

Abstract
The B-cell lymphoma/leukemia 2 (BCL-2) family of proteins has attracted the attention of cancer biologists since the cloning of BCL-2 more than 25 years ago. In the intervening decades, the way the BCL-2 family controls commitment to programmed cell death has been greatly elucidated. Several drugs directed at inhibiting BCL-2 and related antiapoptotic proteins have been tested clinically, with some showing considerable promise, particularly in lymphoid malignancies. A better understanding of the BCL-2 family has also provided insight into how conventional chemotherapy selectively kills cancer cells and why some cancers are more chemosensitive than others. Further exploitation of our understanding of the BCL-2 family promises to offer improved predictive biomarkers for oncologists and improved therapies for patients with cancer.
INTRODUCTION
The discovery of the B-cell lymphoma/leukemia 2 (BCL-2) family of proteins is intimately linked to human cancer. By the mid 1980s, it was well known that many B-cell malignancies possessed translocations involving the immunoglobulin heavy chain locus on chromosome 14.1–4 Identification of the other genes participating in these translocations became an important goal for cancer biologists. In follicular lymphoma, the goal became to identify the partner of t(14;18) on chromosome 18. The cloned and sequenced gene bore no resemblance to known genes and took the name BCL-2.5
Experiments revealed that the BCL-2 protein functioned to oppose the apoptosis pathway of programmed cell death6,7 and was predominantly localized to the mitochondrion,8 at that time both unique findings for an oncogene. Subsequent work has identified many different proteins that are related to BCL-2 by amino acid sequence homology and their participation in the control of apoptosis.9–13 This so-called BCL-2 family of proteins contains pro- as well as antiapoptotic proteins that interact directly with one another to determine whether the cell will commit to apoptosis.
The central event that controls cellular commitment to death via apoptosis is mitochondrial outer membrane permeabilization (MOMP). After MOMP, multiple proapoptotic molecules are released from the mitochondrial intermembrane space to coordinate most of the hallmarks of apoptosis, like nuclear condensation and caspase activation. MOMP is controlled by the BCL-2 family via protein-protein interactions. There are three main subdivisions of the BCL-2 family of proteins (Fig 1). BAX and BAK are proapoptotic effector proteins, so named because when activated, they directly participate in the formation of pores in the mitochondrial outer membrane.15–17 As such, they are essential for apoptosis via the mitochondrial pathway.18 BAX and BAK can be activated by certain members of another subdivision of the BCL-2 family, the proapoptotic BCL-2 homology 3 (BH3) –only proteins. These get their name via possession of the BH3 domain, but no other BH domain. The BH3 domain is required for proapoptotic function of all proapoptotic BCL-2 family proteins. BIM, BID, and perhaps PUMA (known as activator BH3-only proteins) interact with and activate BAX and BAK directly, causing a conformational change that induces BAX and BAK homo-oligomerization, which in turn results in formation of a pore in the mitochondrial outer membrane and MOMP.19–21 Other mechanisms, poorly understood, may also exist for the activation of BAX and BAK.22 For most purposes, MOMP can be considered the point of no return in the pathway to commitment to death via apoptosis.
Fig 1.
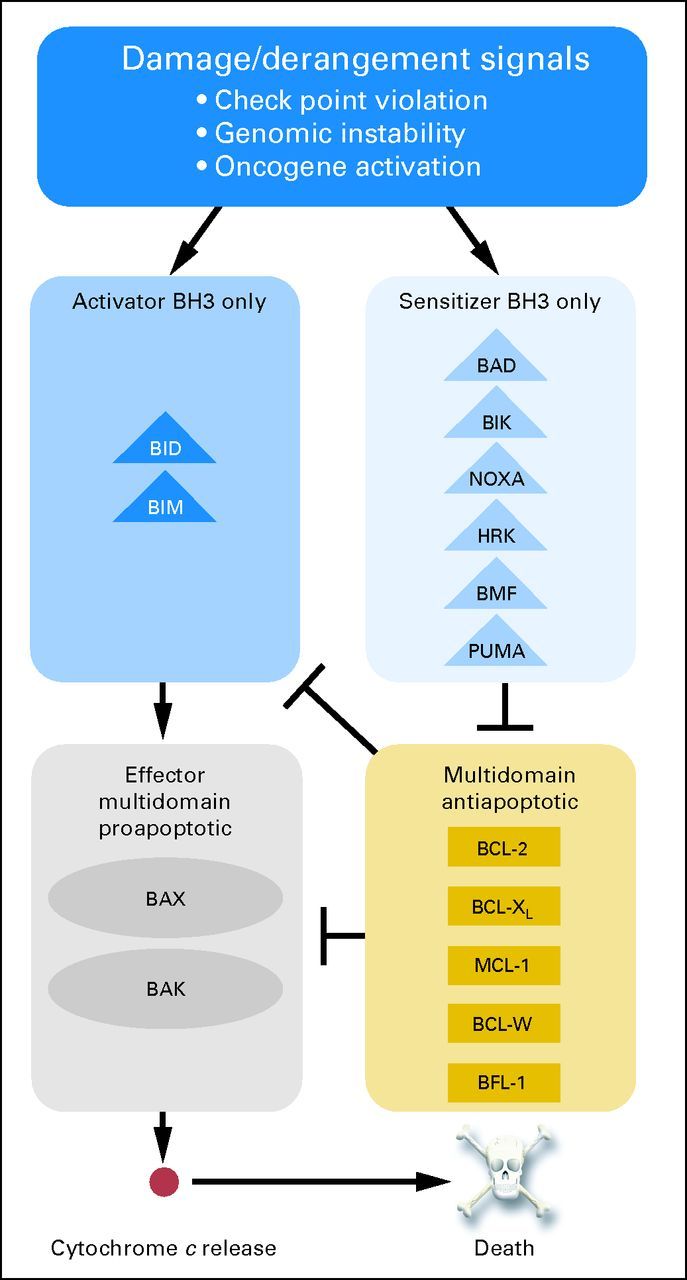
Three families of B-cell lymphoma/leukemia 2 (BCL-2) proteins govern the decision to undergo mitochondrial apoptosis. DNA damage, checkpoint violations, or oncogene activation generates pro-death BCL-2 homology 3 (BH3) –only proteins, which can be subdivided into activator and sensitizer subfamilies based on function. Activator BH3-only proteins such as BID and BIM activate effector multidomain proapoptotic proteins such as BAX and BAK, leading to mitochondrial permeabilization and commitment to apoptotic death. Multidomain antiapoptotic proteins such as BCL-2 oppose this process by sequestering activators and thereby limiting contact with effectors or by sequestering activated effectors. Sensitizer BH3-only proteins such as BAD act as selective inhibitors of antiapoptotic proteins. BCL-w, BCL-2–like protein 2; BCL-XL, BCL extra long; BFL-1, BCL-2–related gene expression in fetal liver; MCL-1, MCL-1, myeloid cell leukemia 1. Data adapted.14
MOMP can be inhibited by the antiapoptotic proteins, which are now known to include BCL-2, BCL extra long (BCL-XL), BCL-2–like protein 2 (BCL-w), MCL-1, myeloid cell leukemia 1 (MCL-1), and (BCL-2–related gene expression in fetal liver (BFL-1; also known as A1).23,24 They inhibit apoptosis by sequestering activator BH3-only proteins, preventing their activation of BAX and BAK.18 Antiapoptotic proteins can also bind monomeric activated BAX and BAK, preventing the formation of oligomers and pores. In all of these cases, the antiapoptotic protein binds the hydrophobic face of the amphipathic alpha-helical BH3 domain of the proapoptotic protein in a hydrophobic groove. Proapoptotic sensitizer BH3-only proteins lack the ability to directly activate BAX and BAK and instead promote cell death by competitively inhibiting the ability of antiapoptotic proteins to bind the BH3 domains of BAX and BAK and activator BH3-only proteins.25
BCL-2 FAMILY AS DIRECT THERAPEUTIC TARGET IN CANCER
As a prosurvival oncogene overexpressed as a result of cancer-specific translocations, BCL-2 clearly deserves consideration for therapeutic inhibition. There are many efforts under way to target BCL-2 as well as other antiapoptotic proteins with a variety of compounds of variable potencies (Table 1). Here, we briefly review the three drugs that have advanced furthest clinically.
Table 1.
Fluorescence Polarization Assays Measuring Displacement of a FITC-BID Peptide From All Six Antiapoptotic Human BCL-2 Family Proteins to Compare IC50 Values of Small-Molecule BCL-2 Antagonists
| Protein | Compound IC50 (uM) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| EGCG | Gossypol | ApoG | Antimycin A | Chelerythrine | ABT-737 | Obatoclax | BH3I-1 | YC137 | |
| BCL-XL | 0.59 | 3.03 | 2.80 | 2.70 | Approximately 10 | 0.064 | 4.69 | 5.86 | > 20 |
| BCL-2 | 0.45 | 0.28 | 0.64 | 2.95 | Approximately 10 | 0.12 | 1.11 | 1.14 | 6.43 |
| BCL-w | 2.33 | 1.4 | 2.10 | 4.57 | > 10 | 0.024 | 7.01 | 2.33 | 2.21 |
| BCL-B | 0.36 | 0.16 | 0.37 | 1.83 | > 10 | > 10 | 2.15 | 1.08 | 3.10 |
| BFL-1 | 1.79 | > 10 | > 10 | > 10 | Approximately 10 | > 20 | 5.00 | 4.65 | > 20 |
| MCL-1 | 0.92 | 1.75 | 3.35 | 2.51 | > 10 | > 20 | 2.90 | 2.17 | 2.47 |
NOTE. Data adapted.26
Abbreviations: ApoG, apogossypol; BCL-2, B-cell lymphoma/leukemia 2; BCL-B, BCL-2–like protein 10; BCL-w, BCL-2–like protein 2; BCL-XL, BCL extra long; BFL-1, BCL-2–related gene expression in fetal liver; BH3I-1, BCL-2 homology 3 domain inhibitor 1; EGCG, epigallocatechin gallate; FITC, fluoresceine isothiocyanate; IC50, half maximal inhibitory concentration; MCL-1, myeloid cell leukemia 1.
Oblimersen Sodium
The first strategy tested in the clinic was inhibiting expression of the BCL-2 protein. Oblimersen sodium (Genasense; Genta, Berkeley Heights, NJ) is a single-stranded, phosphorothioated 18-mer DNA molecule with sequence complementary to that of BCL-2 mRNA.27 The proposed mechanism of action is that oblimersen hybridizes with the BCL-2 mRNA, provoking the hydrolysis of the mRNA by RNAse H, resulting in a decrease in BCL-2 protein production and hence in cellular BCL-2 protein levels.28
Oblimersen has been tested in clinical trials in several solid tumors and hematologic malignancies, alone and in combination with conventional agents. Early-phase clinical trials in chronic lymphocytic leukemia (CLL),29 multiple myeloma,30 and melanoma31 provided some evidence of clinical benefit; however, phase III data have been unconvincing. One study of oblimersen in combination with dexamethasone in advanced multiple myeloma did not show a survival advantage.32 A separate study of fludarabine plus cyclophosphamide with or without oblimersen showed a modest survival benefit in a posthoc analysis of patients with CLL with fludarabine-sensitive disease who achieved complete or partial remission.33 On the basis of the lack of clear clinical benefit, oblimersen has not been approved by the US Food and Drug Administration, and its future development remains uncertain.
The disappointing clinical performance of oblimersen should not, however, be taken as a refutation of the putative benefit of targeting BCL-2 in cancers. Although oblimersen has been found to reduce BCL-2 protein levels in cell lines in vitro,34 its ability to decrease BCL-2 levels in either primary tumor cells or in peripheral blood mononuclear cells in vivo is uncertain at best.35 Therefore, it is entirely possible that the lack of clinical efficacy of oblimersen is simply a result of the inability of the oligonucleotide to reduce BCL-2 protein levels in tumors in vivo.
Obatoclax
Obatoclax (GX-15-070) is a small molecule that has been found to bind to all BCL-2 family proteins, including BCL-2, BCL-XL, and MCL-1,36 albeit with modest affinity (Table 1). It is proposed to operate like a BH3 mimetic, competitively inhibiting binding of proapoptotic proteins to the hydrophobic groove of antiapoptotic proteins. However, preclinical work has supported many different mechanisms of action, some of which do not require BAX and BAK to kill cells, casting doubt on the central mechanism of obatoclax.37 Related to this, treatment with obatoclax does not cause prominent thrombocytopenia, an in vivo pharmacodynamic marker of BCL-XL inhibition. Furthermore, obatoclax causes reversible mental status changes,38 an effect inconsistent with any known function of BCL-2 family proteins.
Obatoclax has shown some modest clinical activity in a variety of early-phase clinical trials, including in CLL and small-cell lung cancer (SCLC). A recently presented phase I study of obatoclax in combination with fludarabine and rituximab in patients with relapsed CLL showed a partial response rate of 54%, with no complete remissions.39 The median time to progression in this relapsed population was reasonably long at 20 months. For SCLC, although early-phase clinical trials looked promising,40 a randomized phase II study in combination with carboplatin and etoposide for patients with extensive-stage SCLC did not show a statistically significant benefit in the primary end point of overall response rate.41
It is notable that serum pharmacodynamic biomarkers used in clinical trials, like oligonucleosomal cleaved DNA, may well indicate the provocation of apoptosis somewhere in the body by obatoclax. However, this should not be construed as evidence that the drug is operating directly on BCL-2 family proteins, because many agents (such as most conventional chemotherapy drugs) are known to induce apoptosis without directly interacting with BCL-2 family proteins.
ABT-737/ABT-263/ABT-199
ABT-737 is a small molecule BH3 mimetic that binds to BCL-2, BCL-XL, and BCL-w.42 Because this matches the binding pattern of the BH3 domain of the BH3-only protein BAD,43,44 ABT-737 is sometimes referred to as a BAD BH3 mimetic. It was developed by Abbott Laboratories (Abbott Park, IL) from a small-molecule screen that identified compounds that bound with modest affinity to distinct but proximal subsites in the BH3 binding groove of BCL-XL. The series of compounds that emerged gave rise to ABT-737. Most of the considerable body of preclinical work performed on this series of compounds has employed ABT-737. ABT-263 (Navitoclax; Abbott Laboratories) is closely related chemically and interacts with BCL-2 proteins with selectivity similar to that of ABT-737.45 A significant difference is that ABT-263 is orally bioavailable, and it is ABT-263 that has been used in initial clinical trials.
It is important to note that the Abbott compounds bind to BCL-2 family proteins with much greater affinity than any other reported small-molecule antagonists of BCL-2 family proteins, generally 100- to 10,000-fold greater (Table 1). In fact, although ABT-263 is indeed selective for BCL-2, BCL-XL, and BCL-w, it still binds MCL-1 with greater affinity than paninhibitors like obatoclax.46 Preclinical studies of both ABT-737 and ABT-263 are consistent with a BH3-mimetic mechanism of killing and have shown that the drugs displace proapoptotic proteins from BCL-2.47 Killing by ABT-737 requires BAX or BAK, and ABT-737 also has direct mitochondrial effects,48 as would be expected from on-target activity. Furthermore, there is good correlation between sensitivity of mitochondria to BAD BH3 peptides and sensitivity of the cells in which they reside to ABT-737.14,49
Treatment with ABT-737 reproducibly causes thrombocytopenia in a dose-dependent fashion in all mammals in which it has been tested.50 Thrombocytopenia can be observed within hours of drug treatment and is not caused by decreased platelet production. Rather, platelets, which contain mitochondria, undergo apoptosis and are rapidly cleared from circulation by the reticuloendothelial system in the spleen and liver. Platelets show increased sensitivity to ABT-737 with increasing age, so there is a selection for younger, larger platelets to persist in circulation.51 Drug treatment with ABT-737 causes increased platelet production in the bone marrow and may also lead to platelet dysfunction.52
Clinically significant thrombocytopenia has been mitigated in clinical trials through institution of a lead-in lower dose of ABT-263 and continuous dosing to stimulate platelet production, so when higher therapeutic doses are administered, the platelet nadir is not as low or prolonged.53 Two different laboratories have demonstrated that thrombocytopenia is an on-target toxicity resulting from the increasing dependence of platelets on BCL-XL for prevention of apoptosis as they age.50,54 The data are strong enough that thrombocytopenia can be viewed as a pharmacodynamic marker of successful inhibition of BCL-XL in vivo. As a consequence, failure to observe significant thrombocytopenia in vivo after treatment with a putative inhibitor of BCL-XL raises doubts about the true mechanism of action of the drug concerned.
Significant clinical benefit has been observed in trials of ABT-263, most notably in CLL. In a phase I single-agent trial in relapsed or refractory lymphoid malignancies, 10 of 46 evaluable patients had a partial response.55 Patients with CLL/small lymphocytic lymphoma received disproportionate benefit, with eight of 16 achieving a partial response and 13 of 16 showing reduction in tumor size. Common adverse events in this population with compromised bone marrow reserve included anemia, thrombocytopenia, lymphopenia, and neutropenia as well as infection and diarrhea. In only one patient was a dose-limiting toxicity of grade 4 thrombocytopenia observed. A follow-up study focusing on only relapsed or refractory CLL in a heavily pretreated population confirmed these initial findings, with nine (31%) of 29 patients achieving a partial response and 18 (62%) of 29 maintaining stable disease; median progression-free survival was 25 months.53
Perhaps unsurprisingly, response was more modest in a phase I single-agent study of relapsed or refractory SCLC and other solid tumors.56 Of 38 evaluable patients, there was only one partial response (in a patient with SCLC) and eight patients with SCLC or carcinoid who had stable disease. The partial response endured for longer than 35 months in a patient who had progressive disease on second-line therapy before enrollment. The drug was well tolerated, with fewer cytopenias than were seen in the lymphoid malignancies trials. Diarrhea, nausea, vomiting, and fatigue were the most common adverse events.
Subsequent trials are under way testing the combination of ABT-263 with standard agents in both solid tumors and lymphoid malignancies, with a particular focus on CLL. A recent preliminary report was presented on a phase I study of ABT-263 in combination with fludarabine, cyclophosphamide, and rituximab or bendamustine and rituximab (BR) in patients with relapsed or refractory CLL.57 Both combinations seemed to be well tolerated and did not exhibit unacceptable myelotoxicity. The most common adverse events were nausea, fatigue, and neutropenia. Grade 3 to 4 thrombocytopenia was seen in 19% of patients. The overall response rate of 81% was significantly better than that observed with ABT-263 monotherapy. Furthermore, six of 16 patients assessed in the BR arm achieved a complete response, a significantly higher rate than would be expected from BR alone in this patient population.
Concern about thrombocytopenia has limited the introduction of ABT-263 into clinical trials of acute leukemias, despite ex vivo evidence of tumor sensitivity. Because in hematologic malignancies it seems that dependence on BCL-2 is much more common than dependence on BCL-XL, Abbott has developed another compound for clinical use: ABT-199. The key feature of this drug is that it is more selective against BCL-2, lacking high affinity binding to BCL-XL. It is anticipated that thrombocytopenia will no longer be a major toxicity, allowing for higher dosing, greater BCL-2 inhibition, and better clinical response. A first-in-human phase I trial in patients with relapsed or refractory CLL or non-Hodgkin's lymphoma is under way for this oral drug. If the results are encouraging, it would facilitate the evaluation of ABT-199 in the acute leukemias, in which in vitro indications of BH3 mimetic efficacy are promising.14,58
DETERMINANTS OF SENSITIVITY AND RESISTANCE TO BCL-2 INHIBITION
For any targeted therapy, it is essential to understand why cancer cells should be selectively sensitive and to identify characteristics of cells that predict response to therapy. Without this information, one cannot be sure that the drug works according to its proposed mechanism. Furthermore, clinical development would be challenging, because it would be difficult to identify the subset of patients who would respond to the drug.
Although many different tissues and cells express BCL-2, not all of them are dependent on BCL-2 for continued survival. A key determinant of whether a cell is dependent on BCL-2 is whether BCL-2 is already occupied by pro-death proteins.43 For instance, BCL-2 in CLL cells is largely occupied by the activator BIM.47 When a BH3 mimetic like ABT-737 competes for this binding, BIM can be displaced to activate BAX and BAK, causing MOMP (Fig 2). Alternatively, BCL-2 might be occupied by activated monomeric BAX. When this is displaced, an activated BAX monomer can oligomerize with similarly displaced activated BAX monomers, resulting in pore formation and MOMP. If BCL-2 is largely unoccupied, the binding of a BH3 mimetic may reduce antiapoptotic reserve, but it is less likely to directly cause MOMP as a single agent.
Fig 2.
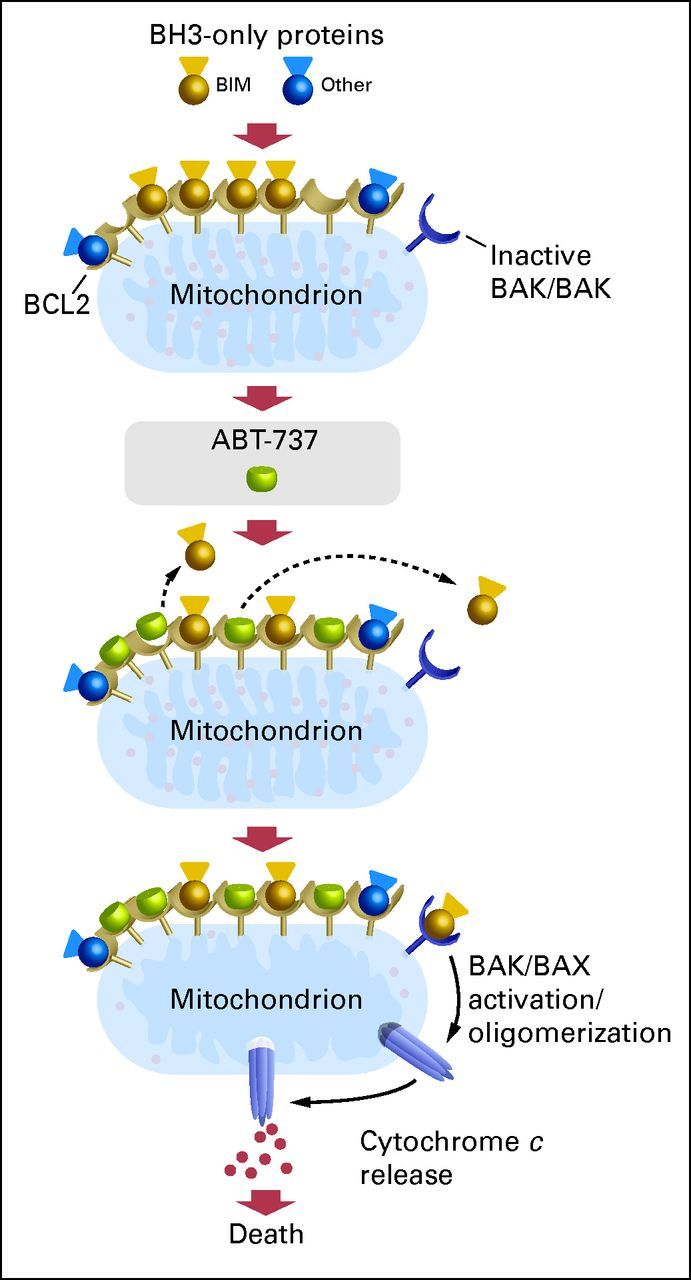
Model of ABT-737–induced death at the mitochondria. Mitochondrial B-cell lymphoma/leukemia 2 (BCL-2) in chronic lymphocytic leukemia cells is typically occupied by BIM. ABT-737 displaces BIM, and BCL-2 becomes occupied by ABT-737. Freed BIM then interacts with BAX or BAK, leading to oligomerization, cytochrome c release and subsequent irreversible commitment to programmed cell death. When BCL-2 is occupied by activator BCL-2 homology 3 (BH3) –only proteins, the cancer cell is sensitive to treatment with ABT-737 and other chemotherapeutic agents. Data adapted.45
This high occupation state of BCL-2 can be identified by probing mitochondria with synthetic BH3 domain peptides that compete for the BH3 binding site on BCL-2.43 There is good correlation between mitochondrial sensitivity to the BAD BH3 peptide and cellular sensitivity to ABT-737.14,47,49 Alternatively, BCL-2–BIM complex abundance predicts response to ABT-737 in lymphoma cell lines.49 Whether either approach can predict individual clinical response remains to be seen.
Resistance to ABT-737 can be caused by high expression of MCL-1 and BFL-1, antiapoptotic proteins that are poorly bound by the drug.48,58–60 Even if proapoptotic proteins are effectively displaced from BCL-2 by ABT-737, they can be sequestered by MCL-1 and BFL-1 before they can cause MOMP. This mechanism likely underlies the finding that in vitro sensitivity of CLL cells to ABT-737 can be predicted by a ratio of (MCL-1 + BFL-1) / BCL-2.61 An additional predictive strategy involving the BCL-2 family was recently described, suggesting that the NOXA/MCL-1 balance may determine sensitivity to ABT-737 in stroma-exposed CLL cells.62 Perhaps most interesting is the recent observation that clinical response to ABT-263 in patients with CLL correlates with low MCL-1 expression, high BIM–MCL-1 ratio, or high BIM–BCL-2 ratio.53
Other pharmacologic approaches directed against BCL-2 family proteins exist, but they are less advanced clinically than the three compounds profiled in this article. Of the compounds currently in clinical development, only the BH3-mimetic drug ABT-263 has published data to confirm high affinity and specificity for BCL-2 family members as well as the mechanistic data to support its potential utility.
BCL-2 FAMILY AND CONVENTIONAL CHEMOTHERAPY
Why Does Chemotherapy Work?
Although the BH3 mimetics are an exciting new class of anticancer agents, it is important to recognize that oncologists have for decades been exploiting important differences in BCL-2 family function with conventional chemotherapy, even if this mechanism was not understood. Conventional chemotherapeutic agents mainly kill cells via the mitochondrial pathway of apoptosis, commitment to which is regulated by the BCL-2 family. Our better understanding of how this family of proteins regulates commitment to cell death has revealed that differences in this family may well underlie the clinical efficacy of conventional chemotherapy, offer predictive power, and explain its poorly understood therapeutic index.
The question of why chemotherapy works (ie, why there should be a useful therapeutic index between normal and malignant cells) has not been clearly answered, despite its centrality to oncologic practice. Given the millions of people who have received conventional chemotherapy, this lack of understanding—and indeed the lack of modern scientific interest in this question—is surprising. The clinical observation is that conventional agents, which largely target DNA or microtubules, often cause reductions in tumor burden by several logs, even sometimes completely eliminating the cancer. At the same time, there is no normal tissue, except for perhaps the bone marrow, that ever sustains even a one-log kill in response to chemotherapy.
Popular explanations are unsatisfying. In pioneering work, Skipper et al63 observed a correlation between proliferation rate of leukemias in vitro and their chemosensitivity in vivo. Increased proliferation rates in cancer cells compared with those in normal cells has henceforth become the prevalent explanation of a therapeutic index for chemotherapy. However, this answer is at best incomplete. Clinical data to support this hypothesis are scant. For instance, there are few data to support the contention that the solid tumors that respond robustly to chemotherapy have higher proliferation rates than those that do not respond.
Stark counterexamples are also readily available in hematologic malignancies. For instance, CLL and follicular lymphoma are slowly dividing tumors. In the case of CLL, the average CLL cell may divide only once every few months.64 Nonetheless, these are generally chemosensitive tumors. Finally, there are many tissues like the epidermis that have a proliferation rate (2.7% in one study65) comparable to those of most solid tumors. Yet direct epidermal toxicity from chemotherapy is exceedingly rare. We do not mean to imply that the toxicity of conventional chemotherapy is always unrelated to cell cycle and proliferation. There is good evidence that certain agents are most toxic to cells in certain phases of the cell cycle, phases that are occupied only by dividing cells.66,67 Furthermore, it may be that loss of cell-cycle checkpoint control governs some aspects of chemosensitivity.68 Once again, however, these principles are rarely examined in patient samples.
Mitochondrial Apoptotic Priming: An Alternative Explanation for Chemosensitivity
Most conventional chemotherapeutic agents kill via the mitochondrial pathway of apoptosis regulated by the BCL-2 family of proteins. Commitment to the mitochondrial pathway of apoptosis behaves as a critical phenomenon, in which there is a rapid, binary transition from a normally functioning cell to the rapid execution of a program of cell death. We hypothesized that some cells might start closer to the point of commitment than others, and these cells would be more sensitive to conventional chemotherapeutic agents that target ubiquitous elements like DNA and microtubules.69 The critical difference thus would reside not in how cells dynamically responded to chemotherapy but rather in their initial position with respect to the point of commitment to apoptosis before treatment. Testing this hypothesis required developing an objective way to measure proximity to the point of commitment to apoptosis, a property we refer to as priming (Figs 3A, 3B).
Fig 3.
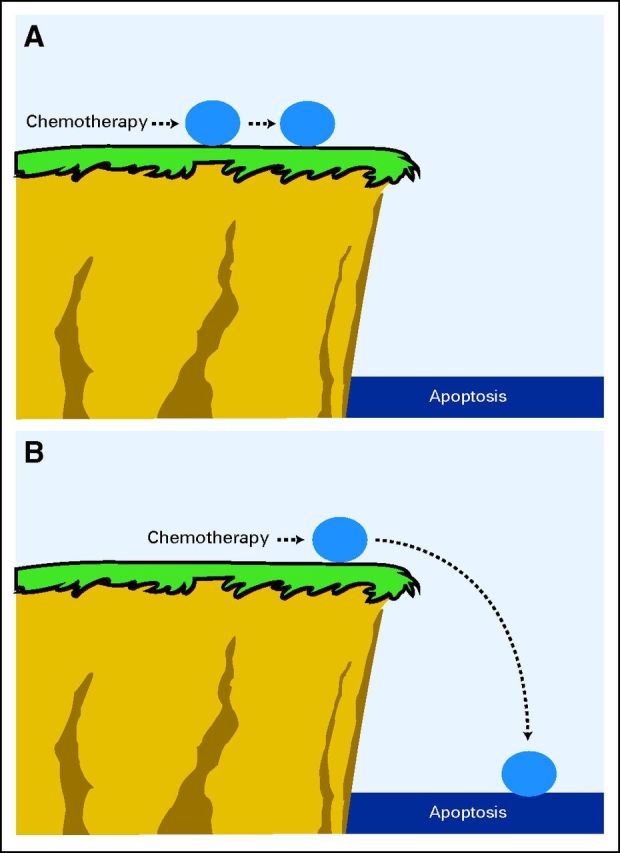
Priming is the proximity of a malignant cell to the threshold of apoptosis. (A) An unprimed cell is exposed to chemotherapy and pushed toward the threshold of apoptosis, but because it starts far from the threshold, this change is not enough to kill the cell. (B) A primed cell exposed to the same chemotherapy agent at the same dose is pushed the same distance toward the threshold of apoptosis, but because it starts closer to this threshold, the change is enough to push the cell over the edge, resulting in apoptotic cell death.
Priming can be measured with a tool known as BH3 profiling (Fig 4A), a functional assay that measures mitochondrial response to measured death signals in the form of synthetic peptides derived from the BH3 domains of BH3-only proteins. These proapoptotic peptides by themselves can activate BAX and BAK or antagonize antiapoptotic BCL-2 proteins much like the intact proteins from which they are derived. Thus, proximity to commitment to apoptosis, or priming, can be compared between cells by comparing mitochondrial sensitivity to certain BH3 peptides. Cells bearing mitochondria that are more sensitive to BH3 peptides are described as being more primed (Fig 3A) than those bearing less sensitive mitochondria, which are described as unprimed (Fig 3B).
Fig 4.

BCL-2 homology 3 (BH3) profiling. (A) Single-cell suspensions derived from peripheral blood, bone marrow, lymph nodes, solid tumors, or normal tissue are permeabilized via digitonin and stained with JC-1. Permeabilized cells are then exposed to BH3-only peptides in a 384-well format, and the decay of ΔΨm is measured as fluorescence at 590 nm, via the JC dye, by plate reader. Note that for heterogeneous clinical samples, this process can be performed with fluorescence activated cell sorting and a BH3 profile derived for a cellular subpopulation of interest. Data adapted.70 (B) Comparison of mitochondrial priming among a variety of primary human cancers and normal tissues. Cancers with clinical follow-up were classified as known chemosensitive or known chemoresistant. Cancers classified as typically chemoresistant or typically chemosensitive lacked individual clinical follow-up data. Data shown are mean ± standard deviation across all specimens tested. Analysis of variance was used to demonstrate statistical significance between the different categories, with a Tukey's multiple comparison post-test. NS, not significant (ie, P > .05). (*) P < .01. (†) P < .001. Data adapted.69
Although the conditions that lead to priming are established by the many members of the BCL-2 family, this tool does not identify the individual contribution of each. Rather, BH3 profiling provides a summary statement about the readiness of a cell to undergo apoptosis via the mitochondrial pathway. The molecular basis to priming seems to reside in a high occupation of antiapoptotic BCL-2 proteins by proapoptotic BCL-2 proteins. This is a likely explanation for the observation that although CLL and acute lymphoblastic leukemia (ALL) consistently express much more BCL-2 than any solid malignancy, they are also much more chemosensitive than almost any solid malignancy. The resolution of this apparent paradox lies in the fact that BCL-2 is highly occupied by proapoptotic proteins in both CLL and ALL.14,47 This mechanism may also be why high BCL-2 expression has not been a consistent predictor of poor prognosis in human cancers.71,72
If differences in mitochondrial priming are responsible for the therapeutic index observed for chemotherapy, then normal chemoresistant tissues should be less primed than chemosensitive cancer cells. We tested this hypothesis on normal mouse and human tissues and numerous primary cancer samples (Fig 4B).69 Consistently, chemoresistant normal mouse and human tissues were less primed than chemosensitive cancer samples. The highest primed normal tissues were hematopoietic, consistent with the well-established clinical observation that the most chemosensitive normal cells are hematopoietic in origin, so myelosuppression is the most common dose-limiting toxicity of cytotoxic chemotherapy. It thus seems that differential mitochondrial priming is an important mechanism underlying the therapeutic index of conventional chemotherapy.
Measuring Priming to Predict Response and Modulating Priming to Increase Response
If differential priming underlies differences in chemosensitivity between normal and malignant cells, might it also underlie differences between chemosensitive and chemoresistant cancers? To answer this question, we performed BH3 profiling on pretreatment samples from patients with four different malignancies: multiple myeloma, acute myeloid leukemia, ALL, and ovarian cancer.69 In all four diseases, we found that higher pretreatment priming predicted better clinical response to chemotherapy and generally more durable responses as well. We are currently exploring whether BH3 profiling might be exploited as a predictive biomarker to aid in therapeutic decision making in the clinic.
If priming is an important determinant of chemosensitivity, it seems a reasonable goal to evaluate whether priming might be selectively modulated in cancer cells. The strategy would be to identify agents that would selectively provoke apoptotic signaling in cancer cells, even if they did not provoke sufficient signaling to kill as single agents. Such agents could be applied in combination with conventional chemotherapy, which could then kill cells whose priming was increased by the targeted agent. BH3 profiling would likely be useful in the identification of such priming agents. We tested a simple proof of principle of this strategy using a cell line and ABT-737 as the priming agent and found that we could indeed increase both priming and chemosensitivity of the cell line.69
THE FUTURE
Research into the BCL-2 family of proteins finally has reached the translational stage in the last few years. It seems likely that the next few years will see the approval of the first agent or agents designed to directly target the BCL-2 family. We know that apoptosis is a final result of many types of chemotherapy. Thus, although agents targeting the BCL-2 family have generally shown only modest clinical benefit as monotherapies, it seems likely that treatment of many different cancers, hematologic and solid, might benefit from the addition of BCL-2 antagonists to combination chemotherapy regimens. There are few, if any, agents in which the mechanism of death is so well understood, from binding of target all the way to commitment to cell death. Such knowledge will likely foster the future development of useful biomarkers to better direct the use of these agents in the clinic.
Acknowledgment
M.S.D. is a Leukemia and Lymphoma Society Special Fellow in Clinical Research, and A.L. is a Leukemia and Lymphoma Society Scholar.
Footnotes
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Anthony Letai, Eutropics Pharmaceuticals (C) Stock Ownership: None Honoraria: None Research Funding: None Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Tsujimoto Y, Finger LR, Yunis J, et al. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 2.Tsujimoto Y, Jaffe E, Cossman J, et al. Clustering of breakpoints on chromosome 11 in human B-cell neoplasms with the t(11;14) chromosome translocation. Nature. 1985;315:340–343. doi: 10.1038/315340a0. [DOI] [PubMed] [Google Scholar]
- 3.Bakhshi A, Jensen JP, Goldman P, et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: Clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 4.Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci U S A. 1985;82:7439–7443. doi: 10.1073/pnas.82.21.7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pegoraro L, Palumbo A, Erikson J, et al. A 14;18 and an 8;14 chromosome translocation in a cell line derived from an acute B-cell leukemia. Proc Natl Acad Sci U S A. 1984;81:7166–7170. doi: 10.1073/pnas.81.22.7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonnell TJ, Deane N, Platt FM, et al. Bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 7.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 8.Hockenbery D, Nuñez G, Milliman C, et al. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 9.Boise LH, González-García M, Postema CE, et al. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 10.Gibson L, Holmgreen SP, Huang DC, et al. Bcl-w, a novel member of the bcl-2 family, promotes cell survival. Oncogene. 1996;13:665–675. [PubMed] [Google Scholar]
- 11.Kozopas KM, Yang T, Buchan HL, et al. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi SS, Park IC, Yun JW, et al. A novel Bcl-2 related gene, Bfl-1, is overexpressed in stomach cancer and preferentially expressed in bone marrow. Oncogene. 1995;11:1693–1698. [PubMed] [Google Scholar]
- 13.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 14.Del Gaizo Moore V, Schlis KD, Sallan SE, et al. BCL-2 dependence and ABT-737 sensitivity in acute lymphoblastic leukemia. Blood. 2008;111:2300–2309. doi: 10.1182/blood-2007-06-098012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuwana T, Mackey MR, Perkins G, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 16.Antonsson B, Montessuit S, Lauper S, et al. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2000;345:271–278. [PMC free article] [PubMed] [Google Scholar]
- 17.Korsmeyer SJ, Wei MC, Saito M, et al. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 18.Lindsten T, Ross AJ, King A, et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cartron PF, Gallenne T, Bougras G, et al. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol Cell. 2004;16:807–818. doi: 10.1016/j.molcel.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 20.Wei MC, Lindsten T, Mootha VK, et al. TBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 21.Kuwana T, Bouchier-Hayes L, Chipuk JE, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Willis SN, Fletcher JI, Kaufmann T, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 23.Cheng EH, Levine B, Boise LH, et al. Bax-independent inhibition of apoptosis by Bcl-XL. Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- 24.Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- 25.Letai A, Bassik MC, Walensky LD, et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 26.Zhai D, Jin C, Satterthwait AC, et al. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ. 2006;13:1419–1421. doi: 10.1038/sj.cdd.4401937. [DOI] [PubMed] [Google Scholar]
- 27.Lopes de Menezes DE, Hudon N, McIntosh N, et al. Molecular and pharmacokinetic properties associated with the therapeutics of bcl-2 antisense oligonucleotide G3139 combined with free and liposomal doxorubicin. Clin Cancer Res. 2000;6:2891–2902. [PubMed] [Google Scholar]
- 28.Cotter FE, Waters J, Cunningham D. Human Bcl-2 antisense therapy for lymphomas. Biochim Biochim Biophys Acta. 1999;1489:97–106. doi: 10.1016/s0167-4781(99)00139-6. [DOI] [PubMed] [Google Scholar]
- 29.O'Brien SM, Cunningham CC, Golenkov AK, et al. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J Clin Oncol. 2005;23:7697–7702. doi: 10.1200/JCO.2005.02.4364. [DOI] [PubMed] [Google Scholar]
- 30.Badros AZ, Goloubeva O, Rapoport AP, et al. Phase II study of G3139, a Bcl-2 antisense oligonucleotide, in combination with dexamethasone and thalidomide in relapsed multiple myeloma patients. J Clin Oncol. 2005;23:4089–4099. doi: 10.1200/JCO.2005.14.381. [DOI] [PubMed] [Google Scholar]
- 31.Bedikian AY, Millward M, Pehamberger H, et al. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: The Oblimersen Melanoma Study Group. J Clin Oncol. 2006;24:4738–4745. doi: 10.1200/JCO.2006.06.0483. [DOI] [PubMed] [Google Scholar]
- 32.Chanan-Khan AA, Niesvizky R, Hohl RJ, et al. Phase III randomised study of dexamethasone with or without oblimersen sodium for patients with advanced multiple myeloma. Leuk Lymphoma. 2009;50:559–565. doi: 10.1080/10428190902748971. [DOI] [PubMed] [Google Scholar]
- 33.O'Brien S, Moore JO, Boyd TE, et al. 5-year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without oblimersen. J Clin Oncol. 2009;27:5208–5212. doi: 10.1200/JCO.2009.22.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benimetskaya L, Miller P, Benimetsky S, et al. Inhibition of potentially anti-apoptotic proteins by antisense protein kinase C-alpha (Isis 3521) and antisense bcl-2 (G3139) phosphorothioate oligodeoxynucleotides: Relationship to the decreased viability of T24 bladder and PC3 prostate cancer cells. Mol Pharmacol. 2001;60:1296–1307. doi: 10.1124/mol.60.6.1296. [DOI] [PubMed] [Google Scholar]
- 35.Dai G, Chan KK, Liu S, et al. Cellular uptake and intracellular levels of the bcl-2 antisense g3139 in cultured cells and treated patients with acute myeloid leukemia. Clin Cancer Res. 2005;11:2998–3008. doi: 10.1158/1078-0432.CCR-04-1505. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen M, Marcellus RC, Roulston A, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konopleva M, Watt J, Contractor R, et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax) Cancer Res. 2008;68:3413–3420. doi: 10.1158/0008-5472.CAN-07-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schimmer AD, O'Brien S, Kantarjian H, et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14:8295–8301. doi: 10.1158/1078-0432.CCR-08-0999. [DOI] [PubMed] [Google Scholar]
- 39.Brown JR, Tesar B, Werner L, et al. Obatoclax in combination with fludarabine and rituximab (FR) is well-tolerated and shows promising clinical activity in relapsed CLL/SLL. Presented at the 53rd Annual Meeting of the American Society of Hematology; December 10-13, 2011; San Diego, CA. [Google Scholar]
- 40.Paik PK, Rudin CM, Brown A, et al. A phase I study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in solid tumor malignancies. Cancer Chemother Pharmacol. 2010;66:1079–1085. doi: 10.1007/s00280-010-1265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langer CJ, Albert P, Kovacs P, et al. A randomized phase II study of carboplatin (C) and etoposide (E) with or without pan-BCL-2 antagonist obatoclax (Ob) in extensive-stage small cell lung cancer (ES-SCLC) J Clin Oncol. 2011;29(suppl 15):453s. abstr 7001. [Google Scholar]
- 42.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 43.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 45.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 46.Vogler M, Weber K, Dinsdale D, et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009;16:1030–1039. doi: 10.1038/cdd.2009.48. [DOI] [PubMed] [Google Scholar]
- 47.Del Gaizo Moore V, Brown JR, Certo M, et al. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng J, Carlson N, Takeyama K, et al. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H, Nimmer PM, Tahir SK, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–951. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- 51.Vogler M, Hamali HA, Sun XM, et al. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood. 2011;117:7145–7154. doi: 10.1182/blood-2011-03-344812. [DOI] [PubMed] [Google Scholar]
- 52.Schoenwaelder SM, Jarman KE, Gardiner EE, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118:1663–1674. doi: 10.1182/blood-2011-04-347849. [DOI] [PubMed] [Google Scholar]
- 53.Roberts AW, Seymour JF, Brown JR, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of Navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mason KD, Carpinelli MR, Fletcher JI, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 55.Wilson WH, O'Connor OA, Czuczman MS, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gandhi L, Camidge DR, Ribeiro de Oliveira M, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kipps TJ, Swinnen LJ, Wierda WG, et al. Navitoclax (ABT-263) plus fludarabine/cyclophosphamide/rituximab (FCR) or bendaymustine/rituximab (BR): A phase I study in patients with relapsed/refractory CLL. Presented at the 53rd Annual Meeting of the American Society of Hematology; December 10-13, 2011; San Diego, CA. [Google Scholar]
- 58.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 59.Vogler M, Butterworth M, Majid A, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113:4403–4413. doi: 10.1182/blood-2008-08-173310. [DOI] [PubMed] [Google Scholar]
- 60.Yecies D, Carlson NE, Deng J, et al. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Al-Harbi S, Hill BT, Mazumder S, et al. An antiapoptotic BCL-2 family expression index predicts the response of chronic lymphocytic leukemia to ABT-737. Blood. 2011;118:3579–3590. doi: 10.1182/blood-2011-03-340364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tromp JM, Geest CR, Breij EC, et al. Tipping the Noxa/Mcl-1 balance overcomes ABT-737 resistance in chronic lymphocytic leukemia. Clin Cancer Res. 2012;18:487–498. doi: 10.1158/1078-0432.CCR-11-1440. [DOI] [PubMed] [Google Scholar]
- 63.Skipper HE, Perry S. Kinetics of normal and leukemic leukocyte populations and relevance to chemotherapy. Cancer Res. 1970;30:1883–1897. [PubMed] [Google Scholar]
- 64.Messmer BT, Messmer D, Allen SL, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest. 2005;115:755–764. doi: 10.1172/JCI23409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weinstein GD, McCullough JL, Ross P. Cell proliferation in normal epidermis. J Invest Dermatol. 1984;82:623–628. doi: 10.1111/1523-1747.ep12261462. [DOI] [PubMed] [Google Scholar]
- 66.Holdaway KM, Finlay GJ, Baguley BC. Relationship of cell cycle parameters to in vitro and in vivo chemosensitivity for a series of Lewis lung carcinoma lines. Eur J Cancer. 1992;28A:1427–1431. doi: 10.1016/0959-8049(92)90537-c. [DOI] [PubMed] [Google Scholar]
- 67.Petru E, Sevin BU, Haas J, et al. A correlation of cell cycle perturbations with chemosensitivity in human ovarian cancer cells exposed to cytotoxic drugs in vitro. Gynecol Oncol. 1995;58:48–57. doi: 10.1006/gyno.1995.1182. [DOI] [PubMed] [Google Scholar]
- 68.Hochhauser D. Modulation of chemosensitivity through altered expression of cell cycle regulatory genes in cancer. Anticancer Drugs. 1997;8:903–910. doi: 10.1097/00001813-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 69.Ni Chonghaile T, Sarosiek KA, Vo TT, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci U S A. 2010;107:12895–12900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dawson SJ, Makretsov N, Blows FM, et al. BCL2 in breast cancer: A favourable prognostic marker across molecular subtypes and independent of adjuvant therapy received. Br J Cancer. 2010;103:668–675. doi: 10.1038/sj.bjc.6605736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hamilton A, Piccart M. The contribution of molecular markers to the prediction of response in the treatment of breast cancer: A review of the literature on HER-2, p53 and BCL-2. Ann Oncol. 2000;11:647–663. doi: 10.1023/a:1008390429428. [DOI] [PubMed] [Google Scholar]