

Abstract
Protein Kinase C (PKC) is a family of Ser/Thr kinases that regulate a multitude of cellular processes through participation in the phosphoinositide signaling pathway. Significant research efforts have been directed at understanding the structure, function, and regulatory modes of the enzyme since its discovery and identification as the first receptor for tumor-promoting phorbol esters. The activation of PKC involves a transition from the cytosolic auto-inhibited latent form to the membrane-associated active form. The membrane recruitment step is accompanied by the conformational rearrangement of the enzyme, which relieves auto-inhibitory interactions and thereby enables PKC to phosphorylate its targets. The multi-domain structure and intrinsic flexibility of PKC present remarkable challenges and opportunities for the biophysical and structural biology studies of this class of enzymes and their interactions with membranes – the major focus of this Current Topics article. I will highlight the recent advances in the field, outline the current challenges, and identify areas where biophysics and structural biology approaches can provide insight into the isoenzyme-specific regulation of PKC activity.
Keywords: protein kinase C, lipid signaling, protein-lipid interactions, diacylglycerol, phosphatidylinositol-4, 5-bisphosphate, phorbol ester, phosphatidylserine, C1 domain, C2 domain, V5 domain, FRET, NMR
TOC image
Scope
The discovery of PKC1,2 and its subsequent identification as the receptor for tumor-promoting phorbol esters3 have spurred the development of a vibrant research field that combines biochemical, genetic, and biophysical approaches. The hallmark of PKC activation is its translocation to cellular endo-membranes that occurs in response to second messengers such as Ca2+, diacylglycerol (DAG), and phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2). This Current Topics article focuses exclusively on the biophysical studies of the PKC isoenzymes and their interactions with membranes. The term “dynamics” is used in two different contexts. “Structural dynamics” refers to the changes in the tertiary structure of the enzyme that accompanies the membrane association step. “Protein dynamics” refers to the motions within the individual domains of PKC that occur on multiple timescales. The intrinsically dynamic nature of PKC is essential for its ability to interact with lipid ligands during the activation process.
Introduction
PKC is a family of multi-modular ~80 kDa Ser/Thr kinases that regulate cell growth, differentiation, apoptosis, and motility.4–6 All members of the PKC family consist of the N-terminal regulatory and C-terminal catalytic domains, connected by a proteolytically sensitive hinge region (Figure 1). Four novel PKC isoenzymes (nPKCs; ε, δ, θ, and η) are activated by DAG alone, whereas the four conventional PKC isoenzymes (cPKCs; α, βI, βII, and γ) require Ca2+ in addition to DAG. The atypical PKCs (aPKCs; ζ and ι/λ) do not bind either second messenger.
Figure 1.
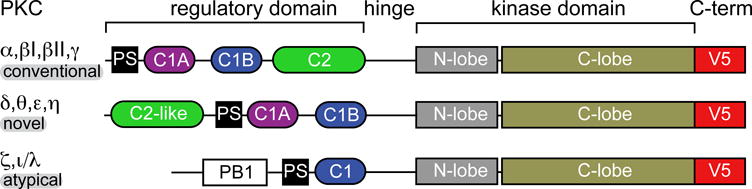
Multi-modular structure of PKC isoenzymes. C1 and C2 are conserved region-1 and conserved region-2 domains, respectively; PS is a pseudo-substrate region; and PB1 is a Phox and Bem1 domain. The most variable PKC regions are the N-terminal regulatory and the C-terminal V5 domains.
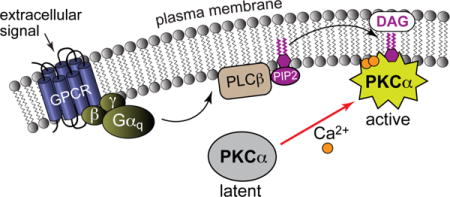
A general overview of the conventional PKC signaling pathway is shown in Figure 2A. G protein-mediated activation of phospholipase Cβ (PLCβ) results in the hydrolysis of lipids bearing the PtdIns(4,5)P2 head group. Two second messengers are generated as a result of this reaction, DAG and inositol 1,4,5-trisphosphate (IP3). The latter induces the release of Ca2+ from the endoplasmic reticulum. The recruitment of PKCs to endo-membranes in response to DAG/Ca2+ relieves the auto-inhibitory interaction within the enzyme thereby activating its kinase function. The regulation of aPKC activity involves protein-protein interactions (reviewed in7,8), but overall, their activation mechanism is not well understood.
Figure 2.
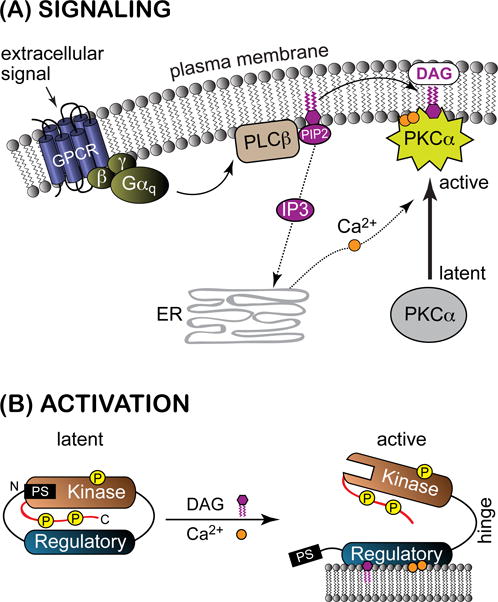
Signaling pathway (A) and activation (B) of PKCα. The abbreviations are given in the text. The C-terminal V5 domain is shown in red. DAG-dependent PKCs are constitutively phosphorylated as part of the maturation process, with one site on the kinase activation loop and two sites on the V5 domain.
Altered levels of expression or activity of DAG-dependent PKC isoenzymes have been implicated in a large number of human diseases such as cardiac disease, cancer, diabetes, and mood disorders.9–12 PKCα, the most predominant isoenzyme in mouse,13 rabbit,14 and human15 hearts, has been identified as a major regulator of heart contractility,15,16 platelet aggregation in thrombosis,17–19 and virtually every development stage of atherosclerotic disease.20,21 Up-regulation of PKCα has been demonstrated in breast cancer cells,22,23 melanoma cells,24 and tissue samples of urinary bladder carcinomas25 and malignant gliomas.26 The opposing roles of PKCδ and PKCε in ischemic disease and reperfusion injury27,28 demonstrated the need for isoenzyme-selective approaches to achieve down-29,30 and up-regulation31–33 of the respective PKC isoenzymes.
Given the differential and sometimes opposing roles of PKC in human disease, there is a need to modulate their function in isoenzyme-selective manner. One of the means to achieve that is to make use of the differences within the most variable regions, such as the N-terminal regulatory and the C-terminal V5 domains. This would also require atomic-level information about PKC in both latent and activated states. However, the structural studies of PKC family, especially at the atomic-level, have not kept pace with the biochemical and genetic advances in the field. This is not due to a lack of effort but rather due to the challenging properties of this class of enzymes, such as their intrinsically dynamic and amphiphilic nature.
These two features are essential for enzyme function, as illustrated in the activation scheme of Figure 2B. The latent cytosolic form, which is auto-inhibited by the N-terminal pseudo substrate region,34–37 is thought to be compact. The auto-inhibitory interaction is released upon interaction of the N-terminal regulatory domain with endo-membranes. The N-terminal domain consists of individual domains, each one being independently folded and having a specialized function. C1 domains interact with membrane-embedded DAG, while the C2 domain of cPKCs interacts with phosphatidylserine (PtdSer) and PtdIns(4,5)P2 in Ca2+-dependent manner. The C2-like domain in nPKCs is not a membrane-binding module. The intrinsic affinities of the C1 and C2 domains to their respective ligands determine the activation threshold of the parent PKC isoenzymes and contribute to the selectivity of the PKC response. It should be noted that PKC activation could be accomplished not only by DAG/Ca2+ driven membrane recruitment, but also by the proteolysis of the hinge region and destabilization of the latent form through interference with intra-molecular interactions. One example of the latter is the activation of PKC by the pseudo-RACK1 peptide reported by the Mochly-Rosen’s group.38
The focus of this article is the biophysical and structural biology studies of conventional and novel PKC isoenzymes and their interactions with membranes. The article follows the structural hierarchy of PKC by starting with the full-length enzymes and then narrowing down the scope to individual domains. For the latter, I will use several examples from the work of my laboratory.
1. Structural dynamics of PKC
The structural dynamics of PKC involves a conformational transition between the latent and activated states. The latent conformation is stabilized by intra-molecular interactions. Identifying the structural basis of these interactions has been challenging because of the multi-domain structure and the associated flexibility of PKCs, the requirement for sample homogeneity with respect to the phosphorylation state, and the sensitivity of the hinge region to proteolysis. This section summarizes the biophysical studies of latent and activated conformations of full-length PKC.
1.1. Latent PKC
In the latent form, the N-terminal pseudo-substrate (PS) region inhibits the active site, which is located between the two lobes of the catalytic domain. The PS sequence has an Ala instead of the phospho-acceptor Ser/Thr site, thereby achieving an auto-inhibitory effect. The structural information about this interaction is currently unavailable for any PKC isoenzyme. Three other intra-molecular interactions have been identified, all of which involve the membrane-binding modules of PKC.
C1B clamp
In 2011, a partial crystal structure of PKCβII from R. norvegicus was determined at 4 Å resolution (PDB ID: 3PFQ).39 One noteworthy feature of the structure is the “C1B clamp” formed by the three domains that are non-adjacent in the amino acid sequence: C1B, the N-terminal lobe of the kinase and V5 (Table 1). It was proposed that this intra-molecular interaction is inhibitory in nature because: (i) the C1B “clamping” onto the conserved Phe629 of V5 prevents this residue from interacting with the adenine of ATP, and (ii) residues 619–621 of V5 occlude the DAG-binding site of C1B. Another feature of the structure is that the C2 domain makes no intra-molecular contacts but is involved in the interactions with the catalytic domain of the neighboring PKC molecules. Understanding how C2 contributes to the stabilization of the latent form at the atomic level will require further studies (vide infra). Three PKCβII regions: the pseudo-substrate, C1A domain, and the hinge region between the regulatory and kinase domains could not be identified in the electron density maps and, as a result, are not present in the structure. Overall, the PKCβII structure was interpreted as that of an “intermediate” (i.e. neither latent nor activated) state on the catalytic pathway.
Table 1.
Intra-molecular interactions in PKC isoenzymes.
| Isoenzy me/Ref. | Interacting domains | Methods | Schematic view of intra-molecular interactions |
|---|---|---|---|
| βII39 | C1B, V5, and N-terminal lobe of kinase | X-ray crystallography, mutagenesis, and phorbol-ester stimulated membrane translocation |
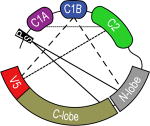 PS denotes the pseudo-substrate region responsible for auto-inhibition. |
| α40,41 | C1A and C2 | Computational modeling, mutagenesis, in-vitro membrane binding by SPR, DAG-stimulated cellular membrane translocation | |
| α42 | C2 and V5 | Mutagenesis, FRET, and DAG-stimulated cellular membrane translocation |
C1 and C2
Using SPR, Slater et al.41 detected a nanomolar-affinity interaction between the chip-immobilized C1 domain consisting of tandem C1A-C1B domains (see Figure 2B) and the full-length PKCα. This interaction was dependent on the presence of PKC activator tetradecanoylphorbol acetate, a phorbol ester. The interaction partner of C1 was identified as the C2 domain by using the C1–C2 construct of PKCα. In addition, extrinsic addition of C1 domain was shown to activate PKCα in a concentration- and phorbol ester-dependent manner. Overall, the data were consistent with C1 and C2 domains being engaged in intra-molecular interactions that maintain PKC in the latent form.
The question of C1–C2 interactions was also explored by Stahelin et al.40 Computational docking procedures were applied to the crystal structure of the C2 domain and the homology model of the C1A to obtain information about the inter-domain interface. From the analysis of the best-scoring docked structures, three putative interactions were identified between the C1A and C2: Asp55-Arg252, Arg42-Glu282, and Phe27-Phe255. Disruption of the electrostatic interactions via mutagenesis resulted in the increase of PKC affinity to DAG-containing membranes, indicating that C1A is less conformationally restricted and therefore has a more accessible DAG-binding site. Simultaneous charge reversal resulted in PKC variants having comparable membrane affinity to that of the wild-type protein. The data support a direct interaction or “tethering” between the C1A and C2 domains, which is released by the phosphatidylserine component of the plasma membrane when it interacts with C2.
C2 and V5
The existence of intra-molecular interactions between the C2 and the C-terminal V5 domains42 (Table 1) was hypothesized based on the observation that V5 binds the Lys-rich region of Syndecan-4, a known activator of PKCα. The C2 domain has a conserved positively charged region referred to as the “Lys-rich cluster” (LRC) that by analogy might be involved in intra-molecular interactions with V5. Indeed, mutations of two V5 phosphorylation sites, Thr638 and S657, and the acidic residues of the V5 hydrophobic motif resulted in the increase of DAG sensitivity, as reported by the DAG-stimulated translocation of PKCα variants to the plasma membrane.42 The DAG sensitivity of PKCα variants correlated with the conformational change that was probed using FRET between the N- and C-termini in the ECFP-PKCα-EYFP fusion protein. Re-introduction of the putative C2-V5 interaction by simultaneous charge reversal of the LRC of C2 and the hydrophobic motif of V5 resulted in membrane translocation behavior similar to that of the wild-type protein. In aggregate, the data support the existence of intra-molecular interaction between the C2 domain and the C-terminal V5 region that contributes to the stabilization of the latent form. The functional interplay between the C2 and V5 domains is also evident in PKCβII, where the phosphorylation of Ser-660 of the hydrophobic motif increases the affinity of the enzyme to Ca2+ and PtdSer.43
Summary
In addition to the auto-inhibition of the catalytic domain by the pseudo-substrate region, several intra-molecular interactions have been identified in the conventional PKC isoenzymes that contribute to the stabilization of the latent form. There is also indirect experimental evidence for the existence of the C1–C2 interactions in novel isoenzymes41 and a mention of the C2-V5 interaction in PKCε.44 To assemble the latent form of PKC in its entirety, more detailed information at the atomic level is needed. Given the complexity of the enzyme and its dynamic nature, structural biology approaches that target “pairwise” interactions between the individual domains is a viable route. The validity of the structural findings can then be tested using mutagenesis and functional assays in the full-length systems.
1.2. Activated PKC
The canonical activation route involves the association of PKC with membranes (Figure 2A). To date, the only source of information about the tertiary structure of lipid-associated PKC is the electron miscroscopy study of PKCδ and its regulatory domain.45 The 2D crystals of both protein species were grown on a lipid monolayer comprising the phosphatidylserine component and DAG with molar concentrations of 45% and 5%, respectively. The 3D image reconstruction revealed the overall shape of the proteins and enabled the tentative assignment of the domains engaged in the interactions with membranes (Figure 3). According to the assignment, a C1 domain – most likely the C1A – is a primary contact of PKCδ with the lipid monolayer. The interactions of the C2 domain with lipids are minor, as expected for a novel PKC isoenzyme. The small protrusion in the structure that contacts the C2 domain was assigned to the C-terminal V5 domain, by comparison with the 3D reconstruction of the regulatory domain alone. The potential role of V5 as a membrane-interaction module is discussed in the section on individual domains.
Figure 3.
3D reconstructions of PKCδ (A, B) and its regulatory domain RDδ (C, D) on lipid monolayers, showing the proposed assignments of individual domains (adapted from45). (A) and (C) represent the views from the membrane surface, which is shown with a textured bar. The catalytic domain is colored green.
Evidence for PKC dimerization
The affinity of individual PKC domains to one another can also manifest itself in inter-molecular interactions, provided that the interaction surfaces are solvent-accessible. The latter can be achieved in either fully or partially activated states of PKC. The homo-dimerization of conventional PKC isoenzymes was first demonstrated using cross-linking with sulfhydryl-selective reagent bis(maleimido)hexane.46 Slater et al.41 inferred dimerization from the concentration dependence of PKCα activity in the presence of enzyme activators. Recently, the homo-dimerization behavior of activated PKCα was characterized in detail using FRET between the donor and acceptor fluorophores that were placed on different PKCα molecules.47 Using deletion and truncation PKCα variants, it was demonstrated that dimer formation is mediated by the interactions between the respective regulatory domains, as well as the regulatory and catalytic domains. For the latter, the C-terminal V5 domain is crucial in stabilizing the dimer interface: its deletion or mutation of Cys619 to Ala abolished dimerization.
Rearrangement of PKC tertiary structure probed by TIRFM and FRET
The spatial rearrangement of the PKC upon maturation and encounter with membrane-embedded activators were recently explored in two separate studies that made elegant use of FRET48 and single-molecule total internal reflection fluorescence microscopy (TIRFM).49 Both studies used conventional PKC isoenzymes that have a Ca2+-responsive C2 domain.
In single-molecule TIRFM experiments, the population-weighted diffusion coefficient of fluorophore-tagged PKCα and its regulatory domain(s) on supported lipid bilayers reported on the protein-membrane interactions. The initial diffusion step for the isolated C2 domain and the full-length PKCα was identical, indicating that C2-membrane interaction is the first step of activation sequence, which is in agreement with previous functional studies.50,51 However, the diffusion of full-length PKCα on lipid bilayers exponentially slowed down with time, compared to the constant diffusion coefficient measured for the isolated C2. By using different truncation variants of the regulatory domain, the authors identified the C1A as the entity involved in the membrane interactions in the absence of DAG. The incorporation of DAG into lipid bilayers resulted in reduction of the diffusion coefficient of both full-length PKCα and C1A-C1B-C2. Frictional drag of both C1A and C1B was needed to quantitatively account for the changes in the diffusion coefficient, suggesting that both domains interact with DAG.
The authors proposed a model where the Ca2+-driven binding of C2 domain to PtdSer- or PtdSer/- containing membranes results in the formation of “pre-DAG intermediate”. In this intermediate, C1A is involved in ligand-independent interactions with membranes, thereby priming itself and the C1B for binding to DAG. The interaction of C1B with DAG can then trigger the release of the pseudo-substrate region from the active site of the kinase.
To probe the conformational rearrangement of PKCs during maturation, activation, and down-regulation in live cells, Newton’s laboratory designed a set of Kinameleon cPKC constructs that had CFP and YFP placed at the N- and C-termini of PKCβII.48 FRET changes in live cells were monitored under both unstimulated and stimulated conditions, with phorbol 12,13-dibutyrate (PDBu) as an agonist. The pattern of FRET ratios characteristic for different conformational states of PKCβII was established. Short stimulation by PDBu resulted in increase of the FRET ratios compared to the latent form of PKCβII, consistent with the pattern observed previously for PKCδ.52 Mutations that impair the kinase function of PKCβII, and prolonged exposure of wild-type species to PDBu – known to cause PKC dephosphorylation and subsequent degradation – resulted in the decrease of FRET ratios. These data correlate well with the kinetics of agonist-stimulated membrane translocation, measured by membrane-to-protein FRET. The kinetics reflects the accessibility of the C1 DAG/phorbol ester-binding site in different PKC states. Wild type PKCβII and PKCα translocate to membranes slower than their kinase-deficient mutants and the isolated C1A-C1B, indicating that the C1 ligand-binding site(s) are masked by the intra-molecular interactions in the latent state. Toggling the DAG affinities of C1A and C1B by mutating a key residue53 enabled the evaluation of their relative function roles. Both C1A and C1B contribute to the agonist-driven membrane binding, with C1B making a predominant contribution.
Summary
According to the current activation model that integrates the results of biophysical and biochemical studies of conventional PKC isoenzymes, the Ca2+-dependent interaction of the C2 domain with PtdSer-containing membranes is the first step in the activation sequence. This interaction facilitates the intra-molecular rearrangement of PKCs that makes the C1A and C1B ligand-binding sites accessible to membrane-embedded DAG. One such mechanism could be the DAG-independent insertion of the C1A domain into the membranes. The engagement of one or more PKC domains with membranes reduces the dimensionality of the search for DAG and thereby increases the effective affinity of C1B and C1A to DAG.
The functional and structural autonomy of individual PKC domains has been instrumental in dissecting the behavior of full-length enzymes in vitro and in live cells. This property also makes these domains amenable to structural biology studies where residue-specific information can be readily obtained. The insights into PKC dynamics and membrane interactions obtained from this “divide-and-conquer” approach are discussed in the next section.
2. Individual domains of PKC
2.1. C2 domains
C2 (conserved region 2) domains are independently folded structural and functional modules comprising ~140 amino acids.54,55 The C2-like domains of novel PKC isoenzymes do not bind Ca2+ or membranes but rather serve as protein-protein interaction modules.56,57 In conventional PKC isoenzymes, the C2 domains target their parent enzymes to the inner leaflet of the PtdSer-containing plasma membrane in response to increase in cytosolic Ca2+ concentration. The functional elements of Ca2+-responsive C2 domains are the Ca2+- and membrane-binding loops (CMBLs) and the LRC, a β3-β4 hairpin region bearing four lysine residues (Figure 4A). The latter are involved in interactions with PtdSer and/or PtdIns(4,5)P2.58,59 Three aspects of Ca2+-dependent C2 domains are discussed here: binding to metal ions, dynamics, and membrane interactions.
Figure 4.
(A) The crystal structure of C2 domain from PKCα complexed to Ca2+ and PtdIns(4,5)P2.60 CMBL regions and the LRC lysine residues, K197, K199, K209, and K211, are highlighted with orange and blue, respectively. Adapted from61: coordination geometries of Pb2+ (B), Ca2+ (C), and Cd2+ (D) ions bound to the C2 domain from PKCα. The ligands are the sidechain oxygens of aspartates, with the exception of W247 and M186, where it is the carbonyl oxygen. The coordination sphere of Pb2 is hemi-directed: all eight ligands are located in one coordination hemisphere that is facing the viewer. The top axial ligands of Ca1 in 1DSY are the phosphoryl oxygens of the short-chain PtdSer analog.
Divalent metal ions
Binding of Ca2+ to C2 domains of conventional PKC isoenzymes is a prerequisite for the association of these domains with membranes. Inspection of the available C2 crystal structures reveals either di- or tri-partite Ca2+ sites. Our solution NMR data indicate that at moderate Ca2+ concentrations and in the absence of membranes, two Ca2+ sites of the C2 domain from PKCα (C2α) are populated.62 The presence of PtdSer-containing membranes increases Ca2+ affinity of C2 domains and results in binding of an additional Ca2+ ion to the protein-membrane complex.63
It has been hypothesized that the functional role of Ca2+ is to alter the electrostatic potential of C2 and thereby enable the domain to interact with anionic lipids.64 Recent work from our laboratory suggests that the chemical identity of a metal ion has profound consequences for the protein-membrane interactions.61,62 All three non-native divalent metal ions that we tested: Pb2+, Cd2+, and Cu2+ bind to C2 with high affinity. However, only Pb2+ is able to act as a functional substituent of Ca2+ by promoting protein-membrane interactions. This is despite drastic differences in metal coordination: Pb2+ at position 2 adopts hemi-directed coordination geometry, where all eight ligands are located in one coordination hemi-sphere, compared to Ca2+ that has a uniform spatial ligand distribution (Figures 4B and C). The ability of Pb2+ to act as a functional substituent of Ca2+ – relevant to the mechanism of Pb(II) toxicity – has been demonstrated in full-length PKCs.65,66
In contrast to Pb2+, the coordination geometry of C2-bound Cd2+ is identical to that of Ca2+ (Figure 4D), yet Cd2+ is unable to facilitate protein-membrane interactions. These data support the idea of specific interactions between protein-bound metal ions and anionic lipids and indicate that altering the electrostatic potential of C2α is not sufficient to drive the membrane association. Indeed, it was shown that charge reversal mutations in the negatively charged loop region of apo C2 domain from PKCβII did not promote its Ca2+-independent membrane recruitment.67 In the crystal structures of C2, Ca2+ coordinates the phosphoryl oxygen(s) of short-chain PtdSer analog (see Figure 4B). In aggregate, these data suggest that functional “competency” of divalent metal ions depends on their ability to expand the coordination sphere in the all-oxygen environment presented by the protein and anionic lipids, rather than their coordination geometry in the membrane-free state of C2 domains.
Dynamics
Ca2+-binding stabilizes conventional C2 domains, resulting in the increase of melting temperature by 30–38 °C.68 A comparison of the apo and metal ion-complexed C2α structures61,62,69 indicates that the average conformation of the protein backbone does not significantly change upon metal ion binding. One notable difference between the apo and metal ion-complexed structures is the conformation of the metal-coordinating sidechains and elevated B-factors for the CMBLs in the apo C2α. This observation prompted us to investigate the backbone dynamics of C2α in different states of metal ligation: apo, C2α·Pb, and C2α·Ca2 using NMR relaxation techniques (K. Morales and T. Igumenova, in preparation).
We found that CMBL1, CMBL3, and LRC in apo C2α have elevated dynamics on the sub-nanosecond timescale. The sub-nanosecond dynamics of C2α is not significantly affected by its interactions with Pb2+ or Ca2+. In addition, CMBL1 and CMBL3 in apo C2α undergo a chemical exchange process that occurs on the microsecond timescale. Binding of a single Pb2+ is sufficient to attenuate this process indicating a significant reduction in conformational flexibility. In the Ca2+-complexed state of C2α, we detected a chemical exchange process that involves the N- and C-terminal regions, particularly a short α-helix H3 close to the C-terminus.
Molecular dynamics simulations conducted on the C2 domain from PKCβ70 (βI and βII are the splice variants that differ only in the V5 domain) revealed that CMBL1, CMBL3, and the C-terminal α-helix H3 undergo significant fluctuations during the 1 ns trajectory. While these fluctuations are present in the Ca2+-bound C2 structures, removal of Ca2+ increases their amplitude. The average position of CMBL1 is similar for the apo and Ca2+-complexed protein; however, CMBL3 moves away from CMBL1 in the apo C2 presumably due to the electrostatic repulsion.
In summary, conventional C2 domains show a range of dynamic behavior on multiple timescales. Ca2+ binding alters the conformational exchange behavior of the loop regions and the termini. The conformational flexibility of loop regions appears to be a shared theme among the lipid-binding modules of PKC (vide infra).
Membrane interactions
High-affinity Ca2+-dependent interaction of C2 domains with PtdSer71 and PtdIns(4,5)P2 is responsible for targeting their parent enzymes to the plasma membrane. According to the stopped-flow fluorescence study of C2α, hydrophobic and electrostatic interactions play a role in membrane recruitment, through their contributions to the “on” and “off”-rates, respectively.72
The first structural information about the geometry of C2 interactions with membranes was obtained by EPR spectroscopy.73 Small unilamellar vesicles (SUVs) composed of either POPC and POPS, or a lipid mixture designed to mimic the inner leaflet of the plasma membrane, were used as membrane mimics. Single-Cys variants of C2α were covalently modified with MTSL, a functional group bearing a stable nitroxide radical. Power saturation EPR measurements were conducted to determine the site-specific membrane depth parameters Φ, which were subsequently used to dock the crystal structure of the Ca2+-complexed C2α onto the membrane. A model that emerged from the EPR restraints has the CMBL3 protruding into the headgroup region, while the CMBL1 and Ca2+ ions are at the headgroup-water interface (Figure 5A). The β3–β4 segment of LRC is positioned almost parallel to the membrane surface, due to the interactions between the LRC residues and the PtdSer head-groups.
Figure 5.
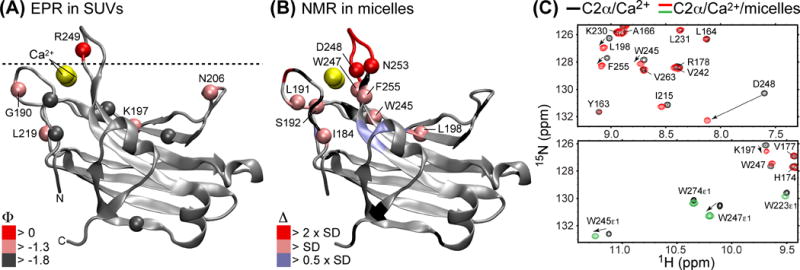
Probing the C2-membrane interactions with EPR and NMR. (A) Adapted from74: depth parameters Φ are mapped onto the structure of the Ca2+-complexed C2α (PDB ID 1DSY69). The spheres correspond to the backbone N-H groups, to facilitate a comparison with the NMR data. The dashed line represents the bilayer phosphate plane. (B) The CSP analysis of C2α in mixed DPS/DPC micelles. The N-H groups are color-coded according to the deviation SD from the mean CSP value. Loop residues that enter an intermediate-exchange regime upon micelle binding (N189, R249, T250, T251, R252) are colored red, with no sphere representation. Prolines and residues whose N-H groups are not spectrally resolved are in black. The CSP values Δ were calculated as described previously75 using 15N-1H HSQC spectra, two expansions of which are given in (C). The data in (C) illustrate the changes in chemical shifts experienced by C2α upon interactions with mixed micelles; the cross-peaks of Nε1-Hε1 groups of the Trp side-chains in the micelle-complexed form are shown in green.
It is instructive to relate this model to the results of NMR experiments that we conducted in a mixed micelle system comprising 1,2-dihexanoyl-sn-glycero-3-[phospho-L-serine] (DPS) and n-dodecylphosphocholine (DPC). The advantage of NMR as applied to these systems is that the stable-isotope enrichment has no effect on the protein structure and its functional properties. Interaction of C2α with micelles changes the electronic environment of the NMR-active nuclei, resulting in the changes of their chemical shifts. The C2-micelle interaction is dependent on the presence of DPS and Ca2+, indicating that mixed micelles are a good membrane mimic for this protein system.
An overlay of the 15N-1H HSQC spectra of the Ca2+-complexed C2α in the absence and presence of mixed DPS/DPC micelles illustrates the chemical shift perturbation (CSP) of the backbone N-H and the Trp sidechain Nε1-Hε1 groups due to binding (Figure 5B and C). For example, the chemical shifts of Trp245 and Trp247, two highly conserved residues that are essential for membrane association50 and control the “on” rate of the process,72 are significantly perturbed, suggesting a direct interaction with micellar environment. Mapping the CSP onto the 3D structure of the Ca2+-complexed C2 domain reveals that the primary membrane interaction site is CMBL3, with a lesser involvement of CMBL1. LRC-harboring face of C2α experiences mild CSPs (color-coded blue, Figure 5B), which is consistent with EPR data and further supports the near-parallel orientation of the domain with respect to the surface of the membrane mimetic. These and other types of experimental data can be further used as ambiguous restraints to dock C2 domains to membrane mimetics76 and obtain the atomic-level information about the geometry of C2-membrane interactions.
C2 and PtdIns(4,5)P2
Another lipid that is essential for targeting conventional isoenzymes to plasma membranes is PtdIns(4,5)P2, whose effective molar concentration in the membrane is ~1%. PtdIns(4,5)P2 contributes to PKC-membrane interactions by increasing the residence time of the protein at the membrane.59 Mutagenesis studies,59,77,78 followed by the determination of the crystal structure of C2α complexed to Ca2+, 1,2-diayl-sn-glycero-3-(phosphoinositol-4,5-bisphosphate), and 1,2-dihexanoyl-sn-glycero-3-(phospho-L-serine)60 demonstrated that C2α interacts with the headgroup of PtdIns(4,5)P2 via the LRC (see Figure 4A). The amine groups of three lysine residues of the LRC, K209, K211, and K197 are engaged in electrostatic interactions with the phosphate groups of PtdIns(4,5)P2. In addition, the sidechains of N153 and Y195 form hydrogen bonds with one of the phosphate groups. It was shown using site-directed EPR methods that PtdIns(4,5)P2 alters the membrane-docking geometry of C2α.74 Through interactions with LRC, the bulky headgroup of PtdIns(4,5)P2 tilts the long axis of C2α by 40±10° degrees towards the bilayer normal, resulting in a more “vertical” domain orientation compared to that shown in Figures 5A and B.
The interaction of C2 domains with PtdIns(4,5)P2 in conventional PKC isoenzymes is enhanced by Ca2+.58,77 Similarly, the presence of PtdIns(4,5)P2 reduces Ca2+ concentration required for membrane binding and thereby increases Ca2+ sensitivity of PKCs.78 To quantitatively characterize the interplay between the metal ions and PtdIns(4,5)P2, we made use of a single high-affinity Pb2+-binding site of C2α and generated three species that differ in the state of metal ligation: apo C2α, C2α·Pb, and C2α·Ca2.79 Using NMR-detected binding experiments with a short-chain PtdIns(4,5)P2, we established that metal ions make individual and comparable contribution to the energetics of the PtdIns(4,5)P2 binding to C2α. Specifically, the affinity of C2α to PtdIns(4,5)P2 increases 4- to 6-fold with progressive saturation of metal ion binding sites. In a complementary NMR-detected binding experiment, we determined that the affinity of the ternary C2α·Pb·C4-PtdIns(4,5)P2 complex to the second Pb2+ ion is 8-fold higher than that of the binary C2α·Pb complex. The synergistic action of these two ligands occurs through modulation of the electrostatic potential of C2α and involves no significant structural changes.
The synergistic effect of the C2 ligands invites a question of whether the recruitment of Ca2+-dependent C2 domains to membranes is target- (PtdSer/PtdIns(4,5)P2)-activated or messenger (Ca2+)-activated.80 Current evidence suggests the dominant role of target lipids in the membrane recruitment process. While the affinity of isolated C2 domains to Ca2+ is too low to respond to physiological Ca2+ concentrations, it increases significantly in the vicinity of target anionic lipids, PtdSer and PtdIns(4,5)P2. Target lipids can then recruit partially metal-bound species of C2 to the membranes, where acquiring an additional metal ion further stabilizes the complex.
Summary and outlook
Originally considered to be non-specific electrostatic switches, conventional C2 domains have emerged as dynamic protein modules that interact with membranes in a nuanced and complex manner. Structural and biophysical studies of Ca2+-dependent C2 domains have greatly advanced our understanding of metal ion-dependent protein-lipid interactions. One question that still remains unanswered is how the binding of divalent metal ions to C2 initiates the activation sequence of conventional PKC isoenzymes.
Another area of interest is the geometry of multivalent interactions between the full-length regulatory domains and membranes. Given the short length of linker regions between C1 and C2, the orientation and position of the domains in the membrane will be interdependent and influenced by membrane composition and chemical identity of lipid ligands. Last but not least, novel PKC isoenzymes rely mostly on C1 domains for membrane recruitment. Do their C2 domains play an indirect role in facilitating membrane binding, or do they simply function as intra- and inter-molecular interaction modules?
2.2. C1 domains
C1 (conserved region 1) domains are independently folded Zn2+ fingers of 50 amino acids. Their function is to target parent PKCs to diacylglycerol (DAG)-containing endo-membranes. The affinity of PKCs to DAG has important implications for the cellular localization of PKC isoenzymes and selectivity of their response.81 The domains occur in tandem, C1A and C1B, and either precede the C2 domain in conventional or follow the C2-like domain in novel isoenzymes (Figure 1). The sequence identity of C1A and C1B domains is typically 36–40% within a given PKC isoenzyme. Eight residues that coordinate two structural Zn2+ ions are strictly conserved across all C1 domains.82 Several three-dimensional structures of C1 domains from PKC isoenzymes have been determined using NMR83,84 and X-ray crystallography.85–87 Only C1B from PKCδ proved to be crystallizable when complexed to the following ligands: a water-soluble phorbol ester, phorbol-13 acetate;85 and general anesthetics, methoxymethylcyclopropane and cyclopropylmethanol that bind to the surface of the domain outside of the canonical DAG pocket.86,87 There are currently no structures of C1 complexed to its native PKC agonist, diacylglycerol.
The features and functional elements of C1 are illustrated using the structure of C1Bδ complexed to phorbol-13 acetate, P13A (Figure 6A). The domain has a treble-clef fold,88 with two Zn2+ coordination sites that are formed by 3 Cys and 1 His sidechains. The secondary structure elements include a short C-terminal helix and 4 β-strands. The ligand-binding pocket is formed by two loops between the β strands 1–2 (β12 loop) and 3–4 (β34 loop). Phorbol-13 acetate, considered a “partial” PKC activator89 compared to DAG and hydrophobic phorbol esters, forms hydrogen bonds exclusively with the backbone groups of the protein with no specific sidechain contacts (Figure 6B).
Figure 6.
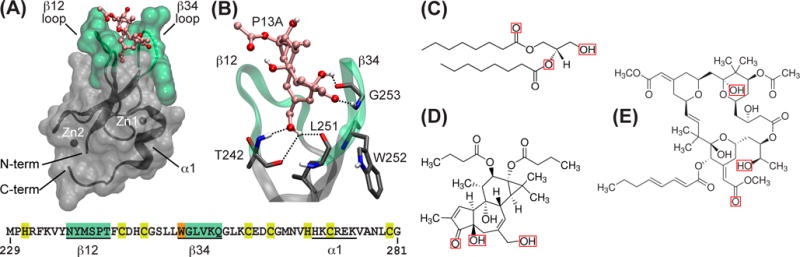
C1 domains and their ligands (adapted from75). (A) Primary and tertiary structure of C1Bδ complexed to phorbol-13 acetate, P13A (PDB ID 1PTR85). The loop regions β12 (residues 237–242) and β34 (residues 252–257) are highlighted in green; α1 is a short α-helix comprising residues 270–275. W252 and Zn2+-coordinating residues are highlighted with orange and yellow, respectively, in the primary structure. (B) Expansion of the C1Bδ ligand-binding site showing hydrogen bonds between P13A and the backbone atoms of C1Bδ. The sidechain of W252 is not involved in direct interactions with the ligand. (C)–(E) Chemical structures of representative C1 ligands: (C) 1,2-dioctanoyl-sn-glycerol, (D) phorbol-12,13-dibutyrate; and (E) Bryostatin-1. Red boxes mark the chemical groups that according to the molecular modeling studies90–92 are involved in hydrogen-bonding interactions with the protein.
C1 domains show considerable promiscuity by binding a wide range of naturally occurring compounds (see e.g., Figure 6D and E), some of which have therapeutic potential.93 One of the most potent C1 ligands are tetracyclic diterpenoids called phorbol esters (PEs).94 Due to their tumor-promoting activity, PEs have found widespread application as pharmacological and research tools for studying the function of PKCs and their role in carcinogenesis.11,95 Another class of C1 ligands, bryostatins, has long been known to antagonize the tumor-promoting effects of PEs. Bryostatins have recently risen to renewed prominence due to their effect on reducing the synaptic loss and plaque formation associated with Alzheimer’s disease in animal models,96 and the development of efficient synthetic routes for potent bryostatin analogs.97–99
Despite the high sequence identity of C1A and C1B domains, their intrinsic affinities to DAG and PE differ, with a notable exception of PKCγ. As demonstrated by the studies in isolated C1 domains,100–102 C1A has a higher affinity to DAG than C1B, and the pattern is reversed for the PEs. Although this pattern generally holds for the full-length enzymes,103–106 additional factors, such as masking of the DAG-binding site by intra-molecular interactions,48 and “priming” steps, such as ligand-independent partitioning of C1A domain into the membrane,49 ultimately determine the individual functional roles of C1A and C1B domains in the cellular context.
Dynamics of β12/β34 loop regions
Inspection of the available NMR structural ensembles of apo C1 domains revealed a significant variability in the conformation of loop regions β12 and β34, suggesting conformational plasticity of the C1 ligand-binding site. We have investigated the dynamics of the C1B domain from PKCα (C1Bα) on multiple time-scales using solution NMR techniques.107 To quantify the sub-nanosecond dynamics of the backbone N-H groups, we measured three relaxation parameters: the 15N transverse and longitudinal relaxation rate constants, and {1H}-15N nuclear Overhauser enhancement. Interpretation of these relaxation parameters using Lipari-Szabo formalism produced a set of residue-specific order parameters S2NH. These parameters describe the spatial restriction of the protein N-H vectors, with 1 corresponding to total rigidity and 0 corresponding to unrestricted motions. The S2NH data revealed an overall increase in the flexibility of ligand-binding loops compared to other regions of the protein, with the most dynamic residues being Ser111 of β12 with an S2NH of 0.73, and Tyr123 of β34 with an S2NH of 0.65.
The C1B residue at position 123 or equivalent has an important functional role: its identity, Trp or Tyr, toggles the domain affinity to DAG. As demonstrated by the Newton’s laboratory,53 C1 domains having either native or mutated Tyr (Trp) at this position have reduced (increased) affinity to DAG. Using 15N rotating-frame relaxation-dispersion NMR experiments, we characterized the dynamics of the N-H backbone groups in the wild-type apo C1Bα and its Y123W variant. We found that loop hinges undergo chemical exchange on the microsecond timescale (Figure 7A).107 As a result of the Tyr→Trp mutation, the kinetics of the exchange – in the form of the sum of the forward and reverse rate constants – is significantly altered, suggesting that the mutation perturbs the conformational equilibrium of apo C1Bα. The same type of correlation between residue identity, DAG affinity, and chemical exchange behavior was observed in another pair of C1 domains, the Trp-containing wild type C1Bδ and its W252Y variant (M. Stewart and T. Igumenova, in preparation).
Figure 7.
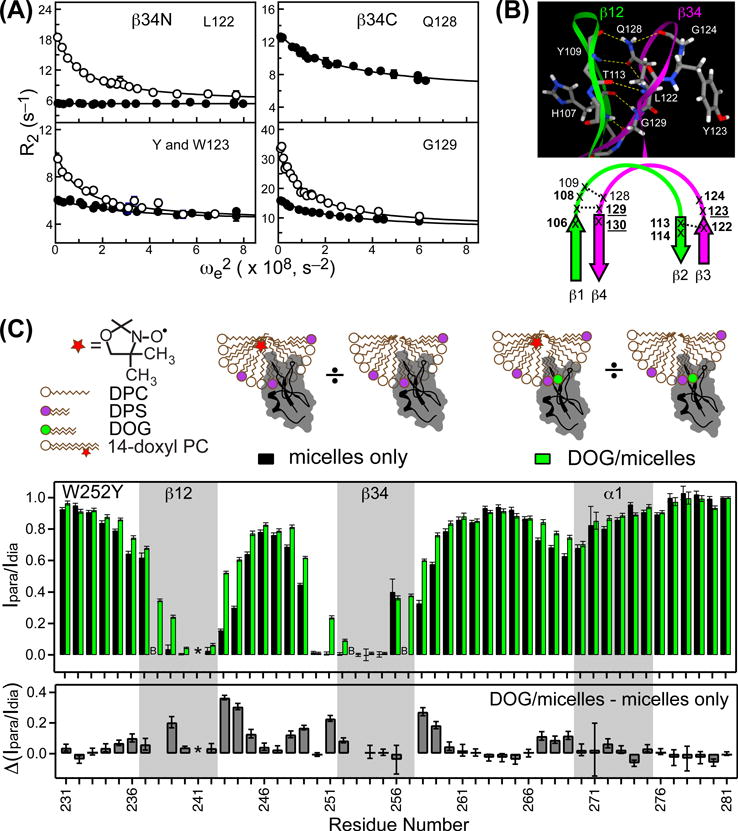
Dynamics of C1 loops and their interaction with mixed micelles. (A) Differences in the μs-timescale dynamics of the wt (solid circles) and Y123W C1Bα (empty circles), illustrated using relaxation-dispersion curves for the N-H backbone groups of L122, Y123, Q128, and G129. (B) Intra- and inter-loop hydrogen bonds that stabilize β12 and β34 loops (top) and summary of conformational dynamics of loop hinges (bottom). Residues with quantifiable dispersion in both wt and mutant are underlined; residues with quantifiable dispersion in either wt or mutant are shown with regular and bold fonts, respectively (adapted from107). (C) Probing the depth of C1Bδ insertion into micellar environment using PRE. The intensity ratios Ipara/Idia of the N-H resonances of W252Y C1Bδ, complexed to paramagnetic and diamagnetic preparations of mixed DPS/DPC micelles, are plotted as a function of amino acid sequence. Data in the absence and presence of DOG are shown with black and green bars, respectively. Residues whose broadening in the micelle-bound state is unrelated to PRE are labeled with “B”. The ratios were normalized to G281, the most C-terminal residue. Δ(Ipara/Idia) is the difference in Ipara/Idia ratios between the DOG-bound and micelle-only W252Y (adapted from75).
Our NMR and computational results107 indicate that, for both wt and Y123W C1Bα, the conformation with open ligand-binding loops is likely to represent the major conformer of apo protein in solution. We speculate that (i) C1 alternates between open- and partially closed-loop conformations, with the latter having higher affinity to DAG, and (ii) the Tyr→Trp mutation contributes to DAG affinity via preferential partitioning of Trp in the interfacial membrane region and possibly by shifting the conformational equilibrium of apo C1 towards the partially closed-loop state. This is in agreement with the recent results from Falke’s group,92 who conducted extensive molecular dynamics simulations of the C1A and C1B domains from PKCα in lipid bilayers. They observed a broad distribution of distances between the tips of β12 and β34 loops in apo C1 domains, with C1A having on average a wider grove. The average loop tip distance increased in the presence of lipid bilayers, whereas interactions with ligands, especially DAG, resulted in its decrease. In addition, the distribution of distances becomes narrower suggesting that ligand binding rigidifies C1 domains. The preferential partitioning of the β34 Trp into the hydrophobic environment was later demonstrated by us quantitatively for the wtC1Bδ/W252Y pair (vide infra).75
In conclusion, C1 domains possess a high degree of conformational plasticity in their ligand-binding loop region. It is plausible that modulation of the binding site geometry through loop dynamics is the property that enables C1 domains to accommodate chemically diverse ligands. Stabilization or pre-selection of a given conformer by a lipid ligand is likely to contribute to both binding affinity and ligand specificity.
Dynamics of structural Zn2+ site
In addition to loop dynamics, we have also detected and characterized the structural dynamics of the Zn(2) coordination sphere in the 50-mer construct of C1Bα.108 The nature of this dynamic process was the Sγ of Cys151, the last Cys residue of the domain, alternating between the Zn2+-bound thiolate and free thiol states. This finding is surprising because structural cysteine-rich Zn2+ sites that stabilize protein folds are considered to be unreactive. We determined the structures of the two exchanging conformations of C1B that differ in zinc coordination using NMR methods and demonstrated the chemical reactivity of Cys151 in longer C1B-containing constructs such as C1B-C2. Our data suggest that Cys151 serves as an entry point for the reactive oxygen species that are known to activate PKCα in a process involving Zn2+ release.109
Interactions with membrane mimics and lipid ligands
Compared to Ca2+-dependent C2 domains, much less is known about the interactions of C1 with membranes at the atomic level. Extensive biophysical and biochemical studies of full-length PKC isoenzymes have provided valuable information on the properties of their respective C1 domains; this is especially relevant to novel PKC isoenzymes, where C1 is responsible for the majority of membrane interactions in the activated form. It was demonstrated using vesicle sedimentation, enzyme activity assays, and SPR methods that PKCα and PKCδ have high specificity for PtdSer,106,110 while for PKCε the specificity is low and other anionic lipids, such as POPG, promote membrane association.110 In all of the referenced studies, mutagenesis was used to assess the role of individual PKC residues in membrane binding and the associated activation of the enzyme. Stopped-flow experiments with fluorescence-based detection provided insight into the kinetics of the membrane-binding process for the Trp-containing variant of C1B from PKCβII. The data led to a model that incorporates an initial membrane pre-association step, which subsequently facilitates the search for a membrane-embedded DAG.111
The extent of PKC interactions with membranes was characterized using a series of lipid monolayer penetration experiments, where the surface pressure change of a lipid monolayer is measured upon injection of protein solution. The conventional PKC isoenzymes, PKCα and PKCγ,101 were found to have higher membrane penetration power than novel isoenzymes, PKCε110 and PKCδ.106 The same type of experiment conducted on the isolated C1 and C2 domains revealed that the extent of C1A-C1B interactions with monolayer lipids is comparable with that of the full-length PKCα and is larger than that of the isolated C2 domain.112 This observation points to the pivotal role of the C1 domain in driving the PKC-membrane interactions.
We used paramagnetic relaxation enhancement (PRE) to probe the depth of insertion of C1Bδ domain into membrane-mimicking micelles at the level of individual residues.75 A paramagnetic agent, the doxyl-labeled lipid, was incorporated into the mixed DPS/DPC micelles either in the absence or presence of short-chain diacylglycerol, 1,2-dioctanoyl-sn-glycerol (DOG). The principle behind PRE-based experiments is that the cross-peaks of protein residues penetrating into the hydrophobic core of the micelles will be broadened, due to the efficient relaxation caused by an unpaired electron. The dependence of peak intensity ratios obtained for the paramagnetic and diamagnetic preparations of micelles indicate that both β12 and β34 loops undergo significant insertion into the micelle core (Figure 7C, top graph). Addition of DOG resulted in a change of C1Bδ-micelle interaction geometry, as evidenced by the differences in the (Ipara/Idia) ratios (Figure 7C, bottom graph). Based on the CSP analysis of the C1Bδ bound to DPC-only and DPC/DPS micelles, we concluded that the C-terminal helix α1 is likely involved in the interactions with the PtdSer headgroup. Using NMR-detected binding experiments, we demonstrated that (i) the presence of PtdSer enhances the affinity of C1Bδ to micelles 2-fold; and (ii) the DAG-sensitizing Trp252 (position equivalent to 123 in conventional PKC isoenzymes) enhances the affinity of the domain to micelles in the absence of ligand. Our data are consistent with the models of Newton111 and Falke49 that include initial ligand-independent partitioning of C1 domains into the lipid bilayer, and point to the pivotal role of aromatic residue at position 123/252 in facilitating this process. The NMR-based restrains, such as CSP data and PRE ratios, can be used as ambiguous restraints to drive the protein-micelle docking and obtain the geometry of protein-micelle interactions, as described by Dancea et al.76
Summary and outlook
C1 domains are highly dynamic hydrophobic membrane-binding domains that can interact with chemically diverse ligands, in addition to their endogenous lipid activator, DAG. High-resolution structures of ligand-complexed C1 domains in the presence of suitable membrane mimetics are needed to fully understand the molecular basis of ligand affinity and specificity. With respect to membrane interactions, much of what was said in the context of future C2 domain studies applies here: the geometry of protein-membrane interactions and the influence of linker regions on these interactions are the topics that require further investigation.
One of the most important questions in the field is how the binding of divalent metal ions initiates the activation sequence of conventional PKC isoenzymes. As described in Section 1.1, C1A has been implicated in intra-molecular auto-inhibitory interactions with C2. What is the structural basis of these interactions and how does Ca2+ binding interferes with them to make the lipid-binding sites on C1 and C2 domains accessible?
2.3. Pseudo-substrate Variable-1 region and C-terminal Variable-5 domain
2.3.1. Pseudo-substrate region
The pseudo-substrate (PS) region, sometimes referred to as Variable-1 (V1), is located at the N-terminus in the conventional PKCs and in between the C2-like (PB1) and C1 domains in novel (atypical) PKCs (Figure 1). The PS region is a short amino acid sequence that is similar to PKC substrates, except that an Ala replaces the Ser or Thr phospho-acceptor sites; it also contains 4 to 6 positively charged amino acids. Mosior and McLaughlin113 demonstrated that the PS-derived peptide from PKCβI/βII binds to LUVs containing anionic lipids, and that the binding curve is sigmoidal with respect to the molar fraction of the anionic lipid. While the energetics of binding would not be sufficient to release the PS from the auto-inhibitory interaction, PS could be involved in the stabilization of the activated PKC by anchoring it to the membrane, in addition to the lipid ligand-dependent interactions of C1 and C2. This is further supported by the observation that the V1-C1-C2-GFP construct from PKCγ partially pre-localizes to the plasma membrane in unstimulated live cells, whereas C1-C2-GFP is uniformly distributed in the cytoplasm.114
2.3.2. C-terminal Variable 5 (V5) domain
V5 is the most C-terminal domain of PKCs comprising 60–80 amino acids that varies in both length and amino-acid composition. V5 has three conserved regions: the “NFD” motif, the turn motif (TM), and the hydrophobic motif (HM) (Figure 8A). Current experimental evidence suggests that V5 plays a role in several processes that regulate PKC activity, such as enzyme maturation, Ca2+ sensitivity, interactions with scaffolding proteins, and down-regulation via interactions with Pin1.
Figure 8.
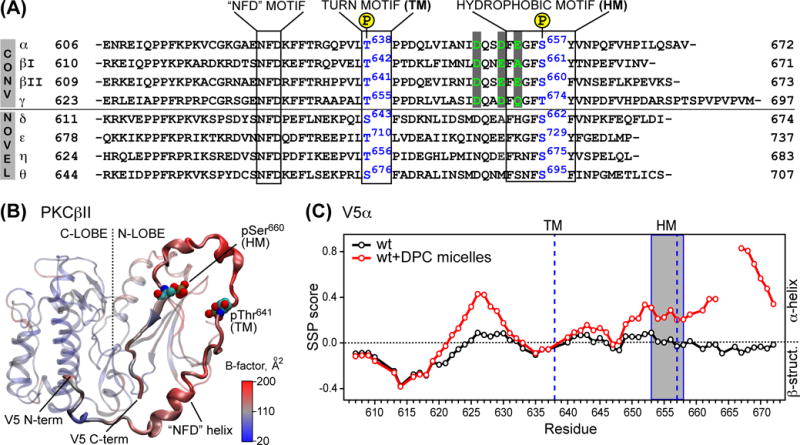
Properties of V5 domain. (A) Alignment of the V5 primary structures of PKC isoenzymes from M. musculus. The conserved motifs are boxed. Acidic residues implicated in the intra-molecular interactions with the C2 domain are shown in green. (B) Catalytic domain (residues 339–679) taken from the partial crystal structure of PKCβII, PDB ID 3PFQ.39 The B-factors of backbone Cα atoms are mapped onto the structure as a color gradient. The N-/C-lobes of the kinase domain and V5 are shown with transparent and opaque representations, respectively. (C) SSP scores calculated using NMR chemical shift data for the free (black) and micelle-bound V5α (red). Upon binding to DPC micelles, V5α acquires a partial α-helical structure, most notably in the NFD region and the most C-terminal residues (adapted from133).
Maturation
Conventional and novel PKCs undergo three ordered phosphorylation reactions to reach catalytic maturity. Two phosphorylation sites belong to the V5 and are the Ser/Thr residues of the HM and TM. In addition, V5 serves as an interaction site between the newly synthesized PKC and phosphoinositide-dependent kinase, PDK-1, which phosphorylates a conserved Thr residue on the activation loop that is not part of the C-terminal V5 region.115 Experimental evidence supports the involvement of the mammalian target of rapamycin complex 2 in the phosphorylation of the TM116,117 and auto-phosphorylation of the HM.118
Ca2+ sensitivity
The negatively charged phosphoryl group at the HM is crucial for the proper Ca2+ response of conventional PKCs. Replacement of Ser660 of the HM with Ala increases Ca2+ concentration required for full membrane binding and activity of PKCβII by almost an order of magnitude.43
Interactions with scaffolding proteins
PKCα interacts with PICK1, a scaffolding protein implicated in synaptic plasticity, using the last four amino acids of V5, QSAV.119 PICK1 was later shown to be a substrate of PKCα.120 V5 also forms part of the PKCβII binding site for RACK1, a scaffolding protein that is critical for proper sub-cellular localization and stabilization of the activated PKC.121
Down-regulation of PKC via interactions with Pin1
A recent addition to the repertoire of cellular mechanisms that control PKC activity was the finding that Pin1, a peptidyl-prolyl isomerase, down-regulates PKCα and PKCβI/βII through its action on the TM of the V5 domain.122 Isomerization of V5 makes the activated (i.e. membrane-bound) enzyme susceptible to dephosphorylation and ubiquitin-mediated degradation.
Structural preferences and dynamics of V5
In the available crystal structures of catalytic domains of PKC,123–132 V5 is wrapped around the N-terminal lobe of the kinase domain. The electron density of V5 is not uniformly well-defined, giving rise to “missing” regions in the crystal structures. Together with the elevated values of B-factors, this indicates a relatively high degree of static or dynamic disorder. The structure of the catalytic domain of PKCβII that has a fully defined V5 domain illustrates the secondary structure elements of V5 (Figure 8B). V5 has two helical segments, one of which is the NFD motif that has been implicated in the auto-inhibitory interactions with the C1B domain.39 The other helical segment is located between the TM and HM. The phosphate group of the TM, pThr641, is engaged in electrostatic interactions with K355, K374, and R415 of the kinase N-lobe. The phosphate group of the HM pSer660 is hydrogen-bonded to the sidechain amide group of Gln411 of the N-lobe.
To determine the conformational preferences and dynamics of the V5 domain in its “free” form, we have prepared and characterized the isolated V5 domain from PKCα (V5α) using NMR spectroscopy and circular dichroism.133 V5α is intrinsically disordered, with a moderate propensity to form extended β-structures in the N-terminal region and weak propensity to form α-helical structures in the NFD regions, and between the TM and HM (Figure 8C). This was shown by calculating the secondary structure propensity (SSP)134 scores that describe the likelihood of a peptide adopting a helical structure (maximum score +1) or an extended β-structure (minimum score −1). Introduction of the phosphorylation-mimicking mutation, T638E/S657E, into V5α did not alter the conformational preferences of the protein, but resulted in the change of the population of the “cis-trans” conformer of the TM segment, Thr638-Pro639-Pro640, which has been implicated in Pin1 interactions.122
The sub-nanosecond dynamics of the V5α backbone was quantified using a set of NMR relaxation parameters that we interpreted using reduced135 spectral density mapping136 formalism. Regions with weak propensity towards α-helical structure formation, such as the region upstream of the NFD motif and the TM-HM segment, show a higher degree of motional restriction than the rest of the protein residues.
These structural and dynamical data indicate that the C-terminal V5 domain has a high degree of conformational flexibility, especially in the isolated form that serves as a model of V5 in the immature and activated states of the kinase. The association of V5α with the N-terminal lobe of the kinase domain during the final step of maturation may include both “conformational selection” and “folding upon binding” mechanisms.137
Interactions with micelles
Using NMR and CD spectroscopy, we demonstrated that V5α has a propensity to partition into the hydrophobic environment and acquire partial helical structure upon doing so.133 This was manifested in a significant change of the V5α chemical shifts upon addition of zwitterionic DPC micelles. The SSP scores of micelle-bound V5 indicate a significant increase in the helical structure content, especially at the C-terminus (Figure 8C). The increase in helical content was also evident in the circular dichroism data.
Given our results on the V5α-DPC micelle interactions, we sought to evaluate the amphiphilicity of the C-terminal segments of V5 domains that follow HM. The hydrophobic moment, <μH>,138 was calculated with the program hmoment of the EMBOSS,139 assuming a helical structure and using a 10-residue window. For the V5α, the μ value of the last 14 amino acids is larger than 0.35, indicating a significant degree of amphiphilicity. Regions with <μH> exceeding 0.35 were present in all PKC isoenzymes except βI. We conclude that the C-terminal segments of V5 have a moderate degree of amphiphilicity. This suggests that V5 may play a role in anchoring immature140,141 and even activated PKC to the membrane surface.
Summary and outlook
The two distinct features of V5 are its conformational plasticity due to a large fraction of unstructured regions and moderate amphiphilicity. These biophysical properties of V5 underlie its multi-faceted function. Further studies are required to understand the structural basis of V5 interactions with scaffolding proteins, Pin1, and membranes. Because V5 is the most variable domain among the PKC isoenzymes, atomic-level information about its interactions with binding partners will provide insight into the isoenzyme specificity and inform the design of isoenzyme-selective agents.
Concluding remarks
This article presented an overview of biophysical studies of dynamics and membrane interactions of PKC. Significant progress has been made in understanding the structural dynamics of full-length PKC isoenzymes. The rearrangement of PKC tertiary structure reveals an intrinsically amphiphilic character of the enzyme, by exposing hydrophobic membrane-interacting regions that were previously sequestered from the solvent in the latent form. Due to the disruption of intra-molecular interactions in the activated PKC, individual membrane-binding domains serve as good models to gain atomic-level information about PKC-membrane interactions.
Despite significant progress, much remains to be learned about these incredibly complex enzymes that regulate signal transduction processes at membrane surfaces. High-resolution structure of latent PKC remains elusive. Several aspects of membrane recruitment process, such as the inter-play between membrane-binding domains, the effect of membrane composition, and the structural basis of interactions with lipid ligands and adaptor/regulatory proteins, require further investigation. The structural and biophysical studies will be deemed successful when the spatiotemporal activation sequence of PKC isoenzymes is characterized at the level of atomic detail. Having this information will provide the molecular framework for understanding the functional response of PKC isoenzymes to different stimuli and inform the development of ways to modulate PKC activity for therapeutic and research purposes.
Acknowledgments
I thank Dr. Krystal A. Morales for collecting the data of Figure 5C.
Funding Sources
The research in my laboratory is supported by the Welch Foundation grant A-1784, NSF CAREER award CHE-1151435, and NIH grant R01 GM108998.
ABBREVIATIONS
- PKC
protein kinase C
- DAG
diacylglycerol
- PtdIns(4,5)P2
phosphatidylinositol 4,5-bisphosphate
- SUVs
small unilamellar vesicles
- LUVs
large unilamellar vesicles
- MTSL
(S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate)
- DPS
1,2-dihexanoyl-sn-glycero-3-[phospho-L-serine]
- DPC
n-dodecylphosphocholine
- CSP
chemical shift perturbation
- LRC
lysine-rich cluster
- CMBL
Ca2+- and membrane-binding loop
- PtdSer
phosphatidylserine
- PDBu
phorbol 12,13-dibutyrate
- DOG
1,2-dioctanoyl-sn-glycerol
- C2α
C2 domain from PKCα
- C1Bα
C1B domain from PKCα
- V5α
V5 domain from PKCα
References
- 1.Inoue M, Kishimoto A, Takai Y, Nishizuka Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. 2. Proenzyme and its actiavtion by calcium-dependent protease from rat brain. J Biol Chem. 1977;252:7610–7616. [PubMed] [Google Scholar]
- 2.Takai Y, Kishimoto A, Inoue M, Nishizuka Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. 1. Purification and characterization of an active enzyme from bovine cerebellum. J Biol Chem. 1977;252:7603–7609. [PubMed] [Google Scholar]
- 3.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct Activation of Calcium-Activated, Phospholipid-Dependent Protein-Kinase by Tumor-Promoting Phorbol Esters. J Biol Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- 4.Newton AC. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 5.Rosse C, Linch M, Kermorgant S, Cameron AJM, Boeckeler K, Parker PJ. PKC and the control of localized signal dynamics. Nature Reviews Molecular Cell Biology. 2010;11:103–112. doi: 10.1038/nrm2847. [DOI] [PubMed] [Google Scholar]
- 6.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moscat J, Diaz-Meco MT. The atypical protein kinase Cs - Functional specificity mediated by specific protein adapters. Embo Reports. 2000;1:399–403. doi: 10.1093/embo-reports/kvd098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki A, Akimoto K, Ohno S. Protein kinase C lambda/iota (PKC lambda/iota): A PKC isotype essential for the development of multicellular organisms. J Biochem. 2003;133:9–16. doi: 10.1093/jb/mvg018. [DOI] [PubMed] [Google Scholar]
- 9.Abrial E, Lucas G, Scarna H, Haddjeri N, Lambas-Senas L. A role for the PKC signaling system in the pathophysiology and treatment of mood disorders: involvement of a functional imbalance? Mol Neurobiol. 2011;44:407–419. doi: 10.1007/s12035-011-8210-4. [DOI] [PubMed] [Google Scholar]
- 10.Churchill E, Budas G, Vallcntin A, Koyanag T, Mochly-Rosen D. PKC Isozymes in chronic cardiac disease: Possible therapeutic targets? Annu Rev Pharmacol Toxicol. 2008;48:569–599. doi: 10.1146/annurev.pharmtox.48.121806.154902. [DOI] [PubMed] [Google Scholar]
- 11.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nature Reviews Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 12.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pass JM, Gao JM, Jones WK, Wead WB, Wu X, Zhang J, Baines CP, Bolli R, Zheng YT, Joshua IG, Ping PP. Enhanced PKC beta II translocation and PKC beta II-RACK1 interactions in PKC epsilon-induced heart failure: a role for RACK1. American Journal of Physiology-Heart and Circulatory Physiology. 2001;281:H2500–H2510. doi: 10.1152/ajpheart.2001.281.6.H2500. [DOI] [PubMed] [Google Scholar]
- 14.Ping PP, Zhang J, Qiu YM, Tang XL, Manchikalapudi S, Cao XN, Bolli R. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circul Res. 1997;81:404–414. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 15.Hambleton M, Hahn H, Pleger ST, Kuhn MC, Klevitsky R, Carr AN, Kimball TF, Hewett TE, Ii GW, Koch WJ, Molkentin JD. Pharmacological- and gene therapy-based inhibition of protein kinase C alpha/beta enhances cardiac contractility and attenuates heart failure. Circulation. 2006;114:574–582. doi: 10.1161/CIRCULATIONAHA.105.592550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 17.Gilio K, Harper MT, Cosemans J, Konopatskaya O, Munnix ICA, Prinzen L, Leitges M, Liu QH, Molkentin JD, Heemskerk JWM, Poole AW. Functional Divergence of Platelet Protein Kinase C (PKC) Isoforms in Thrombus Formation on Collagen. J Biol Chem. 2010;285:23408–23417. doi: 10.1074/jbc.M110.136176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konopatskaya O, Gilio K, Harper MT, Zhao Y, Cosemans J, Karim ZA, Whiteheart SW, Molkentin JD, Verkade P, Watson SP, Heemskerk JWM, Poole AW. PKC alpha regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. 2009;119:399–407. doi: 10.1172/JCI34665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshioka A, Shirakawa R, Nishioka H, Tabuchi A, Higashi T, Ozaki H, Yamamoto A, Kita T, Horiuchi H. Identification of protein kinase C alpha as an essential, but not sufficient, cytosolic factor for Ca2+-induced alpha- and dense-core granule secretion in platelets. J Biol Chem. 2001;276:39379–39385. doi: 10.1074/jbc.M102933200. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Subbulakshmi V, Fields AP, Murray NR, Cathcart MK. Protein kinase calpha regulates human monocyte O-2 production and low density lipoprotein lipid oxidation. J Biol Chem. 1999;274:3764–3771. doi: 10.1074/jbc.274.6.3764. [DOI] [PubMed] [Google Scholar]
- 21.Marino M, Distefano E, Pallottini V, Caporali S, Bruscalupi G, Trentalance A. Activation of IP(3)-protein kinase C-alpha signal transduction pathway precedes the changes of plasma cholesterol, hepatic lipid metabolism and induction of low-density lipoprotein receptor expression in 17-beta-oestradiol-treated rats. Exp Physiol. 2001;86:39–45. doi: 10.1113/eph8602069. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Thorne SH, Sun L, Huang B, Mochly-Rosen D. Sustained inhibition of PKCalpha reduces intravasation and lung seeding during mammary tumor metastasis in an in vivo mouse model. Oncogene. 2011;30:323–333. doi: 10.1038/onc.2010.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lonne GK, Cornmark L, Zahirovic IO, Landberg G, Jirstrom K, Larsson C. PKCalpha expression is a marker for breast cancer aggressiveness. Mol Cancer. 2010;9:76. doi: 10.1186/1476-4598-9-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith SD, Enge M, Bao W, Thullberg M, Costa TD, Olofsson H, Gashi B, Selivanova G, Stromblad S. Protein Kinase Calpha (PKCalpha) Regulates p53 Localization and Melanoma Cell Survival Downstream of Integrin alphav in Three-dimensional Collagen and in Vivo. J Biol Chem. 2012;287:29336–29347. doi: 10.1074/jbc.M112.341917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varga A, Czifra G, Tallai B, Nemeth T, Kovacs I, Kovacs L, Biro T. Tumor grade-dependent alterations in the protein kinase C isoform pattern in urinary bladder carcinomas. Eur Urol. 2004;46:462–465. doi: 10.1016/j.eururo.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Mandil R, Ashkenazi E, Blass M, Kronfeld I, Kazimirsky G, Rosenthal G, Umansky F, Lorenzo PS, Blumberg PM, Brodie C. Protein kinase Calpha and protein kinase Cdelta play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res. 2001;61:4612–4619. [PubMed] [Google Scholar]
- 27.Chen L, Hahn H, Wu GY, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo YR, Bolli R, Dorn GW, Mochly-Rosen D. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci U S A. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorn GW, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, Csukai M, Wu GY, Lorenz JN, Mochly-Rosen D. Sustained in vivo cardiac protection by a rationally designed peptide that causes epsilon protein kinase C translocation. Proc Natl Acad Sci U S A. 1999;96:12798–12803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hahn HS, Yussman MG, Toyokawa T, Marreez Y, Barrett TJ, Hilty KC, Osinska H, Robbins J, Dorn GW. Ischemic protection and myofibrillar cardiomyopathy - Dose-dependent effects of in vivo delta PKC inhibition. Circul Res. 2002;91:741–748. doi: 10.1161/01.res.0000037091.64492.69. [DOI] [PubMed] [Google Scholar]
- 30.Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly-Rosen D. Inhibition of delta-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation. 2003;108:2304–2307. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- 31.Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection by epsilon-protein kinase C activation from ischemia - Continuous delivery and antiarrhythmic effect of an epsilon-protein kinase C-activating peptide. Circulation. 2005;111:44–50. doi: 10.1161/01.CIR.0000151614.22282.F1. [DOI] [PubMed] [Google Scholar]
- 32.Ping PP, Song CX, Zhang J, Guo YR, Cao XN, Li RCX, Wu WJ, Vondriska TM, Pass JM, Tang XL, Pierce WM, Bolli R. Formation of protein kinase C epsilon-Lck signalling modules confers cardioprotection. J Clin Invest. 2002;109:499–507. doi: 10.1172/JCI13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saurin AT, Pennington DJ, Raat NJH, Latchman DS, Owen MJ, Marber MS. Targeted disruption of the protein kinase C epsilon gene abolishes the infarct size reduction that follows ischaemic preconditioning of isolated buffer-perfused mouse hearts. Cardiovasc Res. 2002;55:672–680. doi: 10.1016/s0008-6363(02)00325-5. [DOI] [PubMed] [Google Scholar]
- 34.House C, Kemp BE. Protein-Kinase-C Contains a Pseudosubstrate Prototype in Its Regulatory Domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 35.Nishikawa K, Toker A, Johannes FJ, Zhou SY, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 36.Orr JW, Newton AC. Intrapeptide Regulation of Protein-Kinase-C. J Biol Chem. 1994;269:8383–8387. [PubMed] [Google Scholar]
- 37.Pears CJ, Kour G, House C, Kemp BE, Parker PJ. Mutagenesis of the Pseudosubstrate Site of Protein-Kinase-C Leads to Activation. Eur J Biochem. 1990;194:89–94. doi: 10.1111/j.1432-1033.1990.tb19431.x. [DOI] [PubMed] [Google Scholar]
- 38.Ron D, Mochlyrosen D. An autoregulatory region in protein kinase C - the pseudoanchoring site. Proc Natl Acad Sci U S A. 1995;92:492–496. doi: 10.1073/pnas.92.2.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonard TA, Rozycki B, Saidi LF, Hummer G, Hurley JH. Crystal structure and allosteric activation of protein kinase C betaII. Cell. 2011;144:55–66. doi: 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stahelin RV, Wang JY, Blatner NR, Raftner JD, Murray D, Cho WH. The origin of C1A-C2 interdomain interactions in protein kinase C alpha. J Biol Chem. 2005;280:36452–36463. doi: 10.1074/jbc.M506224200. [DOI] [PubMed] [Google Scholar]
- 41.Slater SJ, Seiz JL, Cook AC, Buzas CJ, Malinowski SA, Kershner JL, Stagliano BA, Stubbs CD. Regulation of PKC alpha activity by C1–C2 domain interactions. J Biol Chem. 2002;277:15277–15285. doi: 10.1074/jbc.M112207200. [DOI] [PubMed] [Google Scholar]
- 42.Stensman H, Larsson C. Identification of acidic amino acid residues in the protein kinase C alpha V5 domain that contribute to its insensitivity to diacylglycerol. J Biol Chem. 2007;282:28627–28638. doi: 10.1074/jbc.M702248200. [DOI] [PubMed] [Google Scholar]
- 43.Edwards AS, Newton AC. Phosphorylation at conserved carboxyl-terminal hydrophobic motif regulates the catalytic and regulatory domains of protein kinase C. J Biol Chem. 1997;272:18382–18390. doi: 10.1074/jbc.272.29.18382. [DOI] [PubMed] [Google Scholar]
- 44.Kheifets V, Mochly-Rosen D. Insight into intra- and inter-molecular interactions of PKC: design of specific modulators of kinase function. Pharmacol Res. 2007;55:467–476. doi: 10.1016/j.phrs.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solodukhin AS, Kretsinger RH, Sando JJ. Initial three-dimensional reconstructions of protein kinase C delta from two-dimensional crystals on lipid monolayers. Cell Signal. 2007;19:2035–2045. doi: 10.1016/j.cellsig.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Huang SM, Leventhal PS, Wiepz GJ, Bertics PJ. Calcium and phosphatidylserine stimulate the self-association of conventional protein kinase C isoforms. Biochemistry. 1999;38:12020–12027. doi: 10.1021/bi990594m. [DOI] [PubMed] [Google Scholar]
- 47.Swanson CJ, Ritt M, Wang W, Lang MJ, Narayan A, Tesmer JJ, Westfall M, Sivaramakrishnan S. Conserved Modular Domains Team up to Latch-open Active Protein Kinase C alpha. J Biol Chem. 2014;289:17812–17829. doi: 10.1074/jbc.M113.534750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antal CE, Violin JD, Kunkel MT, Skovso S, Newton AC. Intramolecular Conformational Changes Optimize Protein Kinase C Signaling. Chem Biol. 2014;21:459–469. doi: 10.1016/j.chembiol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ziemba BP, Li JN, Landgraf KE, Knight JD, Voth GA, Falke JJ. Single-Molecule Studies Reveal a Hidden Key Step in the Activation Mechanism of Membrane-Bound Protein Kinase C-alpha. Biochemistry. 2014;53:1697–1713. doi: 10.1021/bi4016082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Medkova M, Cho W. Mutagenesis of the C2 domain of protein kinase C-alpha. Differential roles of Ca2+ ligands and membrane binding residues. J Biol Chem. 1998;273:17544–17552. doi: 10.1074/jbc.273.28.17544. [DOI] [PubMed] [Google Scholar]
- 51.Nalefski EA, Newton AC. Membrane binding kinetics of protein kinase C betaII mediated by the C2 domain. Biochemistry. 2001;40:13216–13229. doi: 10.1021/bi010761u. [DOI] [PubMed] [Google Scholar]
- 52.Braun DC, Garfield SH, Blumberg PM. Analysis by fluorescence resonance energy transfer of the interaction between ligands and protein kinase C delta in the intact cell. J Biol Chem. 2005;280:8164–8171. doi: 10.1074/jbc.M413896200. [DOI] [PubMed] [Google Scholar]
- 53.Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007;282:826–830. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 54.Cho W, Stahelin RV. Membrane binding and subcellular targeting of C2 domains. Biochimica Et Biophysica Acta-Molecular and Cell Biology of Lipids. 2006;1761:838–849. doi: 10.1016/j.bbalip.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 55.Shao XG, Davletov BA, Sutton RB, Sudhof TC, Rizo J. Bipartite Ca2+-binding motif in C-2 domains of synaptotagmin and protein kinase C. Science. 1996;273:248–251. doi: 10.1126/science.273.5272.248. [DOI] [PubMed] [Google Scholar]
- 56.Benes CH, Wu N, Elia AEH, Dharia T, Cantley LC, Soltoff SP. The C2 domain of PKC delta is a phosphotyrosine binding domain. Cell. 2005;121:271–280. doi: 10.1016/j.cell.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 57.Stahelin RV, Kong KF, Raha S, Tian W, Melowic HR, Ward KE, Murray D, Altman A, Cho W. Protein Kinase C theta C2 Domain Is a Phosphotyrosine Binding Module That Plays a Key Role in Its Activation. J Biol Chem. 2012;287:30518–30528. doi: 10.1074/jbc.M112.391557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guerrero-Valero M, Marin-Vicente C, Gomez-Fernandez JC, Corbalan-Garcia S. The C2 domains of classical PKCs are specific PtdIns(4,5)P-2-sensing domains with different affinities for membrane binding. J Mol Biol. 2007;371:608–621. doi: 10.1016/j.jmb.2007.05.086. [DOI] [PubMed] [Google Scholar]
- 59.Manna D, Bhardwaj N, Vora MS, Stahelin RV, Lu H, Cho WH. Differential roles of phosphatidylserine, PtdIns(4,5)P-2, and PtdIns(3,4,5)P-3 in plasma membrane targeting of C2 domains - Molecular dynamics simulation, membrane binding, and cell translocation studies of the PKC alpha C2 domain. J Biol Chem. 2008;283:26047–26058. doi: 10.1074/jbc.M802617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guerrero-Valero M, Ferrer-Orta C, Querol-Audi J, Marin-Vicente C, Fita I, Gomez-Fernandez JC, Verdaguer N, Corbalan-Garcia S. Structural and mechanistic insights into the association of PKC alpha-C2 domain to PtdIns(4,5)P-2. Proc Natl Acad Sci U S A. 2009;106:6603–6607. doi: 10.1073/pnas.0813099106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morales KA, Yang Y, Long Z, Li P, Taylor AB, Hart PJ, Igumenova TI. Cd2+ as a Ca2+ surrogate in protein-membrane interactions: isostructural but not isofunctional. J Am Chem Soc. 2013;135:12980–12983. doi: 10.1021/ja406958k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morales KA, Lasagna M, Gribenko AV, Yoon Y, Reinhart GD, Lee JC, Cho W, Li P, Igumenova TI. Pb2+ as modulator of protein-membrane interactions. J Am Chem Soc. 2011;133:10599–10611. doi: 10.1021/ja2032772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kohout SC, Corbalan-Garcia S, Torrecillas A, Gomez-Fernandez JC, Falke JJ. C2 domains of protein kinase C isoforms alpha, beta, and gamma: Activation parameters and calcium stoichiometries of the membrane-bound state. Biochemistry. 2002;41:11411–11424. doi: 10.1021/bi026041k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murray D, Honig B. Electrostatic control of the membrane targeting of C2 domains. Mol Cell. 2002;9:145–154. doi: 10.1016/s1097-2765(01)00426-9. [DOI] [PubMed] [Google Scholar]
- 65.Markovac J, Goldstein GW. Picomolar concentrations of lead stimulate brain protein kinase C. Nature. 1988;334:71–73. doi: 10.1038/334071a0. [DOI] [PubMed] [Google Scholar]
- 66.Sun XY, Tian XT, Tomsig JL, Suszkiw JB. Analysis of differential effects of Pb2+ on protein kinase C isozymes. Toxicol Appl Pharmacol. 1999;156:40–45. doi: 10.1006/taap.1999.8622. [DOI] [PubMed] [Google Scholar]
- 67.Edwards AS, Newton AC. Regulation of protein kinase C beta II by its C2 domain. Biochemistry. 1997;36:15615–15623. doi: 10.1021/bi9718752. [DOI] [PubMed] [Google Scholar]
- 68.Torrecillas A, Laynez J, Menendez M, Corbalan-Garcia S, Gomez-Fernandez JC. Calorimetric study of the interaction of the C2 domains of classical protein kinase C isoenzymes with Ca2+ and phospholipids. Biochemistry. 2004;43:11727–11739. doi: 10.1021/bi0489659. [DOI] [PubMed] [Google Scholar]
- 69.Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gomez-Fernandez JC. Ca2+ bridges the C2 membrane-binding domain of protein kinase C alpha directly to phosphatidylserine. EMBO J. 1999;18:6329–6338. doi: 10.1093/emboj/18.22.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Banci L, Cavallaro G, Kheifets V, Mochly-Rosen D. Molecular dynamics characterization of the C2 domain of protein kinase C beta. J Biol Chem. 2002;277:12988–12997. doi: 10.1074/jbc.M106875200. [DOI] [PubMed] [Google Scholar]
- 71.Stahelin RV, Rafter JD, Das S, Cho W. The molecular basis of differential subcellular localization of C2 domains of protein kinase C-alpha and group IVa cytosolic phospholipase A(2) J Biol Chem. 2003;278:12452–12460. doi: 10.1074/jbc.M212864200. [DOI] [PubMed] [Google Scholar]
- 72.Scott AM, Antal CE, Newton AC. Electrostatic and Hydrophobic Interactions Differentially Tune Membrane Binding Kinetics of the C2 Domain of Protein Kinase C alpha. J Biol Chem. 2013;288:16905–16915. doi: 10.1074/jbc.M113.467456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kohout SC, Corbalan-Garcia S, Gomez-Fernandez JC, Falke JJ. C2 domain of protein kinase C alpha: Elucidation of the membrane docking surface by site-directed fluorescence and spin labeling. Biochemistry. 2003;42:1254–1265. doi: 10.1021/bi026596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Landgraf KE, Malmberg NJ, Falke JJ. Effect of PIP(2) binding on the membrane docking geometry of PKC alpha C2 domain: An EPR site-directed spin-labeling and relaxation study. Biochemistry. 2008;47:8301–8316. doi: 10.1021/bi800711t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stewart MD, Cole TR, Igumenova TI. Interfacial Partitioning of a Loop Hinge Residue Contributes to Diacylglycerol Affinity of Conserved Region 1 Domains. J Biol Chem. 2014;289:27653–27664. doi: 10.1074/jbc.M114.585570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dancea F, Kami K, Overduin M. Lipid interaction networks of peripheral membrane proteins revealed by data-driven micelle docking. Biophys J. 2008;94:515–524. doi: 10.1529/biophysj.107.115923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanchez-Bautista S, Marin-Vicente C, Gomez-Fernandez JC, Corbalan-Garcia S. The C2 domain of PKC alpha is a Ca2+-dependent PtdIns(4,5)P-2 sensing domain: A new insight into an old pathway. J Mol Biol. 2006;362:901–914. doi: 10.1016/j.jmb.2006.07.093. [DOI] [PubMed] [Google Scholar]
- 78.Evans JH, Murray D, Leslie CC, Falke JJ. Specific translocation of protein kinase C alpha to the plasma membrane requires both Ca2+ and PIP2 recognition by its C2 domain. Mol Biol Cell. 2006;17:56–66. doi: 10.1091/mbc.E05-06-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morales KA, Igumenova TI. Synergistic Effect of Pb(2+) and Phosphatidylinositol 4,5-Bisphosphate on C2 Domain-Membrane Interactions. Biochemistry. 2012 doi: 10.1021/bi201850h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Corbin JA, Evans JH, Landgraf KE, Falke JJ. Mechanism of specific membrane targeting by C2 domains: Localized pools of target lipids enhance Ca(2+) affinity. Biochemistry. 2007;46:4322–4336. doi: 10.1021/bi062140c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gallegos LL, Newton AC. Spatiotemporal dynamics of lipid signaling: protein kinase C as a paradigm. IUBMB Life. 2008;60:782–789. doi: 10.1002/iub.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hurley JH, Newton AC, Parker PJ, Blumberg PM, Nishizuka Y. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 1997;6:477–480. doi: 10.1002/pro.5560060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu RX, Pawelczyk T, Xia TH, Brown SC. NMR structure of a protein kinase C-gamma phorbol-binding domain and study of protein-lipid micelle interactions. Biochemistry. 1997;36:10709–10717. doi: 10.1021/bi970833a. [DOI] [PubMed] [Google Scholar]
- 84.RIKEN Structural Genomics/Proteomics Initiative, PDB ID: 2E73, 2ENZ, 2ENN, 2ELI, 2YUU.
- 85.Zhang GG, Kazanietz MG, Blumberg PM, Hurley JH. Crystal-Structure of the Cys2 Activator-Binding Domain of Protein-Kinase C-Delta in Complex with Phorbol Ester. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 86.Rahman GM, Shanker S, Lewin NE, Kedei N, Hill CS, Prasad BVV, Blumberg PM, Das J. Identification of the activator-binding residues in the second cysteine-rich regulatory domain of protein kinase C theta (PKC theta) Biochem J. 2013;451:33–44. doi: 10.1042/BJ20121307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shanmugasundararaj S, Das J, Sandberg WS, Zhou XJ, Wang D, Messing RO, Bruzik KS, Stehle T, Miller KW. Structural and Functional Characterization of an Anesthetic Binding Site in the Second Cysteine-Rich Domain of Protein Kinase C delta. Biophys J. 2012;103:2331–2340. doi: 10.1016/j.bpj.2012.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grishin NV. Treble clef finger - a functionally diverse zinc-binding structural motif. Nucleic Acids Res. 2001;29:1703–1714. doi: 10.1093/nar/29.8.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saraiva L, Fresco P, Pinto E, Goncalves J. Characterization of phorbol esters activity on individual mammalian protein kinase C isoforms, using the yeast phenotypic assay. Eur J Pharmacol. 2004;491:101–110. doi: 10.1016/j.ejphar.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 90.Kimura K, Mizutani MY, Tomioka N, Endo Y, Shudo K, Itai A. Docking study of bryostatins to protein kinase C delta Cys2 domain. Chem Pharm Bull (Tokyo) 1999;47:1134–1137. [Google Scholar]
- 91.Pak Y, Enyedy IJ, Varady J, Kung JW, Lorenzo PS, Blumberg PM, Wang SM. Structural basis of binding of high-affinity ligands to protein kinase C: Prediction of the binding modes through a new molecular dynamics method and evaluation by site-directed mutagenesis. J Med Chem. 2001;44:1690–1701. doi: 10.1021/jm000488e. [DOI] [PubMed] [Google Scholar]
- 92.Li JN, Ziemba BP, Falke JJ, Voth GA. Interactions of Protein Kinase C-alpha C1A and C1B Domains with Membranes: A Combined Computational and Experimental Study. J Am Chem Soc. 2014;136:11757–11766. doi: 10.1021/ja505369r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Roffey J, Rosse C, Linch M, Hibbert A, McDonald NQ, Parker PJ. Protein kinase C intervention: the state of play. Curr Opin Cell Biol. 2009;21:268–279. doi: 10.1016/j.ceb.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 94.Goel G, Makkar HP, Francis G, Becker K. Phorbol esters: structure, biological activity, and toxicity in animals. Int J Toxicol. 2007;26:279–288. doi: 10.1080/10915810701464641. [DOI] [PubMed] [Google Scholar]
- 95.Kazanietz MG, Lorenzo PS. Phorbol esters as probes for the study of protein kinase C function. In: Newton AC, editor. Protein Kinase C Protocols. 2003. pp. 423–429. [DOI] [PubMed] [Google Scholar]
- 96.Hongpaisan J, Sun MK, Alkon DL. PKC epsilon activation prevents synaptic loss, Abeta elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J Neurosci. 2011;31:630–643. doi: 10.1523/JNEUROSCI.5209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Keck GE, Poudel YB, Cummins TJ, Rudra A, Covel JA. Total synthesis of bryostatin 1. J Am Chem Soc. 2011;133:744–747. doi: 10.1021/ja110198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu Y, Woo SK, Krische MJ. Total synthesis of bryostatin 7 via C-C bond-forming hydrogenation. J Am Chem Soc. 2011;133:13876–13879. doi: 10.1021/ja205673e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wender PA, Schrier AJ. Total synthesis of bryostatin 9. J Am Chem Soc. 2011;133:9228–9231. doi: 10.1021/ja203034k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Irie K, Oie K, Nakahara A, Yanai Y, Ohigashi H, Wender PA, Fukuda H, Konishi H, Kikkawa U. Molecular basis for protein kinase C isozyme-selective binding: The synthesis, folding, and phorbol ester binding of the cysteine-rich domains of all protein kinase C isozymes. J Am Chem Soc. 1998;120:9159–9167. [Google Scholar]
- 101.Ananthanarayanan B, Stahelin RV, Digman MA, Cho WH. Activation mechanisms of conventional protein kinase C isoforms are determined by the ligand affinity and conformational flexibility of their C1 domains. J Biol Chem. 2003;278:46886–46894. doi: 10.1074/jbc.M307853200. [DOI] [PubMed] [Google Scholar]
- 102.Pu YM, Garfield SH, Kedei N, Blumberg PM. Characterization of the Differential Roles of the Twin C1a and C1b Domains of Protein Kinase C delta. J Biol Chem. 2009;284:1302–1312. doi: 10.1074/jbc.M804796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Slater SJ, Ho C, Kelly MB, Larkin JD, Taddeo FJ, Yeager MD, Stubbs CD. Protein kinase Calpha contains two activator binding sites that bind phorbol esters and diacylglycerols with opposite affinities. J Biol Chem. 1996;271:4627–4631. doi: 10.1074/jbc.271.9.4627. [DOI] [PubMed] [Google Scholar]
- 104.Medkova M, Cho WH. Interplay of C1 and C2 domains of protein kinase C-alpha in its membrane binding and activation. J Biol Chem. 1999;274:19852–19861. doi: 10.1074/jbc.274.28.19852. [DOI] [PubMed] [Google Scholar]
- 105.Szallasi Z, Bogi K, Gohari S, Biro T, Acs P, Blumberg PM. Nonequivalent roles for the first and second zinc fingers of protein kinase C delta - Effect of their mutation on phorbol ester-induced translocation in NIH 3T3 cells. J Biol Chem. 1996;271:18299–18301. doi: 10.1074/jbc.271.31.18299. [DOI] [PubMed] [Google Scholar]
- 106.Stahelin RV, Digman MA, Medkova M, Ananthanarayanan B, Rafter JD, Melowic HR, Cho WH. Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase C delta. J Biol Chem. 2004;279:29501–29512. doi: 10.1074/jbc.M403191200. [DOI] [PubMed] [Google Scholar]
- 107.Stewart MD, Morgan B, Massi F, Igumenova TI. Probing the determinants of diacylglycerol binding affinity in the C1B domain of protein kinase Calpha. J Mol Biol. 2011;408:949–970. doi: 10.1016/j.jmb.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stewart MD, Igumenova TI. Reactive Cysteine in the Structural Zn(2+) Site of the C1B Domain from PKCalpha. Biochemistry. 2012;51:7263–7277. doi: 10.1021/bi300750w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Korichneva I, Hoyos B, Chua R, Levi E, Hammerling U. Zinc release from protein kinase C as the common event during activation by lipid second messenger or reactive oxygen. J Biol Chem. 2002;277:44327–44331. doi: 10.1074/jbc.M205634200. [DOI] [PubMed] [Google Scholar]
- 110.Medkova M, Cho WH. Differential membrane-binding and activation mechanisms of protein kinase C-alpha and -epsilon. Biochemistry. 1998;37:4892–4900. doi: 10.1021/bi972495j. [DOI] [PubMed] [Google Scholar]
- 111.Dries DR, Newton AC. Kinetic analysis of the interaction of the C1 domain of protein kinase C with lipid membranes by stopped-flow spectroscopy. J Biol Chem. 2008;283:7885–7893. doi: 10.1074/jbc.M709943200. [DOI] [PubMed] [Google Scholar]
- 112.Medkova M, Cho W. Interplay of C1 and C2 domains of protein kinase C-alpha in its membrane binding and activation. J Biol Chem. 1999;274:19852–19861. doi: 10.1074/jbc.274.28.19852. [DOI] [PubMed] [Google Scholar]
- 113.Mosior M, McLaughlin S. Peptides that mimic the pseudosubstarte regionof protein kinase C bind to acidic lipids in membranes. Biophys J. 1991;60:149–159. doi: 10.1016/S0006-3495(91)82038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- 115.Gao T, Toker A, Newton AC. The carboxyl terminus of protein kinase c provides a switch to regulate its interaction with the phosphoinositide-dependent kinase, PDK-1. J Biol Chem. 2001;276:19588–19596. doi: 10.1074/jbc.M101357200. [DOI] [PubMed] [Google Scholar]
- 116.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27:1919–1931. doi: 10.1038/emboj.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Behn-Krappa A, Newton AC. The hydrophobic phosphorylation motif of conventional protein kinase C is regulated by autophosphorylation. Curr Biol. 1999;9:728–737. doi: 10.1016/s0960-9822(99)80332-7. [DOI] [PubMed] [Google Scholar]
- 119.Staudinger J, Lu JR, Olson EN. Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-alpha. J Biol Chem. 1997;272:32019–32024. doi: 10.1074/jbc.272.51.32019. [DOI] [PubMed] [Google Scholar]
- 120.Ammendrup-Johnsen I, Thorsen TS, Gether U, Madsen KL. Serine 77 in the PDZ Domain of PICK1 Is a Protein Kinase C alpha Phosphorylation Site Regulated by Lipid Membrane Binding. Biochemistry. 2012;51:586–596. doi: 10.1021/bi2014689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stebbins EG, Mochly-Rosen D. Binding specificity for RACK1 resides in the V5 region of beta II protein kinase C. J Biol Chem. 2001;276:29644–29650. doi: 10.1074/jbc.M101044200. [DOI] [PubMed] [Google Scholar]
- 122.Abrahamsen H, O’Neill AK, Kannan N, Kruse N, Taylor SS, Jennings PA, Newton AC. Peptidyl-prolyl Isomerase Pin1 Controls Down-regulation of Conventional Protein Kinase C Isozymes. J Biol Chem. 2012;287:13262–13278. doi: 10.1074/jbc.M112.349753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xu ZB, Chaudhary D, Olland S, Wolfrom S, Czerwinski R, Malakian K, Lin L, Stahl ML, Joseph-McCarthy D, Benander C, Fitz L, Greco R, Somers WS, Mosyak L. Catalytic domain crystal structure of protein kinase C-theta (PKCtheta) J Biol Chem. 2004;279:50401–50409. doi: 10.1074/jbc.M409216200. [DOI] [PubMed] [Google Scholar]
- 124.Messerschmidt A, Macieira S, Velarde M, Badeker M, Benda C, Jestel A, Brandstetter H, Neuefeind T, Blaesse M. Crystal structure of the catalytic domain of human atypical protein kinase C-iota reveals interaction mode of phosphorylation site in turn motif. J Mol Biol. 2005;352:918–931. doi: 10.1016/j.jmb.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 125.Takimura T, Kamata K, Fukasawa K, Ohsawa H, Komatani H, Yoshizumi T, Takahashi I, Kotani H, Iwasawa Y. Structures of the PKC-iota kinase domain in its ATP-bound and apo forms reveal defined structures of residues 533–551 in the C-terminal tail and their roles in ATP binding. Acta Crystallogr D Biol Crystallogr. 2010;66:577–583. doi: 10.1107/S0907444910005639. [DOI] [PubMed] [Google Scholar]
- 126.Wagner J, von Matt P, Sedrani R, Albert R, Cooke N, Ehrhardt C, Geiser M, Rummel G, Stark W, Strauss A, Cowan-Jacob SW, Beerli C, Weckbecker G, Evenou JP, Zenke G, Cottens S. Discovery of 3-(1H-indol-3-yl)-4-[2-(4-methylpiperazin-1-yl)quinazolin-4-yl]pyrrole-2,5-dione (AEB071), a potent and selective inhibitor of protein kinase C isotypes. J Med Chem. 2009;52:6193–6196. doi: 10.1021/jm901108b. [DOI] [PubMed] [Google Scholar]
- 127.Grodsky N, Li Y, Bouzida D, Love R, Jensen J, Nodes B, Nonomiya J, Grant S. Structure of the catalytic domain of human protein kinase C beta II complexed with a bisindolylmaleimide inhibitor. Biochemistry. 2006;45:13970–13981. doi: 10.1021/bi061128h. [DOI] [PubMed] [Google Scholar]
- 128.George DM, Breinlinger EC, Argiriadi MA, Zhang Y, Wang JF, Bansal-Pakala P, Duignan DB, Honore P, Lang QY, Mittelstadt S, Rundell L, Schwartz A, Sun JK, Edmunds JJ. Optimized Protein Kinase C theta (PKC theta) Inhibitors Reveal Only Modest Anti-inflammatory Efficacy in a Rodent Model of Arthritis. J Med Chem. 2015;58:333–346. doi: 10.1021/jm5013006. [DOI] [PubMed] [Google Scholar]
- 129.George DM, Breinlinger EC, Friedman M, Zhang Y, Wang JF, Argiriadi M, Bansal-Pakala P, Barth M, Duignan DB, Honore P, Lang QY, Mittelstadt S, Potin D, Rundell L, Edmunds JJ. Discovery of Selective and Orally Bioavailable Protein Kinase C theta (PKC theta) Inhibitors from a Fragment Hit. J Med Chem. 2015;58:222–236. doi: 10.1021/jm500669m. [DOI] [PubMed] [Google Scholar]
- 130.Kjaer S, Linch M, Purkiss A, Kostelecky B, Knowles PP, Rosse C, Riou P, Soudy C, Kaye S, Patel B, Soriano E, Murray-Rust J, Barton C, Dillon C, Roffey J, Parker PJ, McDonald NQ. Adenosine-binding motif mimicry and cellular effects of a thieno 2,3-d pyrimidine-based chemical inhibitor of atypical protein kinase C isoenzymes. Biochem J. 2013;451:329–342. doi: 10.1042/BJ20121871. [DOI] [PubMed] [Google Scholar]
- 131.van Eis MJ, Evenou JP, Floersheim P, Gaul C, Cowan-Jacob SW, Monovich L, Rummel G, Schuler W, Stark W, Strauss A, von Matt A, Vangrevelinghe E, Wagner J, Soldermann N. 2,6-Naphthyridines as potent and selective inhibitors of the novel protein kinase C isozymes. Bioorg Med Chem Lett. 2011;21:7367–7372. doi: 10.1016/j.bmcl.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 132.Wang CH, Shang Y, Yu J, Zhang MJ. Substrate Recognition Mechanism of Atypical Protein Kinase Cs Revealed by the Structure of PKC iota in Complex with a Substrate Peptide from Par-3. Structure. 2012;20:791–801. doi: 10.1016/j.str.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 133.Yang Y, Igumenova TI. The C-Terminal V5 Domain of Protein Kinase C alpha Is Intrinsically Disordered, with Propensity to Associate with a Membrane Mimetic. PLoS One. 2013;8 doi: 10.1371/journal.pone.0065699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Farrow NA, Zhang O, Szabo A, Torchia DA, Kay LE. Spectral density function mapping using 15N relaxation data exclusively. J Biomol NMR. 1995;6:153–162. doi: 10.1007/BF00211779. [DOI] [PubMed] [Google Scholar]
- 136.Peng JW, Wagner G. Mapping of spectral density functions using heteronuclear NMR relaxation measurements. J Magn Reson. 1992;98:308–332. [Google Scholar]
- 137.Dyson HJ, Wright PE. Unfolded proteins and protein folding studied by NMR. Chem Rev. 2004;104:3607–3622. doi: 10.1021/cr030403s. [DOI] [PubMed] [Google Scholar]
- 138.Eisenberg D, Weiss RM, Terwilliger TC. The helical hydrophobic moment - a measure of the amphiphilicity of a helix. Nature. 1982;299:371–374. doi: 10.1038/299371a0. [DOI] [PubMed] [Google Scholar]
- 139.Rice P, Longden I, Bleasby A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 140.Sonnenburg ED, Gao T, Newton AC. The phosphoinositide-dependent kinase, PDK-1, phosphorylates conventional protein kinase C isozymes by a mechanism that is independent of phosphoinositide 3-kinase. J Biol Chem. 2001;276:45289–45297. doi: 10.1074/jbc.M107416200. [DOI] [PubMed] [Google Scholar]
- 141.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]