

Abstract
Refractoriness of glioblastoma multiforme (GBM) to current treatment paradigms has necessitated identification of new targets to better the existing therapeutic strategies. One such target is peroxisome proliferator-activated receptor gamma (PPARγ) – a transcription factor involved in regulation of lipid metabolism and inflammation. Expression of PPARγ, a known regulator of cell death-inducing DFFA-like effector (CIDEA), is modulated by hypoxia inducible factor (HIF-1α). While the involvement of CIDEA in lipid metabolism is known, its role in malignancies remains largely unknown. An elevated PPARγ and low CIDEA level was observed in GBM tumors as compared with surrounding non-neoplastic tissue. As reciprocal relation exists between PPAR and HIF-1α: and as HIF-1α is a key component in glioma progression, their role in regulating CIDEA expression in glioblastoma was investigated. Although HIF-1α inhibition had no effect on CIDEA expression, pharmacological inhibition of PPARγ elevated CIDEA levels. PPARγ mediated upregulation of CIDEA was accompanied by decreased recruitment of NFκB and SP1 to their predicted binding sites on CIDEA promoter. Ectopic expression of CIDEA triggered apoptosis, activated JNK, decreased HIF-1α activation and increased PPARγ levels in glioma cells. While CIDEA overexpression induced actin cytoskeletal disruption, cell cycle arrest, release of pro-inflammatory cytokine IL-6 in a JNK-dependent manner; CIDEA mediated apoptotic cell death, decreased STAT3 phosphorylation and increased p53 acetylation was JNK independent. This study highlights for the first time the existence of (i) PPARγ-CIDEA regulatory loop in glioma and (ii) novel function of CIDEA as regulator of glioma cell survival.
Introduction
As dysregulated metabolism promotes malignancy, targeting the regulatory genes of metabolic pathways is emerging as a viable therapeutic approach. Among the three members of cell death-inducing DFFA-like effector (CIDE) protein family, the role of CIDEA in lipid metabolism is documented.1 CIDEA is also known to induce apoptosis.2 Though a low expression level of CIDEA in different malignancies including glioblastoma multiforme (GBM) is documented,3 the mechanisms of its regulation and role in tumor progression remain unexplored.
Proliferator-activated receptor (PPAR) is an important positive regulator of CIDEA expression in murine liver,4 and PPARγ is a hypoxia inducible factor (HIF1α) target. As HIF-1α is associated with poor prognosis in glioblastoma, targeting the HIF pathway is considered as an important therapeutic strategy.5–7 Interestingly, evidences suggest mutual inhibitory mechanisms between PPAR and HIF-1α. Although hypoxic stress-induced HIF-1α negatively regulates PPARγ during adipocytic differentiation,8 pathologic stress-induced PPARγ upregulation involves HIF-1α activation in cardiomyocytes.9 Besides, PPARγ agonist pioglitazone has been reported to inhibit glioma survival.10 Given the importance of HIF-1α in glioma biology, and involvement of PPARγ in CIDEA expression; we investigated the (i) role of HIF-1α and PPARγ in CIDEA expression and (ii) the role of CIDEA in glioma cell survival.
Results
Elevated PPARγ and low CIDEA levels in GBM tumors
CIDEA mRNA expression was found to be significantly lower than PPARγ and HIF-1α in 46 GBM tissue samples from different regions of brain (Gene Expression Omnibus (GEO) data set number: GDS4470, Figure 1a). In addition, a significantly low expression of CIDEA mRNA was also observed in GBM tumors as compared with normal brain tissue (oncomine, TCGA mRNA expression data, Figure 1b). Genes that are tightly co-expressed with CIDEA in glioblastoma were found to be enriched in cytoskeleton pathways (enrichment score: 23%), myosin complex assembly (19%), microtubule functioning (14%), calcium-ion binding (13%), macromolecular complex assembly (11%), cell cycle (10%) and phosphate metabolic process (10%) (Figure 1c). Western blot analysis also revealed an almost undetectable level of CIDEA in GBM tumor samples as well in surrounding normal tissues. This low expression of CIDEA was concomitant with elevated PPARγ levels observed in GBM tumors as compared with the surrounding non-neoplastic tissue (Figure 1d).
Figure 1.

PPARγ and CIDEA expression in glioblastoma. (a) CIDEA expression is significantly low in GBM tumors from different regions of brain as indicated by Gene Expression Omnibus (GEO) database (data set record no. GDS4470). The significance is calculated by two-tailed Mann–Whitney U-test. (b) CIDEA mRNA expression is significantly low in GBM as compared with normal brain tissue. Figure is presented of TCGA expression profiles as retrieved from Oncomine database. (c) Significantly enriched pathways from co-expressed genes of CIDEA. Co-expression data were collected from Oncomine with >80% correlation coefficient in GBM and analyzed using DAVID Bioinformatics Resource 6.7. (d) Western blot analysis demonstrating levels of CIDEA and PPARγ in GBM tumor as compared with surrounding non-neoplastic tissue. The figure shows blots from four independent tumor samples. Blot was re-probed for GAPDH to establish equal loading.
PPARγ regulates CIDEA expression but HIF-1α has no role
HIF-1α and PPARγ are known to negatively regulate each other.11 As PPARγ is known to regulate CIDEA, and as HIF-1α is a potential antiglioma target;6 the involvement of HIF-1α and PPARγ in regulation of CIDEA expression in glioma cells was investigated. Glioma cells were treated with LW6 and/or T0070907 either alone or in combination. LW6 inhibits HIF-1α accumulation and suppresses the expression of hypoxia-induced genes,12 and PPARγ antagonist T0070907 inhibits activation of PPARγ.13 Inhibition of PPARγ elevated CIDEA expression in glioma cells (Figure 2a). However, treatment with HIF-1α had no effect on CIDEA level (Figure 2a). The increased CIDEA levels observed on PPARγ inhibition remained unaffected on co-treatment with HIF-1α inhibitor (Figure 2a). These findings suggested that CIDEA expression in glioma cells is independent of HIF-1α but PPARγ dependent. As PPARγ affected CIDEA protein expression, we determined CIDEA mRNA expression on PPARγ inhibition (Figure 2b). Inhibition of PPARγ increased CIDEA mRNA expression significantly (Figure 2b). The extent of increase in mRNA levels in A172 and U87MG corresponded to the changes in protein expression observed in these two cell lines upon PPARγ inhibition.
Figure 2.

Inter-regulatory relationship between PPARγ and CIDEA. (a) Western blot showing effect of PPARγ and/or HIF-1α inhibition on CIDEA protein expression in glioma cell lines. Blots were re-probed for GAPDH to establish equivalent loading. (b) Real-time PCR indicating elevated CIDEA mRNA expression on PPARγ inhibition. Graph represents fold change of CIDEA total mRNA expression. 18s rRNA was used as control. (c) Ectopic expression of CIDEA elevates PPARγ levels in glioma cell lines. Inset showing heightened CIDEA on transfection with overexpression (OE) construct. Blots were re-probed for GAPDH to established equal loading. (a and c) Representative blot from three independent experiments with identical results. (d) CIDEA negatively regulates HIF-1α transcriptional activation. The graphs represent fold change in HIF-1α luciferase reporter activity over control in cells transfected with CIDEA overexpression construct. Values (b and d) represent the means±S.E.M. from three independent experiments. * denotes significant change from control (P<0.05). LW6 and T007 are HIF-1α and PPARγ inhibitors, respectively.
CIDEA overexpression increases PPARγ expression and decreases HIF-1α activation
We next investigated the consequences of elevated CIDEA expression on HIF-1α and PPARγ expression. This was accomplished by transfecting cells with CIDEA overexpression construct. Increase in CIDEA levels was concomitant with increase in PPARγ expression, albeit to different extent in different cell lines (Figure 2c). This suggested the existence of a PPARγ- CIDEA regulatory loop. Though HIF-1α inhibition had no effect of CIDEA expression, overexpression of CIDEA decreased HIF-1α transcriptional activation (Figure 2d). Thus, CIDEA negatively regulates HIF-1α activation in glioma cells.
PPARγ inhibition decreases recruitment of SP1 and NFκB on CIDEA promoter
Constitutively activated NFκB in GBM tumors promotes their growth and survival.14 PPARγ is known to induce proteosomal degradation of NFκB,15 and NFκB regulates CIDEA expression.16 As there was no significant change in expression of NFκB upon PPARγ inhibition (Figure 3a), we determined whether PPARγ inhibition affects DNA-binding pattern of NFκB on CIDEA promoter. To study the mechanistic detail of PPARγ mediated transcriptional regulation of CIDEA in glioma cell, we chose a 1120 bp genomic region (−1000 to +120) on the CIDEA promoter. All the base pair positions mentioned here are according to the TSS position described by Petterson et al. 16 NFκB-binding sites were located at −839 to −737, −266 to −93 and −26 to +120 regions. Chromatin immunoprecipitation (ChIP) real-time PCR revealed an overall decrease in recruitment of NFκB to these sites on inhibition of PPARγ, as compared with control (Figure 3b). Elevated SP1 levels has been suggested as a prognostic marker in gliomas17 and a strong association of SP1 with NFκB is involved in transcriptional regulation of genes.18 As in silico analysis revealed a SP1 binding site at −26 to +120 on the CIDEA promoter containing the NFκB site, the effect of PPARγ inhibition on SP1 expression and binding to this site on CIDEA promoter was also investigated. While inhibition of PPARγ had no effect on SP1 levels, a decrease in SP1 binding to the CIDEA promoter was observed (Figure 3c). Thus, the decreased recruitment of NFκB to its putative binding sites on CIDEA promoter observed upon PPARγ inhibition was accompanied by a low enrichment of SP1 to its cognate site overlapping the NFκB site.
Figure 3.

Inhibition of PPARγ reduces NFκB and SP1 binding on CIDEA promoter. (a) PPARγ inhibitior T007 has no effect on NFκB and SP1 expression. Blot is representative of two independent experiments. Blots were re-probed for GAPDH to establish equal loading. (b) ChIP-qPCR assays demonstrating decreased binding of NFκB to its cognate sites on CIDEA promoter. DNA isolated from control and PPARγ inhibitor treated A172 glioma cells pre and post immunoprecipitation with anti-NFκB antibody, was amplified using specific primer sets. Binding affinity of NFκB was found to be low at three different putative binding sites on CIDEA promoter on inhibition of PPARγ. (c) PPARγ inhibition decreases SP1 binding to its cognate site on CIDEA promoter at −26 to +120 position, as indicated by ChIP-qPCR assay. Graph (b and c) represents fold change as calculated from Ct values of two independent experiments for a single site.
Role of JNK in CIDEA mediated cell cycle arrest and death
As PPARγ inhibition-induced CIDEA was accompanied by decrease in glioma cell viability (Figure 4a), we next investigated the consequences of CIDEA overexpression on glioma cell survival. Ectopic expression of CIDEA induced cell death (Figure 4b) was concomitant with induction of JNK phosphorylation (Figure 4c). Increased localization of JNK to the mitochondria was observed upon CIDEA overexpression (Figure 4d). As JNK has been implicated as an inducer of apoptosis in glioma cells,19,20 and since JNK mediated apoptotic cell death involves its mitochondrial localization,21 the role of JNK in CIDEA mediated cell death was investigated. However, JNK inhibition failed to rescue CIDEA-mediated glioma cell death (Figure 4e). Although CIDEA-mediated induction of Caspase-3 activation was found to be independent of JNK (Supplementary Figure 1), G2/M phase cell cycle arrest was found to be JNK dependent (Figure 4f). However, the expression of cyclin B1 associated with G2/M phase of cell cycle arrest was elevated only in A172 cells (Supplementary Figure 2). Thus, CIDEA-mediated cell death is JNK independent but cell cycle arrest is JNK mediated.
Figure 4.

CIDEA overexpression induces cell death. (a) PPARγ inhibition reduces glioma cell viability. The graph represents the viable cells, fold change over control, observed when glioma cells were treated with 50 μM of T0070907 for 40 h, as determined by MTS assay. (b) CIDEA overexpression reduces cell viability in glioma cell lines, as determined by MTS. (c) CIDEA overexpression induces phospho-JNK expression in glioma cells. Blot is representative of three independent experiments. Blots were re-probed for β-tubulin to establish equal loading control. (d) CIDEA overexpression induces JNK co-localization into the mitochondria. Representative image showing mitochondria stained with mitotracker green, JNK with Alexa Fluor 594-tagged secondary antibody (red) and nucleus with DAPI (blue). (e) CIDEA-mediated glioma cell death is JNK independent. Graph represents the viable cells, fold change over control, observed when CIDEA overexpressing cells were treated with 10μM JNK inhibitor for 40 h, as measured by MTS assay. The graph (a, b, e) represents the means±S.E.M. from three independent experiments. * Significant decrease from control (P≤0.05). (f) Overexpression of CIDEA induces JNK-mediated G2/M phase cell cycle arrest in A172 glioma cell line. Left panel shows population of cells arrested at G2/M phase, the right panel indicates the fold change in cell population. Graph is representative data of two independent experiments.
CIDEA induces p53 acetylation in glioma cell
As ectopic expression of CIDEA induced cell death, we investigated the status of p53 in CIDEA overexpressing cells. CIDEA induced p53 expression in a JNK-independent manner both in wild-type and mutant p53 glioma cells. Interestingly, p53 was localized in the nucleus of p53 wild-type A172 cells (Figure 5a), whereas a cytosolic localization was observed in p53 mutant cells T98G (Figure 5b). As acetylation of p53 increases its transcriptional activity as well as the transcription independent pro-apoptotic function,22 the status of acetylated p53 in CIDEA overexpressing cells was investigated. Increased acetylation of p53 in cell lines containing transcriptionally active or inactive p53 was observed on CIDEA overexpression (Figures 5a and b).
Figure 5.
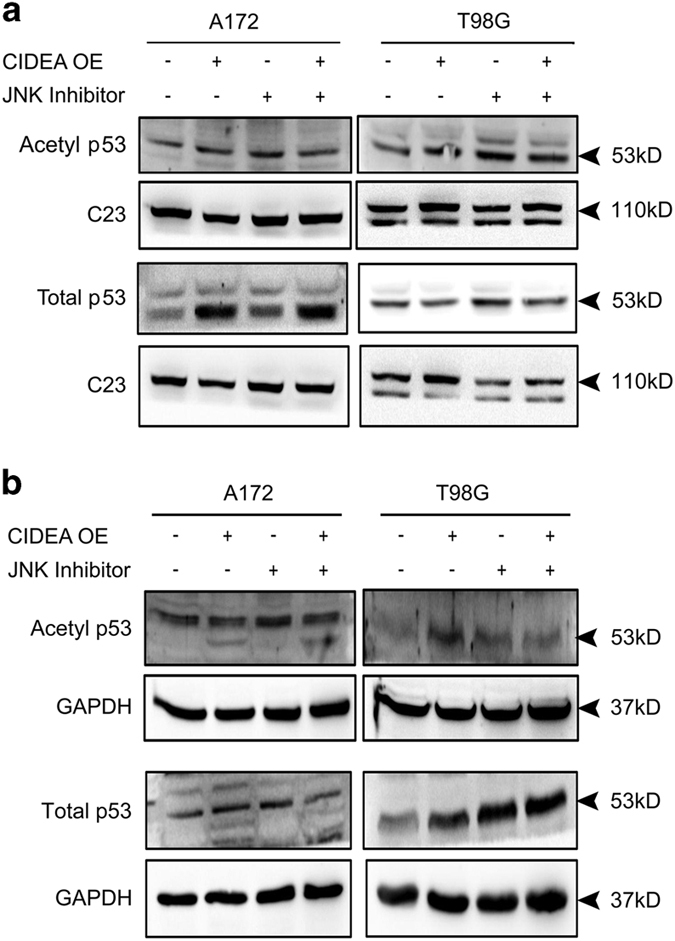
Overexpression of CIDEA increases p53 acetylation in a JNK-independent manner. CIDEA overexpression affects acetylated p53 and total p53 levels in the nucleus (a) and the cytosol (b) of glioma cells in a JNK-independent manner. A representative blot is shown from three independent experiments with identical results. Blots were re-probed with C23 (for nuclear extract) or GAPDH (for cytosolic extract) as loading control.
JNK regulates CIDEA mediated disruption of actin cytoskeletal
CIDEA overexpression was accompanied by altered cell morphology, suggestive of disrupted cytoskeletal architecture (Supplementary Figure 3). Vasodilator-stimulated phosphoprotein (VASP) signaling is critical for dynamic actin reorganization, and VASP phosphorylation controls actin cytoskeleton remodeling.23 Inactivation of cofilin by its phosphorylation leads to accumulation of actin filaments.24 The elevated level of active phosphorylated VASP (Figure 6a) and non-phosphorylated Cofilin (Figure 6b) observed in CIDEA overexpressed cell was abrogated upon JNK inhibition. Immuno-cytochemical studies using antibodies directed against cofilin and direct labelling of actin using fluorophore-tagged Rhodamine further suggested that CIDEA over-expression disrupts polymerized F-actin structure in a JNK dependent manner (Figure 6c). Similar co-localization experiment performed in U87 and T98G glioma cells yielded identical results (data not shown).
Figure 6.

JNK regulates CIDEA mediated actin cytoskeletal disruption. CIDEA overexpression alters the levels of phospho-VASP (a) and phospho-Cofilin (b) in glioma cell lines in a JNK dependent manner. A representative blot is shown from two independent experiments with identical results. Blots were re-probed with β-tubulin to establish loading control. (c) CIDEA overexpression affects actin cytoskeleton architecture in a JNK-dependent manner. Confocal imaging depicts that association between Cofilin (green, Alexa Fluor 488) and actin (red, rhodamine-phalloidin) observed in CIDEA overexpressing cells is reverted by JNK inhibition. Representative images from two independent experiments are shown for indicated conditions.
CIDEA affects IL-6 and STAT3 activation
PPARγ regulates DNA binding and transactivation of STAT3,25 and IL-6 induced STAT3 activation is associated with pro-survival responses in glioma cells.26 CIDEA overexpression increased IL-6 levels in a JNK-dependent manner (Figure 7a). Despite elevating IL-6 levels, CIDEA overexpression abrogated STAT3 phosphorylation in a JNK-independent manner (Figure 7b). Taken together, our findings indicate that CIDEA regulates several pathways associated with glioma cell survival (Figure 7c).
Figure 7.

CIDEA induces IL-6 levels but abrogates STAT3 activation in glioma cells. (a) Ectopic expression of CIDEA elevates IL-6 secretion in a JNK-dependent manner as revealed by Cytokine Bead Array. Graph represents representative data from two independent experiments in three cell lines. (b) CIDEA overexpression abrogates cytosolic and nuclear pSTAT3 (Y705) levels in a JNK independent manner. Blots were re-probed with C23 (for nuclear extract) or β-tubulin (for cytosolic extract) to establish equal loading. Blots shown are representative of three independent experiments. (c) Proposed mechanism of regulation of CIDEA and its role in glioma cell survival.
Discussion
There has been opposing reports regarding the ability of PPARγ to effect tumor progression as both agonists and antagonists have demonstrated anti-tumorigenic properties.27 PPARγ agonists have shown potential anti-glioma effects.10 As low expression of CIDEA in glioma is concomitant with elevated levels of PPARγ and HIF-1α, this study was undertaken to understand the (i) role of PPARγ and HIF-1α in maintaining the low basal expression of CIDEA in GBM, and (ii) the effect of CIDEA overexpression on glioma cell survival. Our findings suggest that inhibition of PPARγ enhances CIDEA expression, whereas HIF-1α inhibition has no effect. Interestingly, increased CIDEA levels triggered glioma cell apoptosis, decreased HIF-1α activation and elevated PPARγ levels.
Although promoter of CIDEA in murine liver cell contains PPARγ inducible PPREs associated with CIDEA expression,4 human CIDEA promoter sequence lacks PPREs. Moreover, PPARγ agonist has failed to induce CIDEA transcriptional activity in human adipocytic cell.16 This suggests that the possibility of direct DNA-binding activity of PPARγ to human CIDEA promoter is negligible. A NFκB site at position −163/−151 serves as an important modulator for TNF-mediated downregulation of CIDEA expression.16 Besides, PPARγ can regulate NFκB activation and DNA binding.15 Also, SP1 and SP3 binding is crucial for positive regulation of non-methylated CIDEA promoter in human adipocytes.28 Our study revealed that PPARγ inhibition affects the recruitment of NFκB and SP1 to their cognate sites on CIDEA promoter. It is possible that increased binding of these two factors on CIDEA promoter in GBM exhibiting elevated NFκB, SP1 and PPARγ levels, could contribute to the low expression of CIDEA in these tumors.
PPARγ agonists inhibit release of pro-inflammatory cytokine IL-6 in monocytes.29 IL-6 elicits survival response in glioma cells through STAT3.26 As heightened STAT3 activation promotes glioma progression, STAT3 inhibitors are regarded as a potential therapeutic target for glioma.6 Depletion of pSTAT3 on CIDEA overexpression possibly prevents IL-6 from exhibiting its pro-survival response despite increase in its levels. Decrease in HIF-1α transcriptional activation and STAT3 phosphorylation following CIDEA overexpression is concomitant with previous findings that STAT3 is a crucial positive regulator of HIF-1α expression.30
Dynamics of actin filaments involving their assembly/disassembly and organization influences cell death through an apoptosis-like pathway.31 In silico analysis predicated that genes associated with cytoskeletal organization are tightly co-expressed with CIDEA in glioblastoma. As de phosphorylated active cofilin is essential for actin cytoskeletal disruption-mediated apoptosis,32 it is possible that CIDEA induces apoptosis by altering actin dynamics. Since acetylation of p53 is correlated with apoptotic responses,22 CIDEA-induced glioma cell death could also be attributed to increased p53 acetylation. Thus, CIDEA induces glioma cell death by affecting different survival pathways. Taken together, this study not only suggests the existence of a CIDEA-PPARγ regulatory loop, but also demonstrates the role of CIDEA as death inducer in glioma cells. By highlighting the clinical relevance of elevated PPARγ levels in regulating expression of pro-apoptotic CIDEA in glioma, this study warrants further investigation directed towards evaluating efficacy of PPARγ inhibitors as effective anti-glioma therapeutic strategy.
Materials and Methods
Processing of patient tissue
Western blot analysis was performed on tissue samples collected from patients with histologically confirmed GBM (n=12) to determine CIDEA and PPARγ expression as described.33 Non-neoplastic brain tissues obtained from margin of the corresponding tumors were used as control. Samples were obtained as per the guidelines of Institutional Human Ethics Committee of NBRC.
In silico analysis
Data set record number GDS4470 from GEO database was queried for CIDEA, PPARγ and HIF-1α. GDS4470 was chosen for its variety of glioblastoma samples from different regions of brain. The expression values for each gene were used to analyze for significant difference by two-tailed Mann–Whitney U-test in Sigma Plot version 10.0. Oncomine database was accessed to obtain cancer versus normal, and co-expression data in glioblastoma samples. The most significant cancer versus normal data was represented. Co-expression data were also obtained from most significant glioblastoma data (TCGA mRNA expression profile) with correlation coefficient of >80%. The co-expressed gene list was then submitted to DAVID Bioinformatics Resources 6.734,35 and significantly enriched pathways were plotted in Microsoft Excel 2007.
Cell culture and treatment
Human glioma cell lines A172, U87MG and T98G obtained from American Type Culture Collection (Manassas, VA, USA), were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS. Semi-confluent cells were transferred to serum-free medium (SFM) for four hours. Cells were then treated with 50 μM of PPARγ inhibitor (T0070907, Tocris Bioscience, Bristol, UK) for 2 h and subsequently co-treated with 20 μM HIF-1α inhibitor (LW6, Calbiochem, Billerica, MA, USA) in SFM. DMSO-treated cells served as controls. Glioma cell lines were transfected with CIDEA overexpression plasmid using Lipofectamine 2000 (Life Technologies, Invitrogen, Carlsbad, CA, USA). After 24 h of transfection, cells were treated with 10 μM of JNK inhibitor (SP600125, Tocris Bioscience) in SFM. Following 48 h of treatment, cells were harvested for subsequent analysis. CIDEA overexpression plasmid was a kind gift from Dr. Peter Arner (Karolinska Institute, Stockholm, Sweden).36
Western blot analysis
Western blot analysis was performed with whole lysates, cytosolic and nuclear protein extracts of cells transfected with CIDEA overexpressing plasmid or treated with inhibitors of PPARγ, HIF-1α or JNK as described,33 using antibodies against CIDEA (Abcam, Cambridge, UK), PPARγ, HIF-1α (Novus Biological, Cambridge, UK), phospho-JNK, JNK, phospho-cofilin, Cofilin, phospho-VASP, VASP, phospho-STAT3 (Y705), STAT3, p53, acetyl-p53 (Lys-373 and Lys-382) (Millipore, Billerica, MA, USA); Cyclin B1, β-tubulin, GAPDH, C-23 and NFκB Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies were purchased from Cell Signaling (Danvers, MA, USA) unless otherwise mentioned. After addition of horse radish peroxidase-conjugated secondary antibodies (Vector Laboratories Inc., Burlingame, CA, USA) blots were exposed to Chemigenius Bioimaging System (Syngene, Cambridge, UK) and images were developed by Gene snap software (Syngene). Reprobing of the blots was performed after stripping to determine the loading control with anti-β-tubulin, GAPDH or C23 antibodies.
Real-time PCR analysis
Real-time PCR was performed for CIDEA and 18S rRNA using ViiA7 Real Time PCR system (Life Technologies, Invitrogen) as per manufacturer’s protocol with default settings of thermal cycles except for custom annealing temperature for each primer pair. Sequences of the primers were as follows: CIDEA forward: 5′-CCAGCACGTCCCCACTTG-3′, CIDEA reverse: 5′-CGTTAAGGCAGCCGATGAAG-3′, 18S rRNA forward: 5′-CAGCCACCCGAGATTGAGCA-3′, 18S rRNA reverse: 5′-TAGTAGCGACGGGCGGTGTG-3′. Data were analyzed with ViiA7 software.
Flow cytometric analysis of cell cycle progression and caspase 3 activation
Cells transfected with CIDEA overexpression construct in the presence or absence of JNK inhibitor were harvested and fixed in 1% paraformaldehyde in PBS. The fixed cells were washed in PBS, resuspended in DHE and propidium iodide solution (BD Biosciences, Franklin Lakes, NJ, USA) for 20 min at room temperature and flow cytometric analysis of 106 cells were performed using Cell Quest program on FACS Calibur (Becton Dickinson, San Diego, CA, USA). The percentage of cells in the G1, S and G2/M phases of the cell cycle was analyzed.37 Active caspase 3 level was also detected by FACS analysis. In brief, 106 cells were incubated at 4 °C for 40 min with anti-active caspase 3 antibody (Santa Cruz Biotechnology), washed, incubated with anti-rabbit conjugated to FITC secondary antibody for 30 min on ice, washed again, resuspended in PBS and analyzed by FACS.
Cytokine bead array
Cytokine bead array kit (Human Inflammation CBA kit, BD Biosciences) was used to quantitatively measure cytokine levels in the supernatant collected from CIDEA overexpressing cells treated with or without JNK inhibitor as described.38
Determination of cell viability
Viability of cells treated with PPARγ inhibitor or transfected with CIDEA overexpression plasmid in the presence or absence of 10 μM of JNK inhibitor was assessed using the MTS assay (Promega, Madison, WI, USA) as described.39 Values were expressed as fold change over control.
Luciferase assay
Semi-confluent cells were transfected with 300 ng of CIDEA plasmid, 300 ng of the HIF-1α responsive element luciferase construct (a kind gift from Chinmay Mukhopadhyay, JNU, India ) and 10 ng of Renilla luciferase vector (pRL-TK, Promega, as transfection control) using Lipofectamine 2000 (Life Technologies, Invitrogen). After 48 h, cells were harvested and luciferase activity was measured using the Dual-Luciferase Reporter Assay System Kit (Promega) according to manufacture protocol in a GloMax 96 microplate luminometer.40
Immuno-cytochemistry
Immuno-cytochemistry was performed to determine actin cytoskeleton architecture in glioma cells transfected with CIDEA overexpression plasmid in the presence and absence of JNK inhibitor. Following 40 h of treatment, cells were fixed with 4% formaldehyde. Fixed cells were then incubated in PBS containing 1% BSA for 30 min, followed by incubation with Cofilin antibody at 4 °C in staining solution (containing 6.25 μl rhodamine labeled phalloidin, 2.5 mg BSA in 250 μl PBS) overnight at 4 °C. Cells were washed and labeled with Alexa fluor 488 secondary antibody, washed, mounted with Vectashield mounting medium with DAPI (Vector Labs, Inc.). CIDEA regulated localization of JNK was determined using Mitotracker green and JNK antibody followed by subsequent incubation with Alexa fluor 594 labeled secondary antibody. Cells were washed with PBS, mounted and immunofluorescence was recorded using Apotome upright fluorescence microscope (Carl Zeiss) as described previously.41
ChIP and ChIP real-time PCR assays
ChIP was performed on glioma cells treated with PPARγ inhibitor for 48 h by Chip-IT Enzymatic DNA shearing Kit (Active Motif, Carlsbad, CA, USA) as described previously.42 Following treatment, cells were fixed with 1% formaldehyde at room temperature for exactly 8 min, and further processed according to manufacturer’s instruction. Anti-NFκB (Santa Cruz Biotechnology) and anti-SP1 (Cell signaling) were used for immunoprecipitation and non-specific IgG antibody (Abcam) was used as control. After reverse cross-linking and DNA purification, DNA from input (1:10 diluted) or immune-precipitated (IP) samples were quantified by real-time PCR using ABI 7500 real-time thermal cycler with Power SYBR green PCR Master Mix (Life Technologies, Invitrogen) for 40 cycles. Threshold cycle number (Ct) values of IP samples were normalized by corresponding Ct of 1% input DNA. The relative fold change value was analyzed relative to the control. Primer sequences of CIDEA promoter region used for ChIP real-time PCR analysis are listed in Supplementary Table 1.
Acknowledgments
The work was supported by research grant from the Department of Biotechnology (DBT, Government of India #BT/PR5818/MED/30/839/2012) to ES.
Glossary
- CIDEA
cell death-inducing DFFA-like effector A
- PPAR gamma
peroxisome proliferator-activated receptor gamma
- HIF-1 alpha
hypoxia inducible factor 1 alpha
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- NFκB
nuclear factor kappa B
- Sp1
specificity protein 1
- c23
nucleolin
- VASP
vasodilator-stimulated phosphoprotein
- STAT3
signal transducer and activator of transcription 3
- JNK
c-Jun N-terminal kinase
- GBM
glioblastoma multiforme
- IL-6
interleukin-6
- TSS
translation start site
- CID/CIDEA OE
CIDEA overexpression
- T007
PPAR gamma inhibitor, commercial name-T0070907
- mg
milligram
- μg
microgram
- mM
milimolar
- μM
micromolar
- hrs
hours.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Cell Death Discovery website (http://www.nature.com/cddiscovery)
Edited by A Rufini
References
- Abreu-Vieira G , Fischer AW , Mattsson C , de Jong JM , Shabalina IG , Ryden M et al. Cidea improves the metabolic profile through expansion of adipose tissue. Nat Commun 2015; 6: 7433. [DOI] [PubMed] [Google Scholar]
- Inohara N , Koseki T , Chen S , Wu X , Nunez G . CIDE, a novel family of cell death activators with homology to the 45 kDa subunit of the DNA fragmentation factor. EMBO J 1998; 17: 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR , Yu J , Shanker K , Deshpande N , Varambally R , Ghosh D et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia 2004; 6: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswakarma N , Yu S , Naik S , Kashireddy P , Matsumoto K , Sarkar J et al. Transcriptional regulation of Cidea, mitochondrial cell death-inducing DNA fragmentation factor alpha-like effector A, in mouse liver by peroxisome proliferator-activated receptor alpha and gamma. J Biol Chem 2007; 282: 18613–18624. [DOI] [PubMed] [Google Scholar]
- Zagzag D , Zhong H , Scalzitti JM , Laughner E , Simons JW , Semenza GL . Expression of hypoxia-inducible factor 1alpha in brain tumors: association with angiogenesis, invasion, and progression. Cancer 2000; 88: 2606–2618. [PubMed] [Google Scholar]
- Sen E . Targeting inflammation-induced transcription factor activation: an open frontier for glioma therapy. Drug Discov Today 2011; 16: 1044–1051. [DOI] [PubMed] [Google Scholar]
- Yin S , Kaluz S , Devi NS , Jabbar AA , de Noronha RG , Mun J et al. Arylsulfonamide KCN1 inhibits in vivo glioma growth and interferes with HIF signaling by disrupting HIF-1alpha interaction with cofactors p300/CBP. Clin Cancer Res 2012; 18: 6623–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun Z , Maecker HL , Johnson RS , Giaccia AJ . Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell 2002; 2: 331–341. [DOI] [PubMed] [Google Scholar]
- Krishnan J , Suter M , Windak R , Krebs T , Felley A , Montessuit C et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab 2009; 9: 512–524. [DOI] [PubMed] [Google Scholar]
- Grommes C , Karlo JC , Caprariello A , Blankenship D , Dechant A , Landreth GE . The PPARgamma agonist pioglitazone crosses the blood-brain barrier and reduces tumor growth in a human xenograft model. Cancer Chemother Pharmacol 2013; 71: 929–936. [DOI] [PubMed] [Google Scholar]
- Yang K , Jiang Q , Wang Z , Li M , Zhang Q , Lu W et al. Mutual Inhibitory Mechanisms between PPARγ and Hif-1α: Implication in Pulmonary Hypertension. Receptors Clin Investig 2015; 2: e626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K , Kang JE , Park SK , Jin Y , Chung KS , Kim HM et al. LW6, a novel HIF-1 inhibitor, promotes proteasomal degradation of HIF-1alpha via upregulation of VHL in a colon cancer cell line. Biochem Pharmacol 2010; 80: 982–989. [DOI] [PubMed] [Google Scholar]
- Qin C , Morrow D , Stewart J , Spencer K , Porter W , Smith R 3rd et al. A new class of peroxisome proliferator-activated receptor gamma (PPARgamma) agonists that inhibit growth of breast cancer cells: 1,1-Bis(3'-indolyl)-1-(p-substituted phenyl)methanes. Mol Cancer Ther 2004; 3: 247–260. [PubMed] [Google Scholar]
- Robe PA , Bentires-Alj M , Bonif M , Rogister B , Deprez M , Haddada H et al. In vitro and in vivo activity of the nuclear factor-kappaB inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res 2004; 10: 5595–5603. [DOI] [PubMed] [Google Scholar]
- Hou Y , Moreau F , Chadee K . PPARgamma is an E3 ligase that induces the degradation of NFkappaB/p65. Nature Commun 2012; 3: 1300. [DOI] [PubMed] [Google Scholar]
- Pettersson AT , Laurencikiene J , Nordstrom EA , Stenson BM , van Harmelen V , Murphy C et al. Characterization of the human CIDEA promoter in fat cells. Int J Obes 2008; 32: 1380–1387. [DOI] [PubMed] [Google Scholar]
- Guan H , Cai J , Zhang N , Wu J , Yuan J , Li J et al. Sp1 is upregulated in human glioma, promotes MMP-2-mediated cell invasion and predicts poor clinical outcome. Int J Cancer 2012; 130: 593–601. [DOI] [PubMed] [Google Scholar]
- Hirano F , Tanaka H , Hirano Y , Hiramoto M , Handa H , Makino I et al. Functional interference of Sp1 and NF-kappaB through the same DNA binding site. Mol Cell Biol 1998; 18: 1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit D , Ghildiyal R , Anto NP , Sen E . Chaetocin-induced ROS-mediated apoptosis involves ATM-YAP1 axis and JNK-dependent inhibition of glucose metabolism. Cell Death Dis 2014; 5: e1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P , Dixit D , Sen E . Oncrasin targets the JNK-NF-kappaB axis to sensitize glioma cells to TNFalpha-induced apoptosis. Carcinogenesis 2013; 34: 388–396. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran DN , Reddy EP . JNK signaling in apoptosis. Oncogene 2008; 27: 6245–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H , Woods NT , Piluso LG , Lee HH , Chen J , Bhalla KN et al. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J Biol Chem 2009; 284: 11171–11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbeck B , Huttelmaier S , Schluter K , Jockusch BM , Illenberger S . Phosphorylation of the vasodilator-stimulated phosphoprotein regulates its interaction with actin. J Biol Chem 2000; 275: 30817–30825. [DOI] [PubMed] [Google Scholar]
- Arber S , Barbayannis FA , Hanser H , Schneider C , Stanyon CA , Bernard O et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 1998; 393: 805–809. [DOI] [PubMed] [Google Scholar]
- Wang LH , Yang XY , Zhang X , Huang J , Hou J , Li J et al. Transcriptional inactivation of STAT3 by PPARgamma suppresses IL-6-responsive multiple myeloma cells. Immunity 2004; 20: 205–218. [DOI] [PubMed] [Google Scholar]
- Rahaman SO , Harbor PC , Chernova O , Barnett GH , Vogelbaum MA , Haque SJ . Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 2002; 21: 8404–8413. [DOI] [PubMed] [Google Scholar]
- Campbell MJ , Carlberg C , Koeffler HP . A Role for the PPARgamma in Cancer Therapy. PPAR Res 2008; 2008: 314974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D , Da L , Tang H , Li T , Zhao M . CpG methylation plays a vital role in determining tissue- and cell-specific expression of the human cell-death-inducing DFF45-like effector A gene through the regulation of Sp1/Sp3 binding. Nucleic Acids Res 2008; 36: 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C , Ting AT , Seed B . PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998; 391: 82–86. [DOI] [PubMed] [Google Scholar]
- Niu G , Briggs J , Deng J , Ma Y , Lee H , Kortylewski M et al. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol Cancer Res 2008; 6: 1099–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursch W , Hochegger K , Torok L , Marian B , Ellinger A , Hermann RS . Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci 2000; 113: 1189–1198. [DOI] [PubMed] [Google Scholar]
- Chua BT , Volbracht C , Tan KO , Li R , Yu VC , Li P . Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat Cell Biol 2003; 5: 1083–1089. [DOI] [PubMed] [Google Scholar]
- Dixit D , Sharma V , Ghosh S , Mehta VS , Sen E . Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFalpha)-induced apoptosis through SIRT1 inhibition. Cell Death Dis 2012; 3: e271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang , da W , Sherman BT , Lempicki RA . Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Huang , da W , Sherman BT , Lempicki RA . Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurencikiene J , Stenson BM , Arvidsson Nordstrom E , Agustsson T , Langin D , Isaksson B et al. Evidence for an important role of CIDEA in human cancer cachexia. Cancer Res 2008; 68: 9247–9254. [DOI] [PubMed] [Google Scholar]
- Alam S , Sen E , Brashear H , Meyers C . Adeno-associated virus type 2 increases proteosome-dependent degradation of p21WAF1 in a human papillomavirus type 31b-positive cervical carcinoma line. J Virol 2006; 80: 4927–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari R , Sharma V , Koul N , Ghosh A , Joseph C , Hossain SkU et al. Ebselen abrogates TNFalpha induced pro-inflammatory response in glioblastoma. Mol Oncol 2009; 3: 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit D , Sharma V , Ghosh S , Koul N , Mishra PK , Sen E . Manumycin inhibits STAT3, telomerase activity, and growth of glioma cells by elevating intracellular reactive oxygen species generation. Free Radic Biol Med 2009; 47: 364–374. [DOI] [PubMed] [Google Scholar]
- Tewari R , Choudhury SR , Ghosh S , Mehta VS , Sen E . Involvement of TNFalpha-induced TLR4-NF-kappaB and TLR4-HIF-1alpha feed-forward loops in the regulation of inflammatory responses in glioma. J Mol Med 2012; 90: 67–80. [DOI] [PubMed] [Google Scholar]
- Ghosh S , Tewari R , Dixit D , Sen E . TNFalpha induced oxidative stress dependent Akt signaling affects actin cytoskeletal organization in glioma cells. Neurochem Int 2010; 56: 194–201. [DOI] [PubMed] [Google Scholar]
- Ghosh S , Paul A , Sen E . Tumor necrosis factor alpha-induced hypoxia-inducible factor 1alpha-beta-catenin axis regulates major histocompatibility complex class I gene activation through chromatin remodeling. Mol Cell Biol 2013; 33: 2718–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.