

Abstract
Background
Feed efficient cattle consume less feed and produce less environmental waste than inefficient cattle. Many factors are known to contribute to differences in feed efficiency, however the underlying molecular mechanisms are largely unknown. Our study aimed to understand how host gene expression in the rumen epithelium contributes to differences in residual feed intake (RFI), a measure of feed efficiency, using a transcriptome profiling based approach.
Results
The rumen epithelial transcriptome from highly efficient (low (L-) RFI, n = 9) and inefficient (high (H-) RFI, n = 9) Hereford x Angus steers was obtained using RNA-sequencing. There were 122 genes differentially expressed between the rumen epithelial tissues of L- and H- RFI steers (p < 0.05) with 85 up-regulated and 37 down-regulated in L-RFI steers. Functional analysis of up-regulated genes revealed their involvement in acetylation, remodeling of adherens junctions, cytoskeletal dynamics, cell migration, and cell turnover. Additionally, a weighted gene co-expression network analysis (WGCNA) identified a significant gene module containing 764 genes that was negatively correlated with RFI (r = −0.5, p = 0.03). Functional analysis revealed significant enrichment of genes involved in modulation of intercellular adhesion through adherens junctions, protein and cell turnover, and cytoskeletal organization that suggest possible increased tissue morphogenesis in the L-RFI steers. Additionally, the L-RFI epithelium had increased expression of genes involved with the mitochondrion, acetylation, and energy generating pathways such as glycolysis, tricarboxylic acid cycle, and oxidative phosphorylation. Further qPCR analysis of steers with different RFI (L-RFI, n = 35; M-RFI, n = 34; H-RFI, n = 35) revealed that the relative mitochondrial genome copy number per cell of the epithelium was positively correlated with RFI (r = 0.21, p = 0.03).
Conclusions
Our results suggest that the rumen epithelium of L-RFI (efficient) steers may have increased tissue morphogenesis that possibly increases paracellular permeability for the absorption of nutrients and increased energy production to support the energetic demands of increased tissue morphogenesis compared to those of H-RFI (inefficient) animals. Greater expression of mitochondrial genes and lower relative mitochondrial genome copy numbers suggest a greater rate of transcription in the rumen epithelial mitochondria of L-RFI steers. Understanding how host gene expression profiles are associated with RFI could potentially lead to identification of mechanisms behind this trait, which are vital to develop strategies for the improvement of cattle feed efficiency.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2935-4) contains supplementary material, which is available to authorized users.
Keywords: Feed efficiency, Residual feed intake, RNA-sequencing, Beef cattle, Rumen epithelium, Transcriptome
Background
Feed is one of the major costs in beef cattle production, accounting for approximately 70 % of total expenditures [1]. As feed price continues to rise due to factors such as human population growth and decreasing amount of arable land [2], beef cattle feed efficiency has become an increasingly important contributor to reduce the cost of production. Recent efforts in the selection of feed efficient cattle have been focused on residual feed intake (RFI), a measure of feed efficiency that is independent of growth and body weight [3]. The RFI of an individual is calculated as the difference between actual feed intake and expected feed intake based on maintenance and growth requirements [4]. Animals with low (negative) RFI (L-RFI) are considered feed efficient because they consume less feed than expected whereas animals with high (positive) RFI (H-RFI) are feed inefficient due to consuming more feed than expected [4]. Selection for L-RFI animals would not only reduce cost of production, but also decrease the environmental footprint because they produce 28 and 15 % less methane [5] and manure [6], respectively, than H-RFI animals.
Variation in RFI may be due to differences in many biological processes that are influenced by genetic and environmental factors. Physiological processes that have been found to contribute to variation in RFI include those associated with feed intake, digestion, metabolism, activity, and stress [7, 8]. Genetic selection and improvement of RFI is possible since RFI is a moderately heritable trait (0.28 to 0.45) [3, 9, 10], however, the molecular mechanisms underlying RFI are largely unknown. Because variation in RFI is due to differences in many processes, multiple genes from various pathways are likely involved [11–13]. Discovering differences at the molecular level may potentially lead to identification of biomarkers for use in the selection of L-RFI (feed efficient) animals.
In ruminant animals, such as cattle, the rumen may play an important role in feed efficiency. It is the main site of microbial feed digestion and fermentation that produces short-chain fatty acids (SCFAs), which are a major energy source for cattle accounting for up to 80 % of total energy requirements [14, 15]. SCFAs are mainly absorbed from the rumen [16] and metabolized within the rumen epithelium, liver and peripheral tissues to generate energy for use in maintenance, growth and activity [17, 18]. Studies have indicated that SCFAs are absorbed by both simple diffusion [16] and protein-mediated transport through bicarbonate - dependent and - independent mechanisms [19]. Moreover, ketogenic enzymes within the mitochondria of the rumen epithelium play a crucial role in the metabolism of SCFAs, to produce ketone bodies that are an important circulating source of energy throughout the body [20, 21].
Differences in the ability of the rumen epithelium to absorb and metabolize SCFAs may be associated with variation in feed efficiency. Therefore, the objective of this study was to investigate differences in gene expression between the rumen epithelial tissues of feed efficient and inefficient beef cattle using RNA-sequencing (RNA-seq) technology and bioinformatics. We hypothesized that the rumen epithelium of L-RFI (efficient) animals would have greater expression of genes involved in absorption and metabolism of energetic substrates such as SCFAs.
Methods
Animals and rumen tissue collection
Hereford x Aberdeen Angus hybrid steers (n = 175) were born in the spring of 2006 and raised at the University of Alberta Roy Berg Kinsella Research Station (Alberta, Canada) in accordance with the Canadian Council of Animal Care guidelines [22]. The University of Alberta Animal Care and Use Committee for Livestock approved the experimental protocols (Moore-55-2006).
Steers were under feedlot conditions while feed intake data was collected using the GrowSafe automated feeding system (GrowSafe Systems Ltd., Airdrie, Alberta, Canada). The finishing diet was fed for 90 days and was composed of 56.7 % barley, 28.3 % oats, 10 % alfalfa pellets, and 5 % feedlot supplement (32 % crude protein beef supplement containing Rumensin (400 mg/kg) and 1.5 % canola oil). After slaughter, a 4-cm2 piece of rumen tissue was obtained from the central region of the ventral sac and rinsed with sterilized PBS buffer (pH = 6.8) before being placed in a 50 mL tube containing RNAlater solution (Invitrogen, Carlsbad, CA). The samples were then stored at −80 °C until further processing.
RFI values of all steers were calculated as described by Nkrumah et al. (2006) [5] and classified as L-RFI (feed efficient; RFI < −0.5), medium RFI (M-RFI; −0.5 ≤ RFI ≤ 0.5), or H-RFI (feed inefficient; RFI > 0.5), which have been reported by Durunna et al. 2011 [23]. The rumen epithelial tissues were collected from all animals when they were slaughtered and the rumen epithelia from the most extreme L-RFI (n = 9; RFI = −1.4 to −2.33 kg/day) and H-RFI (n = 9; RFI = 1.32 to 3.23 kg/day) steers (Additional file 1) were selected for transcriptome analysis using RNA-seq.
RNA extraction and sequencing
After thawing, rumen tissue was placed on a petri dish and papillae (~80 mg) were obtained using sterile scissors and scalpels. The total RNA was extracted from the papillae using mirVana kit (Ambion, Austin, TX) according to the manufacturer’s instructions. RNA integrity of the total RNA samples was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and Nanodrop 2000c spectrophotometer (Thermo Scientific, Wilmington, DE) was used to measure the concentration and purity. Only samples with RNA integrity greater than 7 were subjected to RNA-seq library construction.
For each of the 18 samples, a cDNA library was generated from 100 ng of total RNA following the protocol of the TruSeq RNA Sample Preparation v2 kit (Illumina, San Diego, CA). First, total RNA was fragmented and cDNA was synthesized through reverse transcription. The resulting double-stranded cDNA was then subjected to end repair and 3’-end adenylation before ligation of index adapters. Libraries were amplified by 15 cycles of PCR, then validated with the Agilent 2200 TapeStation (Agilent Technologies) and quantified with a Qubit fluorometer using a Qubit dsDNA HS Assay Kit (Invitrogen, Carlsbad, CA). The individual indexed libraries were then pooled and sequenced in five lanes on the Illumina HiSeq 2000 system at the McGill University and Genome Quebec Innovation Centre (Quebec, Canada) to obtain high-quality, 100-bp paired-end reads (Average Phred quality score ≥ 33).
Transcriptome mapping
We used the software package TopHat2 (v2.0.9) [24] with Bowtie2 (v2.1.0) [25] to align the high-quality RNA-seq reads to the bovine reference genome, UMD3.1 (Ensembl v83.31). Samtools (v1.1) [26] was used to sort the BAM alignment files that were generated from TopHat2 by name and these sorted BAM files were converted to SAM files for input to HTSeq-count (v0.6.1) [27], which is a software tool that counts the number of reads per gene using the Ensembl (v83.31) bovine gene annotation.
Differential gene expression and principal component analysis
Differential gene expression analysis was performed using the Bioconductor (v3.0) [28] package DESeq2 (v1.6.2) [29] in the R statistical software program (v3.1.2) [30]. Firstly, DESeq2 normalized gene count data using the relative log expression method based on “size factors” to account for RNA-seq library size differences and dispersion estimates were calculated. Then, pairwise comparison of expression was made between the L-RFI and H-RFI group for every gene based on a negative binomial model. Fold changes were obtained along with their associated p-values. The Benjamini Hochberg method (B-H) was used to control the false discovery rate (FDR) by adjusting p-values to correct for multiple testing. A gene was defined as differentially expressed (DE) between the L-RFI and H-RFI group if it had a B-H adjusted p-value (FDR) less than 0.05.
DESeq2 was also used for Principal Component Analysis (PCA) to cluster samples based on gene expression data. Firstly, count data was normalized based on library size and regularized log transformed. The top 500 genes with the most variance (i.e. Largest standard deviations) were compared between samples to calculate principal components in order to view clustering in two dimensions.
Weighted Gene Co-expression Network Analysis (WGCNA)
The WGCNA R software package (v1.41.1) [31] was used to identify modules containing genes that are co-expressed and correlated with the RFI trait. Gene expression data was firstly filtered by keeping genes with at least one read in 50 % of the samples, then normalized by count per million reads (CPM) and Log2 transformed (Log2(CPM + 1)). Removal of the very lowly expressed genes resulted in 14,916 genes for input to WGCNA. Unsigned, weighted correlation network construction and module detection was performed using the automatic one-step function, blockwiseModules. The resulting gene modules were assigned color names by the software and Module-Trait relationships were calculated by Pearson correlation. Modules with statistically significant (p < 0.05) correlations were selected for downstream analysis as potential biologically interesting modules associated with the RFI trait. Additionally, a hierarchical clustering dendrogram of samples and trait heatmap was created using unfiltered, Log2CPM data. Sample clustering was based on Euclidean distance and the traits used in the heatmap were residual feed intake (RFI), dry matter intake (DMI), metabolizable energy intake (MEI), birth weight (BirthWT), weaning weight (WeanWT), metabolic mid-weight (MWT), end weight (EndWT), carcass weight (CWT), and average daily gain (ADG).
Functional gene annotation
Ensembl gene ID lists were converted to gene names and symbols using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) [32, 33] and the R/Bioconductor package, biomaRt (v2.22.0) [34]. DAVID and Ingenuity Pathway Analysis (IPA; QIAGEN Redwood City, www.qiagen.com/ingenuity) were used for functional annotation of DE genes and WGCNA gene modules. In DAVID, we examined genes for enrichment in functional categories such as SP_PIR_Keywords and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways [35, 36]. To correct for multiple testing in DAVID, p-values were adjusted using B-H correction. Biological terms were considered significant if the adjusted p-value was less than 0.05. The genes input to IPA were compared to the manually curated Ingenuity knowledge base to find significantly enriched canonical pathways, and cellular and molecular processes. Statistical significance (p < 0.05) was calculated using the right-tailed Fischer’s Exact Test and B-H corrected for multiple testing.
The identified core transcripts (i.e. genes with at least one read in every sample) were input to the Protein Annotation Through Evolutionary Relationship (PANTHER) [37] gene list analysis tool to determine the core functions of the rumen epithelium. Functional classification of genes in categories such as molecular function and biological process was based on Gene Ontology (GO) annotations.
Quantitative real-time PCR (qPCR) validation of selected differentially expressed genes
To validate the identified DE genes from RNA-seq, total RNA was extracted from rumen papillae of 30 L-RFI samples (RFI = −0.51 to −2.33 kg/day) and 27 H-RFI samples (RFI = 0.66 to 3.23 kg/day), respectively. Primers targeting five selected DE genes including Trans-2,3-Enoyl-CoA Reductase (TECR), Cytochrome C Oxidase Subunit VIIIA (COX8A), Solute Carrier Family 25 Member 39 (SLC25A39), Pyruvate Kinase M2 (PKM2) and Triosephosphate Isomerase 1 (TPI1) were designed using Primer-BLAST (NCBI, Bethesda, MD), Primer Express software (Applied Biosystems, Foster City, CA) or obtained through literatures. The specificity of primers was checked with BLAST (NCBI) and the UCSC In-Silico PCR program [38]. Table 1 lists the housekeeping gene (endogenous control) and the five DE genes selected for qPCR validation along with their GenBank accession number, primer sequences, product length and functional pathway.
Table 1.
Gene name, accession number, functional pathway, primer sequences, and product length of qPCR validated genes
| Gene name (symbol) | GenBank accession number | Functional pathway | Forward and reverse primer sequence (5'-3') | Product length (bp) | Reference |
|---|---|---|---|---|---|
| Triosephosphate Isomerase 1 (TPI1) | NM_001013589 | Glycolysis, Gluconeogenesis, Acetylation | Fwd: GGGAGGAAGAACAATCTGGGG | 107 | This study |
| Rev: GCGAAGTCAATGTAGGCGGT | |||||
| Trans-2,3-Enoyl-CoA Reductase (TECR) | NM_001034748 | Biosynthesis of unsaturated fatty acids, Acetylation | Fwd: CCAAGGGCAAGTCCCTGAAG | 82 | This study |
| Rev: AGGTCCCGGAAGTAGAGTGT | |||||
| Cytochrome C Oxidase Subunit VIIIA (COX8A) | NM_174024 | Oxidative Phosphorylation | Fwd: TTTGACTTCGCGACCTTGG | 60 | This study |
| Rev: TTACGGCACGGAGTAGACTG | |||||
| Solute Carrier Family 25 Member 39 (SLC25A39) | NM_001075415 | Mitochondrial substrate/solute carrier | Fwd: AGCTAATGCCTCCCTCCAGA | 54 | This study |
| Rev: GGCACTTCCATTTGGCGTAG | |||||
| Pyruvate Kinase M2 (PKM2) | NM_001205727 | Glycolysis, Gluconeogenesis | Fwd: TGTCACCCATTACCAGCGAC | 130 | This study |
| Rev: TATCTGGCCACCTGATGTGC | |||||
| SUZ12 polycomb repressive complex 2 subunit (SUZ12) | NM_001205587 | Endogenous control | Fwd: CATCCAAAAGGTGCTAGGATAGATG | 160 | [62] |
| Rev: TGGGCCTGCACACAAGAATG |
All samples were analyzed in triplicate during qPCR using the StepOnePlus Real-Time PCR System (Applied Biosystems). Each reaction contained 10 μL Fast SYBR® Green Master Mix (Applied Biosystems), 1 μL forward primer (20 pmol/μL), 1 μL reverse primer (20 pmol/μL), 7 μL nuclease-free water, and 1 μL cDNA template (2.5 ng/μL). The amplification program consisted of preincubation at 95 °C for 20 s followed by 40 cycles of denaturation at 95 °C for 3 s and annealing/extension at 64 °C for 30 s. Melting curve analysis was performed to confirm single product amplification. The expression of three housekeeping genes (RPS18 (Ribosomal Protein S18), GAPDH (Glyceraldehyde 3-phosphate dehydrogenase), SUZ12 (SUZ12 polycomb repressive complex 2 subunit)) was tested in this study using NormFinder [39] and PROC MIXED procedure of SAS (v9.2; SAS Institute Inc., Cary, NC). We chose SUZ12 because it was the most stably expressed between and within the two groups. For each sample, the expression of each targeted gene was presented as ΔCt, which was obtained by subtracting the average Ct (Cycle threshold) of the SUZ12 housekeeping gene from the average Ct of the target gene. A low ∆Ct indicates higher expression and vice versa. An unpaired t-test in SAS (SAS Institute Inc.) was used to analyze whether a gene showed significant difference between RFI groups.
DNA Extraction
Total DNA was extracted and used to quantify the relative mitochondrial genome copy numbers. The DNA was extracted from the rumen papillae (~80 mg) of 104 animals (35 L-RFI, 34 M-RFI, 35 H-RFI) using a bead beating, phenol-chloroform method as described by Li et al., 2012 [40]. Briefly, each sample was placed in a 2-mL microcentrifuge tube containing zirconium beads (0.3 g; 0.1 mm diameter) and washed twice with 1 mL of TN150 buffer (10 mM Tris–HCl [pH 8.0], 150 mM NaCl) by vortexing and centrifugation at 14,600 g for 5 min at 4 °C. The tissue was then resuspended in 1 mL of TN150 and the Mini-BeadBeater-8 (BioSpec products, Bartlesville, OK) was used to mechanically disrupt cells at 5000 rpm for 3 min. After bead beating, samples were immediately placed on ice and washed two times by phenol and chloroform-isoamyl alcohol (24:1). The DNA was then precipitated at −20 °C overnight using cold 100 % ethanol and 3 M sodium acetate. After precipitation, DNA was washed two times with 70 % ethanol and dissolved in 30 μL nuclease-free water. The DNA concentration and quality was measured using Nanodrop (Thermo Scientific).
qPCR determination of relative mitochondrial DNA copy number per cell
The relationship between RFI and the relative copy number of mitochondrial DNA (mtDNA) per cell of the rumen epithelium was performed using correlation analysis. The ratio of mtDNA to nuclear DNA (nDNA) was used to determine the relative copy number of mtDNA per cell. We selected the ND1 (NADH dehydrogenase subunit 1) gene as the mtDNA target (GenBank accession number: NC_006853, Location: 3101–4056) and the single copy, DDX3Y (DEAD box polypeptide 3, Y-linked) gene as the nDNA target (GenBank accession number: AC_000187, Location: 3458510–3468728,). The ND1 primer sequences used to amplify a 200 bp product were 5-GAACCACTACGACCCGCTACA-3 (forward), 5-GAGTTGGAAGCTCAGCCTGATC-3 (reverse) and the DDX3Y primer sequences used to amplify a 136 bp product were 5-ATCGTGGGCGGAATGAGTGT-3 (forward), 5-CTTGGTGGAAGCGGTTTTGA-3 (reverse) [41].
For qPCR, a standard curve was generated for both the mtDNA and nDNA. First, PCR was performed on pooled DNA to amplify mtDNA and nDNA separately. The PCR mix contained 50 ng/μL DNA along with 10X PCR buffer, 50 mM MgCl2, 10 mM dNTP, 20 pmol of each primer, Taq polymerase, and nuclease-free water to a final volume of 50 μL. The PCR program consisted of 1 cycle 94 °C for 5 min, 30 cycles of 94 °C for 30 s, 62 °C for 30 s, 72 °C for 30 s and 1 cycle of 72 °C for 7 min with hold at 4 °C. Each PCR product was then subjected to agarose gel electrophoresis in order to excise DNA bands for purification using the QIAquick Gel Extraction Kit (Qiagen). The concentration of the purified mtDNA and nDNA was measured by Nanodrop (Thermo Scientific) and an eight-point standard curve with 10 fold dilutions was created for each. DNA copy number of the standard curve points was calculated as described by Speicher and Johnson (2012) [42]. Expression of mtDNA and nDNA within 104 samples (35 L-RFI, 34 M-RFI, 35 H-RFI) was then analyzed by qPCR along with their respective standard curve using an annealing temperature of 62 °C. DNA copy number within samples was extrapolated from the standard curve and all samples and standard curves were analyzed in triplicate. A one-way ANOVA was performed in SAS (SAS Institute Inc.) to compare RFI groups. Also, a Pearson correlation analysis of relative mitochondrial DNA copy number per cell and RFI was performed in SAS (SAS Institute Inc.) and R [30].
Results
Identification of the rumen epithelial transcriptome
On average there were 42 million high-quality, paired reads generated per sample (mean ± SD = 41,991,418 ± 6,835,817) from RNA-seq and the overall read alignment rate to the bovine reference genome was 85 ± 3.72 %. The total number of genes expressed (genes with at least one read in at least one sample) within the rumen epithelium was 22,338 with an average of 14,677 ± 1062 genes per L-RFI sample and 16,239 ± 1326 per H-RFI sample. There were 11,284 core genes (genes with at least one read in every sample) among samples and of these core genes, 9700 can be annotated by PANTHER. The top molecular functions for the core genes were catalytic activity and binding, which included 30.7 and 30.1 % of the core genes, respectively. In the biological processes category, 32.6 % of the core genes were involved in cellular process and 47.5 % of the genes were associated with metabolic process (Fig. 1).
Fig. 1.
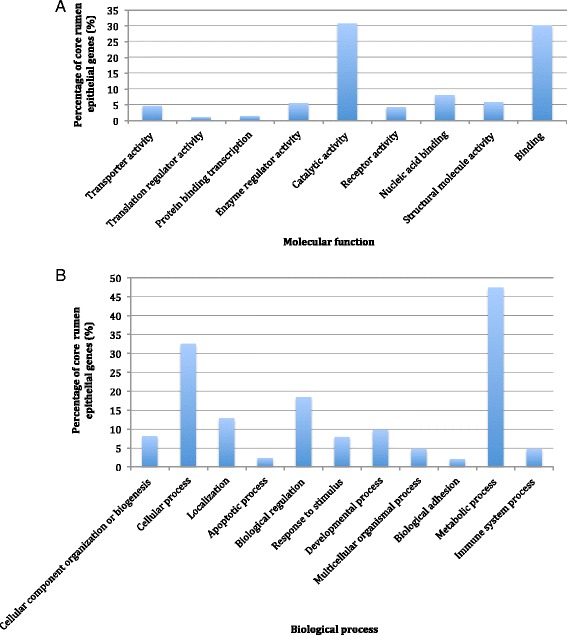
Biological functions of the core rumen epithelial genes as identified by PANTHER. The percentage of 9700 core rumen epithelial genes involved in various (a) Molecular functions and (b) Biological processes
Comparison of transcriptome profiles between two RFI groups
The PCA plot (Fig. 2) shows that L-RFI and H-RFI gene expression profiles seem to fall into distinct clusters, with those from L-RFI animals grouped together. However, there was a L-RFI outlier sample (271 L) that did not cluster with the rest of the samples (Fig. 2), which may possibly be related to the animal’s low ADG and EndWT (Fig. 3). Hierarchical clustering by WGCNA showed that all the L-RFI samples clustered together while the H-RFI samples did not (Fig. 3). When comparing animals with similar ADG and EndWT in the trait heat map, L-RFI individuals had lower RFI, DMI and MEI than H-RFI individuals. The means of the two groups for all traits (Additional file 2) were compared and there was no significant difference in BirthWT, WeanWT, MWT, EndWT, CWT, and ADG. However, the L-RFI group had significantly (p < 0.001) lower RFI, DMI and MEI by 3.82 kg/day, 3.86 kg/day and 4.68 Mcal/day, respectively.
Fig. 2.
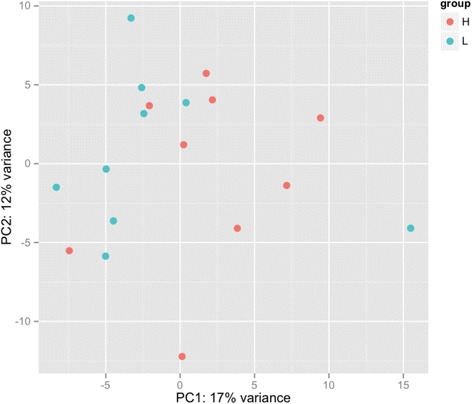
PCA plot of the rumen epithelial transcriptomes from cattle with L- and H- RFI. The L-RFI (Blue, n = 9) and H-RFI (Red, n = 9) samples are plotted along the first two principal components axis (PC1 and PC2)
Fig. 3.
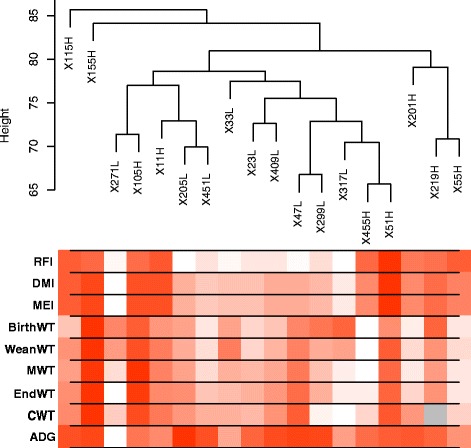
Hierarchical clustering dendrogram of rumen epithelial transcriptomes (9 L-RFI, 9 H-RFI) and trait heat map. The gradient from white to dark red in the trait heat map represents low to high levels of the trait while grey represents unavailable data. Traits examined were residual feed intake (RFI), dry matter intake (DMI), metabolizable energy intake (MEI), birth weight (BirthWT), weaning weight (WeanWT), metabolic mid-weight (MWT), end weight (EndWT), carcass weight (CWT), and average daily gain (ADG)
Differential gene expression in rumen epithelium between low- and high- RFI cattle
There were 122 genes DE in the rumen epithelial tissues between L- and H- RFI animals (Additional file 3). Of these DE genes, 85 were up-regulated and 37 down-regulated in the L-RFI group. DAVID annotation of the 122 DE genes found no significant KEGG pathways, however the biological term acetylation was significantly enriched (FDR < 0.05). A total of 30 DE genes were involved in the process of acetylation, 26 of which were up-regulated and 4 down-regulated in L-RFI animals (Table 2). These genes have functions in protein synthesis (i.e. RPL10, RPS15, RPL36), protein degradation (i.e. PSMB6, UBC/UBA52, UBE2V1), cytoskeletal organization (i.e. DSTN, ACTB, TUBB5, TMSB10), energetic substrate generation (i.e. TECR, TPI1), and stress response (i.e. HSPB1, HSF1).
Table 2.
Rumen epithelial DE genes between L- and H- RFI steers significantly (FDR < 0.05) involved in acetylation
| Ensembl ID | Gene symbol | Gene name | Fold changea | FDR adjusted p-value |
|---|---|---|---|---|
| ENSBTAG00000005654 | TMSB10 | Thymosin beta 10 | 1.44 | 1.28E-02 |
| ENSBTAG00000012632 | TECR | Trans-2,3-enoyl-CoA reductase | 1.43 | 1.02E-05 |
| ENSBTAG00000030974 | TUBA4A | Tubulin, alpha 4a | 1.43 | 5.63E-04 |
| ENSBTAG00000007454 | RPL10 | Ribosomal protein L10 | 1.40 | 2.03E-04 |
| ENSBTAG00000011969 | HSPB1 | Heat shock 27 kDa protein 1 | 1.34 | 1.28E-02 |
| ENSBTAG00000013390 | PSMB6 | Proteasome (prosome, macropain) subunit, beta type, 6 | 1.32 | 1.41E-02 |
| ENSBTAG00000016874 | DNAJB1 | DnaJ (Hsp40) homolog, subfamily B, member 1 | 1.29 | 1.28E-02 |
| ENSBTAG00000019718 | RPS15 | Ribosomal protein S15 | 1.29 | 1.85E-03 |
| ENSBTAG00000006969 | TUBB5 | Tubulin, beta | 1.28 | 4.02E-02 |
| ENSBTAG00000001794 | RPL36 | Ribosomal protein L36 | 1.28 | 2.49E-02 |
| ENSBTAG00000020560 | CLPTM1 | Cleft lip and palate associated transmembrane protein 1 | 1.27 | 1.41E-02 |
| ENSBTAG00000004379 | ETHE1 | Ethylmalonic encephalopathy 1 | 1.26 | 3.75E-02 |
| ENSBTAG00000000411 | HGS | Hepatocyte growth factor-regulated tyrosine kinase substrate | 1.26 | 1.28E-02 |
| ENSBTAG00000020751 | HSF1 | Heat shock transcription factor 1 | 1.26 | 2.84E-02 |
| ENSBTAG00000021455 | CFL1 | Cofilin 1 (non-muscle) | 1.26 | 1.41E-02 |
| ENSBTAG00000014872 | CAPNS1 | Calpain, small subunit 1 | 1.26 | 1.36E-02 |
| ENSBTAG00000016952 | PSMD5 | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 5 | 1.26 | 4.07E-02 |
| ENSBTAG00000026199 | ACTB | Actin, beta | 1.24 | 3.13E-02 |
| ENSBTAG00000017246; ENSBTAG00000007737 |
UBC; UBA52 | Ubiquitin C; Ubiquitin A-52 residue ribosomal protein fusion product 1 | 1.24 | 1.91E-02 |
| ENSBTAG00000016024 | MYL9 | Myosin regulatory light polypeptide 9 | 1.24 | 6.77E-03 |
| ENSBTAG00000019782 | TPI1 | Triosephosphate isomerase 1 | 1.24 | 3.56E-02 |
| ENSBTAG00000006495 | GNB2 | Guanine nucleotide binding protein (G protein), beta polypeptide 2 | 1.24 | 4.89E-02 |
| ENSBTAG00000027316 | UBE2V1 | Ubiquitin-conjugating enzyme E2 variant 1 | 1.23 | 4.02E-02 |
| ENSBTAG00000015434 | DSTN | Destrin (actin depolymerizing factor) | 1.22 | 4.86E-02 |
| ENSBTAG00000000215 | GNB1 | Guanine nucleotide binding protein (G protein), beta polypeptide 1 | 1.18 | 4.06E-02 |
| ENSBTAG00000013362 | DNM2 | Dynamin 2 | 1.17 | 4.86E-02 |
| ENSBTAG00000009780 | GTF2I | General transcription factor II, i | 0.85 | 4.86E-02 |
| ENSBTAG00000009541 | SUCLG2 | Succinate-CoA ligase, GDP-forming, beta subunit | 0.84 | 4.86E-02 |
| ENSBTAG00000038488 | TMSB4 | Thymosin beta 4, X-linked | 0.77 | 1.60E-03 |
| ENSBTAG00000013982 | UACA | Uveal autoantigen with coiled-coil domains and ankyrin repeats | 0.72 | 2.11E-02 |
aFold change is gene expression in L-RFI relative to H-RFI
Ingenuity analysis of the 85 differentially up-regulated genes in L-RFI samples showed that the top significantly enriched canonical pathway was remodeling of the epithelial adherens junctions through endocytosis (i.e. DNM2), degradation (i.e. HGS), and cytoskeletal organization (i.e. ACTB, TUBB, TUBA4A) (Table 3 and Additional file 3). The other enriched canonical pathways were involved in functions such as protein synthesis (eIF2 signaling), cytoskeletal dynamics, cell growth, proliferation, apoptosis, and migration (signaling by Rho family GTPases, RhoGDI signaling, ephrin B signaling) (Table 3). For the 37 DE genes that were down-regulated in L-RFI animals, there were no significant canonical pathways.
Table 3.
Top Ingenuity canonical pathways significantly enriched (p < 0.05) for up- and down- regulated DE genes in L-RFI rumen epithelium
| Ingenuity canonical pathways | FDR adjusted p-value | Genes |
|---|---|---|
| Up-regulated | ||
| Remodeling of Epithelial Adherens Junctions | 1.51E-03 | ACTB, TUBB5, DNM2, TUBA4A, HGS |
| Signaling by Rho Family GTPases | 2.42E-03 | RHOG, CFL1, ACTB, MYL12B, GNB1, MAPK1, GNB2 |
| RhoGDI Signaling | 2.58E-03 | RHOG, CFL1, ACTB, MYL12B, GNB1, GNB2 |
| eIF2 Signaling | 3.11E-03 | UBA52, RPL36, MAPK1, RPL18A, RPS15, RPL10 |
| Ephrin B Signaling | 5.40E-03 | CFL1, GNB1, MAPK1, GNB2 |
qPCR validation of selected DE genes
To show the reliability of the differential gene expression analysis of DESeq2, we further selected five DE genes for validation using qPCR. As shown in Table 4, of these five genes, four (TECR, COX8A, SLC25A39, PKM2) had significantly greater expression (i.e. Lower ∆Ct, p < 0.05) in L-RFI while one (TPI1) tended to have significantly greater expression (p < 0.1). These results were in agreement with the differential expression analysis of DESeq2 in that the genes had greater expression in L-RFI rumen epithelium.
Table 4.
Gene expression comparison between the rumen epithelium of L- and H- RFI animals by qPCR
| Gene | L-RFI ∆Ct (n = 30) | H-RFI ∆Ct (n = 27) | P-value |
|---|---|---|---|
| TPI1 | −3.23 ± 0.57 | −2.99 ± 0.38 | 0.07 |
| TECR | −2.36 ± 0.57 | −2.00 ± 0.51 | 0.01* |
| COX8A | −7.85 ± 1.30 | −6.57 ± 1.89 | 0.004* |
| SLC25A39 | −3.67 ± 0.51 | −0.24 ± 0.60 | 3.8E-30* |
| PKM2 | −6.64 ± 0.58 | −3.78 ± 0.53 | 4.4E-26* |
Data are Mean ± SD
*p-value <0.05
Identification of WGCNA gene co-expression modules correlated with RFI
WGCNA was conducted to identify gene co-expression modules that are correlated with RFI. We identified a total of eight co-expressed gene modules with three of them significantly correlated with RFI (Fig. 4). The Brown (162 genes) and Green (133 genes) modules had a significant positive correlation with RFI (Brown module: r = 0.54, p = 0.02; Green module: r = 0.51, p = 0.03) while the Turquoise module (764 genes) had a significant negative correlation with RFI (r = −0.50, p = 0.03). Functional analysis of the genes enriched in the Green module did not result in any significantly enriched biological functions or pathways while there was only one significant biological term, which was muscle protein containing four genes in the Brown module. Many significant biological functions were identified for the turquoise module with IPA and DAVID analysis (Tables 5 and 6) and therefore this study focused on functional examination of the 764 genes enriched in this module, which had greater expression in the L-RFI epithelium.
Fig. 4.
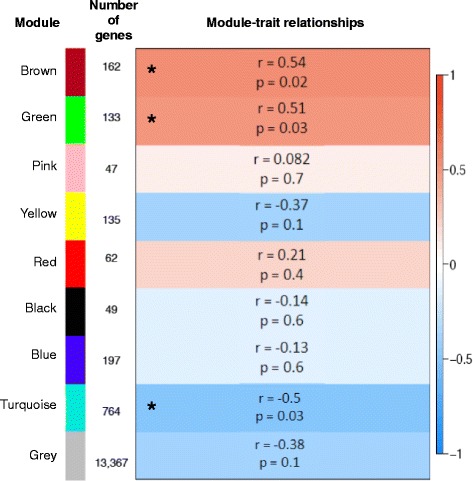
WGCNA identification of rumen epithelial gene modules correlated with the RFI trait. Module names and sizes are shown along with the module-trait relationships indicating the correlation coefficients and p-values. The strength of the correlation is colored by different intensities of red (positive correlation) and blue (negative correlation). Asterisks indicate modules that are significantly (p < 0.05) correlated with RFI
Table 5.
Biological functions and pathways significantly (FDR < 0.05) enriched in the Turquoise module (764 genes) using DAVID
| DAVID functional category | Biological term | Number of genes | FDR adjusted p-value |
|---|---|---|---|
| SP_PIR_Keyword | Acetylation | 195 | 8.22E-44 |
| Phosphoprotein | 210 | 9.16E-19 | |
| Ribonucleoprotein | 51 | 3.63E-17 | |
| Cytoplasm | 138 | 1.17E-16 | |
| Ribosomal protein | 44 | 2.53E-16 | |
| Protein biosynthesis | 20 | 2.24E-06 | |
| Isopeptide bond | 23 | 5.54E-06 | |
| Proteasome | 15 | 8.76E-06 | |
| Ubl conjugation | 29 | 5.80E-05 | |
| Nucleotide-binding | 74 | 3.65E-04 | |
| Wd repeat | 22 | 8.04E-04 | |
| Actin-binding | 17 | 9.48E-04 | |
| Transit peptide | 35 | 4.94E-03 | |
| Chaperone | 17 | 6.61E-03 | |
| Threonine protease | 7 | 8.72E-03 | |
| Methylation | 17 | 1.67E-02 | |
| Initiation factor | 9 | 1.80E-02 | |
| ATP-binding | 50 | 2.43E-02 | |
| Mitochondrion | 47 | 3.18E-02 | |
| Protein transport | 24 | 3.47E-02 | |
| KEGG_Pathway | bta03010:Ribosome | 36 | 3.05E-21 |
| bta03050:Proteasome | 14 | 1.86E-05 | |
| bta04520:Adherens junction | 14 | 1.97E-03 | |
| bta00190:Oxidative phosphorylation | 19 | 5.94E-03 | |
| bta04810:Regulation of actin cytoskeleton | 22 | 2.65E-02 | |
| bta03040:Spliceosome | 16 | 3.95E-02 |
Table 6.
Top biological functions and pathways significantly (p < 0.05) enriched in the Turquoise module with IPA
| IPA category | Biological term | Number of genes | FDR adjusted p-value |
|---|---|---|---|
| Molecular and cellular function | Protein synthesis | 89 | 6.06E-11 |
| Cellular growth and proliferation | 232 | 6.06E-11 | |
| Post-translational modification | 51 | 2.80E-07 | |
| Protein folding | 17 | 7.92E-07 | |
| Cell death and survival | 211 | 1.03E-06 | |
| Canonical pathway | EIF2 signaling | 51 | 5.85E-30 |
| Regulation of eIF4 and p70S6K signaling | 35 | 3.28E-18 | |
| mTOR signaling | 39 | 3.60E-18 | |
| Remodeling of epithelial adherens junctions | 19 | 3.21E-11 | |
| Epithelial adherens junction signaling | 25 | 9.05E-10 |
Functional analysis of gene module negatively associated with RFI
When functional analysis was performed using DAVID, 20 biological functions and 6 KEGG pathways were significantly enriched for genes in the turquoise module (Table 5). The most significant biological functions were acetylation (195 genes) and phosphoprotein (210 genes), which are involved in post-translational modification. Many of the other significant biological functions identified were also involved in protein related processes such as protein synthesis (i.e. ribosomal protein (44 genes), protein biosynthesis (20 genes)), degradation (i.e. proteasome (15 genes), threonine protease (7 genes)), folding (i.e. chaperone (17 genes)), and transport (i.e. transit peptide (35 genes), protein transport (24 genes)). In addition, biological terms associated with energy catabolism (i.e. ATP-binding (50 genes)) and anabolism (i.e. mitochondrion (47 genes)) were also identified. KEGG analysis of the 195 genes within the acetylation category indicated significant involvement of the acetylation related genes in ribosome and proteasome pathways. These pathways were also significantly enriched in the turquoise module along with oxidative phosphorylation, adherens junction, regulation of actin cytoskeleton, and spliceosome (Table 5). In the adherens junction pathway, there were 14 genes from the turquoise module that may regulate both the strong and weak adhesion between cells (Fig. 5).
Fig. 5.
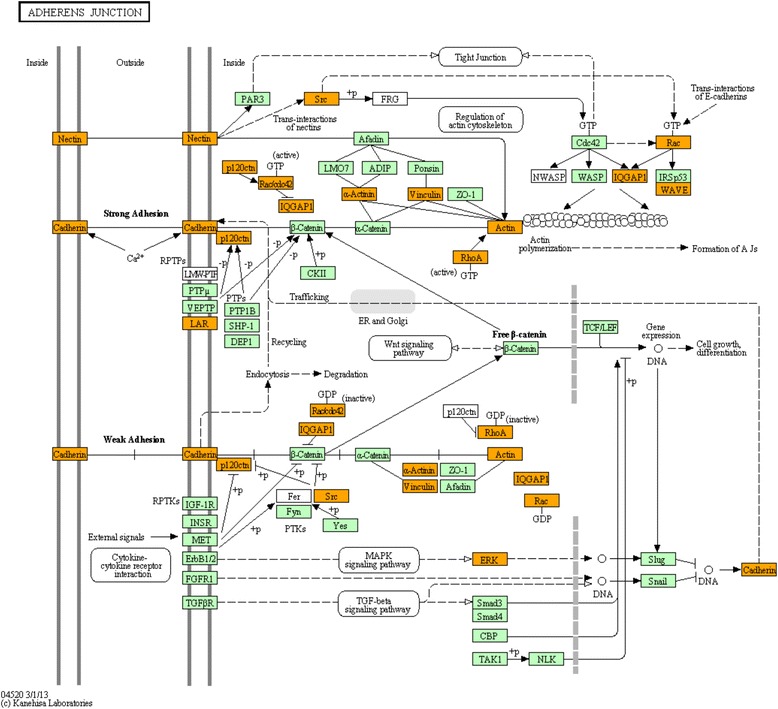
KEGG pathway for adherens junction. Genes within the turquoise module that have increased expression in the L-RFI rumen epithelium are shown in orange
Further functional analysis using IPA showed that the top five most significant molecular and cellular functions for the genes in the turquoise module were protein synthesis, cellular growth and proliferation, post-translational modification, protein folding and cell death and survival (Table 6). Furthermore, the top canonical pathways were eIF2 signaling, regulation of eIF4 and p70S6K signaling, mTOR signaling and epithelial adherens junctions remodeling and signaling (Table 6).
Additionally, many genes from the turquoise module are involved in energy generating pathways such as glycolysis, tricarboxylic acid (TCA) cycle, and oxidative phosphorylation (Table 7). We also found three DE genes involved in glycolysis and three associated with oxidative phosphorylation, which were up-regulated in the L-RFI epithelium (Table 7). The DE genes involved in glycolysis were TPI1 (Fold Change (FC; L-RFI/H-RFI) = 1.24), HK1 (FC = 1.23), and PKM2 (FC = 1.30) while the DE genes involved in oxidative phosphorylation were COX8A (FC = 1.28), ATP6AP1 (FC = 1.39), and ATP6V0D1 (FC = 1.26) (Table 7 and Additional file 3).
Table 7.
Genes involved in major energy generating pathways with increased expression in the L-RFI rumen epithelium
| Energy generating pathway | Gene symbol | Gene name | DE gene (D) and/or within turquoise module (T) |
|---|---|---|---|
| Glycolysis | GPI | Glucose-6-phosphate isomerase | T |
| PFKL | Phosphofructokinase, liver | T | |
| ALDOA | Aldolase A, fructose-bisphosphate | T | |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | T | |
| TPI1 | Triosephosphate isomerase 1 | D, T | |
| HK1 | Hexokinase 1 | D | |
| PKM2 | Pyruvate kinase M2 | D | |
| Tricarboxylic acid cycle | CS | Citrate synthase | T |
| SUCLG1 | Succinate-CoA ligase, alpha subunit | T | |
| ACO2 | Aconitase 2, mitochondrial | T | |
| SDHB | Succinate dehydrogenase complex, subunit B | T | |
| DLAT | Dihydrolipoamide S-acetyltransferase | T | |
| ACLY | ATP citrate lyase | T | |
| Oxidative phosphorylation | ATP5D | ATP synthase, H+ transporting, mitochondrial F1 complex, delta subunit | T |
| COX7A2L | Cytochrome c oxidase subunit VIIa polypeptide 2 like | T | |
| ATP5J2 | ATP synthase, H+ transporting, mitochondrial Fo complex, subunit F2 | T | |
| NDUFA10 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 10, 42 kDa | T | |
| COX8A | Cytochrome c oxidase subunit VIIIA (ubiquitous) | D, T | |
| ATP6V1F | ATPase, H+ transporting, lysosomal 14 kDa, V1 subunit F | T | |
| PPA1 | Pyrophosphatase (inorganic) 1 | T | |
| UQCRQ | Cytochrome b-c1 complex subunit 8 | T | |
| NDUFB10 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 10, 22 kDa | T | |
| NDUFS8 | NADH dehydrogenase (ubiquinone) Fe-S protein 8, 23 kDa (NADH-coenzyme Q reductase) | T | |
| ATP6AP1 | ATPase, H+ transporting, lysosomal accessory protein 1 | D, T | |
| ATP6V0D1 | ATPase, H+ transporting, lysosomal 38 kDa, V0 subunit D1 | D, T | |
| ATP6V0E1 | ATPase, H+ transporting, lysosomal 9 kDa, V0 subunit E1 | T | |
| COX10 | Cytochrome c oxidase assembly protein | T | |
| COX4I1 | Cytochrome c oxidase subunit IV isoform 1 | T | |
| UQCRC1 | Ubiquinol-cytochrome c reductase core protein I | T | |
| ATP6V0A1 | ATPase, H+ transporting, lysosomal V0 subunit A1 | T | |
| NDUFV1 | NADH dehydrogenase (ubiquinone) flavoprotein 1, 51 kDa | T | |
| PPA1 | Pyrophosphatase (inorganic) 1 | T | |
| SDHB | Succinate dehydrogenase complex, subunit B, iron sulfur (Ip) | T |
Relative mtDNA copy number and RFI
The transcriptome analysis results showed up-regulation of genes in the L-RFI epithelium involved in mitochondrial functions such as oxidative phosphorylation. Therefore, we speculated that L-RFI animals would have more relative mtDNA copy numbers (mtDNA/nDNA) in the rumen epithelium than H-RFI animals. To test this hypothesis, we investigated the relationship between RFI and relative mtDNA copy number using a larger population including M-RFI (n = 34) animals along with L-RFI (n = 35) and H-RFI (n = 35) animals. The correlation analysis showed a significant positive relationship (r = 0.21, p = 0.03) between RFI and the relative mtDNA copy number per cell in the rumen epithelium (Fig. 6). The epithelium of L-RFI animals had 741 ± 35 (mean ± SD) relative mtDNA copy numbers per cell while the epithelium of M- and H- RFI animals had 1104 ± 89 and 1000 ± 80, respectively. There was a significantly greater copy number of mtDNA in the rumen epithelium of M- and H- RFI animals compared to L-RFI animals (p < 0.01). However, no significant difference in mtDNA copy number was observed between M- and H- RFI animals (p > 0.05).
Fig. 6.
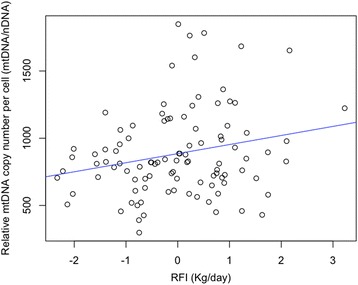
Correlation scatterplot of RFI and relative mtDNA copy number per cell of rumen epithelium (mtDNA/nDNA). Correlation coefficient (r) = 0.21, p-value = 0.03
Discussion
Improving cattle feed efficiency could greatly reduce feed expense and increase profit for producers. Our study of steers divergent in feed efficiency as evaluated by RFI showed that L-RFI individuals are more feed efficient compared to H-RFI individuals because they were able to consume 3.82 kg less dry matter per day for similar growth, body weight, and carcass weight. These results coincide with those of others, which found that selection of L-RFI steers with 3.77 kg per day lower feed intake than H-RFI steers may lead to savings of $45.60 per head over a 120-day finishing period and there was no correlation of RFI with growth, body weight and carcass weight [4]. The difference in feed consumption between L- and H- RFI steers for similar production illustrates that differences may exist in the efficiency of feed conversion into energetic substrates, absorption, energy production, and in the distribution of energy for growth, maintenance, and activity requirements. Ruminants utilize the substrates produced by the rumen microbes for their growth. Studies have shown that the rumen microbial community differs between L- and H- RFI steers and may impact feed digestion and fermentation [43]. Although, the rumen microbial community has been associated with divergence in RFI, it is unknown whether there are differences in the rumen tissue that contribute to variation in RFI.
Our study found that 47.5 % of the core genes among the rumen epithelial tissues of beef steers are involved in metabolic processes. Indeed, the rumen epithelium was known to be highly metabolic because it contains a high density of mitochondria in the stratum basale layer [44] and metabolizes a large proportion of absorbed SCFAs [45]. Through hierarchical clustering, we found that the whole transcriptome profiles of the rumen epithelial tissues of H-RFI (inefficient) steers did not cluster together while all L-RFI (efficient) steers did. This indicates that the rumen epithelial transcriptome of H-RFI animals are more variable and there are similarities among the gene expression profiles of L-RFI animals that may be associated with higher feed efficiency.
We speculated that there was greater expression of genes involved in absorption of SCFAs from the rumen of L-RFI animals. However, we did not find evidence of differences in the expression of known or candidate SCFA transporters belonging to solute carrier (SLC) gene families such as SLC4A, SLC16A (monocarboxylate transporters), SLC21A, SLC22A, or SLC26A [19] between the L- and H- RFI animals in this study (data not shown). Although we did not observe differences in the expression of genes encoding SCFA transporters, our results do show evidence of differences in the expression of genes that are associated with paracellular permeability, which may impact simple diffusion of SCFAs and other nutrients. For example, there were 14 genes up-regulated in the L-RFI epithelium involved in the adherens junction pathway, which is important in modulating cell-cell adhesion [46, 47]. These genes encoded the plasma membrane cell adhesion molecules (CAMs), E-cadherin and nectin, as well as intracellular regulators of these molecules such as p120ctn, v-Src, IQGAP1, and ACTB. CAMs require attachment to the actin cytoskeleton in order to mediate cell adhesion [48]. Additionally, the E-cadherin CAM requires interaction with a key regulator, p120ctn, in order to promote strong adhesion between cells [49]. In our study, the L-RFI epithelium had increased expression of the p120ctn regulator, however, there was also increased expression of negative regulators such as v-Src and IQGAP1 that promote weak adhesion. Furthermore, we found several DE genes that were up-regulated in the L-RFI epithelium encoding Dynamin-2 (DNM2), Hepatocyte growth factor-regulated tyrosine kinase substrate (HGS), and cytoskeletal components (Beta-actin (ACTB), Tubulin beta-5 (TUBB5), and Tubulin alpha-4a (TUBA4A)) that are involved in remodeling of epithelial adherens junctions. This suggests there is increased regulation of intercellular adhesion strength in the L-RFI epithelium. We also found that regulation of the actin cytoskeleton was a pathway that was up-regulated in the L-RFI epithelium. The regulation of intercellular adhesion strength and the actin cytoskeleton are vital to many processes such as cell migration, tissue remodeling, and maintenance of tissue integrity [50]. Indeed, we found up-regulated DE genes (RHOG, CFL1, ACTB, MYL12B, GNB1, GNB2, MAPK1) in the L-RFI epithelium involved in several important signaling pathways regulating cell migration including signaling by Rho family GTPases, RhoGDI, and Ephrin B that mediate their effects on cell migration though modulation of intercellular adhesion and regulation of the actin cytoskeleton [51–53]. Overall, the increased expression of genes related to modulation of intercellular adhesion strength, cytoskeletal organization, and cell migration signaling pathways suggest there is greater cell mobility and dynamic remodeling within the L-RFI epithelium, which may create gaps between cells resulting in greater paracellular permeability for the absorption of nutrients. Therefore, the L-RFI epithelium may potentially have increased paracellular permeability to maximize nutrient absorption in order to compensate for lower feed intake/substrate availability. Future studies are needed to examine the intercellular spaces, cell migration, and nutrient absorption in the rumen epithelium to determine whether they are truly different between steers divergent in RFI.
In addition to greater cell mobility and dynamic remodeling, further evidence from our transcriptome analyses results support that the rumen epithelium of L-RFI animals may have greater tissue morphogenesis compared to H-RFI animals. Tissue morphogenesis is a process that involves changes to the number, shape, size, position, and gene expression of cells in order to develop and maintain a tissue [54]. For instance, we found up-regulation of functions such as cellular growth, proliferation, and death in the L-RFI epithelium, which may indicate increased cell turnover. There was also up-regulation of protein synthesis, degradation, folding, transport, and post-translational modification processes that suggest increased protein turnover. Future studies to measure rumen papillae density/surface area or rumen volume are necessary to determine whether the L-RFI epithelium has increased tissue morphogenesis and permeability due to a larger surface area to volume ratio. Greater tissue morphogenesis suggests that the L-RFI epithelium may have greater cell growth and proliferation. This may be due to increased paracellular permeability causing the epithelial cells to be exposed to nutrients at a faster rate.
It is speculated that the difference in the expression of genes involved in tissue morphogenesis could be also associated with the variation in nutrient availability and flux. L-RFI animals had lower feed intake, suggesting that these animals may have lower microbial products available for the animals. However, it has been reported that L-RFI animals tended to have a greater concentration of total SCFAs (p = 0.059) in the rumen fluid with significantly greater concentrations of butyrate (p < 0.001) and valerate (p = 0.006) [43]. High concentrations of nutrients are associated with changes in the rumen epithelium such as extensive sloughing of surface epithelial cells, and increased cell migration and proliferation [55, 56]. Although the concentration of ruminal SCFAs was not measured for this study, the lack of difference in the expression of genes associated with SCFA oxidation and ketogenesis between L- and H- RFI suggest that the groups of animals may have similar SCFA levels in the rumen, although low RFI animals eat less. On the other hand, in L-RFI animals, nutrient flux may have been increased to maximize absorption because they have less substrate availability by consuming less feed. Regardless, future studies are needed to measure rate of nutrient absorption as well as the concentration of nutrients in the rumen content and epithelium to define their roles in affecting rumen tissue gene expression and in relation to RFI.
While there was no difference in the expression of SCFA metabolism genes, we found up-regulation of genes in the L-RFI epithelium involved in energy generating pathways such as glycolysis, TCA cycle, and oxidative phosphorylation. The majority of cellular energy, in the form of ATP, is generated through oxidative phosphorylation within the mitochondrial inner membrane [57]. Our results on increased expression of genes encoding mitochondrial proteins involved in oxidative phosphorylation in the L-RFI epithelium suggest that efficient steers may have increased energy production in the rumen epithelium. This may also indicate higher mitochondrial activity and/or greater mitochondrial genome copy numbers in the rumen epithelium of L-RFI steers compared to H-RFI steers. RFI was positively correlated (r = 0.21, p = 0.03), although very weakly, to the mitochondrial genome copy number per cell of the epithelium. Although there were fewer mitochondrial genomes in L-RFI animals, we observed increased expression of mitochondrial genes such as oxidative phosphorylation genes. This suggests that there is greater transcription from less genomes in the mitochondria of L-RFI animals. While we could not directly measure mitochondrial activity in our study, others have reported a more rapid rate of oxidative phosphorylation in the mitochondria of skeletal muscle and the liver in L-RFI beef cattle [58, 59]. We speculate that the mitochondria in the L-RFI rumen may also have the same capability, which needs to be further studied. Overall, increased expression of glycolytic and oxidative phosphorylation genes in the L-RFI epithelium suggests increased energy production that provides a resource for the increased tissue morphogenesis occurring to maintain barrier integrity in the L-RFI epithelium, which is an energetically expensive process. Although expensive, we speculate the L-RFI animals have enough energy for other processes throughout the body although they eat less. Future studies on examination of epithelial metabolism and whole body energy balance are needed to support our speculation.
Additionally, our study showed significant enrichment of genes in the L-RFI epithelium associated with acetylation, which is a post-translational modification process. These genes were involved with various processes such as protein synthesis, protein degradation, cytoskeletal organization, metabolism of energetic substrates, and stress response. This suggests that acetylation modulates genes belonging to a wide range of cellular processes. Acetylation is known to be a metabolically sensitive protein modification process [60]. It is known that approximately 35 % of all mitochondrial proteins are acetylated and the majority of these acetylated proteins are involved in energy metabolism [61]. The expression level of genes encoding proteins that have the potential to become acetylated could be an indicator of the level of energy metabolism occurring within animals. Our results on the increased expression of mitochondrial and metabolic genes suggest greater activity of the enzymes and more energy flow in low RFI animals.
Conclusions
The RNA-seq based transcriptome analysis showed increased expression of genes in the L-RFI epithelium involved in modulation of adherens junctions, cytoskeletal organization, and cell migration signaling pathways that may cause large intercellular gaps and result in greater paracellular absorption of nutrients. Also, the L-RFI epithelium showed increased tissue morphogenesis as characterized by increased expression of genes involved in cell and protein turnover, modulation of intercellular adhesion strength, and cell migration, which may be due to increased absorption rate or a larger surface area to volume ratio. There was no difference in SCFA metabolism; however, there was increased expression of genes associated with glycolysis and oxidative phosphorylation in the L-RFI epithelium suggesting greater energy production. The increase in energy production provides a resource for the dynamic processes involved in maintaining tissue barrier integrity. A limitation of this study was that the epithelial tissue used for RNA-seq was obtained from only one location within the rumen. Pooling epithelial samples from multiple locations in the rumen is recommended to provide a better representation of the host transcriptome. Also, the epithelial tissue was acquired from animals only after slaughter. The transcriptome within animals may vary throughout their life and therefore it would be optimal to obtain epithelial samples from live animals through biopsy at various time points throughout the day and at different ages of the animal. Another limitation was that rumen size/volume and tissue morphology (i.e. rumen papillation parameters) was not examined, and the concentration and absorption of ruminal SCFAs could not be measured. Future work is needed to determine whether feed efficient steers truly have increased simple diffusion of SCFAs through the rumen wall, increased tissue morphogenesis, and increased energy production for the epithelium and for whole body energy use.
Regardless, this is one of the first efforts to study rumen epithelium in terms of its function and role in feed efficiency and our results suggest that the rumen epithelium may contribute to variation in RFI through differences in the expression of genes that affect the paracellular transport of nutrients, tissue morphology, and mitochondrial function. This provides a fundamental understanding on how host gene expression profiles are associated with RFI, which could potentially lead to identification of biomarkers for selection and improvement of cattle feed efficiency.
Acknowledgements
The research was funded by the Alberta Livestock and Meat Agency Ltd. (Edmonton, Canada; Project number: 2015P008R) and NSERC discovery grant. RSGK was also partly supported by the Queen Elizabeth II Graduate Scholarship. We thank X. Sun (University of Alberta) for technical support in total RNA library construction.
Availability of data and materials
The raw sequencing dataset supporting the results of this study have been deposited at publicly available NCBI’s Gene Expression Omnibus Database (http://www.ncbi.nlm.nih.gov/geo/). The data are accessible through GEO Series accession number GSE76501 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76501).
Authors’ contributions
RSGK, LLG, and YC designed the experiments. LLG participated in the animal study and collected rumen tissue samples. RSGK performed the experiments, data analysis, interpretation, and wrote the manuscript. YC assisted in RNA extraction and qPCR. GL assisted in transcriptome mapping and functional gene analysis. RSGK, LLG, PS, YC, and GL revised the manuscript and approved the final version for submission.
Competing interests
The authors declare that they have no competing interests.
Additional files
RFI values of animals selected for transcriptome analysis. (DOCX 36 kb)
Phenotypic measures of the animals used for transcriptome profiling analysis. (DOCX 52 kb)
List of 122 genes DE in the rumen epithelial tissues between L- and H- RFI animals. (DOCX 145 kb)
References
- 1.Cottle D, Kahn L. Beef cattle production and trade. Australia: CSIRO Publishing; 2014. p. 221. [Google Scholar]
- 2.Alston JM, Pardey PG. Agriculture in the global economy. J Econ Perspect. 2014;28(1):121–146. doi: 10.1257/jep.28.1.121. [DOI] [Google Scholar]
- 3.Arthur PF, Archer JA, Johnston DJ, Herd RM, Richardson EC, Parnell PF. Genetic and phenotypic variance and covariance components for feed intake, feed efficiency, and other postweaning traits in Angus cattle. J Anim Sci. 2001;79:2805–2811. doi: 10.2527/2001.79112805x. [DOI] [PubMed] [Google Scholar]
- 4.Basarab JA, Price MA, Aalhus JL, Okine EK, Snelling WM, Lyle KL. Residual feed intake and body composition in young growing cattle. Can J Anim Sci. 2003;83:189–204. doi: 10.4141/A02-065. [DOI] [Google Scholar]
- 5.Nkrumah JD, Okine EK, Mathison GW, Schmid K, Li C, Basarab A, Price MA, Wang Z, Moore SS. Relationships of feedlot feed efficiency, performance, and feeding behavior with metabolic rate, methane production, and energy partitioning in beef cattle. J Anim Sci. 2006;84:145–153. doi: 10.2527/2006.841145x. [DOI] [PubMed] [Google Scholar]
- 6.Okine EK, Basarab JA, Baron V, Price MA. Net feed efficiency in young growing cattle: III. Relationships to methane and manure production. Can J Anim Sci. 2001;81:614. [Google Scholar]
- 7.Richardson EC, Herd RM. Biological basis for variation in residual feed intake in beef cattle. 2. Synthesis of results following divergent selection. Aust J Exp Agr. 2004;44:431–440. doi: 10.1071/EA02221. [DOI] [Google Scholar]
- 8.Herd RM, Arthur PF. Physiological basis for residual feed intake. J Anim Sci. 2009;87:E64–E71. doi: 10.2527/jas.2008-1345. [DOI] [PubMed] [Google Scholar]
- 9.Koch RM, Swiger LA, Chambers D, Gregory KE. Efficiency of feed use in beef cattle. J Anim Sci. 1963;22:486–494. doi: 10.2527/jas1963.222486x. [DOI] [Google Scholar]
- 10.Crowley JJ, McGee M, Kenny DA, Crews DH, Jr, Evans RD, Berry DP. Phenotypic and genetic parameters for different measures of feed efficiency in different breeds of Irish performance-tested beef bulls. J Anim Sci. 2010;88:885–894. doi: 10.2527/jas.2009-1852. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Gondro C, Quinn K, Herd RM, Parnell PF, Vanselow B. Global gene expression profiling reveals genes expressed differentially in cattle with high and low residual feed intake. Anim Genet. 2011;42:475–490. doi: 10.1111/j.1365-2052.2011.02182.x. [DOI] [PubMed] [Google Scholar]
- 12.Karisa BK, Thomson J, Wang Z, Stothard P, Moore SS, Plastow GS. Candidate genes and single nucleotide polymorphisms associated with variation in residual feed intake in beef cattle. J Anim Sci. 2013;91:3502–3513. doi: 10.2527/jas.2012-6170. [DOI] [PubMed] [Google Scholar]
- 13.Perkins SD, Key CN, Garrett CF, Foradori CD, Bratcher CL, Kriese-Anderson LA, Brandebourg TD. Residual feed intake studies in Angus-sired cattle reveal a potential role for hypothalamic gene expression in regulating feed efficiency. J Anim Sci. 2014;92:549–560. doi: 10.2527/jas.2013-7019. [DOI] [PubMed] [Google Scholar]
- 14.Wolin MJ. The rumen fermentation: a model for microbial interactions in anaerobic ecosystems. Adv Microb Ecol. 1979;3:49–77. doi: 10.1007/978-1-4615-8279-3_2. [DOI] [Google Scholar]
- 15.Bergman EN. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. 1990;70:567–590. doi: 10.1152/physrev.1990.70.2.567. [DOI] [PubMed] [Google Scholar]
- 16.López S, Hovell FDD, Dijkstra J, France J. Effects of volatile fatty acid supply on their absorption and on water kinetics in the rumen of sheep sustained by intragastric infusions. J Anim Sci. 2003;81:2609–2616. doi: 10.2527/2003.81102609x. [DOI] [PubMed] [Google Scholar]
- 17.Ash R, Baird GD. Activation of volatile fatty acids in bovine liver and rumen epithelium. Biochem J. 1973;136:311–319. doi: 10.1042/bj1360311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nayananjalie WAD, Wiles TR, Gerrard DE, McCann MA, Hanigan MD. Acetate and glucose incorporation into subcutaneous, intramuscular, and visceral fat of finishing steers. J Anim Sci. 2015 doi: 10.2527/jas.2014-8374. [DOI] [PubMed] [Google Scholar]
- 19.Aschenbach JR, Bilk S, Tadesse G, Stumpff F, Gabel G. Bicarbonate-dependent and bicarbonate-independent mechanisms contribute to nondiffusive uptake of acetate in the ruminal epithelium of sheep. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1098–G1107. doi: 10.1152/ajpgi.90442.2008. [DOI] [PubMed] [Google Scholar]
- 20.Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. 1980;60(1):143–187. doi: 10.1152/physrev.1980.60.1.143. [DOI] [PubMed] [Google Scholar]
- 21.Leighton B, Nicholas AR, Pogson CI. The pathway of ketogenesis in rumen epithelium of the sheep. Biochem J. 1983;216:769–772. doi: 10.1042/bj2160769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canadian Council on Animal Care (CCAC) In: Guide to the care and use of experimental animals. 2. Olfert ED, Cross BM, McWilliams AA, editors. Ottawa, ON: CCAC; 1993. [Google Scholar]
- 23.Durunna ON, Mujibi FD, Goonewardene L, Okine EK, Basarab JA, Wang Z, Moore SS. Feed efficiency differences and reranking in beef steers fed grower and finisher diets. J Anim Sci. 2011;89(1):158–67. doi: 10.2527/jas.2009-2514. [DOI] [PubMed] [Google Scholar]
- 24.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriotomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Hansaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinform. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anders S, Pyl PT, Huber W. HTSeq – a Python framework to work with high-throughput sequencing data. Bioinform. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.R Core Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. [Google Scholar]
- 31.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 33.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durinck S, Spellman P, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41(D1):D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Brief Bioinform. 2012;14(2):144–161. doi: 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andersen CL, Ledet-Jensen J, Ørntoft T. Normalization of real-time quantitative RT-PCR data: a model based variance estimation approach to identify genes suited for normalization applied to bladder and colon cancer datasets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- 40.Li M, Zhou M, Adamowicz E, Basarab JA, Guan LL. Characterization of bovine ruminal epithelial bacterial communities using 16S rRNA sequencing, PCR-DGGE, and qRT-PCR analysis. Vet Microbiol. 2012;155(1):72–80. doi: 10.1016/j.vetmic.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 41.Yue X, Dechow C, Chang T, DeJarnette JM, Marshall CE, Lei C, Liu W. Copy number variations of the extensively amplified Y-linked genes, HSFY and ZNF280BY, in cattle and their association with male reproductive traits in Holstein bulls. BMC Genom. 2014;15:113. doi: 10.1186/1471-2164-15-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Speicher DJ, Johnson NW. Detection of human herpesvirus 8 by quantitative polymerase chain reaction: development and standardisation of methods. BMC Infect Dis. 2012;12:210. doi: 10.1186/1471-2334-12-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guan LL, Nkrumah JD, Basarab JA, Moore SS. Linkage of microbial ecology to phenotype: correlation of rumen microbial ecology to cattle’s feed efficiency. FEMS Microbiol Lett. 2008;288:85–91. doi: 10.1111/j.1574-6968.2008.01343.x. [DOI] [PubMed] [Google Scholar]
- 44.Graham C, Simmons NL. Functional organization of the bovine rumen epithelium. Am J Physiol Regul Integr Comp Physiol. 2005;288:R173–R181. doi: 10.1152/ajpregu.00425.2004. [DOI] [PubMed] [Google Scholar]
- 45.Sehested J, Diernæs L, Møller PD, Skadhauge E. Ruminal transport and metabolism of short-chain fatty acids (SCFA) in vitro: effect of SCFA chain length and pH. Comp Biochem Physiol. 1999;123:359–368. doi: 10.1016/S1095-6433(99)00074-4. [DOI] [PubMed] [Google Scholar]
- 46.Takeichi M. Cadherins: a molecular family important in selective cell-cell adhesion. Annu Rev Biochem. 1990;59:237–252. doi: 10.1146/annurev.bi.59.070190.001321. [DOI] [PubMed] [Google Scholar]
- 47.Guo X, Rao JN, Liu L, Zou T, Turner DJ, Bass BL, Wang J. Regulation of adherens junctions and epithelial paracellular permeability: a novel function for polyamines. Am J Physiol Cell Physiol. 2003;285:C1174–C1187. doi: 10.1152/ajpcell.00015.2003. [DOI] [PubMed] [Google Scholar]
- 48.Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–219. doi: 10.1016/S0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- 49.Thoreson MA, Anastasiadis PZ, Daniel JM, Ireton RC, Wheelock MJ, Johnson KR, Hummingbird DK, Reynolds AB. Selective uncoupling of p120ctn from e-cadherin disrupts strong adhesion. J Cell Biol. 2000;148:189–201. doi: 10.1083/jcb.148.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baum B, Georgiou M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J Cell Biol. 2011;192(6):907–917. doi: 10.1083/jcb.201009141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- 52.Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J. 2005;390:1–9. doi: 10.1042/BJ20050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park I, Lee H. EphB/ephrinB signaling in cell adhesion and migration. Mol Cells. 2015;38(1):14–19. doi: 10.14348/molcells.2015.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heisenberg CP, Bellaïche Y. Forces in tissue morphogenesis and patterning. Cell. 2013;153(5):948–962. doi: 10.1016/j.cell.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 55.Steele MA, Croom J, Kahler M, AlZahal O, Hook SE, Plaizier K, McBride BW. Bovine rumen epithelium undergoes rapid structural adaptations during grain-induced subacute ruminal acidosis. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1515–R1523. doi: 10.1152/ajpregu.00120.2010. [DOI] [PubMed] [Google Scholar]
- 56.Goodlad RA. Some effects of diet on the mitotic index and the cell cycle of the ruminal epithelium of sheep. Quart J Exp Physiol. 1981;66:487–499. doi: 10.1113/expphysiol.1981.sp002590. [DOI] [PubMed] [Google Scholar]
- 57.Alberts B, Johnson A, Lewis J, et al. Molecular biology of the cell. 4th edition. The Mitochondrion. New York: Garland Science; 2002. [Google Scholar]
- 58.Kolath WH, Kerley MS, Golden JW, Keisler DH. The relationship between mitochondrial function and residual feed intake in Angus steers. J Anim Sci. 2006;84:861–865. doi: 10.2527/jas.2005-519. [DOI] [PubMed] [Google Scholar]
- 59.Lancaster PA, Carstens GE, Michal JJ, Brennan KM, Johnson KA, Davis ME. Relationships between residual feed intake and hepatic mitochondrial function in growing beef cattle. J Anim Sci. 2014;92:3134–3141. doi: 10.2527/jas.2013-7409. [DOI] [PubMed] [Google Scholar]
- 60.Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40(2):323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson KA, Hirschey MD. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012;52:23–25. doi: 10.1042/bse0520023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rekawiecki R, Rutkowska J, Kotwica J. Identification of optimal housekeeping genes for examination of gene expression in bovine corpus luteum. Reprod Biol. 2012;12:362–367. doi: 10.1016/j.repbio.2012.10.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw sequencing dataset supporting the results of this study have been deposited at publicly available NCBI’s Gene Expression Omnibus Database (http://www.ncbi.nlm.nih.gov/geo/). The data are accessible through GEO Series accession number GSE76501 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76501).