

Abstract
Background
Residual feed intake (RFI) is a powerful indicator for energy utilization efficiency and responds to selection. Low RFI selection enables a reduction in feed intake without affecting growth performance. However, the effective variants or major genes dedicated to phenotypic differences in RFI in quality chickens are unclear. Therefore, a genome-wide association study (GWAS) and RNA sequencing were performed on RFI to identify genetic variants and potential candidate genes associated with energy improvement.
Results
A lower average daily feed intake was found in low-RFI birds compared to high-RFI birds. The heritability of RFI measured from 44 to 83 d of age was 0.35. GWAS showed that 32 of the significant single nucleotide polymorphisms (SNPs) associated with the RFI (P < 10−4) accounted for 53.01 % of the additive genetic variance. More than half of the effective SNPs were located in a 1 Mb region (16.3–17.3 Mb) of chicken (Gallus gallus) chromosome (GGA) 12. Thus, focusing on this region should enable a deeper understanding of energy utilization. RNA sequencing was performed to profile the liver transcriptomes of four male chickens selected from the high and low tails of the RFI. One hundred and sixteen unique genes were identified as differentially expressed genes (DEGs). Some of these genes were relevant to appetite, cell activities, and fat metabolism, such as CCKAR, HSP90B1, and PCK1. Some potential genes within the 500 Kb flanking region of the significant RFI-related SNPs detected in GWAS (i.e., MGP, HIST1H110, HIST1H2A4L3, OC3, NR0B2, PER2, ST6GALNAC2, and G0S2) were also identified as DEGs in chickens with divergent RFIs.
Conclusions
The GWAS findings showed that the 1 Mb narrow region of GGA12 should be important because it contained genes involved in energy-consuming processes, such as lipogenesis, social behavior, and immunity. Similar results were obtained in the transcriptome sequencing experiments. In general, low-RFI birds seemed to optimize energy employment by reducing energy expenditure in cell activities, immune responses, and physical activity compared to eating.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2861-5) contains supplementary material, which is available to authorized users.
Keywords: Residual feed intake, Genome-wide association study, RNA-sequencing, Quality chickens
Background
Residual feed intake (RFI) is an effective indicator for the feed utilization efficiency of animals. Low RFI selection enables a lower energy intake without sacrificing growth performance because it is independent of the metabolic body weight and daily gain regardless of the phenotype or genetic level [1, 2]. The feed conversion rate (FCR) is another synthetic trait that can optimize energy usage and is negatively correlated with growth traits [3, 4]. Chinese consumers prefer quality chickens (also called yellow plumage chickens) with a long feeding time and excellent meat quality but focus less on body weight at the market. Therefore, RFI selection is more applicable to non-fast growing chickens for the genetic improvement of energy metabolism.
To date, up to 60 quantitative trait loci (QTLs) have been reported to have significant associations with chicken feeding traits including dry matter digestibility, dry matter intake, feeding efficiency, feed intake, RFI and FCR, of which 20 were associated with RFI (http://www.animalgenome.org/QTLdb/). However, the above RFI-related QTLs are difficult to be applied to quality chickens due to the differences on breeds and trait measurement time.
Genome-wide association studies (GWAS), at first, are used to screen candidate markers associated with human diseases [5, 6]. With the continuous improvement and reduced cost of genotyping technology, this technique is being more widely applied to animal breeding and genetics. GWAS-related applications in RFI are primarily concentrated on beef cattle [7–9] and pigs [10–12] and have been applied to laying hens [13] and broilers [14]. The development of the high density 600 K genotyping array may provide technical assistance for the identification of causative and credible single nucleotide polymorphisms (SNPs) that affect the chicken RFI [15].
The transcriptome is a full set of the RNAs transcribed in the cell at a certain developmental phase or under a specific stress condition and possesses spatial and temporal expression characteristics [16]. RNA sequencing can be a gateway to success in identifying differentially expressed genes (DEGs), discovering alternative splicing events, and conducting studies on gene evolution [17–20]. Previous studies showed that the RFI was so complex that all relevant genes might not operate in a single physiological pathway [21, 22].
The present study combined GWAS with transcriptome sequencing to identify distinct genes or regions that affected the chicken RFI. Our results lay the foundation for improvements in energy efficiency and reductions in feed cost.
Results
Growth characteristics and genetic parameters
The pure line N301 belongs to the dwarfism yellow-plumage chicken with a medium growth speed. At the beginning of the feeding trial (44 d of age), the average body weights of the male and female chickens were 794 (±124) g and 760 (±98) g, respectively. When the trial was finished (83 d of age), the male chickens reached 1913 (±230) g and the female chickens reached 1780 (±160) g. The average daily feed intakes (ADFIs) of the male and female chickens recorded by the electronic feeding station were 109 (±15) g and 102 (±15) g, respectively. The linear regression equation well described the changes in body weight at different ages. The average R2 across all birds was 98.4 %. Only four birds had an R2 lower than 80 % (Fig. 1), suggesting that the estimates for average daily gain (ADG) and mid-test metabolic body weight (MMBW) for each bird were reliable.
Fig. 1.
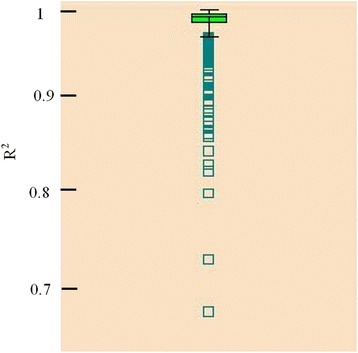
Distributional R2 for regression of body weight against the day of the test for each chicken. The vertical ordinate denotes the R2 for the growth rate
The test of fixed effects showed that the gender effect on RFI was not significant (P > 0.1), but the pen effect was significant (P < 0.01). One explanation for this result may be that the chickens were already separated based on gender when they were arranged in different pens, thereby turning the nested gender effect into the pen effect. Next, we compared the growth traits between the birds with the highest- and lowest-ranked RFIs (Table 1). No significant difference was found for any growth trait, which was similar to the results obtained in cattle [23]. However, the ADFI for the lowest RFI-ranked birds was 39 g lower than the ADFI for the highest RFI-ranked birds (P < 0.01).
Table 1.
Growth characteristics (average ± S.E.) of 10 % of the chickens with the lowest and highest residual feed intake rankings
| Traitsa | Lowest 10 % | Highest 10 % | P-valueb |
|---|---|---|---|
| RFI during 44–83-d old (g) | −17.40 (±0.45) | 20.16 (±0.56) | / |
| ADFI (g) | 87.32 (±0.90) | 126.10 (±1.18) | <0.01 |
| Body weight at 44-d old (g) | 771.60 (±9.31) | 770.62 (±10.78) | 0.94 |
| Body weight at 83-d old (g) | 1832.92 (±16.07) | 1858.03(±16.07) | 0.35 |
| ADG (g) | 27.8 (±0.31) | 27.98 (±0.46) | 0.75 |
| MBW (g) | 1318.3 (±11.69) | 1335.0 (±15.12) | 0.38 |
| FCR during 44–83-d old | 3.31 (±0.03) | 4.76 (±0.09) | <0.01 |
a RFI residual feed intake, ADFI average daily feed intake, ADG average daily gain, MBW mid-test body weight, FCR feed conversion rate
b P-value obtained by t-test
The heritability estimate for the RFI from 44 to 83 d of age was 0.35 (Table 2), which fell into the 0.21 to 0.49 range reported for other populations [1, 2]. This result implied that the RFI was under a moderate level of genetic control. Phenotypic RFI selection provides a method to reduce feed intake without affecting either the ADG (rp = 0.03) or MMBW (rp = 0.01). However, RFI selection had a few consequences over the ADG if only the genetic contribution was considered (rg = 0.27), which was consistent with previous reports [1, 2, 24]. The positive genetic correlation between RFI and FCR found in our population (rg = 0.75) was higher than the correlation reported in other domestic animals [24, 25].
Table 2.
Genetic parameter estimation for the growth and feeding traits
| Traitsc | No.d | Heritability (±S.E.) | RFI during44-83-d old (g) |
|---|---|---|---|
| RFI during44-83-d old (g) | 1158 | 0.3542 ± 0.0701 | / |
| ADFI (g) | 1158 | 0.3950 ± 0.0749 | 0.7266a/0.7544 ± 0.0718b |
| Body weight at 44-d old (g) | 1158 | 0.5598 ± 0.0848 | −0.0293/−0.2059 ± 0.1462 |
| Body weight at 83-d old (g) | 1158 | 0.3683 ± 0.0765 | 0.0247/0.0314 ± 0.1649 |
| ADG (g) | 1158 | 0.3186 ± 0.0723 | 0.0286/0.2725 ± 0.1626 |
| MMBW (g0.75) | 1158 | 0.4706 ± 0.0821 | 0.0126/−0.0233 ± 0.1563 |
| FCR during 44–83-d old | 1158 | 0.2159 ± 0.0594 | 0.6558/0.7465 ± 0.0902 |
aPearson correlation
bGenetic correlation (average ± S.E.)
c RFI residual feed intake, ADFI average daily feed intake, ADG average daily gain, MBW mid-test body weight, FCR feed conversion rate
d Number of samples for estimating heritability
Genome-wide association study
The original RFI values calculated from 426 individuals were examined for compliance with normality prior to the GWAS using the Anderson-Darling test. The results showed that the initial distribution deviated from the normal distribution (P < 0.01) (Additional file 1: Table S1). After Johnson transformation, the phenotypic data were subjected to statistical analysis (Fig. 2).
Fig. 2.
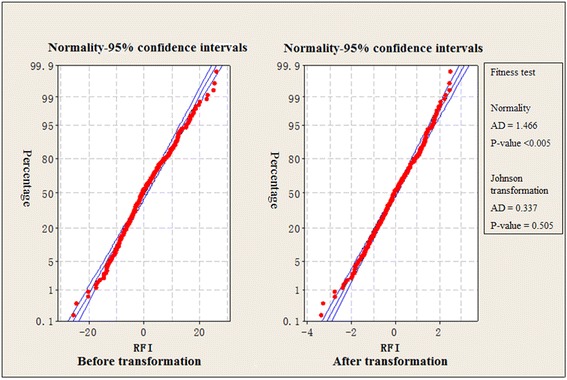
Probability graph of residual feed intake
Because the kinship of all tested individuals could not be entirely eliminated when the IBS matrix was introduced into the mixed model, the genomic control inflation/deflation parameter (λ) was introduced [26]. In total, 32 SNPs were significantly associated with the RFI with P-values below the threshold (10−4) after correction by a λ equal to 1.005 (Fig. 3); these SNPs accounted for 53.01 % of the additive genetic variance. These significant points corresponded to 13 known genes and one unannotated gene in the chicken genome: SLC17A8, COBL, PCDH19, JAKMIP1, ZFYVE28, PPP1R7, SEPT2, RYBP, PDZRN3, CHL1, UTS2R, ZMPSTE24, SYT6 and LOC101749255 (Table 3).
Fig. 3.
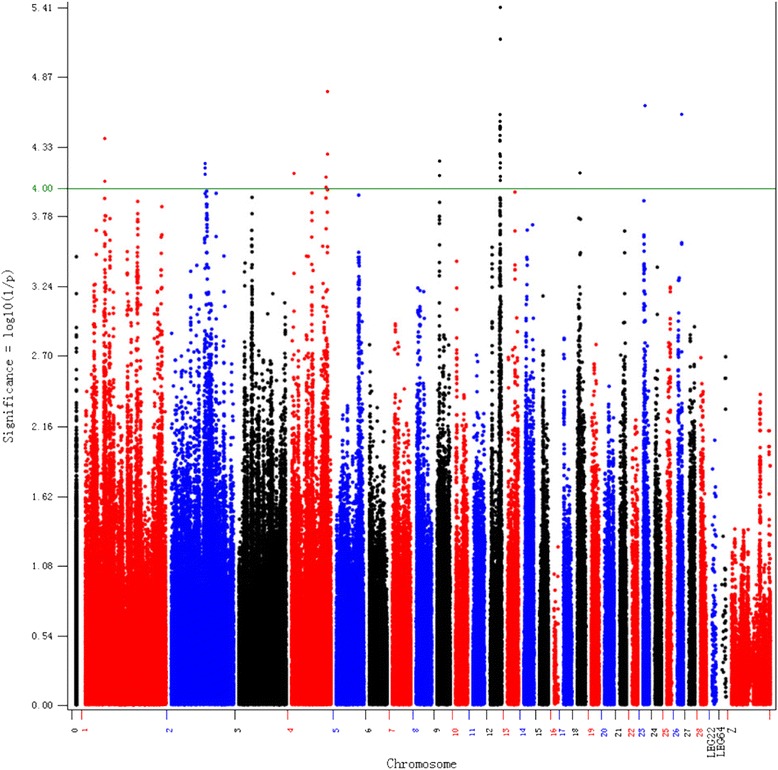
Manhattan plot of the SNP additive effects on residual feed intake (RFI) in chickens. The green line was the genome-wide significance threshold (10−4)
Table 3.
SNPs significantly associated with RFI
| Locus | Chr | Positiona | Effectb | P-valuec | MAFd | Var(%)e | Nearest gene | SNP-gene relation |
|---|---|---|---|---|---|---|---|---|
| rs313001261 | 1 | 47140273 | 0.35 | 4.08E-05 | 0.36 | 9.52 | SLC17A8 | UTR-3 |
| rs14820118 | 1 | 47142175 | 0.34 | 8.75E-05 | 0.35 | 8.53 | SLC17A8 | downstream |
| rs313802356 | 2 | 81015541 | −0.44 | 6.90E-05 | 0.16 | 2.24 | COBL | UTR-3 |
| rs316977875 | 2 | 81047713 | −0.44 | 6.37E-05 | 0.16 | 2.24 | COBL | intron |
| rs312370583 | 2 | 81093795 | −0.44 | 7.73E-05 | 0.16 | 2.42 | COBL | intron |
| rs312326618 | 4 | 5197844 | −0.3 | 7.60E-05 | 0.44 | 4.8 | PCDH19 | intron |
| rs316570003 | 4 | 78962129 | 0.38 | 8.13E-05 | 0.22 | 9.71 | JAKMIP1 | intron |
| rs14494646 | 4 | 79000778 | 0.44 | 9.74E-05 | 0.13 | 6.93 | JAKMIP1 | upstream |
| rs312925954 | 4 | 82293366 | 0.45 | 1.76E-05 | 0.16 | 4.52 | ZFYVE28 | intron |
| rs16445177 | 4 | 82306738 | 0.4 | 5.39E-05 | 0.18 | 4.29 | ZFYVE28 | intron |
| rs312767762 | 9 | 4440306 | −0.53 | 7.90E-05 | 0.1 | 8.43 | PPP1R7 | intron |
| rs16663153 | 9 | 4507514 | −0.51 | 6.10E-05 | 0.11 | 8.13 | SEPT2 | downstream |
| rs14046165 | 12 | 16386012 | 0.33 | 4.20E-05** | 0.38 | 4.15 | RYBP | intron |
| rs312899555 | 12 | 16393667 | 0.33 | 5.37E-05** | 0.31 | 2.63 | RYBP | intron |
| rs313947867 | 12 | 16400923 | 0.33 | 4.28E-05** | 0.31 | 2.63 | RYBP | intron |
| rs317049993 | 12 | 16423468 | 0.32 | 3.39E-05** | 0.4 | 4.38 | RYBP | upstream |
| rs315004580 | 12 | 16640380 | 0.32 | 3.00E-05* | 0.47 | 5.54 | PDZRN3 | intron |
| rs315162282 | 12 | 16648446 | 0.32 | 3.27E-05* | 0.47 | 5.54 | PDZRN3 | intron |
| rs317287197 | 12 | 16660649 | 0.32 | 2.66E-05* | 0.47 | 5.54 | PDZRN3 | intron |
| rs316098097 | 12 | 16673403 | 0.32 | 3.86E-05** | 0.47 | 5.54 | PDZRN3 | intron |
| rs314131263 | 12 | 16681057 | 0.32 | 3.45E-05** | 0.47 | 5.62 | PDZRN3 | intron |
| rs14046530 | 12 | 16758193 | −0.32 | 6.89E-05** | 0.49 | 4.54 | PDZRN3 | upstream |
| rs315238546 | 12 | 17083942 | 0.3 | 5.60E-05** | 0.41 | 5.94 | LOC101749255 | downstream |
| rs315693318 | 12 | 17089689 | 0.3 | 8.62E-05** | 0.41 | 5.94 | LOC101749255 | downstream |
| rs315157887 | 12 | 17160768 | 0.37 | 3.92E-06** | 0.38 | 7.41 | LOC101749255 | upstream |
| rs317584843 | 12 | 17195875 | 0.32 | 6.31E-05** | 0.3 | 5.22 | CHL1 | upstream |
| rs314285248 | 12 | 17230860 | 0.33 | 6.92E-06** | 0.46 | 5.08 | CHL1 | intron |
| rs317278144 | 12 | 17239654 | 0.35 | 3.34E-05* | 0.25 | 7.47 | CHL1 | intron |
| rs317188563 | 12 | 17260556 | 0.28 | 8.02E-05** | 0.5 | 1.88 | CHL1 | intron |
| rs316897066 | 18 | 3599249 | 0.28 | 7.54E-05 | 0.44 | 5.18 | UTS2R | downstream |
| rs315285389 | 23 | 1733290 | −0.38 | 2.27E-05* | 0.24 | 3.39 | ZMPSTE24 | upstream |
| rs315491506 | 26 | 3777283 | −0.38 | 2.65E-05 | 0.21 | 9.28 | SYT6 | intron |
aPhysical position
bAdditive effect of allele B (minor allele)
c P-value corrected for inflation factor λ, *FDR ≤10 %, ** FDR ≤5 %
dMinor allele frequency
eThe proportion of the phenotypic variance accounted by the SNP
These SNPs were widely distributed on eight chicken (Gallus gallus) chromosomes (GGA). GGA18, GGA23 and GGA26 possessed only one significant SNP each. Two of the significant SNPs were located in the 3’ untranslated region (UTR) and downstream of SLC17A8 on GGA1, two were located within a 4.4–4.6 Mb region on GGA9, three were located within a 81.0–81.1 Mb region on GGA2, five were located on GGA4. Notably, more than half of the total significant SNPs were located within a 1 Mb narrow region (16.3–17.3 Mb) on GGA12.
This narrow region contained 555 tested SNPs around eight genes. We observed 99 haplotype blocks extending from 0 to 64 kb (Fig. 4). The significant SNPs were entirely located within the blocks with the exception of SNP rs317049993. Three significant points in the RYBP introns (rs14046165, rs312899555, and rs313947867) had identical additive effects because they were in a state of complete linkage. Six haplotypes were found in a block determined by the SNPs rs14046165 and rs312899555; “GATGA” occurred more commonly in the other three non-significant SNPs with a frequency of 31.2 %. Four haplotypes were found in another block determined by the SNP rs313947867; “GGGC” was the most common haplotype in the other three non-significant SNPs with a frequency of 35 %. Five significant SNPs in complete linkage were found in the introns of PDZRN3, although they were positioned in two different blocks.
Fig. 4.
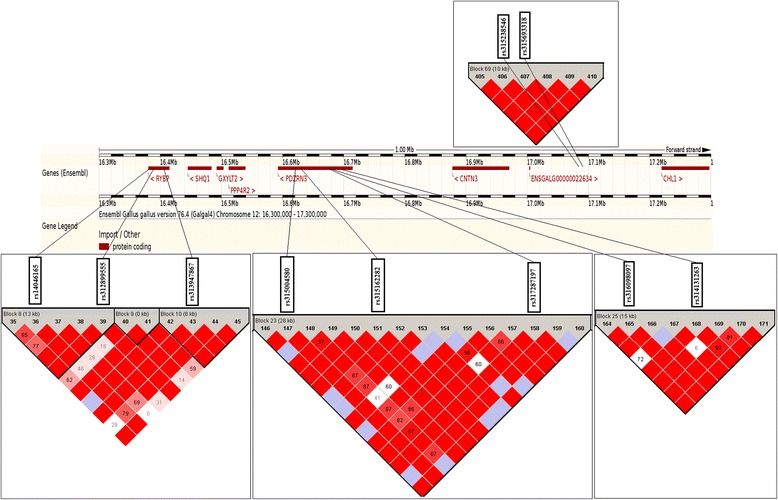
Ensembl genes on the region from 16.3 to 17.3 Mb on chicken chromosome 12 and haplotype blocks constructed using significant RFI-related SNPs
The proportion of phenotypic variance explained by rs316570003 in the intron of JAKMIP1 was highest among the significant SNPs. Because the original RFI variable was transformed into the normal distribution data, the increment of the additive effect accompanied by the minor allele could not accurately reflect its impact on the change in RFI. In contrast, the direction of the additive effect could be used to judge whether the minor allele would be beneficial to the RFI. The results showed that nine wild-type mutations were in favor of decreasing the chicken’s RFI value.
The chip used in the present study had a very high density. The average interval between two SNPs was 1.8 Kb, which resulted in little chance for recombination among neighboring SNPs gathered in a limited range of the chromosome. Linkage disequilibrium (LD) analysis was performed to determine the extent to which the causative SNP could generate linkage to the significant SNP. The pairwise LD measured by R2 values for the present population was calculated for GGA12, where the most significant SNPs were concentrated. The results showed that the LD level was high (R2 = 0.3) at short distances, slightly decreased (R2 = 0.2) when the distances increased to 40–60 kb and low (R2 ≤ 0.15) when the distance increased to 80–100 kb. Similar trends were found in GGA2 (Fig. 5). Therefore, 50 kb was accepted as a reasonable distance to cause moderate LD between two SNPs, and we captured all genes distributed on the 50 kb flanking regions of the significant SNPs for GO analysis.
Fig. 5.
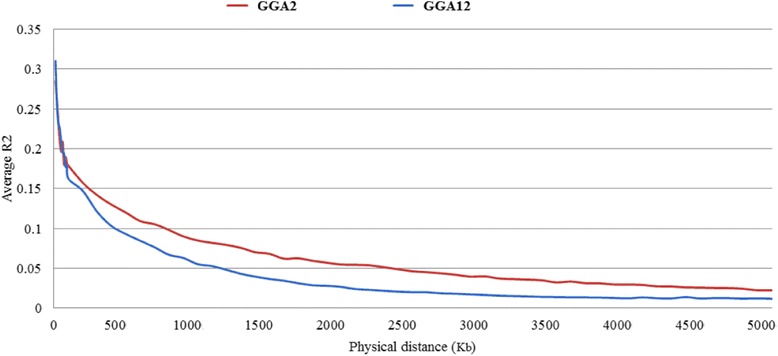
Relationship between the physical distance and LD values between SNP markers in the quality chickens used in the present study
The results from the first level classification revealed that 45 GO terms in molecular function, 40 GO terms in biological process and 27 of GO terms in cellular component corresponded to the input genes (Additional file 2: Table S2). A deeper analysis was conducted in the molecular function GO terms to obtain more information. Protein binding (12.5 %) was reflected by eight different genes and might be the major molecular function that differed in RFI-divergent birds, followed by zinc ion binding (10 %), calcium ion binding (7.5 %), and ATP binding (5 %). Protein binding was significant, including the synthesis of a peptides from amino acid molecules and the formation of a high-grade protein structure by combining polypeptide chains. This result indicated that the difference in protein binding was associated with the rapid growth of the experimental birds, specifically muscle development. The chief role of the calcium ion was to compromise potential on two sides of the cell membranes and to maintain muscle contraction and relaxation, indicating that differences in motor skills would occur in RFI-divergent birds. Therefore, differences in ATP binding should not be overlooked. ATP is the primary source of fuel for every biochemical and physiological process and will induce metabolic disorders in the event of an exception.
RNA sequencing of the transcriptome
Read mapping
RNA sequencing was performed in four individuals, as follows: two chickens with the highest RFI values (H1 and H2) and two with the lowest RFI values (L1 and L2). Among the four samples, H2 had the lowest mapping rate (total mapped reads/total reads) of approximately 88.4 % compared to the rates above 90 % for the other three samples (Table 4). Some of these reads had more than one matching sequence in the genome; these reads were removed from the subsequent analysis. The clean reads were evenly distributed across all chromosomes. Based on the annotation analysis of the clean reads mapped to the genome, we found that 61 % of these reads were derived from exons, 32.62–35.56 % were from intergenic regions, and only a small number were derived from splicing and intronic regions.
Table 4.
Statistics for read mapping
| Sample name | L2 | H1 | H2 | L1 |
|---|---|---|---|---|
| Total readsa | 25,313,336 | 25,731,202 | 20,249,350 | 28,438,634 |
| Total mappedb | 22,702,806 (89.69 %) | 23,216,653 (90.23 %) | 17,916,485 (88.48 %) | 25,601,368 (90.02 %) |
| Multiple mappedc | 681,109 (2.69 %) | 690,852 (2.368 %) | 575,647 (2.84) | 695,892 (2.45 %) |
| Uniquely mappedd | 22,021,697 (87.00 %) | 22,525,801 (87.54 %) | 17,340,838 (85.64 %) | 24,905,476 (87.58 %) |
| Reads map to “+”e | 11,365,825 | 11,622,304 | 8,966,742 | 12,813,376 |
| Reads map to “−”f | 11,336,981 | 11,594,349 | 8,949,743 | 12,787,992 |
aTotal reads: the number of clean reads
bTotal mapped: the number of clean reads that could be mapped to the chicken genome
cMultiple mapped: the number of clean reads corresponding to a plurality of locations in the genome
dUniquely mapped: the number of clean reads corresponding to a unique location in the genome
eReads map to “+”: reads mapped to the sense strand
fReads map to “−”: reads mapped to the antisense strand
DEG analysis
Four samples were classed into two groups based on their RFI rankings prior to the DEG analysis. The average expression of identical genes in the high-RFI group was compared with the low-RFI group to screen for DEGs. Biological replicates of the expressed genes had a high correlation coefficient (0.936) between the high-RFI group and low-RFI group (Additional file 3: Figure S1). As a generalized representation index, RPKM (reads per kilobase of transcript per million mapped reads) was used to denote each transcript’s expression quantity [27]. After bioinformatics assessment and comparison, 119 differentially expressed transcripts were identified between the high- and low-RFI groups that corresponded to 116 known genes. A total of 74 of the DEGs were up-regulated in the low-RFI group and the rest were down-regulated compared with high-RFI group (Additional file 4: Table S3). Interestingly, three genes (CCKAR, LOC395159, and miR-122-1) were only expressed in the low-RFI group. The expression of miR-6705 was significantly lower in the low-RFI group than in the high-RFI group with a fold change of eight. In contrast, the expression of PCK1 was significantly higher in the low-RFI group than in the high-RFI group with a fold change of 39.
MiRDB (http://mirdb.org/miRDB/) and TargetScan (http://www.targetscan.org/) are two effective websites for microRNA (miRNA) target prediction. Based on their prediction results, miR-122-5p and miR-122-3p were predicted to act on 693 target genes. Three targets (COL3A1, PER2, and CAV1) were found to be DEGs. However, these genes were up-regulated in the low-RFI group, which was not consistent with the negative regulatory interaction between the miRNA and its target. MiR-6705-5p was predicted to act on 704 target genes. Nine targets (LPL, GLCCI1, COL3A1, B3GNT2, ABHD17B, CAV1, RHOBTB1, CHAC1, and LUM) were found to be DEGs. Except for LPL, the other eight genes were up-regulated in the low-RFI group, indicating they might be negatively regulated by miR-6705-5p. Of note, the expression of COL3A1 and CAV1 could be impacted by both the miR-122-1 and miR-6705.
Hepatic PCK1 expression was drastically increased by 28-fold compared to its normal expression level. Similarly, the subcutaneous, intercellular, and abdominal fat contents were increased when a simple orchiectomy was implemented in capons, implying negative regulation of hepatic PCK1 expression and abdominal fat content by testosterone [28].
Discussion
In Koch’s model, b2BW0.75 is normally used as metabolic energy (ME) for maintenance. However, b2BW0.75 is most likely to represent only the ME maintenance output for basal metabolism. To determine the maintenance requirement, two non-determinative factors (energy for environmental stress resistance and voluntary activity) should be considered simultaneously. In the present study, RFI was phenotypically independent of MMBW (rp = 0.01) and ADG (rp = 0.03), indicating that there was no difference in the use of the ME for maintenance of the basal metabolism and for growth between high and low RFI individuals [29]. In reality, high-RFI birds tried to replenish their energy by eating more; thus, the redundant energy might be exploited by stress or physical activity [30]. We speculated that the low-RFI birds had less governable overall energy to expend on stress and physical activity. In the present study, the low-RFI birds spent less time eating (rg = 0.49) (data not shown), indicating that they reduced the ME demand at the expense of less physical activity related to feed intake.
Four significant SNPs on GGA1 and GGA9 in the present study did fall into the reported QTLs for chicken RFI [31]. However, only the significant SNPs on GGA12 passed chromosome-wise FDR threshold of 5 %, probably because of limited sample size resulting in low power to detect a QTL by testing the marker effect. Another possible cause was the low RFI phenotypic variability in sex-linked dwarf chicken used in the present study. The RFI values of 426 individuals ranged from −25.6 g to 25.6 g. The standard deviation of the RFI was simply 8.36 g, which was much less than that observed in normal meat-type chickens [2].
In the present study, we identified eight genes located in a 1 Mb narrow region, and almost all of the significant points in GGA12 were located in introns. The SNPs in the introns could play negative, positive, or bidirectional regulatory roles in gene expression and affect alternative splicing [32, 33]. PDZRN3 was reported to exert a negative effect on lipogenesis. In mouse 3 T3-L1 preadipocytes, up-regulation of STAT5b and C/EBPβ was observed in response to PDZRN3 silencing, resulting in increased expression of PPARγ at both the mRNA and protein levels and the promotion of 3 T3-L1 cell differentiation into adipocytes [34]. As a candidate gene for human schizophrenia and mental deficiency, CHL1 is relevant to learning behavior and reorganization of the frame of thinking. The CHL1-deficient mice displayed reduced enthusiasm for fresh food hunting and a blockage in social contact that was attributed to chaos in the neural circuits that connected the brain’s limbic system to the cerebral cortex [35]. JAKMIP1 was of special concern because its intronic SNP rs316570003 explained the highest phenotypic variance. This gene was reported to be associated with microtubule polymers and to participate in cytoskeleton rearrangement, cell polarization, and intracellular trafficking [36].
These findings combined with the implications of the GO analysis provided hints that RFI-related genes improved energy utilization efficiency by adjusting cellular procedural activities, thereby assisting muscle and neural development and growth and eventually improving and enhancing energy efficiency.
Transcriptome sequencing helped identify the DEGs causing the diversity in the RFI phenotypes. In the absence of mRNA expression, we speculate that there should be a role for CCKAR in the constant energy use. Indeed, CCKAR was reported to be associated with appetite control [37, 38]. CCK is a hormone that causes gallbladder contractions and enables the promotion of trypsin secretion [39]. Central CCK could be motivated by a combination of endogenous CCK and its receptor CCKAR to trigger a feeling of satiety. Indeed, an increased feed intake was detected in CCKAR-deficient mice [38], and the risk variants of porcine CCKAR were significantly correlated with feeding traits [40]. The effect of the CCK gene might be weakened by a lack of its receptor in high-RFI individuals, resulting in increased feed intake.
The DEGs were consigned to six different gene-interaction networks predicted by IPA (Additional file 5: Table S4). The networks were more comprehensive and detailed when combined with the function of homologous genes from humans and mice. For this reason, they provided redundant information that could not be applied to chickens, such as cancer or cardiac failure; thus, the related gene functions were simply suggestive in poultry. However, these networks provided important clues to improve the energy balance from the perspectives of genetics, nutrition and bio-pharmaceuticals.
Low RFI selection made dramatic changes in some biological pathways possible (Table 5). For example, the cell death and apoptosis of tumor cell lines, activation of antigen presenting cells (APCs) and cell viability of kidney cell lines were decreased during low RFI selection. Additionally, chickens with low RFI were more prone to vascular lesions. Every change was a result of the mutual action of DEGs.
Table 5.
Predictable changes in biological functioning based on DEG analysis in low-RFI individuals
| Categories | Functions annotation | P-value | Predicted | Molecules |
|---|---|---|---|---|
| Cell Death and Survival | apoptosis of lung cancer cell lines | 5.80E-06 | ↓ | CAMP, EREG, G0S2, HSPA5, MCL1, RASD1, SERPINF1, TNFSF10 |
| apoptosis of tumor cell lines | 2.08E-05 | ↓ | AR, BCL2A1, CAMP, CAV1, COL18A1, CREM, CTSD, EREG, G0S2, HSPA5, IL18, MAOA, MCL1, NFKBIA, NR0B2, PLAU, RASD1, SERPINF1, TNFSF10, UCHL1 | |
| cell death of lung cancer cell lines | 3.21E-06 | ↓ | CAMP, EREG, G0S2, HSPA5, MCL1, NFKBIA, RASD1, SERPINF1, TNFSF10 | |
| cell death of tumor cell lines | 2.75E-05 | ↓ | AR, BCL2A1, BTG1, CAMP, CAV1, COL18A1, CREM, CTSD, EREG, FETUB, G0S2, HSPA5, IL18, MAOA, MCL1, NFKBIA, NR0B2, PLAU, PPAT, RASD1, SERP | |
| cellular degradation | 3.77E-04 | ↓ | AR, CACNA1D, CAV1, CREM, CTSD, LYZ, UCHL1 | |
| Cellular Assembly and Organization | formation of filaments | 1.36E-03 | ↑ | AR, CAV1, COL18A1, HSPA5, PTGDS, SERPINF1, TTR, TUBB |
| Organismal Development | development of body axis | 1.99E-04 | ↑ | ANGPTL3, CCKAR, COL18A1, CREM, CTSD, DIO3, GPR34, HHEX, HSP90B1, INSIG1, LUM, MAOA, NFKBIA, PLAU, SERPINF1 |
| Immune Cell Trafficking | activation of antigen presenting cells | 3.47E-04 | ↓ | CAMP, GJB1, HNF4A, HSP90B1, IL18, LECT2, SERPINF1, TNFSF10 |
| Renal and Urological System Development | cell viability of kidney cell lines | 3.05E-04 | ↓ | CAV1, HSP90B1, HYOU1, NFKBIA |
| Cardiovascular Disease | vascular lesion | 2.94E-07 | ↑ | ACAT2, CACNA1D, COL18A1, IL18, LPL, MGP, mir-221, NFKBIA, PLAU, TNFSF10 |
Apoptosis and APC viability warranted attention because they were tightly linked to immunity.
Apoptosis was effectively under the control of genes. When low-RFI birds suffer from Avian Leukosis Virus, Marek’s Disease or reticuloendotheliosis, related DEGs might be responsible for blocking the energy supply to tumor cells, thereby slowing apoptosis and accelerating tumor formation.
APCs are capable of ingesting and processing pathogenic microorganisms by phagocytosis or pinocytosis, which in turn produces peptide fragments that contain antigenicity domains [41, 42]. Decreased activation of APCs in low-RFI birds might cause a delay in the immune response or a reduced ability to kill pathogens. HSP90B1 is an important immune protein that is involved in cell protection under heat stress. Increased HSP90B expression in response to heat stress causes various reductions in cell damage and even repairs damaged proteins [43]. The reduced HSP90B1 expression in low-RFI birds signified a weaker ability to resist the effect of environmental factors compared to high-RFI birds.
Additionally, the changes in some DEGs were found to function in the regulation of fat metabolism (Fig. 6). For low-RFI birds, 13 DEGs were predicted to facilitate the concentration of lipids, 10 were predicted to play a pivotal role in the activation of lipid synthesis, and 5 were associated with the activation of fatty acid metabolism. Although a few DEGs had inhibitory effects, the above three specific physiological functions seemed to be enhanced overall. The results in the present study showed that low-RFI birds could achieve greater synthetic metabolism of lipids compared to high-RFI birds, whereas the results of another study showed that they displayed weaker catabolic abilities [44]. The faster anabolism of fat than catabolism in low-RFI birds might cause abnormal storage of fat and a decrease in heat release.
Fig. 6.
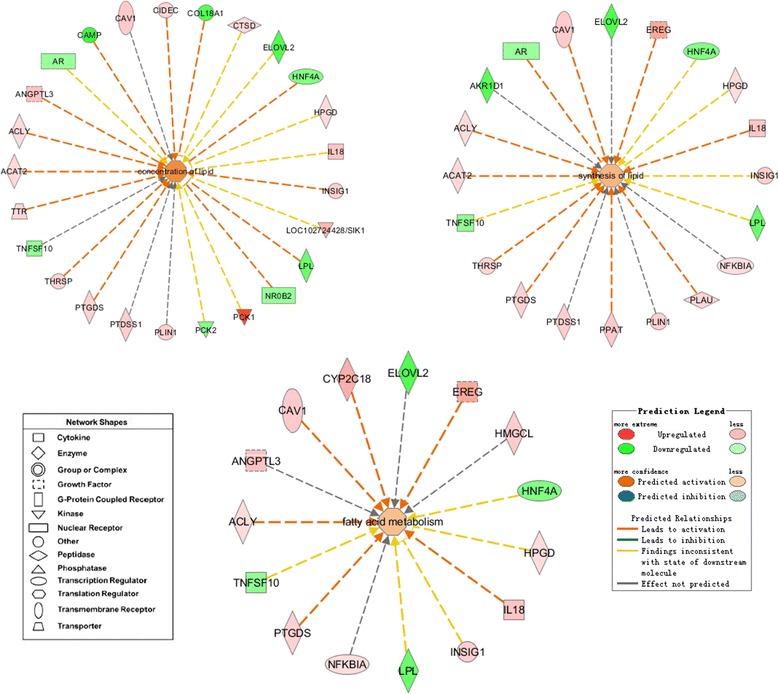
Predictable changes in fat metabolism based on DEG analysis in low-RFI individuals
Among the lipometabolism-related DEGs, PCK1 was particularly noteworthy because it was remarkably increased in the low-RFI group. The PCK1 gene is associated with obesity, insulin resistance, and type II diabetes in mammals [45–47]. Abdominal fat content was assumed to be positively correlated with PCK1 mRNA expression in birds [28]. Thus, high PCK1 expression in low-RFI birds might signal a concurrent increase in the abdominal fat mass. Interestingly, AR expression was significantly lower in the low-RFI birds than in the high-RFI birds. The interaction of androgen with its receptor (AR) was closely related to bone metabolism in birds [48]. The AR in the cytoplasm enters the nucleus by integrating with androgen and then binding to the androgen responsive element, leading to the activation of androgen [49]. Low AR expression interferes with the positive physiological effect of androgen, leading to high PCK1 expression.
A systematic analysis of the GWAS and RNA sequencing results was performed. The results showed there was no overlap of the genes within the 50 kb flanking region containing the significant SNPs detected by GWAS and any DEG detected by RNA sequencing. One explanation is that the actual linkage distance between the QTLs and the significant SNP is larger than 50 kb. However, we stopped measuring the RFI when the chickens were 83 d of age, whereas the liver sample collection was performed at 91 d of age. During this time, the RFI ranking may have slightly changed, resulting in a mismatch. Nonetheless, when the search scope was broadened [11] we still found that some potential genes within the 500-kb flanking region of the significant RFI-related SNPs in GWAS were identified as DEGs in chickens with divergent RFIs, including MGP, HIST1H110, HIST1H2A4L3 and OC3 near two SNPs (rs313001261 and rs14820118) in GGA1, NR0B2 near the SNP rs315285389, PER2 near the SNP rs16663153, ST6GALNAC2 near the SNP rs316897066, and G0S2 near the SNP rs315491506. Notably, the concurrent genes involved in GWAS and RNA sequencing played critical roles in a wide spectrum of biological processes. Therefore, their relationships with RFI need to be verified.
Conclusions
The heritability of RFI during the period from 44 to 83 d of age was moderate. The RFI was particularly affected by 32 significant SNPs that could explain 53.01 % of the additive genetic variance. Seventeen RFI-related SNPs were located in a 1 Mb region (16.3–17.3 Mb) of GGA12. This region was identified as a key candidate region affecting the energy utilization efficiency of chickens because it contained genes associated with lipogenesis, social behavior, and immunity. Although they obtained less metabolic energy, low-RFI birds maintained normal growth that was comparable to that of the high-RFI birds by allocating less energy to cell activity, the immune response, and physical activity in relation to eating and thus optimizing the use of the limited resources. Nevertheless, low-RFI selection cannot mobilize spare energy to withstand environmental stress and causes potential health risks that should not be ignored. At the same time, the low-RFI birds tended to become fatter.
Methods
Animals and measurement of feeding traits
The yellow-plumage dwarf chicken line N301 was used in the present study. Chicks with common characteristics of incompetent physical conditions (chicks that were malnourished or crippled) were eliminated after birth. The chickens were raised in a closed house to control the temperature and illumination during the early growth stage (0–4 wk of age). At the end of week 4, electronic chips were placed below the jaw and in the middle of the wattles. Then, the chickens were transferred to a half-open vertical ventilation hoop house. A total of 620 male chickens were maintained in three fence-separated pens on one side of the house, and 538 female ones were maintained in three fence-separated pens on the other side of the same house. Each pen had 17 feeding stations and 5 hanging water fountains in a 20 × 6 square meter area. The animals were fed a diet containing 2837 kcal/kg ME and 200 g CP/kg during the early growth stage. Then, the diet was switched to a high-energy diet containing 2900 kcal/kg ME and 190 g CP/kg. Daily feed intake and body weight were recorded for each bird throughout the feeding trial from 44 to 83 d of age.
Calculation of residual feed intake
The ADG and MMBW for each individual were estimated based on the traditional model that was previously presented [50] and applied [23]. Age was expected to be linearly related to body weight because the experimental birds were in a period of rapid growth. Thus, their relationship was in compliance with the following equation: BW = μ + a × DOT + e, where μ was the intercept, was the regression coefficient that represented ADG, DOT was the day of the test, and e was the residual. The mid-test body weight (MBW) was the predicted body weight on day 21 of the test, whereas MMBW was the MBW raised to the 0.75th power. ADFI was the mean of the daily feed intake throughout the 40 d of the experiment. The RFI was calculated from the subsequent model when all of the above parameters for each bird were estimated as follows: ADFI = b0 + b1 × MMBW + b2 × ADG + RFI, where b0 was the intercept, b1 and b2 represented the partial regression coefficients for MMBW and ADG, respectively, and RFI was the residual of the model.
To better understand the difference in the growth and feeding characteristics among individuals with divergent RFI values, data from the 10 % highest- and 10 % lowest-ranked RFI birds were pooled and t-tests were performed on the variables. Given the uncertainty of the gender or pen impact on the RFI, the RFI was corrected once the fixed effect was significant (P < 0.01) after it was calculated for the entire population.
The restricted maximum likelihood method implemented by the DMU package was used to obtain estimates of the phenotypic and genetic (co)variance and heritability [51]. The basic model was: y = Xb + Za + e, where y was the vector of the observations, a was the vector of the animal additive genetic effect, b was the vector of the fixed effects, including gender (two levels) and pen (six levels), e was the vector of random residuals, and X and Z were the incidence matrices. 1158 individuals were included.
SNP genotyping and quality check
Blood samples from 435 male chickens randomly selected from the entire population were collected for DNA extraction using the EZNA Blood DNA Kit (Omega Biotek, Doraville, GA). The DNA samples were qualified and standardized into a final concentration of 50 ng/μl. Genotyping was performed with the Affymetrix 600 K genotyping array by Biotechnology Corporation (Shanghai, China). The genotyping quality control was evaluated in the GenABEL package in the R software (Additional file 6: Figure S2). A total of 415,154 SNPs and 426 individuals were involved in the final GWAS.
Statistical analysis of the single marker GWAS
A general linear mixed model was used for the SNP-phenotype association as follows: Y = Xb + Sa + Zu + e, where Y was the vector of the RFI values after normal transformation and b was the estimator of the fixed effect. Because the pen effect did not exert a significant influence on the normal-transformed RFI in the male chickens, the fixed effects were ignored in this model. a was the SNP substitution effect and u was the random additive genetic effect following the multinomial distribution u ~ N (0, ); here, Φ was the relationship matrix and was the polygenetic variance. The genomic kinship matrix was used for the adjustment for population structure due to its better ability to estimate true covariance between individual genomes [52]. S and Z were the incidence matrices for a and u. e was a vector of residuals with a distribution of N (0, ); here, was the residual variance.
The level of significance
Because sub-structures could still exist in the population, the P-value was corrected using the genomic control inflation/deflation parameter (λ) as a simple and rational option to solve the family data issue [26]. The significant threshold P-value after λ correction was 10−4, based on a hint by The Wellcome Trust Case Control Consortium and some articles [53, 54]. A chromosome-wise false discovery rate (FDR) method was used for verifying significant SNPs. The p-values of each SNP were sorted in ascending order, and the following formula was applied to obtain FDR for each SNP: mP(i)/i, in which m is the total number of SNPs, and P is the p-value of the ith SNP [55].
Haplotype analysis
Linkage disequilibrium analysis was implemented in the region where multiple significant SNPs clustered using the Haploview software [56]. A pairwise strong LD was defined as the case when the one-sided upper and lower 95 % confidence limits of D’ exceeded 0.98 and 0.7, respectively. The block was constructed in cases when over 95 % of the informative SNPs in a region displayed a strong LD to identify potential regions of causal mutation for RFI [57].
Phenotypic and additive variants analysis
The fraction of phenotypic variance explained by the significant SNPs was computed by the following model: y = μ + SNP + e, where y was a vector of the RFI values, μ was the population mean, SNP was a vector of the genotypes in different individuals with regards to the significant SNP (fixed effect), and e was a vector of random errors. The proportion of the phenotypic variance explained by the SNP was reflected by the determination coefficient (R2) in the linear model. The proportion of additive variance explained by the SNP was computed in a similar manner; the only difference was that the phenotypic values were substituted for the breeding values.
RNA extraction and RNA sequencing
Total RNA was extracted from the liver samples using the TRI reagent (Ambion, Applied Biosystems) according to the manufacturer’s instructions. The ND-2000 (NanoDrop Technologies) was used to verify the RNA integrity, and the Agilent Bioanalyzer 2100 (Agilent Technologies) was used to measure the concentration of the RNA samples. An RNA integrity number larger than 8.0 was considered to be acceptable for cDNA library construction.
Four samples (two with the highest RFI values and two with the lowest RFI values; Additional file 7: Table S5) were selected for cDNA library construction for RNA sequencing using the TRIzol reagent following the manufacturer’s instructions (Invitrogen). Purified mRNA was fragmented into 200–500-bp fragments and then reverse transcribed into cDNA. The samples were sequenced on a Genome Analyzer IIx (Illumina). The 2 × 100 bp sequencing strategy was adopted.
After sequencing, the generated raw reads were processed to clean reads by filtering low quality reads and adaptor dimers. After presenting the statistical distributions of the GC content and base quality, the filtered reads were mapped to the chicken reference genome using TopHat [58]. The parameters for mapping were as follows: reads-mismatches = 2 and reads-gap-length = 1. The clean reads were assembled and conjoined into contigs using Trinity (http://trinityrnaseq.github.io). The resulting contigs were connected into unigenes and annotated by ANNOVAR [59]. The expression of each gene represented by RPKM was estimated by the DEGseq package in R [60]. Differentially expressed transcripts or genes were identified when the threshold Benjamin q-value was below 0.05 (cut-off at a 5 % false discovery rate) and |log2 (fold change) | ≥1, respectively.
Gene network construction
The DEGs were uploaded to the IPA database (http://www.ingenuity.com) for function and interaction analysis.
Abbreviations
ADFI, average daily feed intake; ADG, average daily gain; APCs, antigen presenting cells; DEGs, differentially expressed genes; FCR, feed conversion rate; FDR, false discovery rate; GGA, chicken (Gallus gallus) chromosome; GWAS, genome-wide association study; LD, linkage disequilibrium; MBW, mid-test body weight; ME, metabolic energy; miRNA, microRNA; MMBW, mid-test metabolic body weight; QTLs, quantitative trait loci; RFI, residual feed intake; RPKM, reads per kilobase of transcript per million mapped reads; SNPs, single nucleotide polymorphisms; UTR, untranslated region
Acknowledgements
We thank Megan M for language editing.
Funding
The authors would like to acknowledge the financial support from the High Technology Research and Development Program of China (2013AA102501), the National Key Technology Research and Development Program (2014BAD08B08), and the China Agriculture Research System (CARS-42-G05, CARS-42-Z17).
Availability of data and materials
The Affymetrix 600 K genotyping data and RNA sequencing data supporting our findings are available at the animal genome website (http://www.animalgenome.org/repository/pub/SCAU2016.0217/). Anyone who wants to take the raw phenotype data can contact the author directly via E-mail: zhenqiangxu@163.com.
Authors’ contributions
ZX, CJ, QN, DZ, XZ conceived and designed the study. ZX, YZ, JX performed the experiments. ZX, ZZ, CJ analyzed the data. CJ, DZ contributed the materials. ZX wrote the manuscript. XZ supervised the study. All authors read, edited and approved the final manuscript.
Authors’ information
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All animal care and handling procedures conformed to the Animal Care Committee of South China Agriculture University (Guangzhou, People’s Republic of China). Animals involved in this study were humanely sacrificed as necessary to ameliorate their suffering.
Additional files
Normality test for residual feed intake. (DOC 28 kb)
GO mapping for genes within the 50 bp flanking regions of the significant RFI-related SNPs. (XLS 31 kb)
{kind=link}
Biological replicates of the expressed genes in the two RFI-divergent groups. (PNG 162 kb)
Differentially expressed genes identified in RNA sequencing in divergent RFI individuals. (XLS 37 kb)
Networks constructed using the differentially expressed genes predicted by IPA. (XLS 1284 kb)
{kind=link}
The flow chart for genotyping quality control for the SNPs and individuals. (PNG 97 kb)
Growth characteristics of the samples used for RNA sequencing. (DOC 27 kb)
Contributor Information
Zhenqiang Xu, Email: zhenqiangxu@163.com.
Congliang Ji, Email: 34954584@qq.com.
Yan Zhang, Email: 782424707@qq.com.
Zhe Zhang, Email: 18848127@qq.com.
Qinghua Nie, Email: 308514138@qq.com.
Jiguo Xu, Email: xujiguo163@163.com.
Dexiang Zhang, Email: zhangdexiang0001@sina.com.
Xiquan Zhang, Email: xqzhang@scau.edu.cn.
References
- 1.Pakdel A, et al. Genetic parameters of ascites-related traits in broilers: correlations with feed efficiency and carcase traits. Br Poult Sci. 2005;46(1):43–53. doi: 10.1080/00071660400023805. [DOI] [PubMed] [Google Scholar]
- 2.Aggrey SE, et al. Genetic properties of feed efficiency parameters in meat-type chickens. Genet Sel Evol. 2010;42:25. doi: 10.1186/1297-9686-42-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leenstra FR, Pit R. Fat deposition in a broiler sire strain. 4. Performance of broiler progeny of four differently selected sire lines. Poult Sci. 1988;67(1):10–5. doi: 10.3382/ps.0670010. [DOI] [PubMed] [Google Scholar]
- 4.Gunsett FC. Merit of utilizing the heritability of a ratio to predict the genetic change of a ratio. J Anim Sci. 1987;65:936–42. [Google Scholar]
- 5.Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haines JL, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 7.Bolormaa S, et al. Detection of quantitative trait loci in Bos indicus and Bos taurus cattle using genome-wide association studies. Genet Sel Evol. 2013;45:43. doi: 10.1186/1297-9686-45-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santana MH, et al. Genome-wide association analysis of feed intake and residual feed intake in Nellore cattle. BMC Genet. 2014;15:21. doi: 10.1186/1471-2156-15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abo-Ismail MK, et al. Single nucleotide polymorphisms for feed efficiency and performance in crossbred beef cattle. BMC Genet. 2014;15:14. doi: 10.1186/1471-2156-15-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahana G, et al. A genome-wide association scan in pig identifies novel regions associated with feed efficiency trait. J Anim Sci. 2013;91(3):1041–50. doi: 10.2527/jas.2012-5643. [DOI] [PubMed] [Google Scholar]
- 11.Do DN, et al. Genome-wide association and systems genetic analyses of residual feed intake, daily feed consumption, backfat and weight gain in pigs. BMC Genet. 2014;15:27. doi: 10.1186/1471-2156-15-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onteru SK, et al. Whole genome association studies of residual feed intake and related traits in the Pig. PLoS One. 2013;8(6):e61756. doi: 10.1371/journal.pone.0061756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan J, et al. Genome-wide association studies for feed intake and efficiency in two laying periods of chickens. Genet Sel Evol. 2015;47:82. doi: 10.1186/s12711-015-0161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, et al. Detection of QTL controlling feed efficiency and excretion in chickens fed a wheat-based diet. Genet Sel Evol. 2015;47:74. doi: 10.1186/s12711-014-0082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kranis A, et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genomics. 2013;14:59. doi: 10.1186/1471-2164-14-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu S, et al. Weak base pairing in both seed and 3’ regions reduces RNAi off-targets and enhances si/shRNA designs. Nucleic Acids Res. 2014;42(19):12169–76. doi: 10.1093/nar/gku854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khalife M, et al. Transcriptomic analysis brings new insight into the biological role of the prion protein during mouse embryogenesis. PLoS One. 2011;6(8):e23253. doi: 10.1371/journal.pone.0023253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mudge JM, et al. The origins, evolution, and functional potential of alternative splicing in vertebrates. Mol Biol Evol. 2011;28(10):2949–59. doi: 10.1093/molbev/msr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilhelm BT, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453(7199):1239–43. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, et al. Global gene expression profiling reveals genes expressed differentially in cattle with high and low residual feed intake. Anim Genet. 2011;42(5):475–90. doi: 10.1111/j.1365-2052.2011.02182.x. [DOI] [PubMed] [Google Scholar]
- 22.Kong BW, et al. Gene expression in breast muscle associated with feed efficiency in a single male broiler line using a chicken 44K oligo microarray. I. Top differentially expressed genes. Poult Sci. 2011;90(11):2535–47. doi: 10.3382/ps.2011-01435. [DOI] [PubMed] [Google Scholar]
- 23.Williams YJ, et al. Variation in residual feed intake in Holstein-Friesian dairy heifers in southern Australia. J Dairy Sci. 2011;94(9):4715–25. doi: 10.3168/jds.2010-4015. [DOI] [PubMed] [Google Scholar]
- 24.Arthur PF, et al. Genetic and phenotypic variance and covariance components for feed intake, feed efficiency, and other postweaning traits in Angus cattle. J Anim Sci. 2001;79(11):2805–11. doi: 10.2527/2001.79112805x. [DOI] [PubMed] [Google Scholar]
- 25.Cai W, Kaiser MS, Dekkers JC. Genetic analysis of longitudinal measurements of performance traits in selection lines for residual feed intake in Yorkshire swine. J Anim Sci. 2011;89(5):1270–80. doi: 10.2527/jas.2010-3107. [DOI] [PubMed] [Google Scholar]
- 26.Amin N, van Duijn CM, Aulchenko YS. A genomic background based method for association analysis in related individuals. PLoS One. 2007;2(12):e1274. doi: 10.1371/journal.pone.0001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mortazavi A, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 28.Duan J, et al. Androgen inhibits abdominal fat accumulation and negatively regulates the PCK1 gene in male chickens. PLoS One. 2013;8(3):e59636. doi: 10.1371/journal.pone.0059636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luiting P, et al. Metabolic differences between white leghorns selected for high and low residual food consumption. Br Poult Sci. 1991;32(4):763–82. doi: 10.1080/00071669108417402. [DOI] [PubMed] [Google Scholar]
- 30.van Eerden E, et al. Residual feed intake and its effect on salmonella enteritidis infection in growing layer hens. Poult Sci. 2004;83(11):1904–10. doi: 10.1093/ps/83.11.1904. [DOI] [PubMed] [Google Scholar]
- 31.De Koning DJ, et al. Segregation of QTL for production traits in commercial meat-type chickens. Genet Res. 2004;83(3):211–20. doi: 10.1017/S0016672304006846. [DOI] [PubMed] [Google Scholar]
- 32.Lu S, Cullen BR. Analysis of the stimulatory effect of splicing on mRNA production and utilization in mammalian cells. RNA. 2003;9(5):618–30. doi: 10.1261/rna.5260303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parikh I, et al. An intronic PICALM polymorphism, rs588076, is associated with allelic expression of a PICALM isoform. Mol Neurodegener. 2014;9:32. doi: 10.1186/1750-1326-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honda T, Ishii A, Inui M. Regulation of adipocyte differentiation of 3T3-L1 cells by PDZRN3. Am J Physiol Cell Physiol. 2013;304(11):C1091–7. doi: 10.1152/ajpcell.00343.2012. [DOI] [PubMed] [Google Scholar]
- 35.Morellini F, et al. Reduced reactivity to novelty, impaired social behavior, and enhanced basal synaptic excitatory activity in perforant path projections to the dentate gyrus in young adult mice deficient in the neural cell adhesion molecule CHL1. Mol Cell Neurosci. 2007;34(2):121–36. doi: 10.1016/j.mcn.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 36.Steindler C, et al. Jamip1 (marlin-1) defines a family of proteins interacting with janus kinases and microtubules. J Biol Chem. 2004;279(41):43168–77. doi: 10.1074/jbc.M401915200. [DOI] [PubMed] [Google Scholar]
- 37.Dunn IC, et al. Decreased expression of the satiety signal receptor CCKAR is responsible for increased growth and body weight during the domestication of chickens. Am J Physiol Endocrinol Metab. 2013;304(9):E909–21. doi: 10.1152/ajpendo.00580.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moran TH, et al. Disordered food intake and obesity in rats lacking cholecystokinin a receptors. Am J Physiol. 1998;274(3 Pt 2):R618–25. doi: 10.1152/ajpregu.1998.274.3.R618. [DOI] [PubMed] [Google Scholar]
- 39.Gibbs J, Young RC, Smith GP. Cholecystokinin decreases food intake in rats. Obes Res. 1997;5(3):284–90. doi: 10.1002/j.1550-8528.1997.tb00305.x. [DOI] [PubMed] [Google Scholar]
- 40.Houston RD, et al. The cholecystokinin type a receptor g.179A > G polymorphism affects feeding rate. Anim Genet. 2008;39(2):187–8. doi: 10.1111/j.1365-2052.2007.01692.x. [DOI] [PubMed] [Google Scholar]
- 41.Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8(8):607–18. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuo J, Rowe M. Herpesviruses placating the unwilling host: manipulation of the MHC class II antigen presentation pathway. Viruses. 2012;4(8):1335–53. doi: 10.3390/v4081335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J Biol Chem. 2008;283(27):18473–7. doi: 10.1074/jbc.R800007200. [DOI] [PubMed] [Google Scholar]
- 44.Gabarrou JF, et al. Energy balance of laying hens selected on residual food consumption. Br Poult Sci. 1998;39(1):79–89. doi: 10.1080/00071669889439. [DOI] [PubMed] [Google Scholar]
- 45.Beale EG, Harvey BJ, Forest C. PCK1 and PCK2 as candidate diabetes and obesity genes. Cell Biochem Biophys. 2007;48(2–3):89–95. doi: 10.1007/s12013-007-0025-6. [DOI] [PubMed] [Google Scholar]
- 46.Millward CA, et al. Phosphoenolpyruvate carboxykinase (Pck1) helps regulate the triglyceride/fatty acid cycle and development of insulin resistance in mice. J Lipid Res. 2010;51(6):1452–63. doi: 10.1194/jlr.M005363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rees SD, et al. The promoter polymorphism -232C/G of the PCK1 gene is associated with type 2 diabetes in a UK-resident South Asian population. BMC Med Genet. 2009;10:83. doi: 10.1186/1471-2350-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Syed F, Khosla S. Mechanisms of sex steroid effects on bone. Biochem Biophys Res Commun. 2005;328(3):688–96. doi: 10.1016/j.bbrc.2004.11.097. [DOI] [PubMed] [Google Scholar]
- 49.Qiu YQ, Leuschner I, Braun PM. Androgen receptor expression in clinically localized prostate cancer: immunohistochemistry study and literature review. Asian J Androl. 2008;10(6):855–63. doi: 10.1111/j.1745-7262.2008.00428.x. [DOI] [PubMed] [Google Scholar]
- 50.Koch RM, et al. Efficiency of feed use in beef cattle. J Anim Sc. 1963;22:486–94. doi: 10.2527/jas1963.222486x. [DOI] [Google Scholar]
- 51.Madsen, P., et al. DMU – a package for analyzing multivariate mixed models. In Proc. 8th World Congr.Genet. Appl. Livest. Prod. Belo Horizonte, Minas Gerais, Brazil, 2006:11–27.
- 52.Eding H. Marker-based estimates of between and within population kinships for the conservation of genetic diversity. J Anim Breed Genet. 2001;118(3):141–59. doi: 10.1046/j.1439-0388.2001.00290.x. [DOI] [Google Scholar]
- 53.Barton AW, et al. Identification of novel RA susceptibility loci at chromosomes 10p15, 12q13 and 22q13. Nat Genet. 2008;40(10):1156–59. doi: 10.1038/ng.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayes BJ, et al. A validated genome wide association study to breed cattle adapted to an environment altered by climate change. PLoS One. 2009;4(8):1050. doi: 10.1371/journal.pone.0006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benjamini Y, Hochberg Y. Controlling the false discovery rate-A practical and powerful approach to multiple testing. J R Stat Soc. 1995;57(57):289–300. [Google Scholar]
- 56.Barrett JC, et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 57.Gabriel SB, et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 58.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):110–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang L, et al. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–8. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The Affymetrix 600 K genotyping data and RNA sequencing data supporting our findings are available at the animal genome website (http://www.animalgenome.org/repository/pub/SCAU2016.0217/). Anyone who wants to take the raw phenotype data can contact the author directly via E-mail: zhenqiangxu@163.com.