

Abstract
Context:
The disorder currently accepted as Pierre Robin syndrome/anomaly/sequence (PRS) has been plagued by controversy ever since initially being described. Controversy exists not only about the appropriate terminology and etiopathogenesis of the disorder but also about its management. Clinical findings and treatment outcomes of a large database of 266 PRS cases were compared with the current state of knowledge in the scientific literature, relating to history, clinical description, diagnostic criteria, epidemiology, theories of oligohydramnios, mandibular catch-up growth, midfacial hyperplasia, and the early management.
Aims of Part 2:
Contribute to the sparse scientific knowledge about pathogenesis and involved genetics.
Subjects and Methods:
An analysis of this large database was conducted focusing on genetic involvement, family history, and the incidence of additional syndromes.
Results:
Beside of differences related to clinical signs of dyspnea, feeding problems and mortality rates, various concomitant syndromes, and genetic abnormalities were found in cases of Fairbairn–Robin triad (FRT) and Siebold–Robin sequence (SRS), in addition to differences in relation to clinical signs of dyspnea, feeding problems, and mortality rates.
Conclusion:
Multiple FRT cases presented with various concomitant syndromes and genetic abnormalities, but only one type occurred in two SRS cases. The latter presented a significantly different mortality rate when compared to the FRT subgroup.
Keywords: Fairbairn–Robin triad, glossoptosis, micrognathia, pathogenesis, Pierre Robin sequence, Siebold–Robin sequence
INTRODUCTION
To date, it is understood that around half of the patients suffering from isolated palatal clefts may exhibit additional clinical signs on examination, thus being regarded as syndromic.[1] Particular syndromes might be present in Pierre Robin sequence (PRS) children presenting with isolated cleft palates and, thus, the clinical picture of PRS therefore needs to be further subdivided into syndromic and into nonsyndromic.
Due to coexisting anomalies of various organs and structures as well as disorders of the metabolism and/or the immune system, syndromic PRS patients present particular problems to their interdisciplinary treatment team, especially with regard to surgical reconstructive procedures and perioperative management.
The second part of this series addresses the incidence of syndromic and nonsyndromic cases, feeding, and breathing problems as well as the mortality rate associated with a series of 266 PRS patients and 1518 isolated hard and/or soft palate cleft cases.
SUBJECTS AND METHODS
A total of 266 PRS cases were identified and their charts were extracted for investigation, from an overall database of 4185 cleft cases at the cleft clinic. In addition to the PRS cases, a total of 1518 isolated cleft palate (ICP) cases were found in the database. The remainder of the ICP did not exhibit any other distinguishing signs, other than a hard and/or soft palate cleft and, therefore, represented a specific cleft division of their own. For further in-depth analysis, the PRS cases were subdivided into 21 Siebold–Robin sequence (SRS) cases and 245 Fairbairn–Robin triad (FRT) cases according to their specific clinical signs.[2] Both subgroups, together with the ICP cases, were subsequently investigated for being either syndromic or nonsyndromic based on the clinical presence of additional signs on examination. As the mortality rate in PRS patients is considered to be significant due to apnea and/or aspiration of fluid or food, the charts were screened for continuous feeding and breathing problems due to glossoptosis. The family history was carefully examined for commonalities, searching for genetic causality in addition to that of environmental exposure.
RESULTS
A total of 266 PRS and 1518 ICP cases were included in this retrospective analysis. Among the 266 PRS cases, 7.9% presented without a cleft palate on examination, therefore being diagnosed as SRS. As shown in Table 1, 90.5% of SRS cases were nonsyndromic and 9.5% were diagnosed to be syndromic SRS revealing additional clinical signs such as congenital heart defects or myopathy, as well as two cases of spondyloepimetaphyseal dysplasia.
Table 1.
Syndromic versus nonsyndromic cases and occurrence of genetic findings not allocable to any known syndromes
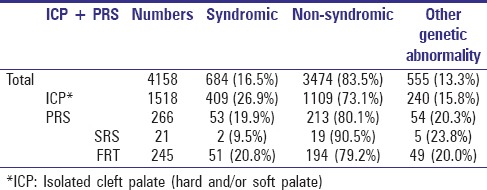
Of PRS group, 92.1% disclosed a cleft palate on examination, in addition to micrognathia and a compromised airway due to glossoptosis. These cases were allocated to the subgroup of FRT and 20.8% of them presented with a syndrome, which is more than double that of the SRS subgroup. An overall of 21 different syndromes in combination with FRT could be detected. The following syndromes occurred in descending order: Stickler (22); Binder (9), among which Stickler and Binder were combined (4); van der Woude (6); Moebius (3); oromandibular hypogenesis (1); Hirschsprung (1); Miller (1); Catel–Manzke (1); fetal alcohol (1); Klippel–Feil (1); oro-palatal digital (1); otomandibular dysostosis (1); popliteal pterygium (1); Wolf–Hirschhorn (1); chromosome 5 + 11 (1); Richter-Costa-Perreira (1); Spondyloepimetaphyseal dysplasia with joint laxity (SEMDL) (1); atelosteogenesis (1); and congenital myopathy (1). In addition, FRT occurred with other genetic abnormalities not being allocable to known syndromes, mostly presenting with additional symptoms related to congenital heart, gastrointestinal, and urinary tract defects, as well as ear and musculoskeletal system defects such as club feet and various hand deformities, including syndactyly. Therefore, roughly 20% of those initially nonsyndromic FRT-labeled patients disclosed genetic abnormalities with no association to already established syndromes.
Of the 1518 ICP cases, 26.9% were syndromic. The most common coexisting syndromes in ICP patients in this database were the Demarquay-van der Woude, Treacher-Collins, Binder, and Sphrintzen (22q11.2-deletion) syndromes. Compared to SRS and FRT, however, slightly less nonsyndromic ICP cases (15.8%) revealed genetic abnormalities not being allocable to known syndromes. Table 1 summarizes the numbers and percentages of syndromic and nonsyndromic cases of ICP and PRS and includes those nonsyndromic cases where genetic abnormalities occurred that were not connected to specific syndromes.
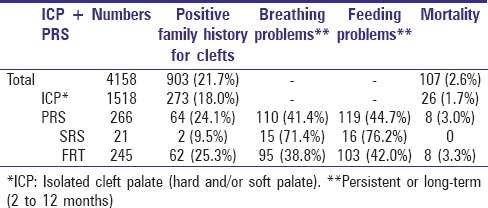
A positive family history for clefts may indicate that an environmental exposure is not the only contributing factor in the development of FRT and SRS. Whereas a positive family history for clefts was detected in over 20% of the total cleft database and within the FRT subgroup, it was not so frequent in ICP cases (18%) and rather rare in SRS cases with only 9.5%.
While around 40% of FRT cases suffered from long-term or persistent breathing and feeding difficulties, the figure was nearly doubled in the SRS subgroup.
Analysis of the mortality rate detected that no patient of the SRS subgroup demised whereas the mortality rate in the FRT subgroup was even higher than the one in the overall cleft database. Table 2 provides the summary of the results discussed above.
Table 2.
Family history, breathing and feeding problems, mortality rate
DISCUSSION
A database of 266 cases, initially labeled as PRS, was analyzed in detail, resulting in a subsequent subdivision of a major FRT and minor SRS group where clinically no soft and/or hard palate cleft was found.
Due to the amount of cases in which concomitant known syndromes could be detected in this database, a further subclassification of the two groups into syndromic and nonsyndromic has been performed. In 1911, Shukowsky[3] described a deformational PRS group including isolated cases, without any associations to syndromes. He further pointed out that the deficient mandibular growth in those cases might be attributed to physical or mechanical restraint. He then suggested to denominate PRS cases associated with syndromes, chromosomal abnormalities such as deletions or duplications, teratogens, or neuromuscular diseases, and these accounted for 60% of his cases as deformational. A similar attempt to classify PRS cases into “isolated PRS = IRS” and “PRS as part of a syndrome = RSS” has been proposed in 1984 by Pasyayan and Lewis.[4] They distinguished between nonsyndromic (probably originating from various etiologies) and syndromic cases that may be inherited by similar ways as the concomitant syndromes. They further suggested the use of two terminologies: (1) Sequence, indicating the sequence of events during the pathogenesis in nonsyndromic cases and (2) syndrome, for cases with an accompanying known syndrome.
In a more recent analysis of 117 PRS cases in 2001, Holder-Espinasse et al. reported around 35% of their PRS cases as syndromic.[5] They suggested a future subclassification of syndromic and nonsyndromic PRS cases to be of practical and clinical importance. This, aside from a different etiopathogenesis, might further allude to the prognosis and/or the potential of mandibular catch-up growth.[6] Compared to those results, the database presented here revealed an average of 80.1% nonsyndromic FRT (79.2%) and SRS (90.5%) patients. Both SRS (23.8%) and FRT (20%) cases showed similar percentages relating to genetic findings that were not allocable to any known syndromes. FRT cases (20.8%) presented with more than double as many known associated syndromes compared to SRS cases (9.5%), among which Stickler as described in other patient series[7,8,9] and Binder were the most common.
Family members of FRT patients revealed nearly three times more clefts than those of SRS patients, keeping in mind the fact that SRS patients themselves lack the clinical sign of a cleft palate on examination. Somewhat more astonishing are the results that nearly double the amount of long-term feeding, and breathing difficulties can be found in SRS patients when compared to FRT patients. However, none of these SRS patients demised whereas the mortality rate in FRT patients (3.3%) is almost twice the amount of that in ICP patients. The latter might be explained by the possibility that in case of glossoptosis in FRT patients, the tongue might become trapped in the cleft palate leading to critical airway obstruction with potential fatality. Of major concern is that most fatal cases related to aspiration which occurred at home. The majority of the fatalities presented in this cleft database was patients suffering from holoprosencephaly (1.3%), Patau syndromes (0.5%), and FRT cases (0.2%).
Most of the long-term breathing and feeding difficulties in SRS patients are probably due to an early cleft repair in FRT patients which corrects feeding disorders, installing a separation between the oral and nasal cavity and due to a more severe micrognathia in SRS patients where neuromuscular reasons[3] eventually play a role.
The patient group labeled as “PRS” contains a much wider spectrum of diversity than previously described. Cases with soft and/or hard palate clefts, in addition to micrognathia, glossoptosis, and breathing disorders, should be classified as FRT. Whereas breathing and feeding difficulties prevailed more often in SRS cases, and rather higher mortality rate was demonstrated among FRT cases, compared to no deaths in the SRS subgroup. Furthermore, the previous means of classification of PRS cases into syndromic and nonsyndromic remains accurate and applicable, given that 21 identified syndromes were associated with mainly the FRT subgroup.
CONCLUSION
A future refinement in the classification of PRS cases is important. Both a subdivision into FRT and SRS and into syndromic and nonsyndromic cases will result in an adequate and appropriate early and subsequent definite individual treatment plan, thereby preventing unnecessary surgery and/or adverse comorbidities.
Multiple FRT cases presented with various concomitant syndromes and genetic abnormalities, but only one type occurred in two SRS cases. The latter presented a significantly different mortality rate when compared to the FRT subgroup
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
The authors would like to thank Mrs Jennilee Blom for her invaluable job as research assistant.
REFERENCES
- 1.Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: Understanding genetic and environmental influences. Nat Rev Genet. 2011;12:167–78. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bütow KW, Zwahlen RA, editors. Cleft – Ultimate Treatment. 2nd ed. Durban: Reach Publisher; 2016. Pierre Robin sequence treatment; pp. 337–65. [Google Scholar]
- 3.Shukowsky WP. Zur ätiologie des stridor inspiratorius congenitus [“The ethiology of congenital inspirational stridor”] Jahrb Kinderheilk. 1911;73:459–74. [Google Scholar]
- 4.Pasyayan HM, Lewis MB. Clinical experience with the Robin sequence. Cleft Palate J. 1984;21:270–6. [PubMed] [Google Scholar]
- 5.Holder-Espinasse M, Abadie V, Cormier-Daire V, Beyler C, Manach Y, Munnich A, et al. Pierre Robin sequence: A series of 117 consecutive cases. J Pediatr. 2001;139:588–90. doi: 10.1067/mpd.2001.117784. [DOI] [PubMed] [Google Scholar]
- 6.Rintala A, Ranta R, Stegars T. On the pathogenesis of cleft palate in the Pierre Robin syndrome. Scand J Plast Reconstr Surg. 1984;18:237–40. doi: 10.3109/02844318409052844. [DOI] [PubMed] [Google Scholar]
- 7.van den Elzen AP, Semmekrot BA, Bongers EM, Huygen PL, Marres HA. Diagnosis and treatment of the Pierre Robin sequence: Results of a retrospective clinical study and review of the literature. Eur J Pediatr. 2001;160:47–53. doi: 10.1007/s004310000646. [DOI] [PubMed] [Google Scholar]
- 8.Hennekam RC, Krantz ID, Allanson JE, editors. Gorlin's Syndromes of the Head and Neck. 5th ed. New York, Oxford: University Press; 2010. Well known miscellaneous syndromes; pp. 1258–94. [Google Scholar]
- 9.Evans AK, Rahbar R, Rogers GF, Mulliken JB, Volk MS. Robin sequence: A retrospective review of 115 patients. Int J Pediatr Otorhinolaryngol. 2006;70:973–80. doi: 10.1016/j.ijporl.2005.10.016. [DOI] [PubMed] [Google Scholar]