority, these criteria were: i) genome coverage; ii) segregation; iii) linkage statistics; and iv) RR. Markers with weak linkage statistics were excluded, with the exception of a few markers which were included to maximise genome coverage, provided their map order matched the physical map. Uniparentally inherited markers segregating 1:1 were selected preferentially to biparentally inherited markers segregating 3:1 (preventing auto-correlations when comparing recombination estimates in parental maps from the same pedigree), although markers segregating 3:1 were selected in cases where they increased map coverage. Linkage analysis was performed using the maximum likelihood algorithm in Join-Map v.4 [70]. Map distance in cM was calculated using Haldane’s mapping function. To estimate RR (cM/Mb), we aligned linkage groups (LGs) against the 11 main scaffolds of BRASUZ1. MapChart 2.2 software [71] was used to compare physical and genetic maps. Markers were accepted when the RR between adjacent markers was less than 25 cM/Mb, since greater RR probably reflects genotyping error(s).
Variation in RR and relationships with genome features and population genetic parameters
RR was estimated by comparing 2,452 meioses (i.e., 1,226 progenies in total across the 5 pedigrees × 2 parents in each pedigree; see above) in E. globulus over the 640 Mb of the reference genome of E. grandis. Variation in RR was studied at two different levels: within chromosomes and genome-wide. At the chromosome level, two estimates of RR were used: i) a RR for each chromosome of each parent, calculated as the ratio of the total LG length in cM to the corresponding physical size per scaffold in Mb; and ii) an average chromosome RR (n = 11) across all 10 parents. The RR for each chromosome of each parent (n = 110; Table 2) was used to test the significance of the parent and chromosome main effects, using a fixed effect model with no interaction and Type 3 Sum of Squares. The chromosome averages (n =11) were used for studying the (Pearson’s) correlation with other chromosomes features (chromosome average data for the main features are presented in Table 4). These analyses were undertaken using PROC MIXED and PROC CORR of SAS respectively. At the genome-wide level, for each parent, we used the total genetic distance and the corresponding physical size, to estimate an average RR.
Heterozygosity, diversity and divergence indexes were correlated against RR at the chromosome level. Relative heterozygosity was estimated for each individual and at the chromosome level for each individual, from the total number of polymorphic markers derived from the DArT assay. Diversity and divergence indexes were derived from independent samples [34]. These diversity indexes were calculated from six Eucalyptus species from subgenus Symphyomyrtus, using ~ 90 range-wide samples per species. The six species belong to three sections within Symphyomyrtus: section Latoangulatae (E. grandis and E. urophylla), section Maidenaria (E. globulus, E. nitens and E. dunnii) and section Exsertaria (E. camaldulensis). These species are among the most important hardwood plantations species world-wide and are representative of the sections (lineages) that contain the other economically important eucalypt species [29]. We used the average diversity (HHw) in each species, as well as the average diversity within, and divergence (Fst) between, the six species in our correlation analyses at the chromosome level. The ‘global’ estimates (i.e., those averaged across six species) of diversity and divergence were also analysed after partitioning the markers into those within genes and those >5 kb from a gene (i.e., intra- versus intergenic markers; following [34]).
We also tested for relationships between RR in E. globulus and genomic features of the Eucalyptus reference genome [19], based on our estimates of average RR for each chromosome, using Pearson’s correlation. These chromosomal features included: physical size of each E. grandis chromosome scaffold (in Mb), the GC content, average gene density, the number of tandem duplicated genes and the density of transposable elements (also broken into subcategories of DNA transposable elements, retrotransposons and uncategorised elements, and superfamilies within the DNA transposable elements and retrotransposon subcategories).
Abbreviations
AL, the sum of all HSP lengths; BLAST, basic local alignment search tool; CALP, the cumulative alignment percentage; CIP, the cumulative percentage of sequence identity; cM, centimorgan; CV, coefficient of variation; DArT, diversity array technology; Fst, the proportion of the genetic variance in a sub population relative to the total genetic variance; HHw, genetic diversity assuming Hard-Weinberg equilibrium; HSP, the length of high-scoring segment pairs; LGs, linkage groups; LINE, long interspersed nuclear element; Mb, megabase; QTL, quantitative trait loci; RR, recombination rate; SINE, short interspersed nuclear element
Acknowledgements
The authors thank the anonymous reviewers for their comments, which helped improve the manuscript.
Funding
This work was supported by grants from the Australian Research Council (DP110101621, DP130104220 and DP140102552) and the TRANZFOR project (ref 230793; funded under FP7-people), which supported scientific transfers between the European Union and Australia-New Zealand.
Availability of data and materials
The data supporting our findings are presented in the manuscript. The genetic length (cM) and physical size (Mb) used to calculate RR for each chromosome and individual is provided in Table 2. The genomic attributes which were used to calculate correlations with these RR estimates are provided in Table 4. Raw data will be provided on request to the corresponding author.
Authors’ contributions
JMG, RV, BP and JF were involved in the design of the study, data analysis and interpretation. JMG conceived the study. JF wrote the manuscript. CH contributed to data acquisition and analysis. IL performed bioinformatics analysis. All authors contributed to drafting, and read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Contributor Information
Jean-Marc Gion, Email: jean-marc.gion@cirad.fr.
Corey J. Hudson, Email: chudso18@its.jnj.com
Isabelle Lesur, Email: ilesur@pierroton.inra.fr.
René E. Vaillancourt, Email: rene.vaillancourt@utas.edu.au
Brad M. Potts, Email: B.M.Potts@utas.edu.au
Jules S. Freeman, Email: Jules.Freeman@utas.edu.au
References
- 1.Webster MT, Hurst LD. Direct and indirect consequences of meiotic recombination: implications for genome evolution. Trends Genet. 2012;28:101–9. doi: 10.1016/j.tig.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Sun P, Zhang R, Jiang Y, Wang X, Li J, Lv H, et al. Assessing the patterns of linkage disequilibrium in genic regions of the human genome. FEBS J. 2011;278:3748–55. doi: 10.1111/j.1742-4658.2011.08293.x. [DOI] [PubMed] [Google Scholar]
- 3.Spencer CCA, Deloukas P, Hunt S, Mullikin J, Myers S, Silverman B, et al. The Influence of recombination on human genetic diversity. PLoS Genet. 2006;2:9. doi: 10.1371/journal.pgen.0020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeffreys AJ, Neumann R. Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat Genet. 2002;31:267–71. doi: 10.1038/ng910. [DOI] [PubMed] [Google Scholar]
- 5.Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, et al. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;12:876–9. doi: 10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrov DA, Lozovskaya ER, Hartl DL. High intrinsic rate of DNA loss in Drosophila. Nature. 1996;384:346–9. doi: 10.1038/384346a0. [DOI] [PubMed] [Google Scholar]
- 7.Nam K, Ellegren H. Recombination drives vertebrate genome evolution. PLoS Genet. 2012;8:5. doi: 10.1371/journal.pgen.1002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Creighton H, McClintock B. A correlation of cytological and genetical crossing-over in Zea mays. Proc Natl Acad Sci U S A. 1931;17:492–7. doi: 10.1073/pnas.17.8.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smukowski CS, Noor MAF. Recombination rate variation in closely related species. Heredity. 2011;107:496–508. doi: 10.1038/hdy.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, et al. Fine scale recombination rate differences between sexes, populations and individuals. Nature. 2010;467:1099–103. doi: 10.1038/nature09525. [DOI] [PubMed] [Google Scholar]
- 11.Heffner E, Sorrells M, Jannink J. Genomic selection for crop improvement. Crop Sci. 2009;49:1–12. doi: 10.2135/cropsci2008.08.0512. [DOI] [Google Scholar]
- 12.Goddard ME, Hayes BJ. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet. 2009;10:381–91. doi: 10.1038/nrg2575. [DOI] [PubMed] [Google Scholar]
- 13.Rodgers-Melnick E, Bradbury PJ, Elshire RJ, Glaubitz JC, Acharya CB, Mitchell SE, et al. Recombination in diverse maize is stable, predictable, and associated with genetic load. Proc Natl Acad Sci U S A. 2015;112:3823–8. doi: 10.1073/pnas.1413864112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bauer E, Falque M, Walter H, Bauland C, Camisan C, Campo L, et al. Intraspecific variation of recombination rate in maize. Genome Biol. 2013;14:R103. doi: 10.1186/gb-2013-14-9-r103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levin DA. Pest pressure and recombination systems in plants. Amer Nat. 1975;109:437–51. doi: 10.1086/283012. [DOI] [Google Scholar]
- 16.Petit RJ, Hampe A. Some evolutionary consequences of being a tree. Annu Rev Ecol Evol Syst. 2006;37:187–214. doi: 10.1146/annurev.ecolsys.37.091305.110215. [DOI] [Google Scholar]
- 17.Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 18.Neale DB, Kremer A. Forest tree genomics: growing resources and applications. Nature Rev Genet. 2011;12:111–22. doi: 10.1038/nrg2931. [DOI] [PubMed] [Google Scholar]
- 19.Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, et al. The genome of Eucalyptus grandis. Nature. 2014;510:356–62. doi: 10.1038/nature13308. [DOI] [PubMed] [Google Scholar]
- 20.Slavov GT, DiFazio SP, Martin J, Schackwitz W, Muchero W, Rodgers-Melnick E, et al. Genome resequencing reveals multiscale geographic structure and extensive linkage disequilibrium in the forest tree Populus trichocarpa. New Phytol. 2012;196:3. doi: 10.1111/j.1469-8137.2012.04258.x. [DOI] [PubMed] [Google Scholar]
- 21.Thavamanikumar S, Southerton SG, Bossinger G, Thumma BR. Dissection of complex traits in forest trees — opportunities for marker-assisted selection. Tree Genet Genomes. 2013;9:627–39. doi: 10.1007/s11295-013-0594-z. [DOI] [Google Scholar]
- 22.Chancerel E, Lamy J-B, Lesur I, Noirot C, Klopp C, Ehrenmann F. High-density linkage mapping in a pine tree reveals a genomic region associated with inbreeding depression and provides clues to the extent and distribution of meiotic recombination. BMC Biol. 2013;11:50. doi: 10.1186/1741-7007-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plomion C, Aury J-M, Amselem J, Alaeitabar T, Barbe V, Belser C, et al. Decoding the oak Genome: Public Release of Sequence Data, Assembly, Annotation and Publication Strategies. 2015. [DOI] [PubMed] [Google Scholar]
- 24.The Hardwood Genomics Project . Chinese Chestnut Genome and QTL Sequences V1.1. 2015. [Google Scholar]
- 25.Kullan ARK, van Dyk MM, Jones N, Kanzler A, Bayley A, Myburg AA. High density genetic linkage maps with over 2,400 sequence-anchored DArT markers for genetic dissection in an F2 pseudo-backcross of Eucalyptus grandis x E. urophylla. Tree Genet Genomes. 2012;8:163–75. doi: 10.1007/s11295-011-0430-2. [DOI] [Google Scholar]
- 26.Petroli CD, Sansaloni CP, Carling J, Steane DA, Vaillancourt RE, Myburg AA, et al. Genomic characterization of DArT markers based on high-density linkage analysis and physical mapping to the Eucalyptus genome. PLoS ONE. 2012;7:9. doi: 10.1371/journal.pone.0044684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva-Junior OB, Grattapaglia D. Genome-wide patterns of recombination, linkage disequilibrium and nucleotide diversity from pooled resequencing and single nucleotide polymorphism genotyping unlock the evolutionary history of Eucalyptus grandis. New Phytol. 2015 doi: 10.1111/nph.13505. [DOI] [PubMed] [Google Scholar]
- 28.Brooker MIH. A new classification of the genus Eucalyptus. Aust Syst Bot. 2000;13:79–148. doi: 10.1071/SB98008. [DOI] [Google Scholar]
- 29.Grattapaglia D, Vaillancourt RE, Shepherd M, Thumma BR, Foley W, Kulheim C, et al. Progress in Myrtaceae genetics and genomics: Eucalyptus as the pivotal genus. Tree Genet Genomes. 2012;8:463–508. doi: 10.1007/s11295-012-0491-x. [DOI] [Google Scholar]
- 30.Byrne M . Phylogeny, diversity and evolution of eucalypts. In: Sharma AK, Sharma A, editors. Plant Genome: Biodiversity and Evolution. Enfield: Science Publishers; 2008. pp. 303–46. [Google Scholar]
- 31.Ribeiro T, Barrela RM, Bergès H, Marques C, Loureiro J, Morais-Cecilio L, et al. Advancing Eucalyptus genomics: cytogenomics reveals conservation of Eucalyptus genomes. Front Plant Sci. 2016;7:510. doi: 10.3389/fpls.2016.00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shepherd M, Kasem S, Lee D, Henry R. Construction of microsatellite linkage maps for Corymbia. Silvae Genet. 2006;55:228–38. [Google Scholar]
- 33.Hudson CJ, Kullan ARK, Freeman JS, Faria DA, Grattapaglia D, Kilian A, et al. High synteny and colinearity among Eucalyptus genomes revealed by high-density comparative genetic mapping. Tree Genet Genomes. 2012;8:339–52. doi: 10.1007/s11295-011-0444-9. [DOI] [Google Scholar]
- 34.Hudson CJ, Freeman JS, Myburg AA, Potts BM, Vaillancourt RE. Genomic patterns of species diversity and divergence in Eucalyptus. New Phytol. 2015;206:1378–90. doi: 10.1111/nph.13316. [DOI] [PubMed] [Google Scholar]
- 35.Brondani RPV, Williams ER, Brondani C, Grattapaglia D. A microsatellite-based consensus linkage map for species of Eucalyptus and a novel set of 230 microsatellite markers for the genus. BMC Plant Biol. 2006;6:20. doi: 10.1186/1471-2229-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cutter AD, Payseur BA. Genomic signatures of selection at linked sites: unifying the disparity among species. Nat Rev Genet. 2013;14:262–74. doi: 10.1038/nrg3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salomé PA, Bomblies K, Fitz J, Laitinen RAE, Warthmann N, Yant L, et al. The recombination landscape in Arabidopsis thaliana F2 populations. Heredity. 2011;108:447–55. doi: 10.1038/hdy.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Comeron JM, Ratnappan R, Bailin S. The many landscapes of recombination in Drosophila melanogaster. PLoS Genet. 2012;8:10. doi: 10.1371/journal.pgen.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koren A, Ben-Aroya S, Kupiec M. Control of meiotic recombination initiation: a role for the environment? Curr Genet. 2002;42:129–39. doi: 10.1007/s00294-002-0340-9. [DOI] [PubMed] [Google Scholar]
- 40.John B. Meiosis. Cambridge: Cambridge University Press; 2005. [Google Scholar]
- 41.Francis KE, Lam SY, Harrison BD, Bey AL, Berchowitz LE, Copenhaver GP. Pollen tetrad-based visual assay for meiotic recombination in Arabidopsis. Proc Natl Acad Sci U S A. 2007;104:3913–8. doi: 10.1073/pnas.0608936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lenormand T, Dutheil J. Recombination differences between sexes: a role for haploid selection. PLoS Biol. 2005;3:3. doi: 10.1371/journal.pbio.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Os H, Stam P, Visser RGF, Van Eck HJ. RECORD: a novel method for ordering loci on a genetic linkage map. Theor Appl Genet. 2005;112:30. doi: 10.1007/s00122-005-0097-x. [DOI] [PubMed] [Google Scholar]
- 44.Bartholomé J, Mandrou E, Mabiala A, Jenkins J, Nabihoudine I, Klopp C, Schmutz J, Plomion C, Gion J-M. High-resolution genetic maps of Eucalyptus improve Eucalyptus grandis genome assembly. New Phytol. 2014;206:4. doi: 10.1111/nph.13150. [DOI] [PubMed] [Google Scholar]
- 45.Li W, Freudenberg J. Two-parameter characterization of chromosome-scale recombination rate. Genome Res. 2009;19:2300–7. doi: 10.1101/gr.092676.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mugal CF, Nabholz B, Ellegren H. Genome-wide analysis in chicken reveals that local levels of genetic diversity are mainly governed by the rate of recombination. BMC Genomics. 2013;14:86. doi: 10.1186/1471-2164-14-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crisp MD, Burrows GE, Cook LG, Thornhill AH, Bowman DMJS. Flammable biomes dominated by eucalypts originated at the Cretaceous-Palaeogene boundary. Nat Commun. 2011;2:193. doi: 10.1038/ncomms1191. [DOI] [PubMed] [Google Scholar]
- 48.Serres-Giardi L, Belkhir K, David J, Glémin S. Patterns and evolution of nucleotide landscapes in seed plants. Plant Cell. 2012;24:1379–97. doi: 10.1105/tpc.111.093674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaut BS, Wright SI, Rizzon C, Dvorak J, Anderson LK. Recombination and underappreciated factor in the evolution of plant genomes. Nat Rev Genet. 2007;8:77–84. doi: 10.1038/nrg1970. [DOI] [PubMed] [Google Scholar]
- 50.Barnes TM, Kohara Y, Coulsen A, Hekimi S. Meiotic recombination, noncoding DNA and genomic organization in Caenorhabditis elegans. Genetics. 1995;141:1. doi: 10.1093/genetics/141.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Federoff NV. Transposable elements, epigenetics, and genome evolution. Science. 2012;338:758–67. doi: 10.1126/science.338.6108.758. [DOI] [PubMed] [Google Scholar]
- 52.Oliver KR, McComb JA, Greene WK. Transposable elements: powerful contributors to angiosperm evolution and diversity. Genome Biol Evol. 2013;5:1886–901. doi: 10.1093/gbe/evt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennetzen JL, Ma J, Devos KM. Mechanisms of recent genome size variation in flowering plants. Ann Bot. 2005;95:127–32. doi: 10.1093/aob/mci008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grover CE, Wendel JF. Recent insights into mechanisms of genome size change in plants. J Botany. 2010 [Google Scholar]
- 55.Dolgin ES, Charlesworth B. The effect of recombination rate on the distribution and abundance of transposable elements. Genetics. 2008;178:2169–77. doi: 10.1534/genetics.107.082743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright SI, Agrawal N, Bureau TE. Effects of recombination and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 2003;13:1897–903. doi: 10.1101/gr.1281503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flowers JM, Molina J, Rubinstein S, Huang P, Schaal BA, Purugganan MD. Natural selection in gene-dense regions shapes the pattern of polymorphism in wild and domesticated rice. Mol Biol Evol. 2012;29:675–87. doi: 10.1093/molbev/msr225. [DOI] [PubMed] [Google Scholar]
- 58.Begun DJ, Aquadro CF. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature. 1992;9:519–20. [DOI] [PubMed]
- 59.Payseur BA, Nachman MW. Gene density and human nucleotide polymorphism. Mol Biol Evol. 2002;19:336–40. [DOI] [PubMed]
- 60.Slotte T. The impact of linked selection on plant genomic variation. Brief Funct Genomics. 2014;13:268–75. doi: 10.1093/bfgp/elu009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Street NR, Scofield DG, Invarsson PK. Natural selection and recombination rate variation shape nucleotide polymorphism across the genomes of three related Populus species. Genetics. 2016;202:1185–200. doi: 10.1534/genetics.115.183152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nordborg M, Hu TT, Ishino Y, Jhaveri J, Toomajian C, Zheng H, et al. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 2005;3:7. doi: 10.1371/journal.pbio.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Opperman R, Emmanuel E, Levy AA. The effect of sequence divergence on recombination between direct repeats in Arabidopsis. Genetics. 2004;168:2207–15. doi: 10.1534/genetics.104.032896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–33. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 65.Freeman JS, Potts BM, Downes GM, Pilbeam D, Thavamanikumar S, Vaillancourt RE. Stability of quantitative trait loci for growth and wood properties across multiple pedigrees and environments in Eucalyptus globulus. New Phytol. 2013;198:1121–34. [DOI] [PubMed]
- 66.Sansaloni CP, Petroli CD, Carling J, Hudson CJ, Steane DA, Myburg AA, et al. A high-density Diversity Arrays Technology (DArT) microarray for genome-wide genotyping in Eucalyptus. Plant Methods. 2010;6:16. doi: 10.1186/1746-4811-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steane DA, Myburg AA, Sansaloni C, Petroli C, Grattapaglia D, Kilian A, et al. DArT arrays for genetic mapping and diversity analysis of Eucalyptus. Mol Phylogenet Evol. 2011;59:206–24. doi: 10.1016/j.ympev.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 68.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. 2011;17:10–2. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 69.Salse J, Bolot S, Throude M, Jouffe V, Piegu B, Quraishi UM, et al. Identification and characterization of shared duplications between rice and wheat provide new insight into grass genome evolution. Plant Cell. 2008;20:11–24. doi: 10.1105/tpc.107.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Van Ooijen J. JoinMap 4, software for the calculation of genetic linkage maps in experimental populations. Wageningen, Netherlands: Kyazma B.V; 2006. [Google Scholar]
- 71.Voorrips RE. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93:77–8. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting our findings are presented in the manuscript. The genetic length (cM) and physical size (Mb) used to calculate RR for each chromosome and individual is provided in Table 2. The genomic attributes which were used to calculate correlations with these RR estimates are provided in Table 4. Raw data will be provided on request to the corresponding author.