

Abstract
Background
In inflammatory bowel disease (IBD), an inappropriate immune response leads to chronic mucosal inflammation. This response may be partly due to dysregulation of defensins, which are endogenously produced antimicrobial peptides. This study determined whether microRNAs (miRNAs) regulate α-defensin 5 (DEFA5), which could further implicate both in IBD pathogenesis.
Methods
Induction of DEFA5 mRNA and protein expression was determined in Caco-2 cells. An in silico analysis identified putative miRNA binding sites of DEFA5. Expression of these miRNAs was assessed in Caco-2 cells. Regulation of DEFA5 expression by these miRNAs was measured by luciferase assays. Caco-2 cells were transfected with miR-124 and miR-924 mimics, and DEFA5 mRNA and protein expression was measured.
Results
DEFA5 mRNA and protein expression was inducible in Caco-2 cells. Fifteen putative miRNA binding sites were found in DEFA5. The expression of miR-124 and miR-924 decreased following induction. Transfection of a luciferase construct containing the DEFA5 miRNA binding sites resulted in a decrease in luciferase activity compared to transfection of the empty vector. Transfection of a reporter construct containing mismatched miRNA binding sites resulted in restoration of luciferase activities. Transfection of miRNA mimics decreased DEFA5 mRNA expression and protein expression.
Conclusions
miR-124 and miR-924 negatively regulate DEFA5 mRNA and protein expression. These data implicate miRNAs in intestinal innate immune regulation and IBD pathogenesis.
Keywords: Defensin, Inflammatory bowel disease, MicroRNA regulation
Introduction
Crohn's disease (CD), one of the two types of inflammatory bowel disease (IBD), is characterized by inappropriate and continuous activation of the mucosal immune system [1,2]. In the normal small intestine, Paneth cells, an important part of the innate mucosal immune system, maintain microbial homeostasis by secreting defensins, a class of cationic peptides with a broad spectrum of antimicrobial activity [3]. The two broad categories of defensins include α and β-defensins. In the small intestine, α-defensin 5 (DEFA5) is secreted into the lumen of intestinal crypts by Paneth cells after activation of the NOD2 receptor by bacterial endotoxins [4-6]. The α-defensins are chemotactic for cells of both innate and adaptive immune systems, including macrophages and T cells [7]. They are involved in several processes that maintain intestinal homeostasis, including regulation of gut flora, intestinal inflammation, stem cell protection, and crypt development [8-10]. In genetically susceptible individuals, changes in intestinal bacterial flora, including commensal bacteria, play a role in IBD pathogenesis. These changes contribute to the initiation and perpetuation of chronic mucosal inflammation [11-14]. Since defensins maintain the balance between commensal microbes and intestinal mucosa, their dysregulation could contribute to IBD pathogenesis. Defensin expression has been shown to be altered in the setting of NOD2 mutations and changes in certain signaling pathways, but no research has demonstrated the role of microRNAs in defensin regulation. However, SNPs within the 3′ UTR of DEFA5, have been linked with increased susceptibility to IBD, suggesting the possible role of miRNA regulation [15]. Additionally, post-transcriptional regulation of gene expression, such as that by microRNAs, has been shown to play an important role in IBD pathogenesis [16-18]. Altered miRNA expression profiles exist in active IBD indicating that changes in miRNA expression are associated with disease [17-21]. Therefore, the purpose of this study is to examine the role of miRNAs in regulating DEFA5 expression using Caco-2 cells as a model of Paneth cells.
Materials and Methods
Cell lines and cell culture
Human colonic epithelial cell lines (Caco-2, HT29, and HCT116 cells) were previously obtained and used for this study. The Caco-2 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Cellgro, Manassas, VA), while the HT29 and HCT116 cells were cultured in McCoy's 5A Medium (Cellgro, Manassas, VA). The culture medium was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. All cell lines were cultured at 37°C in a humidified atmosphere containing 5% CO2 and the medium was changed every two days. FGF-2 (10 ng/ml, R&D Systems, Minneapolis, MN) was solubilized in culture medium containing heparin sodium salt (1.25 ug/ml; Sigma, St. Louis, MO). FGF-2 was added daily beginning at 24 hours post-plating and until cells were harvested for assay, as described previously [22]. Caco-2 cells have previously been used as a model of Paneth cells, and differentiation into a Paneth cell phenotype has been confirmed with Paneth cell markers, including expression of alpha-defensin 5, alpha-defensin 6, lysozyme, and sPLA2 in previous studies [22-24]. For experiments in which Caco-2 cells were stimulated with TNFα, Caco-2 cells were first treated with FGF-2 for 72 hours, followed by addition of medium alone (control) or medium containing TNFα (R&D Systems, Minneapolis, MN) at 50 ng/mL. Cells were harvested at four hours for mRNA and 8 hours for protein analyses. Prior to cell harvesting for Western blot, Caco-2 cells were treated with Brefeldin-A (Sigma) at 5 ug/ml for a total of 8 hours (replenished every four hours) to prevent secretion of DEFA5 into the cellular medium.
Total RNA
Total RNA was isolated using TRIzol reagent (Life Technologies, Grand Island, NY) according to the manufacturer's protocol. RNA concentrations were determined using a Nano-Drop 1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). The isolated RNA was stored at -80°C until use in Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR).
Quantitative reverse transcription-polymerase chain reaction for mRNA
One microgram of total RNA was converted to complementary DNA (cDNA) using the qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD). qRT-PCR was performed using SYBR Green Power PCR Master Mix (Applied Biosystems, Foster City, CA). The qRT-PCR amplifications were performed on the LightCycler 480 realtime PCR system (Roche, Indianapolis, IN). The cycles passing threshold (Ct) were recorded and the expression of GAPDH was used as an internal control. The primers for DEFA5 were 5′-ACCCAGAAGCAGTCTGGGGAAGA-3′ (forward) and 5′ – GGTGGCTCTTGCCTGAGAACCTGA-3′ (reverse). The primers for GAPDH were 5′ – CGACCACTTTGTCAAGCTCA-3′ (forward) and 5′-AGGGGAGATTCAGTGTGGTG-3′ (reverse).
Quantitative reverse transcription-polymerase chain reaction for miRNA
One microgram of total RNA was converted to cDNA using the NCode VILO miRNA cDNA Synthesis Kit (Life Technologies)followed by qPCR using the NCode EXPRESS SYBR GreenER miRNA qRT-PCR kit (Life Technologies). The cycles passing threshold were recorded and the expression of miRNAs was calculated relative to U6B, a ubiquitously expressed small nuclear RNA. The forward primer for miR-124 was 5′- TAAGGCACGCGGTGAATGCC-3′. The forward primer for miR-924 was 5′-CCTCTGCCCTCTAAAGGTTTGC-3′. The forward primer for U6B was 5′-CGCAAGGATGACACGCAAATTCG-3′. The reverse primer was the NCode miRNA universal qPCR primer (Invitrogen). Data are presented as target miRNA or mRNA expression=2ΔCt, with ΔCt=(U6B or GAPDH Ct – target miRNA or mRNA Ct). qRT-PCR was carried out in triplicate for each sample for both the U6B or GAPDH control and each miRNA or mRNA.
Western blot
Cells were lysed in cold RIPA buffer (Thermo Fisher Scientific, Rockford, IL) supplemented with 1% protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Protein concentrations were determined with the BCA protein assay (Thermo Fisher Scientific). Cell lysates were suspended in × Laemmli sample buffer (Bio-Rad, Hercules, CA) containing 5% 2-mercaptoethanol and boiled for 5 minutes. After heat denaturation, total protein lysates (30 ug/lane) were subjected to tricine-SDS-PAGE [25]. The proteins were then transferred electrophoretically to 0.2 um PVDF membranes. Membranes were blocked in blocking buffer (LI-COR Biosciences, Lincoln, NE) diluted 1:1 in × PBS for 1 hour at room temperature. The blots were incubated with mouse anti-DEFA5 (Santa Cruz Biotechnologies, Santa Cruz, CA; 1:200 dilution), goat anti-actin (Sigma; 1:1000 dilution), and mouse anti-GAPDH (Life Technologies; 1:1000 dilution) overnight at 4°C. After washing with PBST, blots were incubated with Alexa Fluor 680 (Life Technologies) and IRDye 800CW (LI-COR Biosciences) conjugated secondary antibodies (1:10,000 dilution) for 1 hour at room temperature. The band intensities were quantified using an Odyssey infrared imaging system (LI-COR Biosciences) and analyzed using Image Studio Lite 3.1 (LI-COR Biosciences).
DEFA5 3′ UTR construct
The full-length 3′ UTR of DEFA5 (nucleotides 328-451 of NM_021010) was cloned into the PmeI and SacI sites downstream of the dual firefly and Renilla luciferase reporter vector, pmirGLO construct (Promega, Madison, WI) by GenScript. The full length 3′ UTR was also used as a template to mutate the entire seed region of the putative miR-124 and miR-924 binding sites.
miRNA mimic and luciferase construct transfection
Synthesized RNA duplexes of miRNA mimics (agomiRs) to miR-124 and miR-924 and the negative control were obtained from Sigma (St. Louis, MO). A miRNA mimic or a luciferase construct was transfected into Caco-2 cells using Lipofectamine 2000 reagent (Life Technologies) according to the manufacturer's guidelines. At 24 hours post-transfection, cells were harvested for RNA and protein analyses or were harvested for measurement of luciferase activities.
Luciferase reporter assay
Caco-2 cells were lysed in passive lysis buffer and then analyzed for the firefly and Renilla luciferase activities using the Dual-Luciferase Reporter Assay System (Promega) on the GloMax-Multi Detection System Luminometer (Promega) according to the manufacturer's instructions. The firefly luciferase activity was normalized to the renilla luciferase activity.
Statistical analysis
All experiments were performed with four biological repeats in triplicate. R (R Development Core Team, Vienna, Austria) was used for statistical analysis [26]. Statistical significance was determined by 2-tailed Student's t tests (for comparison of two conditions) and one-way ANOVA for comparison of multiple groups. Data are presented as mean ± standard error of the mean. P< 0.05 was considered significant.
Results
DEFA5 expression in colonic epithelial cells
Alpha defensin expression and inducible expression by FGF-2 were ascertained in three different colonic epithelial cell lines: Caco-2, HCT116, and HT29. Both the Caco-2 and HCT116 cell lines expressed DEFA5, while the HT29 cell lines did not express DEFA5. DEFA5 expression was significantly increased in Caco-2 cells following treatment for 72 hours with FGF-2, as previously demonstrated by Brodrick et al. and Tan et al. (Figure 1) [22-24]. Because Caco-2 cells expressed the α-defensins at a highly inducible level, all subsequent work was conducted in that cell line. Next, we confirmed that DEFA5 mRNA expression was inducible in Caco-2 cells by both FGF-2 and TNFα. Specifically, DEFA5 mRNA expression after treatment with FGF-2 (10 ng/ml) for 72 hours and FGF-2 for 72 hours plus TNFα (50 ng/ml) for 4 hours was measured (Figure 2A). Caco-2 cells stimulated with FGF-2 resulted in a statistically significant 5.3 fold increase and those stimulated with FGF-2 followed by TNFα resulted in a statistically significant 17.5 fold increase (Figure 2A). Treatment of Caco-2 cells with FGF-2 in combination with TNFα for 8 hours resulted in a statistically significant 1.8 fold increase in protein expression as measured by densitometry performed on Western blots (Figure 2B). An example Western blot of Actin and DEFA5 is shown (Figure 2C).
Figure 1.
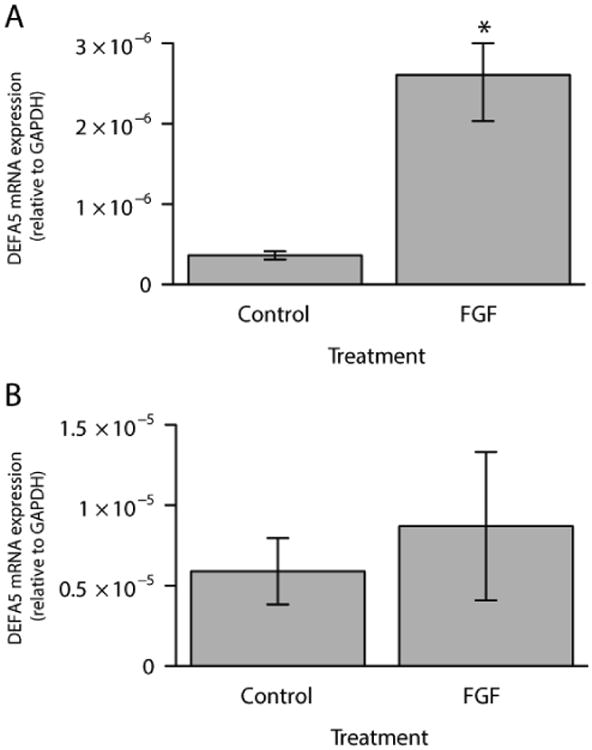
Endogenous DEFA5 expression (control) and following 72 hours treatment with FGF-2 (FGF) in Caco-2 (A) and HCT116 (B) cell lines. Results are mean ± standard error, n=4. *P<0.05
Figure 2.
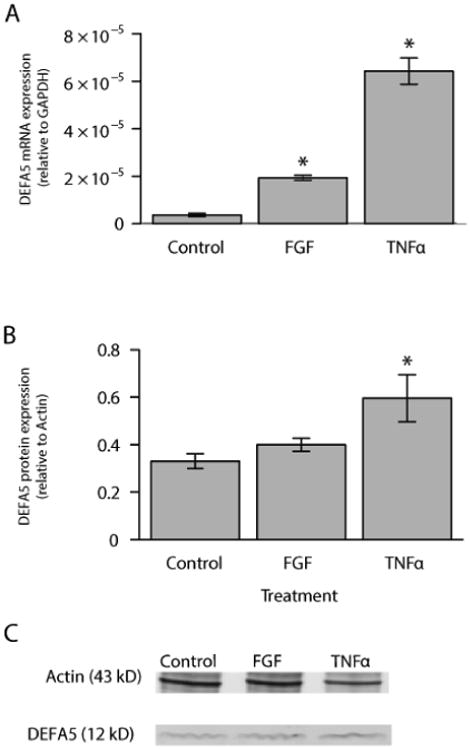
DEFA5 mRNA and protein expression in Control, FGF-2, and FGF-2/TNFα-stimulated Caco-2 colonic epithelial cells. (A) The mRNA expression of DEFA5 was assessed at 72 hours after FGF-2 stimulation or at 72 hours of FGF-2 plus 4 hours of TNFα stimulation. (B) Densitometric analysis of protein expression following induction of Caco-2 cells presented in graphical form. (C) Assessment of DEFA5 protein expression by Western blot. Actin served as a loading control. Results are mean ± standard error, n=4, *P<0.05.
Expressions of miRNAs that bind to DEFA5 3′ UTR in Caco-2 cells
An in silico analysis utilizing miRBase and TargetScan identified fifteen putative miRNA binding sites in the 3′ UTR of DEFA5. We identified the miRNAs with putative binding sites in the DEFA5 3′UTR and searched for miRNAs that were down-regulated in response to FGF-2 and TNFα. Of those miRNAs, we identified miR-124 and mi-924 (Figure 3) [27-30]. The other thirteen miRNAs were not examined further due to low relative expression. Because we hypothesized that the FGF- and TNFα- induced increase in DEFA5 expression may be influenced by a corresponding down-regulation of regulatory miRNAs, we examined the expression of miR-124 and miR-924 in response to FGF-2 and TNFα (Figure 4). DEFA5 induction was accompanied by a significant decrease in expression of miR-124 and miR-924 compared to unstimulated cells (Figures 4A and 4B). Our data demonstrate an inverse correlation between DEFA5 expression and the expression of miR-124 and miR-924, suggesting that DEFA5 may be regulated by miRNAs. Additionally, these data raise the possibility that the effect of FGF-2 and TNFα may be at least in part mediated by a down-regulation of DEFA5-associated miRNAs. To determine the effect of endogenous miRNAs on DEFA5 expression, a luciferase reporter construct containing the wild-type DEFA5 3′UTR was transfected into unstimulated Caco-2 cells. Transfecting this construct resulted in a 76% decrease in relative luciferase activity compared to transfection of the empty vector, which contained no 3′ UTR (Figure 5). These data indicate that the DEFA5 3′ UTR has functional miRNA binding sites to which endogenous miRNAs can bind and negatively regulate DEFA5 gene expression. Next, a luciferase reporter construct containing the DEFA5 3′ UTR with mutated binding sites of either miR-124 or miR-924 were transfected into Caco-2 cells. Transfection of the mismatched miR-124 construct resulted in a restoration to 66% of the relative luciferase activities of the empty vector (Figure 5). Transfection of the mismatched miR-924 construct restored relative luciferase activities to 85% of the empty vector (Figure 5). These data indicate that mutation of the binding sites led to a loss of regulation by each miRNA. This increase in relative luciferase activity compared to transfection of the empty vector was statistically significant in both miRNAs tested. We next tested whether miR-124 and miR-924 can negatively regulate basal DEFA5 expression. A miR-124 or miR-924 mimic was transiently transfected into Caco-2 cells and DEFA5 mRNA expression assessed (Figure 6). The miR-124 and miR-924 mimics significantly decreased DEFA5 mRNA expression by 47% and 50%, respectively. In addition, transfection of the miR-124 mimic resulted in a 75% decrease in DEFA5 protein expression as demonstrated by densitometry and transfection of the miR-924 mimic resulted in a 60% decrease in DEFA5 protein expression. Overall, these data suggest that miRNAs negatively regulate the mRNA and protein expression of DEFA5.
Figure 3.
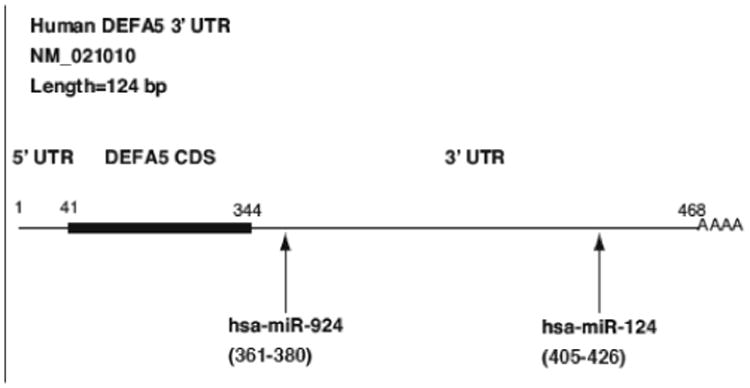
Schematic representation of DEFA5 mRNA with putative miRNA binding sites in 3′ UTR
Figure 4.
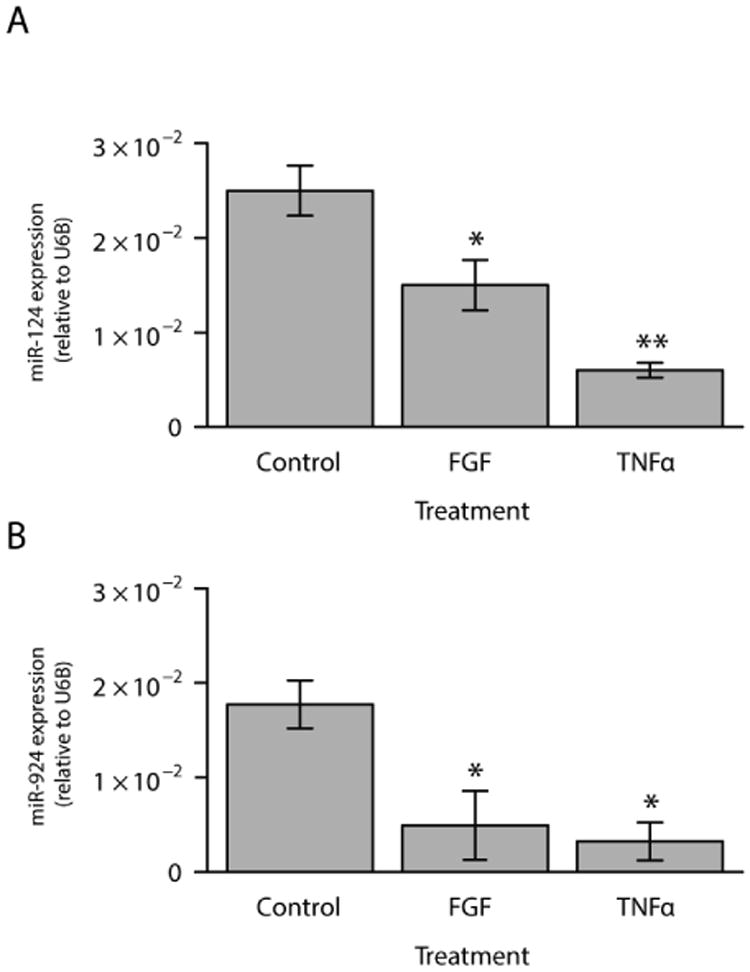
DEFA5-3′-UTR-associated miRNA expression in stimulated Caco-2 colonic epithelial cells. The expression of DEFA5-associated miRNAs was assessed at 72 hours after FGF-2 stimulation or at 72 hours of FGF-2 plus 4 hours of TNFα stimulation. (A) demonstrates the expression of miR-124 and (B) indicates the expression of miR-924. Results are mean ± standard error, n=4, *P<0.05, **P<0.01
Figure 5.
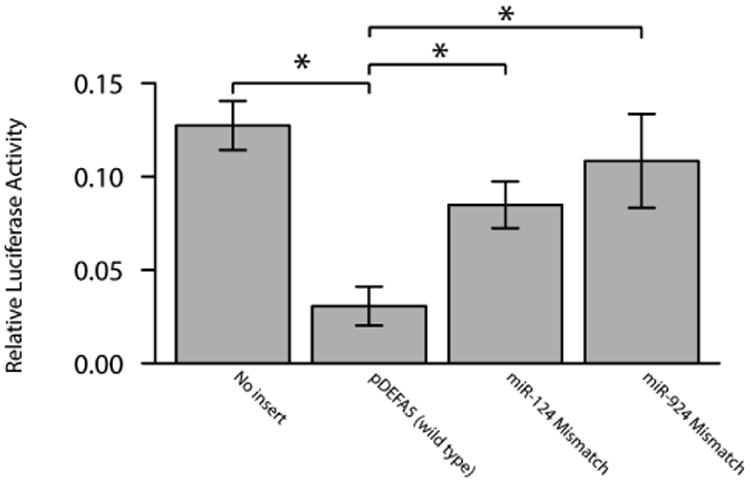
Luciferase reporter activity in the pMIR-DEFA5-3′ UTR reporter construct. Luciferase activity (normalized to Renilla luciferase activity) data is presented relative to the pMIR Reporter. Results are mean ± standard error, n=4, *P<0.05.
Figure 6.
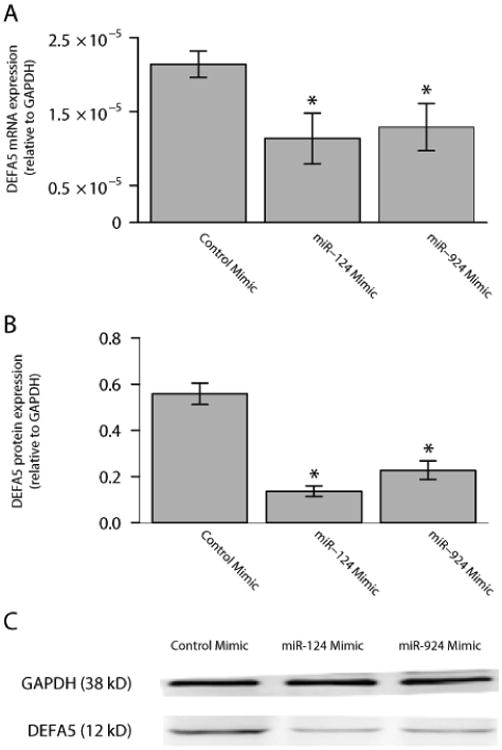
miR-124 and miR-924 inhibition of DEFA5 mRNA and protein expression. (A) DEFA5 mRNA expression was significantly reduced in Caco-2 cells transfected with either miR-124 or miR-924 mimics. * P<0.05 (B) DEFA5 protein expression was significantly reduced in Caco-2 cells transfected with either miR-124 or miR-924 mimics. (C) Assessment of DEFA5 protein expression by Western blot. GAPDH served as a loading control. Results are mean ± standard error, n=4, *P<0.05.
Discussion
To the best of our knowledge, this is the first study to show miRNA regulation of DEFA5. We hypothesized that miRNAs regulate DEFA5 expression. We compared the expression of DEFA5 and miRNAs with putative binding sites in the 3′ UTR of DEFA5 in Caco-2 cells. We found that the expression of miR-124 and miR-924 was inversely correlated with that of DEFA5. Additionally, transfecting luciferase constructs containing the DEFA5 3′ UTR into Caco-2 cells resulted in decreased relative luciferase activity compared to transfecting empty vectors. Furthermore, creating a mismatch in the seed region of the miR-124 and miR-924 in the 3′UTR of DEFA5 resulted in restoration of luciferase activity. Finally, transfection of the miR-124 and miR-924 mimics significantly decreased DEFA5 mRNA expression and protein expression.
MicroRNAs regulate many processes, including differentiation and activation of the cells of the immune system. They have been demonstrated to play a role in a number of autoimmune diseases, including systemic lupus erythematosus, rheumatoid arthritis, and IBD [31-34]. Overall, the role of miRNAs as key negative regulators of inflammation, innate immunity, and epithelial function is being increasingly recognized [35]. A number of changes in miRNA expression in IBD and inflammation have been described [16-18]. As a result, miRNA regulation as an important component of intestinal epithelial innate immunity in epithelial cells is logical [36,37]. In a murine model, miR-146a regulated gut inflammation via the NOD2-sonic hedgehog (SHH) signaling pathway by suppressing SHH and ultimately resulting in increased pro-inflammatory cytokine expression [38]. Conversely, in HT29 cells, miR-122 decreased pro-inflammatory cytokines by downregulating LPS-induced NOD2 expression [39].
Specifically, we demonstrated that miRs-124 and -924 influence DEFA5 gene expression. Our data identifies an additional role for miR-124, which previously had been shown to be involved in IBD by regulation of STAT3 and regulation of CNS macrophages [40,41]. In children with UC and mice with experimental colitis, miR-124 levels were significantly decreased while STAT3 and downstream genes were up-regulated [40]. The IL6/STAT3 pathway activation has been shown to play a role in colitis by promoting inappropriate survival of T cells [42]. In a study examining CNS inflammation in vivo and in an experimental model, miR-124 was shown to play a central role in regulating microglial and macrophage quiescence [41]. miR-124 inhibited macrophage activation by binding the C/EBPα transcription factor [41]. This study defines a novel role for miR-924, which has not been previously described to regulate any genes.
A growing body of research demonstrates that defensins play a central role in IBD pathogenesis, in part related to changes in Wnt-signaling and mutations in NOD2 [43-45]. However, these mutations do not explain changes in defensin expression in a large proportion of patients, and these changes in defensin expression could perhaps be accounted for by dysregulation of miRNA regulation, as demonstrated in this study.
These changes in miRNA expression may explain alterations in α-defensin expression in IBD. Crohn's disease of the terminal ileum is unique because of the large number of Paneth cells and the highest density of microbes in the distal small intestine, which is otherwise low in the normal proximal small intestine [46,47].
In the ileal mucosa of CD, a decrease in antimicrobial activity could be attributed to decreased Paneth cell α-defensin expression [13]. The decreased levels of expression have been suggested to be predisposing factor for development of CD [13]. Additionally, these decreased levels in terminal ileum of CD patients with ileal disease involvement were noted to occur regardless of the presence of inflammation [14,43,48]. Levels of other Paneth cell products were unchanged or increased, indicating that the decrease in DEFA5 could be due to a defect in Paneth cell α-defensin regulation [14]. Alterations in miRNA regulation in IBD may explain defective α-defensin regulation in patients' whose disease cannot be attributed to mutations in NOD2.
One limitation of this study was the absence of an ideal experimental model. Colonic epithelial cell lines express α-defensin at relatively low levels. Because human Paneth cells do not survive under in vitro culture conditions, Caco-2 intestinal epithelial cells were selected since they share characteristics with small intestinal epithelial differentiation in vitro and constitutively express NOD2, much like Paneth cells [5,49]. Caco-2 cells also express fibroblast growth factor receptor-3, which is a critical regulator of Paneth cell differentiation during gut development [22,50]. Additionally, Caco-2 cells treated with FGF-2 provides the most suitable in vitro model available, as demonstrated in previous studies [22-24]. While the use of Caco-2 cells confirmed the regulation of DEFA5 by miRNAs, it may not exactly reflect small intestine physiology.
Overall, this study demonstrates that miRNAs are an important negative regulator of DEFA5. Our data establishes miRNA regulation of defensins and raises the possibility of dysfunctional miRNA regulation contributing to reduced DEFA5 levels seen in CD. Further examination of miRNA regulation of defensins, especially in inflammatory states, may contribute to a better understanding of the pathogenesis and may lead to the development of new diagnostic and therapeutic strategies for IBD patients.
Acknowledgments
We acknowledge Mark Musch PhD for assistance with tissue culture, and Edith Porter MD for assistance in measuring DEFA5 by Western blot. We acknowledge the following sources of support: the National Institute of Health (K08 DK078046 to J.H.K.), the NIH DDRCC core grant (P30 DK42086), and the NIH NIDDK training grant (T32 DK007074 to D.R.B.M.)
References
- 1.Dalal SR, Kwon JH. The Role of MicroRNA in Inflammatory Bowel Disease. Gastroenterol Hepatol. 2010;6:714–722. [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F, Dassopoulos T, Cope L, Maitra A, Brant SR, et al. Genome-wide gene expression differences in Crohn's disease and ulcerative colitis from endoscopic pinch biopsies: insights into distinctive pathogenesis. Inflamm Bowel Dis. 2007;13:807–821. doi: 10.1002/ibd.20110. [DOI] [PubMed] [Google Scholar]
- 3.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 5.Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, et al. Crohn's disease and the NOD2 gene: a role for paneth cells. Gastroenterology. 2003;125:47–57. doi: 10.1016/s0016-5085(03)00661-9. [DOI] [PubMed] [Google Scholar]
- 6.Ouellette AJ. Paneth cell alpha-defensin synthesis and function. Curr Top Microbiol Immunol. 2006;306:1–25. doi: 10.1007/3-540-29916-5_1. [DOI] [PubMed] [Google Scholar]
- 7.Grigat J, Soruri A, Forssmann U, Riggert J, Zwirner J, et al. Chemoattraction of macrophages, T lymphocytes, and mast cells is evolutionarily conserved within the human alpha-defensin family. J Immunol Baltim Md 1950. 2007;179:3958–3965. doi: 10.4049/jimmunol.179.6.3958. [DOI] [PubMed] [Google Scholar]
- 8.Ouellette AJ. Paneth cells and innate mucosal immunity. Curr Opin Gastroenterol. 2010;26:547–553. doi: 10.1097/MOG.0b013e32833dccde. [DOI] [PubMed] [Google Scholar]
- 9.Salzman NH. Paneth cell defensins and the regulation of the microbiome: détente at mucosal surfaces. Gut Microbes. 2010;1:401–406. doi: 10.4161/gmic.1.6.14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salzman NH, Chou MM, de Jong H, Liu L, Porter EM, et al. Enteric salmonella infection inhibits Paneth cell antimicrobial peptide expression. Infect Immun. 2003;71:1109–1115. doi: 10.1128/IAI.71.3.1109-1115.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- 12.Wehkamp J, Fellermann K, Herrlinger KR, Bevins CL, Stange EF, et al. Mechanisms of disease: defensins in gastrointestinal diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2:406–415. doi: 10.1038/ncpgasthep0265. [DOI] [PubMed] [Google Scholar]
- 13.Wehkamp J, Schmid M, Fellermann K, Stange EF. Defensin deficiency, intestinal microbes, and the clinical phenotypes of Crohn's disease. J Leukoc Biol. 2005;77:460–465. doi: 10.1189/jlb.0904543. [DOI] [PubMed] [Google Scholar]
- 14.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, et al. Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc Natl Acad Sci USA. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferguson LR, Browning BL, Huebner C, Petermann I, Shelling AN, et al. Single nucleotide polymorphisms in human Paneth cell defensin A5 may confer susceptibility to inflammatory bowel disease in a New Zealand Caucasian population. Dig Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver. 2008;40:723–730. doi: 10.1016/j.dld.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Fasseu M, Tréton X, Guichard C, Pedruzzi E, Cazals-Hatem D, et al. Identification of restricted subsets of mature microRNA abnormally expressed in inactive colonic mucosa of patients with inflammatory bowel disease. 2010 doi: 10.1371/journal.pone.0013160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu F, Zikusoka M, Trindade A, Dassopoulos T, Harris ML, et al. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology. 2008;135:1624–1635. doi: 10.1053/j.gastro.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 18.Wu F, Zhang S, Dassopoulos T, Harris ML, Bayless TM, et al. Identification of microRNAs associated with ileal and colonic Crohn's disease. Inflamm Bowel Dis. 2010;16:1729–1738. doi: 10.1002/ibd.21267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olaru AV, Selaru FM, Mori Y, Vazquez C, David S, et al. Dynamic changes in the expression of MicroRNA-31 during inflammatory bowel disease-associated neoplastic transformation. Inflamm Bowel Dis. 2011;17:221–231. doi: 10.1002/ibd.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takagi T, Naito Y, Mizushima K, Hirata I, Yagi N, et al. Increased expression of microRNA in the inflamed colonic mucosa of patients with active ulcerative colitis. J Gastroenterol Hepatol. 2010;1:129–133. doi: 10.1111/j.1440-1746.2009.06216.x. [DOI] [PubMed] [Google Scholar]
- 21.Wu F, Guo NJ, Tian H, Marohn M, Gearhart S, et al. Peripheral blood microRNAs distinguish active ulcerative colitis and Crohn's disease. Inflamm Bowel Dis. 2011;17:241–250. doi: 10.1002/ibd.21450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brodrick B, Vidrich A, Porter E, Bradley L, Buzan JM, et al. Fibroblast growth factor receptor-3 (FGFR-3) regulates expression of paneth cell lineage-specific genes in intestinal epithelial cells through both TCF4/beta-catenin-dependent and -independent signaling pathways. J Biol Chem. 2011;286:18515–18525. doi: 10.1074/jbc.M111.229252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan G, Li R, Li C, Wu F, Zhao X, et al. Down-regulation of human enteric antimicrobial peptides by NOD2 during differentiation of the paneth cell lineage. 2015;5:8383. doi: 10.1038/srep08383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tan G, Zeng B. Regulation of human enteric α-defensins by NOD2 in the Paneth cell lineage. Eur J Cell Biol. 2015;94:60–66. doi: 10.1016/j.ejcb.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Haider SR, Reid HJ, Sharp BL. Protein Electrophoresis: Methods and Protocols. Springer Science; 2012. Tricine-SDS-PAGE. [Google Scholar]
- 26.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. [Google Scholar]
- 27.Grifths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32:D109–D111. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 31.Schneider MR. MicroRNAs as novel players in skin development, homeostasis and disease. Br J Dermatol. 2012;166:22–28. doi: 10.1111/j.1365-2133.2011.10568.x. [DOI] [PubMed] [Google Scholar]
- 32.Pekow JR, Kwon JH. MicroRNAs in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:187–193. doi: 10.1002/ibd.21691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filková M, Jüngel A, Gay RE, Gay S. MicroRNAs in rheumatoid arthritis: potential role in diagnosis and therapy. BioDrugs Clin Immunother Biopharm Gene Ther. 2012;26:131–141. doi: 10.2165/11631480-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 34.Amarilyo G, La Cava A. miRNA in systemic lupus erythematosus. Clin Immunol Orlando Fla. 2012;144:26–31. doi: 10.1016/j.clim.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Zhai Z, Wu F, Dong F, Chuang AY, Messer JS, et al. Human autophagy gene ATG16L1 is post-transcriptionally regulated by MIR142-3p. Autophagy. 2014;10:468–479. doi: 10.4161/auto.27553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu LF, Liston A. MicroRNA in the immune system, microRNA as an immune system. Immunology. 2009;127:291–298. doi: 10.1111/j.1365-2567.2009.03092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D, et al. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci USA. 2007;104:1604–1609. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghorpade DS, Sinha AY, Holla S, Singh V, Balaji KN, et al. NOD2-nitric oxide-responsive microRNA-146a activates Sonic hedgehog signaling to orchestrate inflammatory responses in murine model of inflammatory bowel disease. J Biol Chem. 2013;288:33037–33048. doi: 10.1074/jbc.M113.492496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y, Wang C, Liu Y, Tang L, Zheng M, et al. miR-122 targets NOD2 to decrease intestinal epithelial cell injury in Crohn's disease. Biochem Biophys Res Commun. 2013;438:133–139. doi: 10.1016/j.bbrc.2013.07.040. [DOI] [PubMed] [Google Scholar]
- 40.Koukos G, Polytarchou C, Kaplan JL, Morley-Fletcher A, Gras-Miralles B, et al. MicroRNA-124 regulates STAT3 expression and is down-regulated in colon tissues of pediatric patients with ulcerative colitis. Gastroenterology. 2013;145:842–852. doi: 10.1053/j.gastro.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL, et al. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. Nat Med. 2011;17:64–70. doi: 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugimoto K. Role of STAT3 in inflammatory bowel disease. World J Gastroenterol. 2008;14:5110–5114. doi: 10.3748/wjg.14.5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bevins CL, Stange EF, Wehkamp J. Decreased Paneth cell defensin expression in ileal Crohn's disease is independent of inflammation, but linked to the NOD2 1007fs genotype. Gut. 2009;58:882–883. discussion 883-884. [PubMed] [Google Scholar]
- 44.Perminow G, Beisner J, Koslowski M, Lyckander LG, Stange E, et al. Defective paneth cell-mediated host defense in pediatric ileal Crohn's disease. Am J Gastroenterol. 2010;105:452–459. doi: 10.1038/ajg.2009.643. [DOI] [PubMed] [Google Scholar]
- 45.Wehkamp J, Wang G, Kübler I, Nuding S, Gregorieff A, et al. The Paneth cell alpha-defensin deficiency of ileal Crohn's disease is linked to Wnt/Tcf-4. J Immunol Baltim Md 1950. 2007;179:3109–3118. doi: 10.4049/jimmunol.179.5.3109. [DOI] [PubMed] [Google Scholar]
- 46.Porter EM, Bevins CL, Ghosh D, Ganz T. The multifaceted Paneth cell. Cell Mol Life Sci CMLS. 2002;59:156–170. doi: 10.1007/s00018-002-8412-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramasundara M, Leach ST, Lemberg DA, Day AS. Defensins and inflammation: the role of defensins in inflammatory bowel disease. J Gastroenterol Hepatol. 2009;24:202–208. doi: 10.1111/j.1440-1746.2008.05772.x. [DOI] [PubMed] [Google Scholar]
- 48.Satsangi J, Silverberg MS, Vermeire S, Colombel JF. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut. 2006;55:749–753. doi: 10.1136/gut.2005.082909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Molmenti EP, Perlmutter DH, Rubin DC. Cell-specific expression of alpha 1-antitrypsin in human intestinal epithelium. J Clin Invest. 1993;92:2022–2034. doi: 10.1172/JCI116797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vidrich A, Buzan JM, Brodrick B, Ilo C, Bradley L, et al. Fibroblast growth factor receptor-3 regulates Paneth cell lineage allocation and accrual of epithelial stem cells during murine intestinal development. Am J Physiol Gastrointest Liver Physiol. 2009;297:G168–G178. doi: 10.1152/ajpgi.90589.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]