

Abstract
Purpose
Other malignancies have been reported to occur with increased frequency in chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). The aim of this study was to determine the frequency, outcomes, and factors associated with other cancers in patients with CLL/SLL.
Patients and Methods
We reviewed the records of consecutive patients with previously untreated CLL/SLL seen at The University of Texas M. D. Anderson Cancer Center from 1985 to 2005. The number of second cancers observed was compared with the number expected from the Surveillance, Epidemiology, and End Results database.
Results
Among 2,028 patients, 324 (16%) had a history of other cancers and 227 (11.2%) developed other malignancies during the follow-up period. Overall, 625 cancers were observed in 551 patients, including skin (30%), prostate (13%), breast (9%), melanoma (8%), lymphoma (8%), gastrointestinal (9%), lung (6%), and other cancers (17%). The risk of a second cancer was 2.2 times higher than the expected risk. The response rates in patients with and without a history of other cancers were 86% and 92%, respectively (P = .04), and the 5-year survival rates were 70% and 82%, respectively (P < .001). In Cox analysis, independent factors predicting development of new cancers were older age, male sex, and elevated levels of β2-microglobulin, lactate dehydrogenase, and creatinine. In patients who were treated for CLL/SLL, the treatment regimen did not affect the risk of subsequent cancer (P = .49).
Conclusion
Patients with CLL/SLL have more than twice the risk of developing a second cancer and an increased frequency of certain cancer types. Awareness of risk factors could permit early detection.
INTRODUCTION
An increased incidence of other malignant neoplasms has been reported in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and is attributed to disease- or therapy-related immunosuppression. In a retrospective analysis, it was found that in patients with CLL, the risk of all cancers was three-fold, the risk of skin cancer was eight-fold, and the risk of all cancers excluding skin cancer was two-fold higher than that for an age- and sex-matched control population.1 In another study of 4,869 patients with CLL from a series in the Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute (NCI), patients with CLL were at increased risk of developing the following secondary cancers: malignant melanoma, soft-tissue sarcomas, and lung cancer. Because the sites of the second tumors were similar to those in renal transplant recipients, the increased risk of other malignancies in patients with CLL/SLL was attributed to immunologic defects associated with disease.1,2 In addition, concerns have been raised about the carcinogenic or immunosuppressive effects of nucleoside analogs in CLL therapy. However, in a large analysis of second cancers in patients treated under the NCI Group C protocols with fludarabine for relapsed or refractory CLL, the risk of other cancers was higher than in healthy individuals but consistent with rates expected in patients with CLL.3
In the current report, we examine the frequency, characteristics, and clinical outcomes of other malignant neoplasms in patients with CLL/SLL seen at The University of Texas M. D. Anderson Cancer Center. We also assess factors predicting the development of other malignancies, and we compare the risk of second cancers in patients with CLL with the expected risk in healthy individuals.
PATIENTS AND METHODS
An electronic database of consecutive patients with CLL/SLL who presented at M. D. Anderson Cancer Center, Department of Leukemia or Lymphoma/Myeloma, between 1985 and 2005 was searched. Patient records were reviewed to determine presenting features, treatment, and outcomes. A waiver of informed consent and a waiver of authorization were granted by the M. D. Anderson institutional review board. Criteria used for the diagnosis of CLL/SLL and other malignancies are defined in the Appendix (online only).
Therapy
Disease stage and treatment were determined after all clinical, laboratory, and pathologic data were reviewed in a multidisciplinary conference. Patients were treated systemically if they had the standard indications for therapeutic intervention for CLL/SLL.4,5 Regimens varied over the years and according to the department in which the patient was seen. Assignment to different treatment regimens was based on therapy availability, patient preference, and physician discretion. Treatment regimens included in the analysis were fludarabine, with or without prednisone (F ± P)6–8; fludarabine and cyclophosphamide (FC)9; fludarabine and mitoxantrone (FM)10; fludarabine, cyclophosphamide, and rituximab (FCR)11; fludarabine, mitoxantrone, dexamethasone, and rituximab (R-FND)12; fludarabine, cyclophosphamide, mitoxantrone, and rituximab (R-FCM)13; and rituximab, with or without granulocyte-macrophage colony-stimulating factor (R ± GM-CSF).14 Progressive or relapsed disease was treated with salvage regimens, followed by stem-cell transplantation in selected patients.
End Points and Statistical Methods
Analysis of clinical outcomes.
In patients with CLL, the response criteria were those defined by the NCI-sponsored Working Group.15 In patients with SLL, the response and end point assessments conformed to the published NCI International Working Group response criteria.16 Survival was measured from the time of diagnosis of CLL/SLL until death from any cause or last follow-up. Failure-free survival (FFS) was measured from the start of CLL/SLL treatment until disease progression, relapse, or death. To assess CLL/SLL treatment outcomes, survival was measured from the start of treatment until death from any cause or last follow-up. In many patients, follow-up data were obtained from referring physicians.
The χ2 and t tests were used in univariate analyses of categoric and continuous variables, respectively. Survival and hazard functions were estimated using the Kaplan-Meier method, and survival between groups was compared using the two-sided log-rank test. The multivariate Cox proportional hazards regression model was used to examine risk factors related to survival after adjusting for other factors (P ≤ .05 was considered to be statistically significant). All cancers, including nonmelanoma skin cancer, were considered for the analyses presented here, except for the comparison with the SEER database, which does not include data for nonmelanoma skin cancers.
Cancer and mortality risk analysis.
To determine whether a higher than expected number of second primary tumors occurred after the diagnosis of CLL/SLL, we computed standardized incidence ratios (SIRs). The SIRs were calculated by determining the ratio of the observed to the expected number of individuals with second malignancies. The expected number of individuals with secondary malignancies (excluding nonmelanoma skin cancers) was determined using age, sex, and calendar year–specific incidence rates from the SEER database. The program Cohort Analysis for Genetic Epidemiology was used to calculate the SIRs with 95% CIs, assuming that the observed number of malignancies followed a Poisson distribution.17
For patients with multiple malignancies, only the first cancer other than CLL/SLL was considered. A two-sided test was used to test the equality of the observed and expected (O/E) number of cancers. Statistical analyses were carried out using SAS 8.2 and S-Plus, version 2000 software (Insightful Corp, Seattle, WA).
RESULTS
Patient Characteristics
Of the 2,359 consecutive patients identified in the database search, 233 patients were excluded for the following reasons: (1) on subsequent assessment, the diagnosis of CLL/SLL was incorrect, and the disease was better characterized as mantle-cell lymphoma, marginal zone B-cell lymphoma, or hairy cell leukemia; (2) data were incomplete; or (3) the patient was lost to follow-up. The 2,126 remaining patients were included in a previously published analysis.5 In 98 patients, no data regarding other malignant neoplasms were recorded, leaving 2,028 patients in the current analysis.
The presenting characteristics of the 2,028 patients analyzed are listed in Table 1. The median age was 58 years (range, 17 to 90 years). The median absolute lymphocyte count was 17.5 × 109/L. Conventional cytogenetic analysis was performed in 1,551 patients. Most patients (75%) had normal conventional cytogenetics. Fluorescence in situ hybridization analysis was performed in 345 patients. The most common abnormalities are reported in Table 1.
Table 1.
Presenting Characteristics of 2,028 Patients With Untreated CLL/SLL
| Variable | No. of Patients | Total Patients | % |
|---|---|---|---|
| Age ≥ 60 years | 864 | 2,028 | 43 |
| Male sex | 1,240 | 2,028 | 61 |
| Zubrod performance status 0 to 1 | 1,992 | 2,028 | 98 |
| ALC ≥ 30 × 109/L | 669 | 2,028 | 33 |
| Hemoglobin ≥ 11 g/dL | 1,851 | 2,010 | 92 |
| Platelets ≥ 100 × 109/L | 1,863 | 2,010 | 93 |
| Creatinine ≥ 1.5× normal | 44 | 1,994 | 2 |
| Albumin > 3.5 g/dL | 1,904 | 1,985 | 96 |
| LDH ≥ 1× normal | 323 | 1,974 | 16 |
| β2-microglobulin ≥ 3 mg/L | 560 | 1,899 | 29 |
| Bone marrow lymphocytes > 30% | 1,703 | 1,914 | 89 |
| Lymphadenopathy | 1,407 | 2,028 | 69 |
| Hepatomegaly | 133 | 2,007 | 7 |
| Splenomegaly | 351 | 1,995 | 18 |
| Rai stage III to IV | 249 | 2,028 | 12 |
| Cytogenetics | |||
| 17p deletion or 6q deletion | 43 | 1,551 | 3 |
| 11q deletion | 52 | 1,551 | 3 |
| Trisomy 12 | 81 | 1,551 | 5 |
| Fluorescence in situ hybridization | |||
| 17p deletion | 20 | 345 | 6 |
| 11q deletion | 45 | 345 | 13 |
| Trisomy 12 | 55 | 345 | 16 |
| 13q deletion | 122 | 345 | 35 |
| Negative | 103 | 345 | 30 |
| IgVH, unmutated | 213 | 490 | 43 |
| ZAP70-positive | 185 | 479 | 39 |
| CD38-positive | 251 | 1,041 | 24 |
Abbreviations: CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; ALC, absolute lymphocyte count; LDH, lactate dehydrogenase.
Survival
The median follow-up duration for all 2,028 patients was 4.14 years (range, 0 to 20.5 years). The median duration of survival was 10.9 years (95% CI, 10.2 to 11.9) for patients without a history of other malignant neoplasms and 7.6 years (95% CI, 6.4 to 10.0) for patients with a history of other malignancies at the time of presentation at M. D. Anderson. The actuarial 5-year survival rate of the 1,704 patients without a history of other malignant neoplasms was 82% (95% CI, 80% to 85%), compared with 70% (95% CI, 63% to 76%) for the 324 patients with a history of other malignancies at the time of presentation at M. D. Anderson (P < .0001; Fig 1A). Overall, 459 patients have died. The causes of death were as follows: progressive CLL/SLL, n = 110 (24%); infectious complications of CLL/SLL, n = 112 (24.4%); other malignancies, n = 67 (14.6%); cardiopulmonary failure, n = 16 (3.5%); renal failure, n = 11 (2.4%); other comorbidities (diabetes, liver disease, neurologic disease), n = 4 (0.9%); graft-versus-host disease, n = 3 (0.7%); accident/fall, n = 3 (0.7%); thromboembolic episodes, n = 2 (0.4%); and unknown, n = 131 (28.5%). Independent prognostic factors for survival are found in the Appendix (online only).
Fig 1.
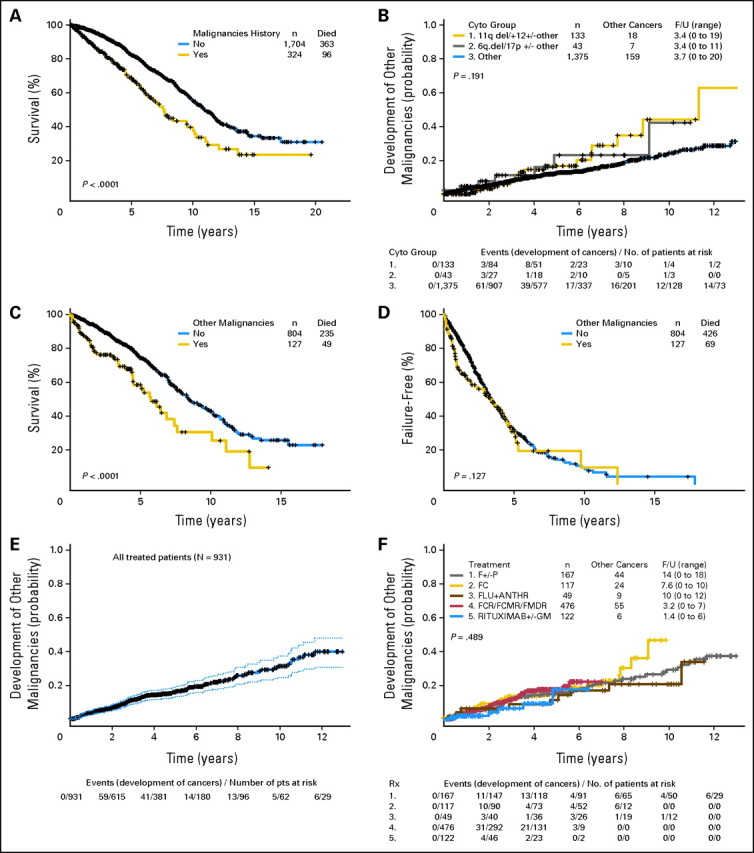
(A) Overall survival of 2,028 patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) by history of other malignancies (from The University of Texas M. D. Anderson Cancer Center presentation date to death or last follow-up). (B) Time from M. D. Anderson Cancer Center presentation date to development of other malignancies by cytogenetic group. (C) Overall survival of 931 patients who required treatment for CLL/SLL, by presence of other malignancies (from date of initiation of therapy). (D) Failure-free survival of 931 patients with CLL/SLL by history of other malignancies at the time of initiation of CLL/SLL therapy. (E) Overall survival of patients who required CLL/SLL treatment (from initiation of CLL/SLL treatment) and 95% CI. (F) Time to development of other malignancies (from initiation of CLL treatment) by treatment regimen. F/U, follow-up; F, fludarabine; P, prednisone; FLU, fludarabine; ANTHR, anthracycline; FCR, fludarabine, cyclophosphamide, and rituximab; FCMR, fludarabine, cyclophosphamide, mitoxantrone and rituximab; FMDR, fludarabine, mitoxantrone, dexamethasone, and rituximab; GM, granulocyte-macrophage colony-stimulating factor.
Incidence of Other Malignancies
A total of 625 cancers were reported or observed in the 551 patients (27.5%) who had a history of another malignant neoplasm at the time of their first visit to M. D. Anderson or developed a second cancer during the study period. The types of cancers were as follows: skin carcinomas (n = 187, 29.9%), prostate (n = 80, 12.8%), breast (n = 58, 9.3%), gastrointestinal (n = 56, 9%), melanoma (n = 53, 8.4%), lymphoma (n = 51, 8.1%), lung (n = 38, 6.1%), urinary tract (n = 23, 3.7%), genital (n = 23, 3.7%), myeloid tumors (n = 16, 2.6%), endocrine (n = 19, 3%), brain (n = 6, 1%), sarcoma (n = 6, 1%), and other cancers (n = 9, 1.4%). Overall, 487 patients (88.4%) had one other malignant neoplasm, 60 patients (10.9%) had two, three patients (0.5%) had three, and one patient (0.02%) had four other malignancies.
Comparison With SEER Data
A second cancer developed in 216 patients after the diagnosis of CLL/SLL. The median follow-up duration among patients who developed a second cancer was 6.3 years (range, 0 to 20.5 years). Among our patients with CLL/SLL, the overall risk of having a second cancer was 2.2 times higher than the expected risk (calculated from the SEER data) and was higher in male patients and in patients ≥ 60 years (Table 2). Eleven patients with nonmelanoma skin cancer were excluded from this analysis.
Table 2.
Comparison of Frequency of Other Cancers in CLL/SLL Patients at The University of Texas M. D. Anderson Cancer Center With SEER Data
| Variable | Observed No. of Cancers | Expected No. of Cancers | O/E | 95% CI for O/E |
|---|---|---|---|---|
| Total | 216 | 98.03 | 2.20 | 1.93 to 2.51 |
| Sex | ||||
| Male | 142 | 63.51 | 2.24 | 1.90 to 2.62 |
| Female | 74 | 34.5 | 2.14 | 1.71 to 2.66 |
| Age, years | ||||
| < 60 | 79 | 39.18 | 2.02 | 1.62 to 2.48 |
| ≥ 60 | 137 | 58.83 | 2.33 | 1.97 to 2.73 |
| Site | ||||
| Non-Hodgkin's lymphoma | 42 | 4.46 | 9.42 | 6.98 to 12.47 |
| Hodgkin's lymphoma | 5 | 0.34 | 14.71 | 6.48 to 30.12 |
| Acute myeloid leukemia | 6 | 0.76 | 7.09 | 3.7 to 15.35 |
| Prostate | 32 | 23.68 | 1.35 | 0.96 to 1.98 |
| Lung | 23 | 20.47 | 1.12 | 0.75 to 1.63 |
| Breast (female) | 20 | 12.56 | 1.59 | 1.03 to 2.36 |
| Melanoma | 19 | 3.08 | 6.17 | 3.97 to 9.24 |
| Other myeloid leukemias* | 10 | 0 | 0 | N/A |
Abbreviations: CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; SEER, Surveillance, Epidemiology, and End Results; O/E, observed/expected.
Includes esophageal, lower gastrointestinal, liver, pancreas, stomach, small intestine, and/or any site of the digestive system.
Our patients with CLL/SLL had a significantly higher observed/expected (O/E) ratio of Hodgkin's lymphoma, non-Hodgkin's lymphoma, acute myeloid leukemia, other myeloid (including monocytic) leukemias, melanoma, and female breast cancers compared with the SEER data (Table 2). In contrast, the O/E ratio was lower for urinary bladder cancer (0.73; 95% CI, 0.32 to 1.49) and all gastrointestinal cancers (0.74; 95% CI, 0.53 to 1.02). Our patients with CLL/SLL had also lower O/E ratios for cancers of the female genital system, brain, esophagus, colon excluding rectum, rectum and rectosigmoid junction, lung, ovary, thymus, and pancreas compared with the SEER data.
Correlation Between History of Other Malignancies and Patient Characteristics
Three hundred twenty-four patients (16%) had a history of other malignant neoplasms at the time of presentation to M. D. Anderson. Patients with a history of other malignancies were more commonly diagnosed with CLL/SLL at an older age and typically had higher serum β2-microglobulin and creatinine levels and lower levels of albumin at the time of the CLL/SLL diagnosis (Appendix Table A1, online only). A history of other malignant neoplasms was also associated with a smoking history (Table 3).
Table 3.
Presenting Characteristics of Patients With CLL/SLL by History of Other Cancers at The University of Texas M. D. Anderson Cancer Center Presentation Date
| Characteristic | History of Other Cancers |
No History of Other Cancers |
P* | ||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Age, years | |||||
| < 60 | 100 | 9 | 1,064 | 91 | < .001 |
| ≥ 60 | 224 | 26 | 640 | 74 | |
| Sex | |||||
| Female | 117 | 15 | 671 | 85 | .297 |
| Male | 207 | 17 | 1,033 | 83 | |
| Smoking history, n = 1,709 | |||||
| Yes | 148 | 18 | 656 | 82 | .007 |
| No | 122 | 14 | 783 | 86 | |
| β2M, mg/L, n = 1,899 | |||||
| < 2 | 82 | 13 | 547 | 87 | .008 |
| ≥ 2 to < 3 | 114 | 16 | 596 | 84 | |
| ≥ 3 | 110 | 20 | 450 | 80 | |
| Creatinine, mg/dL, n = 1,994 | |||||
| < 1.5 | 305 | 16 | 1,645 | 84 | .007 |
| ≥ 1.5 | 14 | 32 | 30 | 68 | |
| Albumin, g/dL, n = 1,985 | |||||
| ≤ 3.5 | 20 | 25 | 61 | 75 | .042 |
| > 3.5 | 297 | 16 | 1,607 | 84 | |
| Cytogenetics, n = 1,551 | |||||
| del 17p or 6q | 7 | 16 | 36 | 84 | .590 |
| 11q del or +12 | 25 | 19 | 108 | 81 | |
| Others | 212 | 15 | 1,163 | 85 | |
Abbreviation: CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma.
P values are derived from χ2 test to assess association between categorical data.
Factors Predicting Development of Second Cancers After CLL/SLL Diagnosis
We assessed, by baseline characteristics, the hazard functions for developing other second cancers from the time of presentation at M. D. Anderson until last follow-up. Figure 1B shows the proportion of patients who developed second malignant neoplasms after presentation at M. D. Anderson stratified by conventional cytogenetics. At 2 years, 8% of patients with 17p deletion and/or 6q deletion developed other cancers, compared with 3% of patients with 11q deletion and/or trisomy 12, and 5% of patients with other cytogenetic abnormalities. The percentages at 4 years were 12%, 14%, and 10%, respectively. There was no significant difference in risk of other cancers among the three cytogenetic groups (P = .191; Fig 1B). However, patients with 17p deletion, 6q deletion, or 11q deletion and/or trisomy 12 were at a marginally increased risk of developing other cancers (P = .07) compared with other patients. Patients older than 60 years (P < .0001), male sex (P = .006), β2-microglobulin higher than 3 mg/L (P < .0001), creatinine ≥ 1.6 mg/dL (P = .019), lactate dehydrogenase higher than the upper limit of normal (P = .0001), hemoglobin lower than 11 g/dL (P = .003), and splenomegaly (P = .008) were also at increased risk of developing second cancers, but a history of smoking was only marginally associated with increased risk (P = .059).
In Cox analysis, independent factors predicting the development of second cancers were older age, male sex, and elevated levels of β2-microglobulin (> 3 mg/L), lactate dehydrogenase (> 618 U/L), and creatinine (> 1.6 mg/dL; Table 4). Other factors were not statistically significant (see online only Appendix).
Table 4.
Independent Factors Predicting Development of Other Cancers (Cox analysis)
| Factor | RR | 95% CI | Z Score | P |
|---|---|---|---|---|
| Older age | 2.13 | 1.60 to 2.85 | 5.12 | < .001 |
| Male sex | 1.62 | 1.20 to 2.20 | 3.14 | .002 |
| β2-microglobulin > 3 mg/L | 1.54 | 1.10 to 2.17 | 2.49 | .01 |
| Lactate dehydrogenase > 618 U/L | 1.48 | 1.05 to 2.09 | 2.25 | .025 |
| Creatinine > 1.6 mg/dL | 1.92 | 0.94 to 3.95 | 1.79 | .07 |
Abbreviation: RR, relative risk.
Clinical Outcomes in Patients Requiring Therapy for CLL/SLL
Overall, 931 (45.9%) of 2,028 patients required therapy for CLL/SLL, including 127 patients (13.6%) who had other preexisting malignancies at the time of initiation of CLL therapy. The complete response rates in patients with and without a history of other malignancies were 46.5% (59 of 127) and 48.9% (393 of 804), respectively (P = .61). The overall response rates were 85.8% (109 of 127) and 91.5% (736 of 804), respectively (P = .04; χ2 test).
The median follow-up duration for patients requiring therapy was 5.29 years (range, 0 to 21 years). Patients with a history of other cancers had a longer period of time to treatment for CLL/SLL than other patients (Appendix Figs A1 and A2, online only). Survival rates were lower in patients treated for CLL/SLL who had a history of other malignant neoplasms than in those who did not. Forty-nine (39%) of 127 patients with preexisting other malignancies have died, compared with 235 (29%) of 804 without preexisting malignancies. The median survival duration of patients with a history of other malignancies was 5.7 years (95% CI, 4.5 to 7.6 years), compared with 8.4 years (95% CI, 7.6 to 9.9) for those without a history of other malignancies at the time of initiation of CLL/SLL therapy, and the 5-year survival rates were 58% and 74%, respectively (P < .0001; Fig 1C). Respective 5-year survival rates were 53% and 68% for patients ≥ 60 years of age and 60% and 82% for patients younger than 60 years.
The median FFS durations in patients treated for CLL/SLL who did or did not have a history of other malignancies were 3.36 years (95% CI, 1.9 to 4.5) and 3.37 years (95% CI, 3.1 to 3.7), respectively (P = .127). Notably, the 2-year FFS rates were 58% (95% CI, 50% to 69%) and 71% (95% CI, 68% to 75%), respectively, but the curves started to overlap after 2.5 years (P = .127; Fig 1D).
Development of Second Malignant Neoplasm After CLL/SLL Therapy
One hundred thirty-eight (14.8%) of 931 treated patients developed another cancer during the follow-up period (median follow-up duration, 5.3 years; Fig 1E). The number and ratio of other cancers that developed after initiation of CLL/SLL therapy by regimen are listed in Table 5 and Figure 1F. The median follow-up of patients treated with F ± P was 14 years (range, 0 to 18 years); for patients treated with FC, 7.6 years (range, 0 to 10 years); for patients treated with FM, 10 years (range, 0 to 12 years); for patients treated with FCR, R-FND, or R-FCM, 3.2 years (range, 0 to 7 years); and for patients treated with R ± GM-CSF, 1.4 years (range, 0 to 6 years).
Table 5.
Response of CLL/SLL by Treatment in Patients With or Without Other Malignant Neoplasms
| Variable | Treated Patients | F ± P |
FM |
FC |
FCR/R-FCM/R-FND |
R ± GM |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | No. | % | No. | % | ||
| No. of patients treated | 931 | 167 | 49 | 117 | 476 | 122 | |||||
| CR | 452 | 52 | 31 | 17 | 35 | 44 | 38 | 328 | 69 | 11 | 9 |
| Overall response | 845 | 150 | 90 | 42 | 86 | 103 | 88 | 455 | 96 | 95 | 78 |
| Median follow-up, years | 4.3 | 14.3 | 10.3 | 7.6 | 3.2 | 1.4 | |||||
| Had other cancer before CLL/SLL therapy | 127 | 16 | 9.6 | 7 | 14.3 | 17 | 14.5 | 66 | 13.9 | 21 | 17.2 |
| Developed other cancer only after CLL/SLL therapy | 138 | 44 | 26.3 | 9 | 18.4 | 24 | 20.5 | 55 | 11.6 | 6 | 4.9 |
| No other cancer | 666 | 107 | 64.1 | 33 | 67.4 | 76 | 65.0 | 355 | 74.6 | 95 | 77.9 |
Abbreviations: CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; F ± P, fludarabine, with or without prednisone; FM, fludarabine and mitoxantrone; FC, fludarabine and cyclophosphamide; FCR, fludarabine, cyclophosphamide, and rituximab; R-FCM, fludarabine, cyclophosphamide, mitoxantrone, and rituximab; R-FND, fludarabine, mitoxantrone, dexamethasone, and rituximab; R ± GM, rituximab, with or without granulocyte-macrophage colony-stimulating factor; CR, complete response.
At 2 years, the percentage of patients who developed other cancers, according to treatment regimen, was 5.5% for F ± P; 9.2% for FC; 6.3% for FM; 7.7% for FCR, R-FND, or R-FCM; and 4% for R ± GM-CSF. Patients who had a longer follow-up after treatment with a specific regimen were more likely to develop a second cancer. There was no statistically significant difference in the proportion of patients who developed other cancers by regimen (log-rank test, P = .489). However, the shorter follow-up for patients on some regimens precludes definitive conclusions at this time.
DISCUSSION
We found that other malignant neoplasms occurred 2.2 times more frequently in our patients with CLL/SLL than in the general population. Sixteen percent of patients had a history of other malignancies at the time of their presentation at M. D. Anderson with CLL/SLL; this high fraction probably reflects a selection bias for referral to our institution. Among our cohort of patients with CLL/SLL without an antecedent malignancy, 11.2% subsequently developed other cancers, at a median of 2.9 years (range, 0 to 17 years) after the initial visit to M. D. Anderson.
A fundamental question that arises is what factors contribute to the development of second cancers in patients with CLL/SLL. The majority of our patients were treated with fludarabine-containing regimens. Some investigators argue that purine analogs have carcinogenic effects or that they can increase the risk of Richter's transformation in patients with CLL/SLL.18 However, a recent NCI study demonstrated that the risk of other cancers in fludarabine-treated patients with relapsed/refractory CLL was similar to that expected in all patients with CLL and was not attributed to fludarabine.3 Others have raised concerns about 2′-chloro-2′-deoxyadenosine and cyclophosphamide combination therapy, on the basis of a 13% incidence of other cancers in patients with advanced lymphoproliferative malignancies after a median of 16 months.19 Several classes of chemotherapeutic agents are clearly carcinogenic, including alkylating agents, which have been related to the induction of bone cancer in children.20 Controversy about the carcinogenic potential of therapy is further compounded when successful combinations such as FCR result in the prolongation of FFS and overall survival.5,18–21
In the current study, lower rates of response and survival were noted in patients with a history of other cancers. This may be attributed to factors associated with the other malignant neoplasms, including the effects of therapy for the other malignancy. However, a history of other cancers was not an independent factor for survival in multivariate analysis. It became significant only when cytogenetic analysis was excluded from the model, indicating a possible interaction between cytogenetic characteristics and prior malignancies. This finding, together with the marginally increased risk of developing other malignancies in patients with 17p, 6q, or 11q deletion or trisomy 12 (P = .07), suggests that certain cytogenetic abnormalities may be associated with an increased risk of other malignancies in CLL/SLL.
Genetic predisposition has been considered to be a possible contributory factor for the preponderance of second cancers in CLL/SLL.22 The NCI has implemented new research strategies to advance the study of the role of genetic susceptibility to second primary cancers and to implement a national research infrastructure for studies of cancer survivorship.22
The current study shows that the proportion of other malignant neoplasms in patients who required CLL/SLL treatment increased proportionally with duration of follow-up (Fig 1F), and our analysis also demonstrated that a history of other cancers before the CLL/SLL diagnosis was associated with older age; elevated levels of serum β2-microglobulin, creatinine, and albumin; and a smoking history. Independent factors predicting development of other cancers were older age, male sex, and elevated levels of β2-microglobulin, lactate dehydrogenase, and creatinine. Overall, the risk of developing second cancers in patients with CLL in our series (O/E ratio, 2.2; 95% CI, 1.93 to 2.51) was higher than the risk reported by other investigators (O/E ratio, 1.28; 95% CI, 1.19 to 1.37).23 A higher than expected number of hematologic malignancies, melanoma, and female breast cancers were noted in our patients with CLL/SLL. However, a lower than expected number of urinary bladder cancers and all gastrointestinal cancers was observed.
In conclusion, our study emphasizes that other malignancies frequently coexist with CLL/SLL. An association between CLL/SLL treatment and the development of other cancers is not evident, partly because of our observation that patients who required CLL/SLL therapy were not comparable to those who did not need treatment in terms of their presenting features. Further investigation of genetic features that predispose patients with CLL/SLL to develop other malignant neoplasms is warranted. Even with our current imperfect understanding of the predisposition for second cancers, awareness of the risk could permit early detection of these malignancies.
Appendix
The Appendix is included in the full-text version of this article, available online at www.jco.org. It is not included in the PDF version (via Adobe® Reader®).
Patients and Methods
Diagnosis of chronic lymphocytic leukemia/small lymphocytic lymphoma.
The diagnosis of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) was made by morphologic examination with ancillary immunophenotyping. Patients with CLL presented with involvement of blood (absolute lymphocyte count ≥ 5 × 109/L) and bone marrow, and a subset also had lymphadenopathy. Morphologically, most of the neoplastic lymphocytes were small, with condensed chromatin. Patients with SLL had a blood absolute lymphocyte count of less than 5 × 109/L, presented with lymphadenopathy, and underwent tissue biopsy. Biopsy specimens were diffusely characterized by predominantly small lymphocytes, and pseudofollicles (proliferation centers) were identified in most cases. All cases of CLL/SLL had a B-cell immunophenotype (CD19 or CD20 expression), with aberrant expression of CD5. In most cases more recently assessed, surface immunoglobulin and CD20 expression was dim, CD23 was usually positive, and FMC-7 and CD79b were usually negative. All cases assessed were negative for cyclin D1 and lacked cytogenetic or molecular evidence of t(11;14)(q13;q32). All cases assessed also lacked evidence of t(14;18)(q32;q21) involving bcl-2.
Diagnosis of other malignant neoplasms.
Once patients presented to The University of Texas M. D. Anderson Cancer Center and the diagnosis of CLL/SLL was established, an attempt was made to review the pathologic findings of antecedent other malignant neoplasms that arose in this patient group. This review, which is standard procedure at our institution, included examination of diagnostic histologic slides and the accompanying pathology report that often included confirmatory immunophenotypic data. In some cases, only pathology reports or documents that summarized the pathology findings were available for review. Virtually all of the second cancers that arose after the diagnosis of CLL/SLL were reviewed. These patients were observed at M. D. Anderson Cancer Center, and many of these patients underwent biopsy and pathologic examination at our institution.
Results
When only patients who developed other cancers were considered, the median time from diagnosis of a prior malignancy to CLL/SLL was 5.9 years (25th and 75th percentiles, 1.0 to 12.8 years). The median time from diagnosis of CLL/SLL to another malignancy was 3.4 years (25th and 75th percentiles, 1.5 to 7.3 years). There was no apparent window in the development of other cancers.
Independent prognostic factors for survival.
Factors that independently predicted shorter survival in CLL/SLL patients included 17p or 6q deletion, with or without other cytogenetic abnormalities, age at least 60 years, β2-microglobulin level at least 2 mg/L, albumin level less than 3.5 g/dL, creatinine level at least 1.6 mg/dL, absolute lymphocyte count higher than 30 × 109/L, male sex, and hepatomegaly, as previously published.5 A history of other cancers was not significant (P = .21), but when cytogenetics were excluded from the model, a history of other malignancies at the time of presentation at M. D. Anderson became an independent adverse factor predicting overall survival (P = .009).
Factors predicting development of other cancers after CLL/SLL diagnosis.
Absolute lymphocyte count (P = .10), WBC count (P = .13), platelet count (P = .35), albumin level (P = .10), hepatomegaly (P = .33), and performance status (P = .32) were not significantly associated with risk of other malignancies.
In patients with Rai stage II, III, or IV CLL, there was no difference in the incidence of second cancers between those who required and those who did not require CLL treatment (P = .47, P = .21, and P = .15, respectively). Among patients with Rai stage 0 to I disease, those who required CLL treatment had a higher risk of developing another cancer (after presentation at M. D. Anderson) compared with those who did not require treatment (P = .004).
Multivariate analysis: Presenting characteristics of patients with CLL/SLL by history of other cancers.
We fitted a multivariate logistic model using a stepwise variable selection for the presenting characteristics of patients with CLL/SLL by history of other cancers (at M. D. Anderson presentation date). Results are shown in Appendix Table A1. Age 60 years or older was an independent factor associated with history of other cancers (relative risk [RR] = 3.52; P < .001), whereas elevated creatinine (RR = 2.15; P = .055) and smoking history (RR = 1.30; P = .061) were marginally associated with history of other cancers.
Time to CLL/SLL treatment.
When only patients who required CLL/SLL therapy were considered, the median time from first visit at M. D. Anderson (Leukemia or Lymphoma clinic) to CLL/SLL treatment was 0.172 years (95% CI, 0.14 to 0.24 years) for patients without a history of other cancers and 0.175 years (95% CI, 0.14 to 0.31 years) for patients with a history of other cancers (log-rank test, P = .676; Appendix Fig A1). When all 2,028 patients with CLL/SLL were considered, the median time from first visit at M. D. Anderson (Leukemia or Lymphoma clinic) to CLL/SLL treatment for patients without a history of other cancers was 3.0 years (95% CI, 2.5 to 3.4 years) compared with 8.9 years (95% CI, 5.7 to 20+ years) for patients with a history of other cancers (log-rank test, P = .0002; Appendix Fig A2). The longer period of time to treatment in patients with a history of other cancers is likely attributed to lead-time bias (ie, patients with a history of other cancers are commonly observed more aggressively), and their CLL/SLL was diagnosed earlier than that of those referred to our institution with CLL/SLL.
Discussion
Other investigators have reported on the increased incidence of B-cell malignancies in CLL; they found that the familial component of CLL is shared with other lymphoproliferative malignances, suggesting common genetic pathways (Goldin LR, Pfeiffer RM, Li X, et al. Blood 104:1850-1854, 2004).
Figure A1.

Time to treatment from first visit to The University of Texas M. D. Anderson Cancer Center until last follow-up in 931 treated patients by history of other malignancies.
Figure A2.

Time to treatment from first visit to The University of Texas M. D. Anderson Cancer Center until last follow-up in 2,028 patients with chronic lymphocytic leukemia/small lymphocytic lymphoma regardless of requirement for treatment.
Table A1.
Multivariate Analysis: Presenting Characteristics of Patients With CLL/SLL and History of Other Cancers
| Variable | RR | 95% CI | Z Score | P |
|---|---|---|---|---|
| Age ≥ 60 years | 3.52 | 2.64 to 4.69 | 8.56 | < .001 |
| Creatinine > 1.6 mg/dL | 2.15 | 0.98 to 4.68 | 1.92 | .055 |
| Smoking history | 1.30 | 0.99 to 1.72 | 1.87 | .061 |
Abbreviations: CLL/SLL, chronic lymphocytic leukemia/small lymphocytic lymphoma; RR, relative risk.
Footnotes
Supported in part by a Career Development Award from the American Society of Clinical Oncology (A.-M.T.).
Presented in part at the 48th Annual Meeting of the American Society of Hematology, December 9-12, 2006, Orlando, FL; and at the 10th International Conference on Malignant Lymphoma, June 4-7, 2008, Lugano, Switzerland.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Peter McLaughlin, Millennium Pharmaceuticals (U), Berlex (U); Susan O'Brien, Genta (C), Geminx (C), Biogen Idec (C), Eli Lilly (C); Michael J. Keating, Genentech (U) Stock Ownership: None Honoraria: Peter McLaughlin, Co-Med Communications, Physicians Educations Resource, Cogenix, Health Science Communications; Michael J. Keating, Bayer Oncology, Genentech Research Funding: Peter McLaughlin, Berlex, Biogen IDEC Pharmaceuticals, Genentech, Schering-Plough, Integrated Therapeutics, OSI Pharmaceutical, Millenium Pharmaceuticals, Bayer Pharmaceuticals; Susan O'Brien, Genentech, Berlex, Genta, Biogen Idec, Eli Lilly, Novartis, Bristol-Myers Squibb, GeminX; Michael J. Keating, Bayer Oncology, Genentech Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Apostolia-Maria Tsimberidou
Financial support: Apostolia-Maria Tsimberidou, Michael J. Keating
Administrative support: Apostolia-Maria Tsimberidou, Peter McLaughlin, Susan Lerner, Michael J. Keating
Provision of study materials or patients: Peter McLaughlin, Susan O'Brien, William G. Wierda, L. Jeffrey Medeiros, Hagop M. Kantarjian, Michael J. Keating
Collection and assembly of data: Apostolia-Maria Tsimberidou, Sijin Wen, Susan Lerner, Michael J. Keating
Data analysis and interpretation: Apostolia-Maria Tsimberidou, Sijin Wen, Sara Strom, Emil J. Freireich, Michael J. Keating
Manuscript writing: Apostolia-Maria Tsimberidou, Sijin Wen, Peter McLaughlin, L. Jeffrey Medeiros
Final approval of manuscript: Apostolia-Maria Tsimberidou, Sijin Wen, Peter McLaughlin, Susan O'Brien, William G. Wierda, Susan Lerner, Sara Strom, Emil J. Freireich, L. Jeffrey Medeiros, Hagop M. Kantarjian, Michael J. Keating
References
- 1.Manusow D, Weinerman BH. Subsequent neoplasia in chronic lymphocytic leukemia. JAMA. 1975;232:267–269. [PubMed] [Google Scholar]
- 2.Greene MH, Hoover RN, Fraumeni JF., Jr Subsequent cancer in patients with chronic lymphocytic leukemia: A possible immunologic mechanism. J Natl Cancer Inst. 1978;61:337–340. [PubMed] [Google Scholar]
- 3.Cheson BD, Vena DA, Barrett J, et al. Second malignancies as a consequence of nucleoside analog therapy for chronic lymphoid leukemias. J Clin Oncol. 1999;17:2454–2460. doi: 10.1200/JCO.1999.17.8.2454. [DOI] [PubMed] [Google Scholar]
- 4.Rai K, Patel DV. Chronic lymphocytic leukemia. In: Hoffman R, editor. Hematology: Basic Principles and Practice. ed 3. Philadelphia, PA: Churchill Livingstone; 2000. pp. 1350–1363. [Google Scholar]
- 5.Tsimberidou AM, Wen S, O'Brien S, et al. Assessment of chronic lymphocytic leukemia and small lymphocytic lymphoma by absolute lymphocyte counts in 2,126 patients: 20 years of experience at the University of Texas M. D. Anderson Cancer Center. J Clin Oncol. 2007;25:4648–4656. doi: 10.1200/JCO.2006.09.4508. [DOI] [PubMed] [Google Scholar]
- 6.Keating MJ, Kantarjian H, O'Brien S, et al. Fludarabine: A new agent with marked cytoreductive activity in untreated chronic lymphocytic leukemia. J Clin Oncol. 1991;9:44–49. doi: 10.1200/JCO.1991.9.1.44. [DOI] [PubMed] [Google Scholar]
- 7.Keating MJ, O'Brien S, Kantarjian H, et al. Long-term follow-up of patients with chronic lymphocytic leukemia treated with fludarabine as a single agent. Blood. 1993;81:2878–2884. [PubMed] [Google Scholar]
- 8.O'Brien S, Kantarjian H, Beran M, et al. Results of fludarabine and prednisone therapy in 264 patients with chronic lymphocytic leukemia with multivariate analysis-derived prognostic model for response to treatment. Blood. 1993;82:1695–1700. [PubMed] [Google Scholar]
- 9.O'Brien SM, Kantarjian HM, Cortes J, et al. Results of the fludarabine and cyclophosphamide combination regimen in chronic lymphocytic leukemia. J Clin Oncol. 2001;19:1414–1420. doi: 10.1200/JCO.2001.19.5.1414. [DOI] [PubMed] [Google Scholar]
- 10.Tsimberidou AM, Keating MJ, Giles FJ, et al. Fludarabine and mitoxantrone for patients with chronic lymphocytic leukemia. Cancer. 2004;100:2583–2591. doi: 10.1002/cncr.20264. [DOI] [PubMed] [Google Scholar]
- 11.Keating MJ, O'Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol. 2005;23:4079–4088. doi: 10.1200/JCO.2005.12.051. [DOI] [PubMed] [Google Scholar]
- 12.McLaughlin P, Hagemeister FB, Rodriguez MA, et al. Safety of fludarabine, mitoxantrone, and dexamethasone combined with rituximab in the treatment of stage IV indolent lymphoma. Semin Oncol. 2000;27:37–41. [PubMed] [Google Scholar]
- 13.Forstpointner R, Dreyling M, Repp R, et al. The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: Results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood. 2004;104:3064–3071. doi: 10.1182/blood-2004-04-1323. [DOI] [PubMed] [Google Scholar]
- 14.McLaughlin P, Liu N, Poindexter N, et al. Rituximab plus GM-CSF for indolent lymphoma. Ann Oncol; Proceedings of the 9th International Conference on Malignant Lymphomas; Lugano, Switzerland. 2005. abstr 104. [Google Scholar]
- 15.Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: Revised guidelines for diagnosis and treatment. Blood. 1996;87:4990–4997. [PubMed] [Google Scholar]
- 16.Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non-Hodgkin's's lymphomas: NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 17.Liu M, Scheurer ME, Bondy ML, et al. Extension of the cohort analysis for genetic epidemiology program to assess excess risk of cancer. Proceedings of the Joint Statistical Meetings American Statistical Association 2546-2550; 2007. abstr. [Google Scholar]
- 18.Hamblin TJ. Richter's syndrome: The downside of fludarabine? Leuk Res. 2005;29:1103–1104. doi: 10.1016/j.leukres.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 19.Van Den Neste E, Michaux L, Layios N, et al. High incidence of complications after 2-chloro-2′-deoxyadenosine combined with cyclophosphamide in patients with advanced lymphoproliferative malignancies. Ann Hematol. 2004;83:356–363. doi: 10.1007/s00277-004-0858-7. [DOI] [PubMed] [Google Scholar]
- 20.Hawkins MM, Wilson LM, Burton HS, et al. Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst. 1996;88:270–278. doi: 10.1093/jnci/88.5.270. [DOI] [PubMed] [Google Scholar]
- 21.Tam CS, O'Brien S, Wierda W, et al. Seventy percent of complete responders remain in continuous remission: Five-year follow-up of 300 patients treated with fludarabine, cyclophosphamide, and rituximab (FCR) as initial therapy of CLL. J Clin Oncol. 2007;25(suppl):359s. abstr 7008. [Google Scholar]
- 22.Travis LB, Rabkin CS, Brown LM, et al. Cancer survivorship: Genetic susceptibility and second primary cancers—Research strategies and recommendations. J Natl Cancer Inst. 2006;98:15–25. doi: 10.1093/jnci/djj001. [DOI] [PubMed] [Google Scholar]
- 23.Travis LB, Curtis RE, Hankey BF, et al. Second cancers in patients with chronic lymphocytic leukemia. J Natl Cancer Inst. 1992;84:1422–1427. doi: 10.1093/jnci/84.18.1422. [DOI] [PubMed] [Google Scholar]