

Abstract
This review is the first to collate and summarize main data on named and unnamed rearrangement reactions of peroxides. It should be noted, that in the chemistry of peroxides two types of processes are considered under the term rearrangements. These are conventional rearrangements occurring with the retention of the molecular weight and transformations of one of the peroxide moieties after O–O-bond cleavage. Detailed information about the Baeyer−Villiger, Criegee, Hock, Kornblum−DeLaMare, Dakin, Elbs, Schenck, Smith, Wieland, and Story reactions is given. Unnamed rearrangements of organic peroxides and related processes are also analyzed. The rearrangements and related processes of important natural and synthetic peroxides are discussed separately.
Keywords: artemisinin, Baeyer−Villiger, Criegee, Hock, peroxide, rearrangement
Introduction
The chemistry of organic peroxides has more than a hundred-year history. Currently, organic peroxides are widely used as oxidizing agents and initiators for free-radical reactions both in industry and in laboratory. These compounds are produced and involved in various natural and biological processes and were explored extensively as antimalarial agents, anthelmintics, and anticancer drugs.
Organic peroxides, such as alkyl hydroperoxides, aryl hydroperoxides, ketone peroxides, dialkyl peroxides, diacyl peroxides, peroxy esters, peroxydicarbonates, peroxyacetals, and inorganic peroxides are the most important radical initiators that are widely used in industrial processes in the manufacture of polymers from unsaturated monomers [1–9].
Nowadays, the progress in the chemistry of organic peroxides is mainly a result of their biological activity and pharmaceutical application. The search of effective antimalarial and antihelminthic drugs is the main challenge of medicinal chemistry of peroxides. According to the World Health Organization (WHO) malaria is a widely distributed illness. About 3.2 billion people remain at risk of malaria and in 2015 214 million cases of malaria and 438 thousands deaths from it have been registered [10]. Compounds with high antimalarial [11–23], antihelminthic [24–28], and antitumor activities [29–34] were found among natural, semisynthetic, and synthetic peroxides. The main biologically active frame of these compounds includes five-membered 1,2-dioxolane [35–37], 1,2,4-trioxolane [38–39], and six-membered 1,2-dioxane [40–42], 1,2-dioxene [43], 1,2,4-trioxane [22,44–45] cycles. The naturally occuring peroxide artemisinin and its semisynthetic derivatives, artemether, arteether, and artesunate, are applied in large scale for malaria treatment [46–47].
Organic peroxides, their rearrangements and related processes play an important role in the chemistry of oxidation processes. Thus, the key reagent in the Sharpless epoxidation of allylic alcohols [48] and in the manufacture of propylene oxide via the Prilezhaev reaction [49–51] is tert-butyl hydroperoxide. In industry, phenol and acetone are mainly produced by the Hock process, which is based on the rearrangement of cumene hydroperoxide. In 2003, phenol was produced to more than 95% by this oxidation process [52–54]. Another important application of organic peroxides is the synthesis of lactones from cyclic ketones via the Baeyer−Villiger oxidation and it is one of the methods for the synthesis of commercially important caprolactone from cyclohexanone with peracetic acid [55–56].
Autoxidation processes with formation of hydroperoxides and their subsequent free-radical transformations with generation of carbon- and oxygen-centered radicals are key reactions in the drying process of oil-based and alkyd paints containing double bonds [57–61].
Organic peroxides and their transformation play an important role not only in industrial but also in biological processes. Thus, the firefly luciferase-catalyzed oxidation of luciferin yields the peroxy compound 1,2-dioxetane. This four-membered peroxide cycle is unstable and spontaneously decays to carbon dioxide and excited ketones, which release excess energy through light emission (bioluminescence) [62–65]. The in vivo oxidation of cholesterol by singlet oxygen produces the hydroperoxide cholesterol-5α-OOH, which undergoes a Hock oxidation to form atheronal A. The latter possesses proatherogenic effects and triggers the development of cardiovascular diseases [66–71].
The development of the chemistry of organic peroxides is closely related to the application and preparation of unsaturated compounds, such as epoxides, aldehydes, ketones, carboxylic acids, and their derivatives [72–113]. Organic peroxides are widely used as oxidants in oxidative coupling processes [114–120].
Industrial-scale production of readily available and efficient initiators of free radical polymerization and effective biologically active compounds promotes the search for new synthetic methods for peroxides starting from carbonyl compounds, hydrogen peroxide, and hydroperoxides [121–182].
In many cases, rearrangements and related reactions of peroxides are key pathways in laboratory, industrial, and biological processes. The rearrangements of organic peroxides are covered in the literature in hundreds of publications and in several specialized and partial reviews [183–188]. The present review is the first to combine the key data on both, name rearrangements and less well-known rearrangements and related oxidative processes, and to summarize systematically related and different features of these reactions, compares their mechanisms, and assesses the prospects of their application.
By definition, a rearrangement is a migration of an atom or a group of atoms from one atom to another within the same molecule [189]. In contrast, a rearrangement of organic peroxides means a change in the structure of the starting molecule to form an isomeric compound without a peroxy group [183]. The terminology of rearrangements of organic peroxides and related processes encountered in the literature shows that this definition is not generally applicable as rearrangements of peroxides can give both isomeric and non-isomeric compounds either containing a peroxy group or without the latter. In most cases, a rearrangement involves the migration or cleavage of the peroxide group in an intermediate molecule, and the stability of the latter is responsible for the further pathway of the process.
The review covers main studies published over the last 15–20 years with a brief excursion to the history of the development of various reactions and transformations. The review consists of three parts: the first part considers named transformations of organic peroxides (Figure 1), the second one deals with unnamed reactions, and the third part covers transformations of some important natural and synthetic peroxides. Since the term “rearrangements”, as applied to transformations of peroxides, is not clearly defined all parts of the review include processes related to rearrangements.
Figure 1.

The named transformations considered in this review.
Review
1. Named rearrangements of organic peroxides
Rearrangements of organic peroxides are the key steps in many well-known processes such as the Baeyer–Villiger (BV), the Criegee and Hock reactions, the Kornblum–DeLaMare rearrangement, Dakin, and Elbs oxidation.
The BV oxidation is widely used in organic synthesis for the preparation of esters and lactones and the Criegee reaction is applied to transform tertiary alcohols into ketones and aldehydes. The Hock rearrangement is a key step in the cumene (cumene–phenol) process and the Kornblum–DeLaMare is an important tool in the synthesis of functionalized ketones and alcohols, including γ-hydroxy enones. The Dakin oxidation finds application for the synthesis of phenols from arylaldehydes or aryl ketones and the Elbs persulfate oxidation allows the preparation of hydroxyphenols from phenols. Finally, the Schenck and Smith rearrangements are of interest in allyl hydroperoxide transformations.
1.1. Baeyer–Villiger oxidation
The BV reaction is the oxidation of ketones or aldehydes A under the action of hydrogen peroxide, hydroperoxides, Caro’s acid (H2SO5), or organic peracids to yield esters, lactones, or carboxylic acids B (Scheme 1) [190–191].
Scheme 1.
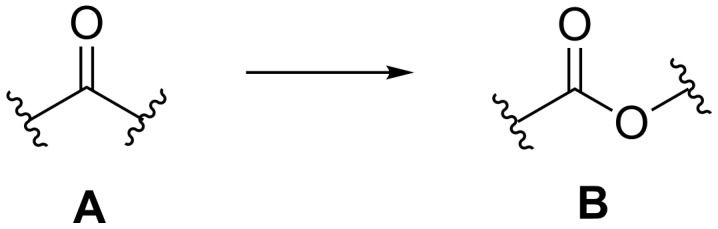
The Baeyer–Villiger oxidation.
Baeyer and Villiger accomplished the oxidation of ketones to esters for the first time in 1899 while they attempted the reaction of Caro’s acid (H2SO5) with menthone, tetrahydrocarvone, and camphor to transform these compounds into the corresponding lactones [192–194].
Since that time, this reaction has shown to be of general applicability and it has gained wide application for the oxidation of carbonyl compounds of different structures. In this reaction, cyclic ketones are transformed into lactones, acyclic ketones, into esters and aldehydes into carboxylic acids. The BV oxidation is one of the most important reactions in organic chemistry because it produces lactones, which are useful synthetic products in polymer, agrochemical, and pharmaceutical industry.
m-Chloroperbenzoic, peracetic, and perfluoroacetic acids, as well as hydrogen peroxide/protic acid, hydrogen peroxide/Lewis acid, and hydrogen peroxide/base systems are widely employed in the Baeyer–Villiger oxidation [185,194–195].
The general mechanism of the peracid-promoted Baeyer–Villiger oxidation involves two main steps. In the first step, the oxygen atom of the peroxide moiety of the peracid 2 binds to the carbonyl group of ketone 1 to form the tetrahedral intermediate 3 which is referred to as the Criegee intermediate. The next step involves the concerted migration of the R2 group to the peroxide oxygen atom, resulting in the formation of ester 4 and carboxylic acid 5 (Scheme 2).
Scheme 2.

The general mechanism of the peracid-promoted Baeyer–Villiger oxidation.
The ability of peracids to oxidize cyclic and acyclic ketones and aldehydes to the corresponding lactones, esters, and carboxylic acids decreases in the series peroxotrifluoroacetic acid > monopermaleic acid > mono-o-perphthalic acid > 3,5-dinitroperbenzoic acid > p-nitroperbenzoic acid > MCPBA ≈ performic acid > perbenzoic acid > peracetic acid >> H2O2 > t-BuOOH [196].
The migratory ability of substituents in the Criegee intermediate decreases in the following series: tertiary alkyl > cyclohexyl > secondary alkyl > benzyl > phenyl > primary alkyl > cyclopentyl, cyclopropyl > methyl. In some cases, stereoelectronical effects strongly influence the regioselectivity of the reaction, specifically the ability of the migrating C–C to align with the back of the breaking O–O bond, and the presence or absence of strain in cyclic ketone substrates [197–198]. The strongest electron-donating group migrates in unsymmetrical ketones [199].
There are thousands of publications on the Baeyer–Villiger reaction. In the latest reviews published by Krow [195] in 1993 and by Renz and Meunier [185] in 1999, the field of application, the reactivity of substrates, and the reaction kinetics and mechanisms are considered in detail. In the review by Strukul, special emphasis was placed on metal-catalyzed Baeyer–Villiger oxidations [196]. Green approaches in the Baeyer–Villiger reaction were highlighted by another review [200].
The present review covers a more modern aspect of this reaction, viz., the performance of the process using hydrogen peroxide. Oxidizing systems containing hydrogen peroxide as the oxidizing agent allow the usual and asymmetric oxidation of the substrate to the target product with high conversion and yield. In recent years, the inexpensive, commercially available, and environmentally friendly H2O2 was utilized in the Baeyer–Villiger reaction with increasing frequency. Various catalysts that activate hydrogen peroxide, such as heterogeneous catalysts based on solid acids [201], zeolites [202–203], Se [204], As [205], Co [206], sulfonated organic ion exchange resins [203,207], and homogeneous catalysts based on Pt [208], Zr [209], Re [210–211], Se [212–213], As [205], Mo [214], Co [215], Brønsted [216], and Lewis acids [217] are described in the literature. The general mechanism of a Lewis acid-catalyzed Baeyer–Villiger rearrangement is presented in Scheme 3 [200,218].
Scheme 3.

General mechanism of the Lewis acid-catalyzed Baeyer–Villiger rearrangement.
Scheme 4 shows the theoretically studied mechanism of the oxidation reaction promoted by H2O2 and the Lewis acid BF3 [217,219]. In the first step, the hydrogen peroxide–boron trifluoride complex 8 reacts with ketone 9 to form adduct 10. The latter intermediate rearranges through transition state 11 into the tetrahedral peroxyacetal intermediate 12. Then BF3 migrates to another oxygen atom through transition state 13 to give the second Criegee intermediate 14. The decomposition of intermediate 14 finally produces 15, hydrogen fluoride (16) and ester 17.
Scheme 4.

The theoretically studied mechanism of the BV oxidation reaction promoted by H2O2 and the Lewis acid BF3.
Despite the fact that the Baeyer–Villiger reaction is known since 1899, the mechanism of this reaction is still not fully understood. The nature of the acid catalyst [220] and the type of O–O-bond cleavage in the Criegee intermediate [221] were found to play an important role in this reaction. Probably the hydrogen bonds in Baeyer–Villiger reactions play an important role [222]. The tetramolecular transition states TS1 and TS2 are considered to be the two key steps determining the course of the oxidation: the nucleophilic addition of a peroxy acid molecule to ketone (TS1) and the migration of R and cleavage of O–O bond (TS2). Thus, electrophilic substrates favor TS1 and nucleophilic migrating groups prefer TS2 (Scheme 5).
Scheme 5.

Proton movements in the transition states of the Baeyer–Villiger oxidation.
The dependence of the course of the Baeyer–Villiger oxidation on the type of O−O-bond cleavage in the Criegee intermediate was studied in the oxidation reaction of 1,2-quinone 18 with perbenzoic acid [221]. The reaction gave two oxidation products – anhydride 20 and the seven-membered α-ketolactone 21. The investigation of the reaction mechanism demonstrated that the formation of the seven-membered α-ketolactone 21 proceeds through the heterolytic O–O-bond cleavage in Criegee intermediate 19, whereas the homolytic O–O cleavage affords anhydride 20 (Scheme 6).
Scheme 6.

The dependence of the course of the Baeyer–Villiger oxidation on the type of O–O-bond cleavage in the Criegee intermediate.
The acid-catalyzed Baeyer–Villiger oxidation of cyclic epoxy ketones 22 produces lactones of type 23, which convert into carbenium ions 24 in the presence of the acid. Subsequently, these ions can be transformed with participation of H2O2 through three different pathways into dihydroperoxides 25, dicarboxylic acids 28, carboxylic acids 26, and keto carboxylic acids 27 (Scheme 7, Table 1) [223].
Scheme 7.

The acid-catalyzed Baeyer–Villiger oxidation of cyclic epoxy ketones 22.
Table 1.
Oxidation of cyclic epoxy ketones 22a–c by H2O2.
| Epoxy ketone | R | 25, % | 26, % | 27, % | 28, % |
| 22a | Me | 25a, 12 | 26a, 6 | 27a, 15 | 28a, 53 |
| 22b | Et | 25b, 19 | a | a | a |
| 22c | Ph | 25c, 19 | – | 27c, 35 | 28c, 18 |
aThe aqueous phase consisted of a complex mixture and could not be analyzed.
The oxidation of isophorone oxide (29) is an industrial process for the production of dimethylglutaric acid 30 (Scheme 8) [223].
Scheme 8.

Oxidation of isophorone oxide 29.
Acyl phosphate 32 can be synthesized from acyl phosphonate 31 in high yield by oxidation with H2O2 (Scheme 9) [224].
Scheme 9.

Synthesis of acyl phosphate 32 from acyl phosphonate 31.
The Baeyer–Villiger oxidation provides a valuable tool for the synthesis of oxygenated natural products [218,225–226] as exemplified by the synthesis of aflatoxin B2 (36, Scheme 10) [227].
Scheme 10.

Synthesis of aflatoxin B2 (36).
The Baeyer–Villiger reaction is also a key step in the multistep synthesis of cannabinergic lactones from dimethylheptylresorcinol. Two regioisomeric cannabinergic lactones were obtained, one of which possessed pronounced affinity towards the CB1 receptor and lower affinities for mCB2 and hCB2 receptors [228].
Oxidation with H2O2–acid systems: With in situ generated peracids from carbodiimide, hydrogen peroxide, and carboxylic acids as catalysts ketones 37 are rearranged to lactones 38 (Scheme 11) [229].
Scheme 11.

The Baeyer–Villiger rearrangement of ketones 37 to lactones 38.
3,4-Dimethoxybenzoic acid (40) was prepared with 78% yield by a Baeyer–Villiger reaction of substrate 39 with 30% H2O2, HCOOH and 1,2-dichloroethane at 50 °C for 24 h (Scheme 12) [230].
Scheme 12.

Synthesis of 3,4-dimethoxybenzoic acid (40) via Baeyer–Villiger oxidation.
Oxone is a convenient reagent for the transformation of α,β-unsaturated ketones 43 of determined stereochemistry into vinyl acetates 44 via the Baeyer–Villiger reaction in dry DMF for 7–39 h (Scheme 13) [231].
Scheme 13.

Oxone transforms α,β-unsaturated ketones 43 into vinyl acetates 44.
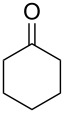
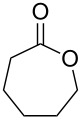
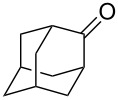
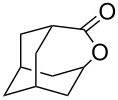
Oxidation with H2O2–heteroorganic catalyst systems: The activity of oxidizing systems such as H2O2/aryl benzyl selenoxide and H2O2/diaryl diselenide is similar to that of m-chloroperbenzoic acid [212,232–233]. The main advantage of these selenium-containing systems is that the catalysts are regenerated and can therefore be used at low loadings [234–236]. Some results of the oxidation of ketones and aldehydes 45a–c to the corresponding esters 46a–c using the H2O2/aryl benzyl selenoxide system are collected in Table 2 [232].
Table 2.
Baeyer–Villiger oxidation of aldehyde 45a and ketones 45b,c using the H2O2/aryl benzyl selenoxide system.
 | |||
| Substrate | Time, h | Product | Yield, % |
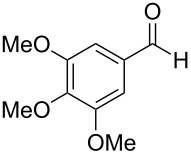 45a |
8 |
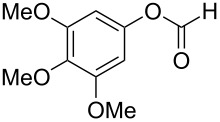 46a |
96 |
 45b |
24 |
 46b |
94 |
 45c |
18 |
 46c |
98 |
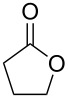
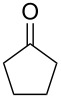
The oxidation results of ketones 47a,b and aldehydes 47c–e to lactones 48a,b and carboxylic acids 49a–c promoted by the H2O2/diaryl diselenide system is presented in Table 3 [212].
Table 3.
Baeyer–Villiger oxidation of ketones 47a,b and aldehydes 47c–e promoted by the H2O2/diaryl diselenide system.
 | ||||
| Ketone | Time, h | Product | Conversion, %a | Selectivity (BV product), %a |
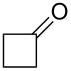 47a |
1 |
 48a |
99 | 90 |
 47b |
8 |
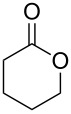 48b |
95 | 94 |
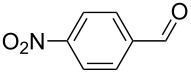 47cb |
2 |
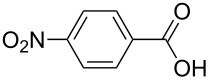 49a |
98 | 98 |
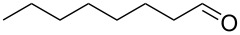 47db |
3 |
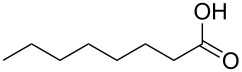 49b |
88 | 96 |
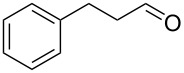 47eb |
3 |
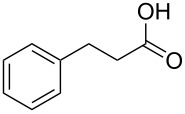 49c |
>90 | 99 |
aDetermined by GC; b60 °C.
In the first step of the catalytic cycle of the Baeyer–Villiger oxidation using diaryl diselenide 50 and hydrogen peroxide seleninic acid 51 is generated, which is then oxidized to perseleninic acid 52. Oxidation of the ketone 45 by perseleninic acid 52 involves the intermediate peroxide 53 (Scheme 14) [235].
Scheme 14.

The Baeyer–Villiger oxidation of ketones 45 using diaryl diselenide and hydrogen peroxide.
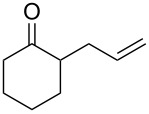
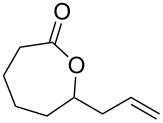
Similarly, the versatile 4-methylenebutanolides 55 can be prepared from (E)-2-methylenecyclobutanones 54 in the presence of (PhSe)2/H2O2 at room temperature (Scheme 15). Likely the Baeyer–Villiger reaction proceeds through the formation of benzeneseleninoperoxoic anhydride [PhSe(O)O]2O in the first step, which then transforms to the active oxidant benzeneseleninoperoxoic acid PhSe(O)OOH [233].
Scheme 15.

Baeyer–Villiger oxidation of (E)-2-methylenecyclobutanones.
The Baeyer–Villiger oxidation of (E)-α,β-unsaturated ketones to (E)-vinyl esters was performed with hydrogen peroxide and dibenzyl diselenide as pre-catalyst at room temperature [236]. Catalyzed by the dibenzyl diselenide, β-ionone (56) was oxidized by H2O2 with formation of (E)-2-(2,6,6-trimethylcyclohex-1-en-1-yl)vinyl acetate (57) with 91% yield (Scheme 16) [237].
Scheme 16.

Oxidation of β-ionone (56) by H2O2/(BnSe)2 with formation of (E)-2-(2,6,6-trimethylcyclohex-1-en-1-yl)vinyl acetate (57).
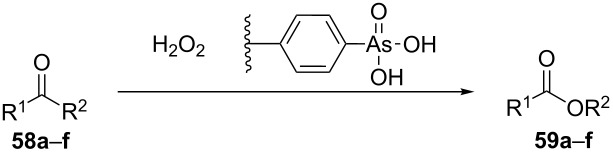
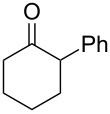
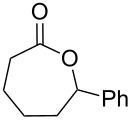
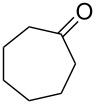
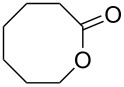
The Baeyer–Villiger oxidation of ketones 58a–f to form esters 59a–f can be accomplished in good yields in the presence of H2O2 and arsenic-containing ion exchange resins on polystyrene as the catalyst (Table 4) [203,205].
Table 4.
Oxidation of ketones 58a–f with 90% H2O2 catalyzed by arsonated polystyrene.
 | ||||
| Ketone | Time, h | Ketone/H2O2, ratio | Product | Yield based on the ketone consumed, % |
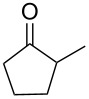 58a |
11 | 1 |
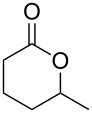 59a |
92 |
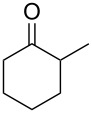 58b |
5 | 5 |
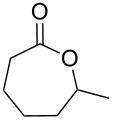 59b |
100 |
 58c |
15 | 1 |
 59c |
100 |
 58d |
23 | 5 |
 59d |
29 |
 58e |
9 | 1 |
 59e |
70 |
 58f |
25 | 5 |
 59f |
100 |
The mechanism of the oxidation of ketones 58a–f by hydrogen peroxide in the presence of arsonated polystyrene 60 as the catalyst is shown in Scheme 17. First, hydrogen peroxide reacts with the arsonic acid 60 to form peroxyarsonic acid 61 or it adds to ketones 58a–f to form vicinal hydroperoxyalkanols 63. In the second step the peroxyarsonic acid 61 adds to ketones 58a–f or the vicinal hydroperoxyalkanols 63 interact with arsonated polystyrene 60 under formation of perester 62. Finally, the decomposition of 62 gives esters 59a–f.
Scheme 17.

The mechanism of oxidation of ketones 58a–f by hydrogen peroxide in the presence of arsonated polystyrene 60.
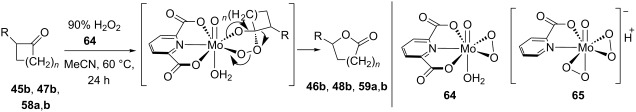
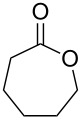
A number of other modern oxidizing systems are based on transition metal-peroxo complexes. The use of transition metal complexes were also used as catalysts for the Baeyer–Villiger reaction and the first example was documented in 1978 [196,214]. For example, Mo(VI) peroxo complexes 64 and 65 were employed as the catalysts and 90% H2O2 served as the oxidizer (Table 5).
Table 5.
Oxidation of cyclic ketones 45b, 47b and 58a,b by H2O2 in the presence of Mo(VI) peroxo complex 64 as the catalyst.
 | ||
| Ketone | Product | Yield, % |
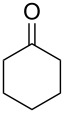 45b |
 46b |
10 |
 47b |
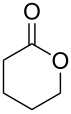 48b |
40 |
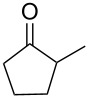 58a |
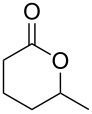 59a |
82 |
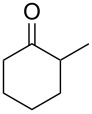 58b |
 59b |
10 |
The results obtained from the reactions using molybdenum systems have stimulated the search for new catalysts based on transition metal complexes. The usage of the platimum complex [(dppe)Pt(CF3(CH2Cl2)]BF4 (66·BF4) allowed the oxidation of 2-methylcyclohexanone (58b) in the presence of 32% H2O2 at room temperature to form 6-methylcaprolactone (59b) in 22% yield (Scheme 18) [238].
Scheme 18.

Oxidation of ketone (58b) by H2O2 to 6-methylcaprolactone (59b) catalyzed by Pt complex 66·BF4.
Acyclic ketones 67 could be oxidized to the corresponding esters 68 in the presence of the catalyst [(dppb}Pt(µ-OH)]22+, where dppb is butane-1,4-diylbis(diphenylphosphane) (Scheme 19) [208].
Scheme 19.

Oxidation of ketones 67 with H2O2 in the presence of [(dppb}Pt(µ-OH)]22+.
The oxidation mechanism of ketones 67 is displayed in Scheme 20.
Scheme 20.

The mechanism of oxidation of ketones 67 in the presence of [(dppb}Pt(µ-OH)]22+ and H2O2.
The use of variable-valence metal complexes opened up a new field of application of the Baeyer–Villiger oxidation and there are now dozens of studies on this topic [196,239–246].
Hydroxylated and methoxylated benzaldehydes 69 (Scheme 21) and acetophenones 72 (Scheme 22) can be oxidized to the corresponding phenols 70a–d, and 73 in good yields in the presence of the H2O2/MeReO3 system in ionic liquids [bmim]BF4 or [bmim]PF6 [247]. Benzoic acids 71a–d, 74 and phenyl esters 75a–d were reported as oxidation byproducts.
Scheme 21.

Oxidation of benzaldehydes 69 in the presence of the H2O2/MeReO3 system.
Scheme 22.

Oxidation of acetophenones 72 in the presence of the H2O2/MeReO3 system.
Sn-containing mesoporous silica nanospheres (Sn-MSNSs) with uniform crater-like mesopores exhibited high activities in the Baeyer–Villiger oxidation of 2-adamantanone (45c) (Scheme 23) [248].
Scheme 23.

Baeyer–Villiger oxidation of 2-adamantanone (45c) in the presence of Sn-containing mesoporous silica nanospheres (Sn-MSNSs).
The Baeyer–Villiger rearrangement of 2-adamantanone (45с) was performed using hydrogen peroxide (H2O2) and stannosilicate zeolites with nanosheet morphology and MFI topology (Sn-MFI-ns) as highly efficient catalysts [249]. The Sn-beta zeolites prepared by a steam-assisted conversion method are efficient catalysts for the Baeyer–Villiger reaction of cyclohexanone to ε-caprolactone [250]. A mesoporous Mg–Al-mixed oxide showed good catalytic efficiency in the Baeyer–Villiger oxidation of a series of ketones to the corresponding lactones and esters in the presence of diluted aqueous H2O2 and benzonitrile [251].
The Baeyer–Villiger oxidation of ketones 76 under the action of oxygen to the related esters 77 was performed using metal-free carbon (Ketjen Black) as a solid catalyst and benzaldehyde as the sacrificing agent. This metal-free carbon catalyst showed excellent catalytic activity and can be recycled after the reaction under oxygen atmosphere at 50 °C (Scheme 24) [252].
Scheme 24.

Aerobic Baeyer–Villiger oxidation of ketones 76 using metal-free carbon.
The boron-containing catalysts LiB(C6F5)4 or Ca[B(C6F5)4]2 were developed for the Baeyer–Villiger oxidation of ketones with aqueous H2O2 to give the lactones in high yields [253–254].
A regioselective Baeyer–Villiger oxidation of functionalized cyclohexenones 78 lead to dihydrooxepine structures 79. Here, the combination of SnCl4 and bis(trimethylsilyl)peroxide (BTSP), in the presence of trans-1,2-diaminocyclohexane as the ligand, generated the desired products 79 in high yields (Scheme 25) [255].
Scheme 25.

A regioselective Baeyer-Villiger oxidation of functionalized cyclohexenones 78 into a dihydrooxepine structures 79.
The Co4HP2Mo15V3O62-catalyzed oxidation of aldehydes and ketones 80 by hydrogen peroxide in ionic liquid [TEBSA][BF4] resulted in carboxylic acids and esters 81 in good to high yields (Scheme 26) [256].
Scheme 26.

The oxidation of aldehydes and ketones 80 by H2O2 catalyzed by Co4HP2Mo15V3O62.
Oxidation with H2O2–base systems: The oxidative cleavage of ketones 82 with hydrogen peroxide in alkaline solution yielded carboxylic acids 84. The authors suggested that the reaction of a ketone with the hydroperoxide anion resulted in the intermediate esters 83, which hydrolyzed in the basic reaction medium with formation of acids 84 (Scheme 27) [257].
Scheme 27.

The cleavage of ketones 82 with hydrogen peroxide in alkaline solution.
The use of hydrotalcites in the Baeyer–Villiger oxidation of various ketones resulted in high yields of the corresponding lactones or esters [258–260]. The esters 86 were synthesized by the reaction of ketones 85 with H2O2 and benzonitrile under basic reaction conditions (KHCO3) with the intermediate generation of peroxyimidic acids. This oxidation can be successfully applied to alkyl-containing ketones to give the target products in yields of 30–91% and good regioselectivity 7:1 to 20:1 (Scheme 28) [261].
Scheme 28.

Oxidation of ketones 85 to esters 86 with H2O2–urea in the presence of KHCO3.
Asymmetric oxidation: Asymmetric Baeyer–Villiger oxidation reactions can be performed using chiral acetals, organic hydroperoxides, chiral metal complexes and organocatalysts [262–263]. There are also Green chemistry approaches for Baeyer−Villiger oxidations based on enzyme-mediated processes, which are used for the preparation of chiral lactones. This type of biocatalysis is useful in synthetic chemistry and either isolated enzymes or living whole cells are applied for the oxidative production of valuable intermediates [264–269].
The asymmetric oxidation of 3-substituted cyclopentane-1,2-diones 87a–f is an efficient tool in organic synthesis for the preparation of unsymmetrical γ-lactone acids 88a–f with high optical purity and good yields (Table 6). These γ-lactone acids are valuable substrates for the synthesis of compounds with potentially useful pharmacological properties, such as homocitrates, alkyl- and aryl-substituted nucleosides [270–272].
Table 6.
Asymmetric oxidation of 3-substituted cyclopentane-1,2-diones 87a–f with the Ti(O-iPr)4/(+)DET/t-BuOOH system.
 | |||
| γ-Lactone acid | R | Yield, % | ee, % |
| 88a | -CH3 | 75 | 93 |
| 88b | -C2H5 | 72 | 93 |
| 88c | -CH2-OBn | 75 | 96 |
| 88d | -Bn | 83 | 96 |
| 88e | -C6H5 | 38 | 86 |
| 88f | 4-F-C6H4- | 43 | 86 |
The reaction starts with an asymmetric epoxidation of the substituted cyclopentane-1,2-dione 87a to form epoxide 89a. The second step involves the Baeyer–Villiger oxidation of epoxide 89a to peroxide 90a followed by the rearrangement into intermediate 91a. The latter is hydrolyzed by H2O to form dicarboxylic acid 92a, which is cyclized under the acidic conditions to γ-lactone acid 88a (Scheme 29) [270].
Scheme 29.

Mechanism of the asymmetric oxidation of cyclopentane-1,2-dione 87a with the Ti(OiPr)4/(+)DET/t-BuOOH system.
In most cases, the Baeyer–Villiger oxidation is a stereospecific and regioselective process with retention of the configuration. The oxidation of cis-4-tert-butyl-2-fluorocyclohexanone (93) with m-chloroperbenzoic acid in the presence of NaHCO3 affords fluorolactones 94 and 95 in 91% and 9% yields, respectively (Scheme 30) [273].
Scheme 30.

The oxidation of cis-4-tert-butyl-2-fluorocyclohexanone (93) with m-chloroperbenzoic acid.
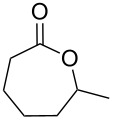
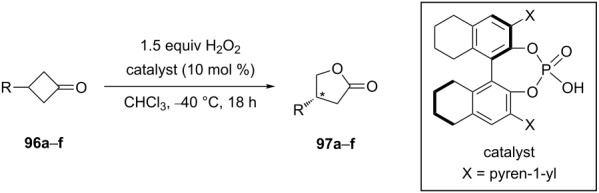
However, in order to perform the asymmetric oxidation of 3-substituted cyclobutanones 96a–f to the corresponding lactones 97a–f (Table 7) [274], it is necessary to employ chiral Brønsted acids [274–277], organocatalysts [278–279] or enzymes [280–282] as the catalyst. The obtained asymmetric oxidation products can be used in the multistep synthesis of new biologically active compounds.
Table 7.
Asymmetric oxidation of 3-substituted cyclobutanones 96a–f.
 | |||
| Ketone | R | Yield, % | ee,% (conf.) |
| 96a | C6H5 | 99 | 88 (R) |
| 96b | 4-MeC6H4 | 99 | 93 (R) |
| 96c | 4-FC6H4 | 99 | 84 (R) |
| 96d | 2-naphthyl | 91 | 86 (R) |
| 96e | C6H5CH2 | 99 | 58 (S) |
| 96f | 4-MeOC6H4CH2 | 99 | 57 (S) |
Possible mechanisms for the asymmetric oxidation of 3-substituted cyclobutanone 96a with H2O2 catalyzed by chiral phosphoric acid are presented in Scheme 31 [275].
Scheme 31.

The mechanism of the asymmetric oxidation of 3-substituted cyclobutanone 96a in the presence of chiral phosphoric acid.
A number of optically active ε- and γ-lactones 99, 100 was prepared by the enantioselective Baeyer–Villiger oxidation of racemic cyclic ketones 98 in up to 99% yield and 95% ee using the chiral N,N′-dioxide–Sc(III) complex as catalyst (Scheme 32) [283].
Scheme 32.

Enantioselective Baeyer–Villiger oxidation of cyclic ketones 98.
In another work, a chiral N,N′-dioxide–Sc(III) complex promoted Baeyer–Villiger oxidation was applied as instrument for a kinetic resolution of racemic 2-substituted cyclopentanones with formation of the 6-substituted δ-lactones in up to 98% ee and >95% regioselectivity [284].
A highly regio- and enantioselective Baeyer–Villiger oxidation of cyclic ketones 101 bearing amido, ureido, or sulfonamido functional groups to lactones 102 and 103 was carried out using the peptide-based catalyst 104. Hydrogen-bonding interactions are responsible for both types of selectivity. Notably, a reversal of the typically seen selectivity was observed with the peptide catalyst (Scheme 33) [285].
Scheme 33.

Regio- and enantioselective Baeyer–Villiger oxidation of cyclic ketones 101.
Versatility of the Baeyer–Villiger reaction with respect to starting reactants: The Baeyer–Villiger reaction cannot only be performed with ketones but also with acetals and aldimines as the starting substrates. The oxidation of cycloalkanone acetals 105a–g with performic acid generated in situ provides a new route to dicarboxylic acids 106a–g and hydroxycarboxylic acids 107a–g (Table 8) [286].
Table 8.
Oxidation of cycloalkanone acetals 105a–g.
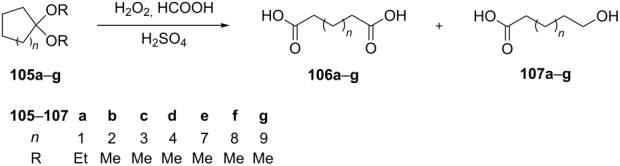 | ||||
| Ketal | H2O2 (6% ethereal solution) | H2O2 (30% aqueous solution) | ||
| Yield of 106, % | Yield of 107, % | Yield of 106, % | Yield of 107, % | |
| 105a | 14 | 51 | 11 | 61 |
| 105b | 6 | 68 | traces | 61 |
| 105c | 63 | 15 | 44 | 17 |
| 105d | 77 | 16 | 57 | 21 |
| 105e | 74 | 14 | 69 | 21 |
| 105f | 62 | 22 | 72 | 15 |
| 105g | 72 | 19 | 65 | 23 |
The proposed mechanism of the oxidation of acetal 105f is shown in Scheme 34.
Scheme 34.

The proposed mechanism of the Baeyer–Villiger oxidation of acetal 105f.
In the first step of the reaction, the elimination of methanol from 105f and formation of 108 takes place. Probably perester 109 is formed alongside of 108. After formation of 109, the reaction proceeds by two different routes A and B (second stage). The first route A leads to formation of epoxide 110, whereas the second route (B) proceeds through the Baeyer–Villiger reaction with formation of lactone 111 and subsequent acid hydrolysis to give 107f. At the third stage (route A), ether 112 is formed from 110 and subsequently rearranged by a Baeyer–Villiger reaction into 113, which is oxidized to form 106f.
This method can be applied to the synthesis of dodecanedioic acid, which is used in anticorrosive composites, polyester and polyamide threads, and lubricants, for the synthesis of tridecanedioic acid, and as a component of perfume formulations.
Scheme 35 presents the synthesis of hydroxy-10H-acridin-9-one 117 starting from tetramethoxyanthracene 114 through the formation of peroxide 115, which rearranges through an acid-catalyzed Baeyer–Villiger-type rearrangement into 116. Hydroxy-10H-acridin-9-ones 117 proved to be promising antipsoriatic agents [287].
Scheme 35.

Synthesis of hydroxy-10H-acridin-9-one 117 from tetramethoxyanthracene 114.
The oxidation of aldimines 118a–f with m-chloroperbenzoic acid in the presence of boron trifluoride etherate produces amides 119a–f in good yields (Table 9). The products of this transformation are strongly dependent on the electronic properties of the aromatic substituents at the carbon atom of the aldimines [288]. In the case of electron-donating substituents on the aryl fragment (Ar), formamides 119a–c are obtained as the result of imine oxidation and aryl migration. On the other hand, electron-withdrawing substituents on the aryl group (Ar) promote the formation of amides 119d–f as result of hydride migration.
Table 9.
Oxidation of aldimines 118a-f to amides by m-CPBA-BF3·Et2O system.
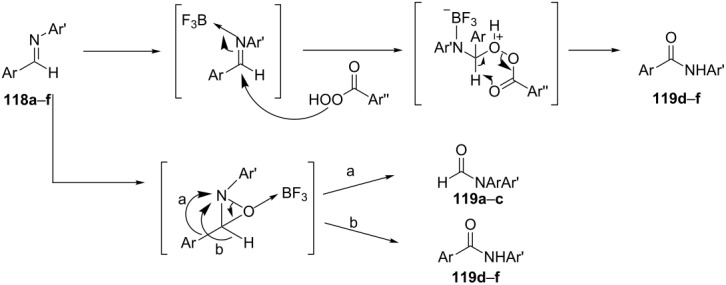 | |||
| Compound | Imine | Product | Yield, % |
| 118a | C6H5CH=NC6H5 | HCON(C6H5)2 | 82 |
| 118b | p-Me-C6H4CH=NC6H5 | HCONC6H5 p-Me-C6H4 | 90 |
| 118c | p-MeO-C6H4CH=NC6H5 | HCONC6H5 p-MeO-C6H4 | 91 |
| 118d | p-NO2-C6H4CH=NC6H5 | p-NO2-C6H4CONHC6H5 | 71 |
| 118e | p-NC-C6H4CH=NC6H5 | p-NC-C6H4CONHC6H5 | 79 |
| 118f | p-F3C-C6H4CH=NC6H5 | p-F3C-C6H4CONHC6H5 | 75 |
The sterically hindered and fully substituted pyrrole 120 underwent a Baeyer–Villiger reaction to yield a 4,5-dihydro-1H-ketopyrrole 121 (Scheme 36) [289].
Scheme 36.

The Baeyer–Villiger oxidation of the fully substituted pyrrole 120.
1.2. Criegee rearrangement
The Criegee rearrangement involves the transformation of a peroxide, mainly peroxyesters B, into carbonates, esters, or ketones C and alcohols D through an oxygen insertion or consecutive oxygen insertions. The peroxyester B is initially prepared from a tertiary alcohol A and a peracid. In addition, the peroxy ester can also be prepared via the reaction of a ketone and a peracid (i.e., through a Baeyer–Villiger oxidation); the additional product of peracid to ketone is often referred to as the Criegee intermediate. From this point of view, the Baeyer–Villiger oxidation is a subset of the Criegee rearrangement (Scheme 37) [290].
Scheme 37.

The Criegee rearrangement.
As mentioned above the Criegee reaction and the Baeyer–Villiger oxidation are related processes and both reactions involve the formation of the Criegee intermediate. The distinguishing feature of the Criegee rearrangement is that the Criegee intermediate rearranges into a carbocation. The mechanism of the Criegee reaction is presented in Scheme 38.
Scheme 38.

The mechanism of the Criegee reaction of a peracid with a tertiary alcohol 122.
Initially the reaction of the peracid with the tertiary alcohol 122 produces perester (Criegee intermediate) 123. One alkyl substituent migrates from the carbon atom to the adjacent oxygen atom and replaces the carboxylic acid moiety to form carbocation 124. Then, the addition of water to carbocation 124 affords ketone 125 and alcohol 126. p-Nitroperbenzoic acid is usually used to oxidize tertiary alcohols because the anion of this acid is a good leaving group.
The Criegee rearrangement was discovered in 1944 in the reaction of decaline ethylperoxoate 127 that rearranged into isomeric ester ketal 128 (Scheme 39) [291].
Scheme 39.

Criegee rearrangement of decaline ethylperoxoate 127 into ketal 128.
The mechanism of the Criegee rearrangement was studied using 2-alkoxy-2-propyl per-4-nitrobenzoates [292]. It was shown that the ionic cleavage of 2-methoxy-2-propyl perester 129 to p-nitrobenzoic acid (132), methyl acetate (133) and dimethyl ether (134) occurred through transition state 130 with generation of dimethoxycarbonium ion 131 (Scheme 40).
Scheme 40.

The ionic cleavage of 2-methoxy-2-propyl perester 129.
Investigations using aromatic peroxy esters 129 demonstrated that the migratory ability of the migrating group R decreases in the series t-Bu > C6H5 > iPr > OEt > OMe > Et > Me [293–294].
The Criegee rearrangement of α-methoxy hydroperoxide 136 obtained from (+)-trans-dihydrocarvone 135 produces trans-5-acetoxy-2-methylcyclohexanone 137 and intermediate peroxyacetate 138 (Scheme 41) [295].
Scheme 41.

The Criegee rearrangement of α-methoxy hydroperoxide 136.
Later on, the Criegee rearrangement was extended [296] to peroxides 139, 142, and 145 which made it possible to selectively synthesize both cyclic 140, 141, 144 and acyclic enol esters 146 and acetal 143 (Scheme 42).
Scheme 42.

Synthesis of enol esters and acetals via the Criegee rearrangement.
The Criegee rearrangement of 1-hydroperoxy-2-oxabicycloalkanes 147a–d in formic or acetic acid containing catalytic amounts of sulfuric acid affords ω-alkoxy-(ω-3)-hydroxyalkanoic acid lactones 148a–d and 149a–d (Table 10) [297].
Table 10.
Synthesis of ω-alkoxy-(ω-3)-hydroxyalkanoic acid lactones 148a–d and 149a–d from 1-hydroperoxy-2-oxabicycloalkanones 147a–d.
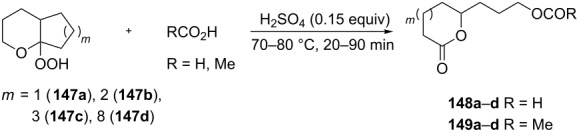 | ||||
| Substrate | RCOOH | Time (min) | Lactone | Yield, % |
| 147a | HCOOH | 20 | 148a | 64 |
| 147a | AcOH | 20 | 149a | 65 |
| 147b | HCOOH | 20 | 148b | 68 |
| 147b | AcOH | 20 | 149b | 70 |
| 147c | HCOOH | 30 | 148c | 57 |
| 147c | AcOH | 30 | 149c | 65 |
| 147d | HCOOH | 90 | 148d | 68 |
| 147d | AcOH | 90 | 149d | 53 |
The transformation of 1-hydroperoxy-2-oxabicycloalkanones 147a–d into ω-alkoxy-(ω-3)-hydroxyalkanoic acid lactones 148a–d and 149a–d is proposed to occur through intermediate peroxy ester 150 (Scheme 43).
Scheme 43.

Proposed mechanism of the transformation of 1-hydroperoxy-2-oxabicycloalkanones 147a–d.
1,2-Dioxolanes and related cyclic systems have attracted considerable attention from synthetic chemists as they may be used for the preparation of biologically active compounds. Under acidic conditions, 3-hydroxy-1,2-dioxolanes 151 are rearranged similarly to the Criegee mechanism into diketone derivatives 152 (Scheme 44) [298].
Scheme 44.

Transformation of 3-hydroxy-1,2-dioxolanes 151 into diketone derivatives 152.
Unlike the Baeyer–Villiger rearrangement, in which only mono-O-insertion can take place, the Criegee rearrangement of peroxide 153 in an acidic medium and under solvent-free conditions does not have such limitations. Thus, the latter reaction can proceed sequentially through the mono-, di-, and tri-O-insertion steps with formation of ketone 154, ester 155 and carbonate ester 156 (Scheme 45) [299–300].
Scheme 45.

Criegee rearrangement of peroxide 153 with the mono-, di-, and tri-O-insertion.
The selective double Criegee rearrangement next to a tertiary carbon was shown in the oxidative fragmentation at the bridgehead position of adamantanes 157a,b. The reaction employed the trifluoroperacetic acid (TFPAA)/trifluoroacetic acid (TFAA) system and afforded compounds 158a,b in high yields (Scheme 46) [300].
Scheme 46.

The sequential Criegee rearrangements of adamantanes 157a,b.
This method for the insertion of an oxygen atom was applied to the oxidation of triarylmethanols 159a–d [299]. The successive insertion of oxygen atoms gave rise to diaryl carbonates 160a–d in good yields (Scheme 47).
Scheme 47.

Synthesis of diaryl carbonates 160a–d from triarylmethanols 159a–d through successive oxygen insertion.
In the last years, new enantiospecific approaches for the synthesis of sesquiterpenes 162 from ketone 161 were developed [301–307]. In these methods, the Criegee rearrangement represents one key step and one example is presented in Scheme 48 [303].
Scheme 48.

The synthesis of sesquiterpenes 162 from ketone 161 with a Criegee rearrangement as one key step.
A method for the large-scale synthesis of a trans-hydrindan derivatives 164, 165 related to vitamin D, based on the Criegee rearrangement of alkene 163 was realized (Scheme 49) [308].
Scheme 49.

Synthesis of trans-hydrindan derivatives 164, 165.
Carbonyl oxides (Criegee intermediates) are one of the most important compounds in tropospheric chemistry [309]. Direct investigations of formaldehyde oxide (CH2OO) or acetaldehyde oxide (CH3CHOO) reactions with water vapor, SO2, NO2 were carried out [310–312].
1.3. Hock rearrangement
The Hock rearrangement is a protic or Lewis acid-promoted rearrangement of hydroperoxides A resulting in a C–C bond cleavage to form alcohol B and carbonyl compound C (Scheme 50) [313].
Scheme 50.

The Hock rearrangement.
The Hock rearrangement is a key step in the cumene process, which is used for the industrial production of phenol (170) and acetone (171) from benzene (166) and propylene (167) in the presence of air and radical initiators. The cumene process was described by Udris and Sergeev in 1947 [314–315] and independently by Hock in 1944 [316–317]. The general scheme of the cumene process, involving the formation of cumene hydroperoxide is shown in Scheme 51.
Scheme 51.

The general scheme of the cumene process.
The cumene process involves the acid-catalyzed rearrangement of cumene hydroperoxide (168) as a key step. The reaction starts with the protonation of the terminal oxygen atom of cumene hydroperoxide (168) followed by the migration of the phenyl group from the benzylic carbon atom to the peroxide oxygen atom and the elimination of a water molecule to form carbocation 169. The carbocation 169 is attacked by a water molecule, a proton is transferred to the oxygen atom attached to the phenyl group, and finally the cleavage of the adduct yields phenol (170) and acetone (171).
The Hock rearrangement of aliphatic hydroperoxides proceeds quite readily in concentrated H2SO4 [318] or superacids [319] (Scheme 52). This is associated with higher resistance of these compounds toward acid-catalyzed rearrangements compared with benzylic or allylic hydroperoxides. For example, aliphatic hydroperoxides are not cleaved in 5–50% aqueous H2SO4 but on the contrary, these compounds are produced under these conditions. More efficient catalysts are the compounds Sn(OTf)2 and La(OTf)3 which can be used for the transformation of 2-hydroperoxy-2,4,4-trimethylpentane (172) into neopentyl alcohol (173) and acetone (171). The Sn(OTf)2 and La(OTf)3-catalyzed reaction afforded neopentyl alcohol (173) in 62 and 70% yield, respectively [320].
Scheme 52.

The Hock rearrangement of aliphatic hydroperoxides.
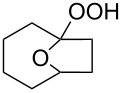
The hydrogen peroxide promoted ring expansion for the synthesis of oxabicycles 176a–c was described for the first time in 1985 [321]. The reaction involved the solvolysis of homoallylic brosylates 174a–c or spiro cyclopropyl carbinols 175a–c in the THF/H2O2 system, resulting in the increase in the ring size by two atoms and the formation of hydroperoxy oxabicyclo derivatives 176a–c (Table 11).
Table 11.
Solvolysis of brosylates 174a–c and spiro cyclopropyl carbinols 175a–c in the THF/H2O2 system.
| Substrate | Product (yield, %) | Substrate | Product (yield, %) |
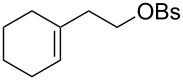 174a |
 176a (78%) |
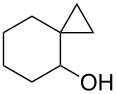 175a |
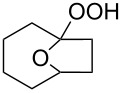 176a (90%) |
 174b |
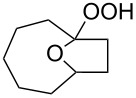 176b (73%) |
 175b |
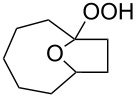 176b (91%) |
 174c |
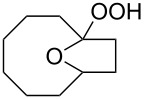 176c (84%) |
 175c |
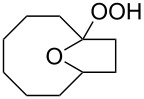 176c (91%) |
The mechanism of the solvolysis of 174 or 175 in the THF/H2O2 system involves the formation of solvolytically generated cyclobutyl hydroperoxides 177 followed by the rearrangement of the latter into oxa-bridged, hydroperoxyhemiketals 176 (Scheme 53).
Scheme 53.

The mechanism of solvolysis of brosylates 174a–c and spiro cyclopropyl carbinols 175a–c in THF/H2O2.
The fragmentation of hydroperoxy acetals 178a–e in the presence of Ca(OCl)2 or t-BuOCl as the catalysts in CH3CN generating esters 179a–e proceeds through the Hock-like rearrangement mechanism (Table 12) [322].
Table 12.
Fragmentation of hydroperoxy acetals 178a–e catalyzed by Ca(OCl)2 or t-BuOCl.
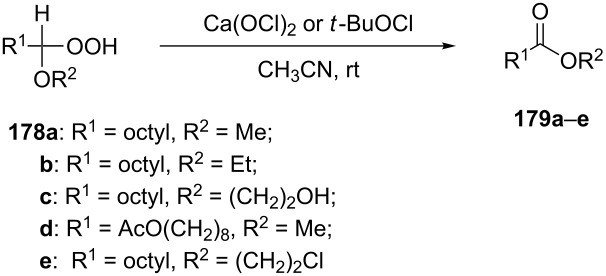 | |||||||
| Substrate | Product | Ca(OCl)2 (equiv) | Time (min) | Yield, % | t-BuOCl (equiv) | Time (min) | Yield, % |
| 178a | 179a | 1.3 | 10 | 75 | 0.25 | 15 | 78 |
| 178b | 179b | 1.3 | 10 | 86 | 1.2 | 10 | 85 |
| 178c | 179c | 1.3 | 10 | 83 | 1.2 | 10 | 84 |
| 178d | 179d | 1.3 | 10 | 85 | 0.25 | 15 | 85 |
| 178e | 179e | 1.3 | 10 | 82 | 1.2 | 10 | 84 |
The fragmentation of hydroperoxy acetals 178 to esters 179 involves the formation and heterolytic fragmentation of intermediate secondary chloroperoxides 180. The possible mechanism of the process is presented in Scheme 54.
Scheme 54.

The fragmentation mechanism of hydroperoxy acetals 178 to esters 179.
The acid-catalyzed rearrangement of phenylcyclopentyl hydroperoxide 181, involving the Hock reaction, is accompanied by the formation of a series of products: 1-phenylcyclopentene (182), phenol (170), cyclopentanone (183), and 5-acetoxyvalerophenone (184) (Scheme 55) [323].
Scheme 55.

The acid-catalyzed rearrangement of phenylcyclopentyl hydroperoxide 181.
An attempt was made [324] to synthesize hydroperoxides through the peroxidation of tertiary alcohols in the presence of a catalytic amount of acid. The treatment of 185 with H2O2 in the presence of a catalytic amount of H2SO4 for 72 hours did not lead to the formation of products via the Hock rearrangement of hydroperoxides, bicyclic hydroperoxides and о-hydroxyphenyl alkyl ketones. Instead, cyclic 2-methylchroman-2-yl hydroperoxide 188, geminal bishydroperoxides 190, and condensation products of peroxides such as 191 were isolated (Scheme 56).
Scheme 56.

The peroxidation of tertiary alcohols in the presence of a catalytic amount of acid.
The reaction mechanism presumably involves the following steps: the replacement of the hydroxy group by hydrogen peroxide to form tertiary hydroperoxides 186, the acid-catalyzed rearrangement of compounds 186 into cyclic phenoxycarbenium ions 187, and the addition of the second hydrogen peroxide molecule to 187 resulting in the formation of cyclic phenoxy hydroperoxide 188. The latter was isolated as the major product in the case of the six-membered ring (n = 1). In the case of the seven-membered ring (n = 2), geminal dihydroperoxide 190 and bridged bis(hydroxy)dialkyl peroxide 191 were obtained instead of 188. In case of the eight-membered ring (n = 3) an exclusive transformation into geminal dihydroperoxide 190 was observed (Table 13).
Table 13.
Yields of products 188, 190, and 191.
| Entry | R | n | 185 | (185:H2O2) | Yield, % | ||
| 188 | 190 | 191 | |||||
| 1 | Me | 1 | a | (1:10) | a (65) | ||
| 2 | Me | 2 | b | (1:10) | b (30) | b (48) | |
| 3 | Et | 2 | b | (1:10) | c (26) | c (36) | |
| 4 | Me | 3 | c | (1:10) | d (12) | ||
The formation of geminal dihydroperoxides 195 was also observed in the acid-catalyzed reaction of bicyclic secondary alcohols 192 with hydrogen peroxide. This reaction starts with the formation of bicyclic hydroperoxides 193 followed by the acid-catalyzed rearrangement with intermediate formation of peroxy hemiacetal 194. The latter is finally transformed into primary geminal bishydroperoxides 195 (Scheme 57) [325].
Scheme 57.

The acid-catalyzed reaction of bicyclic secondary alcohols 192 with hydrogen peroxide.
The photooxidation of 5,6-disubstituted 3,4-dihydro-2H-pyrans 196 generates the stable hydroperoxide 197 as the major product, which rearranges into dioxetane 198 at 28 °C in CCl4 within 13 h. Compounds 198 can be further transformed into keto esters 199 by treatment for 24 h with triphenylphosphine in CCl4 or concentrated HCl in CCl4. When compound 197 is heated at 70 °C its rearrangement into 199 occurs very rapidly and dioxetane 198 was not detected (Scheme 58) [326–327].
Scheme 58.

The photooxidation of 5,6-disubstituted 3,4-dihydro-2H-pyrans 196.
The oxidation of tertiary alcohols 200a–g, 203a,b, and 206, involving the rearrangement of hydroperoxides 201a–g, 204a,b, and 207, occurs in good yields in the presence of such systems as NaBO3·4H2O/BF3·Et2O [328], H2O2/BF3·Et2O, and H2O2/p-TsOH [329] (Scheme 59). The Hock rearrangement can be used to prepare alcohols 202a–g, 205, and 208 containing electron-donating substituents.
Scheme 59.

The oxidation of tertiary alcohols 200a–g, 203a,b, and 206.
The intramolecular capture of the cationic intermediate derived from the Hock rearrangement of peroxyketone 209 provides a direct and efficient one-step synthesis of 2,3-disubstituted furans 210 (Scheme 60) [330].
Scheme 60.

Transformation of functional peroxide 209 leading to 2,3-disubstitued furans 210 in one step.
The benzannulation of indoles 211 can be performed with γ-carbonyl tert-butyl peroxides 212 catalyzed by trifluoromethanesulfonic acid to give carbazoles 213. The key step of this approach is based on the acid-catalyzed rearrangement of tert-butyl peroxides (Scheme 61) [331].
Scheme 61.

The synthesis of carbazoles 213 via peroxide rearrangement.
The direct dehydrogenative construction of C–N bonds between unprotected phenols 215 and a series of 10H-phenoxazines and 10H-phenothiazines 214 with formation of 216 was carried out using a Hock-like activation with O2 followed by amine oxidation (Scheme 62) [332].
Scheme 62.

The construction of C–N bonds using the Hock rearrangement.
The Hock rearrangement plays an important role not only in fine organic synthesis but also in biological processes. Scheme 63 shows the proposed mechanism for the biosynthetic conversion of 217 to 218, which is an important component of the structural skeleton of the antitumor–antibiotic CC-1065 [333].
Scheme 63.

The synthesis of moiety 218 from 217 which is a structural motif in the antitumor–antibiotic of CC-1065.
The synthetic model of the in vivo oxidation of cholesterol (219) by singlet oxygen produces cholesterol-5α-OOH 220, which is subjected to a Hock reaction to form the aldolization product 221 and keto aldehyde (atheronal A, 222) (Scheme 64) [67].
Scheme 64.

The in vivo oxidation steps of cholesterol (219) by singlet oxygen.
Keto aldehyde (atheronal A, 222) exhibits proatherogenic activity and plays a causal role in the development of cardiovascular diseases [66]. The proposed mechanism of the rearrangement of cholesterol-5α-OOH 220 is presented in Scheme 65.
Scheme 65.

The proposed mechanism of the rearrangement of cholesterol-5α-OOH 220.
Therefore, the acid-catalyzed Hock rearrangement of hydroperoxide 220 is a key step in the oxidation of cholesterol (219).
In a photochemical route developed for the synthesis of artemisinin the Hock rearrangement of hydroperoxide 223 selectively affords enol 224. This reactive intermediate 224 is then finally oxidized into artemisinin (Scheme 66) [334].
Scheme 66.

Photochemical route to artemisinin via Hock rearrangement of 223.
1.4. Kornblum−DeLaMare rearrangement
The Kornblum−DeLaMare rearrangement (KDLM) is a rearrangement of organic peroxides A containing a primary or secondary carbon atom into ketones B and alcohols C mainly under base-catalyzed reaction conditions (Scheme 67) [335].
Scheme 67.

The Kornblum–DeLaMare rearrangement.
In 1951, Kornblum and DeLaMare observed that the treatment of 1-phenylethyl tert-butyl peroxide (225) with KOH, NaOEt, or pyridine resulted in the decomposition of 225 to give acetophenone (227) and tert-butanol (228). A three-step mechanism for this reaction was proposed (Scheme 68) [336–337].
Scheme 68.

Kornblum–DeLaMare transformation of 1-phenylethyl tert-butyl peroxide (225).
The reaction commences with a base-mediated α-proton abstraction from 225 to form carbanion 226 and the latter decomposes to yield the tert-butoxide anion and acetophenone (227). These steps occur presumably in a concerted manner. Finally, the protonation of the tert-butoxide anion results in the formation of tert-butanol (228). As alternative bases Et3N [338–339], phosphorus ylides [340] and LiOH [341–342] can be used and the Kornblum–DeLaMare rearrangement proceeds also on SiO2 [343].
The Kornblum–DeLaMare rearrangement is a convenient tool in organic chemistry for the conversion of monocyclic endoperoxides. These compounds are discussed in this review in the order of increasing ring size and the number of the starting substrates.
The treatment of unsubstituted bicyclic endoperoxides 229 by bases affords 4-hydroxyenones 230 [344] which are useful precursors in asymmetric organic syntheses. Alternative synthetic methods towards this class of compounds normally require a metal-catalyzed or biocatalyzed oxidation of diols 231 in an additional reaction step [345] (Scheme 69).
Scheme 69.

The synthesis 4-hydroxyenones 230 from peroxide 229.
The treatment of endoperoxide 232 with triethylamine in ethanol at room temperature results in the O–O-bond cleavage to form 5-hydroxytropolone (233) (Scheme 70) [346].
Scheme 70.
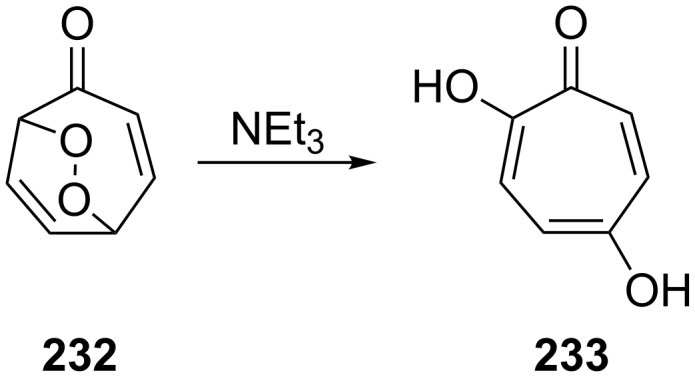
The Kornblum–DeLaMare rearrangement of peroxide 232.
It is interesting to note, that a reduction of the bicyclic endoperoxide 234 with thiourea in methanol at 10 °C produces similar to KDLM product tropolone 235 in 94% yield (Scheme 71) [347].
Scheme 71.

The reduction of peroxide 234.
The Kornblum–DeLaMare reaction of the endoperoxide 236 with triethylamine in chloroform at −30 °C affords tropolone 237 in 97% yield (Scheme 72) [347].
Scheme 72.

The Kornblum–DeLaMare rearrangement of endoperoxide 236.
Tropolones exhibit a broad spectrum of biological activities, including antibacterial, antiviral, antifungal, anti-allergic, anti-oxidant, and anti-inflammatory [348–349].
The treatment of endoperoxide 238 with Et3N gave 1,4-diketone 240 in quantitative yield instead of expected hydroxy ketone 239 (Scheme 73) [350–352].
Scheme 73.

The rearrangement of peroxide 238 under Kornblum–DeLaMare conditions.
The endoperoxide 238 is presumably converted into hemiketal 241, which is rearranged in several steps into diketone 240 (Scheme 74) [351].
Scheme 74.

The proposed mechanism of rearrangement of peroxide 238.
The reaction of endoperoxide 242a containing an electron-donating substituent at the double bond with bases results in the rearrangement product diketone 243. Under the same conditions, the base-catalyzed rearrangement of endoperoxide 242b containing an electron-withdrawing substituent leads to a product mixture of hydroxy ketone 244, and diketones 245 and 246 (Scheme 75) [353].
Scheme 75.

The Kornblum–DeLaMare rearrangement of peroxides 242a,b.
A further study [352] on the base-catalyzed rearrangements of substituted bicyclic endoperoxides showed that the pathway of the rearrangement is largely determined by the position of the substituent. The rearrangement of endoperoxides 247a,b containing an electron-withdrawing substituent in the seven-membered ring occurs mainly via a retro-aldol cleavage giving rise to formyl benzoates 248a,b (Scheme 76).
Scheme 76.

The base-catalyzed rearrangements of bicyclic endoperoxides having electron-withdrawing substituents.
On the other hand, endoperoxides 249a,b bearing electron-withdrawing groups (ester, acetyl) attached to the seven-membered ring are isomerized to diketones 250a,b (Scheme 77) [345].
Scheme 77.

The base-catalyzed rearrangements of bicyclic endoperoxides 249a,b having electron-donating substituents.
The Kornblum–DeLaMare reaction of endoperoxide 251a containing an electron-withdrawing substituent at the bridge head atom lead to the 1,2-dicarbonyl compound 252a whereas the ester 251b polymerized upon treatment with triethylamine (Scheme 78).
Scheme 78.

The base-catalyzed rearrangements of bridge-head substituted bicyclic endoperoxides 251a,b.
The disproportionation of endoperoxide 253 promoted by triethylamine affords β- and γ-hydroxy hydroperoxides 254 and 256. Under these conditions, the reaction afforded oxodiol 255 and diketone 257, which cyclized to hemiketal 258 as the products (Scheme 79) [354].
Scheme 79.

The Kornblum–DeLaMare rearrangement of hydroperoxide 253.
As the above reaction did not allow the isolation of hydroperoxide 254, an alternative strategy towards this compound was developed. The introduction of a protecting group into endoperoxide 253 using 2-methoxypropene gave protected peroxide 259. The subsequent triethylamine-catalyzed rearrangement of 259 leads to protected intermediate 260 the treatment of which under acidic conditions afforded hydroperoxide 254 in 70% yield (Scheme 80).
Scheme 80.

Synthesis of β-hydroxy hydroperoxide 254 from endoperoxide 253.
One approach to the enantioselective synthesis of 4-hydroxyenones 262 is based on the Kornblum–DeLaMare rearrangement of meso-endoperoxides 261 catalyzed by a chiral base [345] (Table 14).
Table 14.
Enantioselective rearrangement of meso-endoperoxides 261a–f into 4-hydroxy enones 262a–f.
| No | Endoperoxide | Reaction conditionsa | No | Product | Yield,% | eе, % | |
| 261a |  |
R = H | 5 mol % cat., rt, 6 h | 262a |  |
97 | 99 |
| 261b | R,R = OC(Me)2O | 5 mol % cat., rt, 10 h | 262b | 99 | 99 | ||
| 261c |  |
R = H | 5 mol % cat., 0 °C, 24 h |
262c |  |
99 | 87 |
| 261d | R = TBS | 5 mol % cat., rt, 12 h | 262d | 83 | 99 | ||
| 261e |  |
R = Bn | 10 mol % cat., rt, 24 h | 262e | 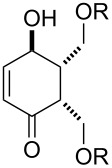 |
90 | 96 |
| 261f | R = -C(Me)2- | 10 mol % cat., rt, 36 h | 262f | 76 | 89 | ||
acat. =
The amine-catalyzed rearrangement of bicyclic endoperoxide 263 produced (S)-(+)-4-hydroxycyclohept-2-en-1-one (264), which was oxidized to bicyclic ketone 265. The synthetic value of chiral bicyclic ketone 265 was demonstrated by the transformation of this compound into (+)-sundiversifolide (266) (Scheme 81) [355].
Scheme 81.

The amine-catalyzed rearrangement of bicyclic endoperoxide 263.
The photooxidation of diene 267 followed by the base-catalyzed rearrangement of meso-endoperoxide 268 lead to (±)-trans,cis-4-hydroxy-5,6-di-O-isopropylidenecyclohex-2-en-1-one (269). The protection of the hydroxy group in compound 269 provides an efficient route to functionalized 4-hydroxy-2-cyclohexene-1-ones 270 (Scheme 82) [356].
Scheme 82.

The base-catalyzed rearrangement of meso-endoperoxide 268 into 269.
The photooxidation of 271 in the presence of tetraphenylporphyrin produces endoperoxide 272, which undergoes a Kornblum–DeLaMare transformation when treated with triethylamine. The obtained product 4-hydroxycyclohexen-2-one 273 releases benzoic acid through β-elimination under the basic conditions to give cyclohexadienone 274 (Scheme 83) [357].
Scheme 83.

The photooxidation of 271 and subsequent Kornblum–DeLaMare reaction.
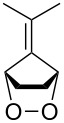
The base-catalyzed isomerization of bicyclic saturated fulvene endoperoxides 275 is employed as one approach to the preparation of 2-alkenylcyclopentanones 276 and cyclopentenones 277 [358]. Thus, the treatment of a solution of endoperoxides 275 in CH2Cl2 with triethylamine while increasing the temperature from 0 °C to room temperature affords hydroxyketone 276. The use of the stronger base DBU results in the formation of 2-vinyl-2-cyclopentenones 277 in high yield (Table 15).
Table 15.
DBU-catalyzed isomerization−dehydration of saturated fulvene endoperoxides 275 to form 2-vinyl-2-cyclopentenones 277.
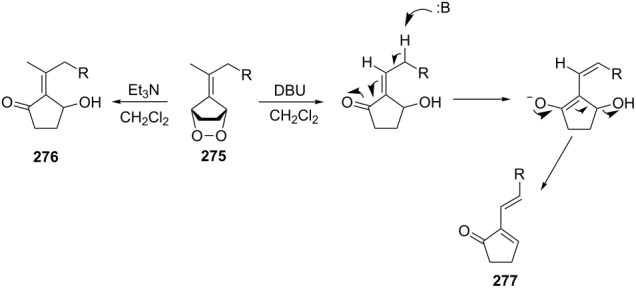 | ||
| Endoperoxide | Cyclopentenone | Yield, % |
 275a |
 277a |
76 |
 275b |
 277b |
83 |
 275c |
 277c |
82 |
 275d |
 277d |
68 |
In the case of acyclic enamine 278, the initial dioxetane product from the photochemical oxidation of 279 rearranged to amide 280. The reactions using cyclic enamines 281 involve the Kornblum–DeLaMare rearrangement of dioxetanes 282 into 1,2-diketones 283 (Scheme 84) [359–360].
Scheme 84.

The Kornblum–DeLaMare rearrangement as one step in the oxidation reaction of enamines.
The Kornblum–DeLaMare rearrangement of 1,2-dioxenes 284 [361], 1,2-dioxanes 286 [362], and tert-butyl peroxides 288 [330,363] produces 1,4-dicarbonyl compounds 285, 287, and 289, respectively (Scheme 85). These compounds are versatile starting substrates for the synthesis of various heterocyclic systems, such as furan, thiophene, and pyrrole derivatives.
Scheme 85.

The Kornblum–DeLaMare rearrangement of 3,5-dihydro-1,2-dioxenes 284, 1,2-dioxanes 286, and tert-butyl peroxides 288.
The reaction of unsymmetrical epoxy dioxanes 290a–d with triethylamine is accompanied by the 1,2-dioxane-ring opening to form 4-hydroxy-2,3-epoxy ketones 291a–d in high yields. The base catalysis involves the abstraction of the most acidic α-proton in the vicinity of the O–O bond followed by the rearrangement accompanied by the O–O-bond cleavage to form 4-hydroxy-2,3-epoxy ketones (Scheme 86) [364].
Scheme 86.

The Kornblum–DeLaMare rearrangement of epoxy dioxanes 290a–d.
The Kornblum–DeLaMare rearrangement is of special synthetic value in view of the synthesis of biologically active compounds. For instance, prostaglandin H2 (292) containing the bicyclic [2.2.1]endoperoxide moiety is rearranged in situ into prostaglandin Е2 (293) (Scheme 87) [365–366].
Scheme 87.

Rearrangement of prostaglandin H2 292.
Nicolaou et al. [367] described the synthesis of epicoccin G (297) and related diketopiperazines 296 through the photooxidation of 294 and the Kornblum–DeLaMare rearrangement of peroxide 295 (Scheme 88).
Scheme 88.

The synthesis of epicoccin G (297).
The base-catalyzed transformation of organic peroxide 298 was used to synthesize compound 299, a precursor for the synthesis of the natural compound phomactin A (300). Phomactin A is a representative of a new class of platelet-activating factor (PAF) antagonists (Scheme 89) [368].
Scheme 89.

The Kornblum–DeLaMare rearrangement used in the synthesis of phomactin A.
In another study [369], the transformation of peroxide 302, produced from 301, was applied to prepare compounds such as 3H-quinazolin-4-one 303, which is a core subunit of some important quinazolinone-based drugs (Scheme 90).
Scheme 90.

The Kornblum–DeLaMare rearrangement in the synthesis of 3H-quinazolin-4-one 303.
The Kornblum–DeLaMare rearrangement is one of the steps in the synthesis of the natural compound angelone from Nauclea, a plant species widely acclaimed for its anti-inflammatory and antibacterial utilities in traditional Chinese herbal medical formulations [370]. A Kornblum–DeLaMare enantiomeric resolution was also used to obtain both fragments of the polypropionate metabolite dolabriferol from a common precursor. The endoperoxide 304 was converted into ketone 305 with the help of the pseudo-enantiomeric quinine-derived catalyst (deMeQ-Ac) in toluene with moderate 47% yield. The peroxide 306 was transformed into ketone 307 with good 92% yield by using Et3N (Scheme 91) [371].
Scheme 91.

The Kornblum–DeLaMare rearrangement in the synthesis of dolabriferol (308).
A sequence consisting of a template-mediated photooxygenation and an acid-catalyzed Kornblum−DeLaMare rearrangement of the intermediate endo-peroxides 310 was used in a one-pot transformation of 3-substituted 2-pyridones 309 into the respective 3-hydroxypyridine-2,6-diones 311 with good enantioselectivity (69–86% ee) (Scheme 92) [372].
Scheme 92.

Sequential transformation of 3-substituted 2-pyridones 309 into 3-hydroxypyridine-2,6-diones 311 in one pot.
The Kornblum–DeLaMare rearrangement of peroxide 312 into hydroxy enone 313 with high yields and regioselectivity has been reported in the total synthesis of (+)-zeylenol and its congeners (Scheme 93) [373].
Scheme 93.

The Kornblum–DeLaMare rearrangement of peroxide 312 into hydroxy enone 313.
The polyfunctionalized carbonyl compounds 317 were prepared via crossover oxidative coupling of ethers 316 with electron-deficient alkenes 315 and vinylarenes 314 in the presence of Co(salen) and TBHP under mild conditions. The transformation involved the combination of a tandem radical reaction and a Kornblum−DeLaMare rearrangement in a one-pot process (Scheme 94) [374].
Scheme 94.

The Kornblum–DeLaMare rearrangement in the synthesis of polyfunctionalized carbonyl compounds 317.
The readily available compounds styrenes 314, amines 318 and perfluoroalkyl iodides 319 were transformed into (Z)-β-perfluoroalkylenaminones 320 via a Co(acac)2/TBHP-promoted multicomponent radical reaction involving sequential fluoroalkylation and Kornblum-DeLaMare rearrangement (Scheme 95) [375].
Scheme 95.

The Kornblum–DeLaMare rearrangement in the synthesis of (Z)-β-perfluoroalkylenaminones 320.
Peroxy products resulted from the reaction of styrenes 314, ethyl diazoacetate (321), and TBHP underwent a Kornblum–DeLaMare rearrangement with formation of γ-ketoester 322 (Scheme 96) [376].
Scheme 96.

The Kornblum–DeLaMare rearrangement in the synthesis of γ-ketoester 322.
The Kornblum–DeLaMare rearrangement is a final step in the total synthesis of the diterpenoids amphilectolide (326) and sandresolide B (328) from a common furan building block 324, which was synthesized from 323. Amphilectolide was obtained through a photooxygenation of 325 in the presence of diisopropylethylamine (DIEA), followed by a one-pot reduction of the intermediate peroxide with sodium borohydride. Sandresolide B was prepared from 327 using tetraphenylporphyrin as a photosensitizer and DBU as a base in 51% yield over two steps (Scheme 97) [377].
Scheme 97.

The Kornblum–DeLaMare rearrangement in the synthesis of diterpenoids 326 and 328.
The total synthesis of the natural products hainanolidol (331) and harringtonolide (332) includes a DBU-promoted Kornblum–DeLaMare rearrangement of endoperoxide 329 to ketone 330 (Scheme 98) [378].
Scheme 98.

The synthesis of natural products hainanolidol (331) and harringtonolide (332) from peroxide 329.
The reaction of the sodium salts of 1,3-dicarbonyl compounds 333, 334 with endoperoxides 263 and 261a in the presence of an organocatalyst affords the trans-fused butyrolactones 339 and 340 in high yield. The reaction proceeds via the formation of bicycles 335, 336 in the case of method A and 337, 338 in the case of method B (Scheme 99) [379].
Scheme 99.

The synthesis of trans-fused butyrolactones 339 and 340.
The leucosceptroid A (341) produced leucosceptroid C (343) and its diastereomer in 78% yield (1:1 dr) under the base-induced reduction of the initial endoperoxide intermediate. Irradiation of a solution of leucosceptroid A (341) in an oxygen-saturated dichloromethane solution containing a catalytic amount of tetraphenylporphyrin (TPP) and N,N-diisopropylethylamine cleanly produced 344 (85% yield). The latter compound represents the base-promoted Kornblum–DeLaMare rearrangement product of endoperoxide 342 (Scheme 100) [380].
Scheme 100.

The synthesis of leucosceptroid C (343) and leucosceptroid P (344) via the Kornblum–DeLaMare rearrangement.
It is worth mentioning that the synthesis of 4-hydroxycyclopentenone 343 and litsaverticillols was achieved in a similar way in other works [381–384].
1.5. Dakin oxidation of arylaldehydes or acetophenones
Generally, the Dakin oxidation is a reaction, in which o- or p-hydroxylated benzaldehydes or acetophenones 345 react with hydrogen peroxide in the presence of a base to form o- or p-dihydroxybenzene 346 and carboxylate 347 (Scheme 101) [385–386].
Scheme 101.

The Dakin oxidation of arylaldehydes or acetophenones.
Actually, the Dakin oxidation is a special case of the Baeyer–Villiger oxidation. Mechanistically, the Dakin oxidation starts with the nucleophilic addition of a hydroperoxide anion to the carbonyl carbon atom of benzaldehyde (348) to form intermediate 349 followed by its rearrangement to phenyl ester 350. The subsequent nucleophilic addition of a hydroxide anion to the carbonyl group of phenyl ester 350 yields intermediate 351, which undergoes a rearrangement accompanied by the elimination of phenoxide anion 352 and carboxylic acid 353. Then, the phenoxide anion 352 deprotonates the carboxylic acid 353 to produce p-dihydroxybenzene (354) and the corresponding carboxylate anion 355 (Scheme 102) [385,387].
Scheme 102.

The mechanism of the Dakin oxidation.
The nucleophilic addition of the hydroperoxide to the carbon atom of a carbonyl group and the [1,2]-aryl migration are the two rate-determining reaction steps in the Dakin oxidation process [387]. The total rate of the Dakin oxidation depends on the nucleophilicity of the hydroperoxide, the electrophilicity of the carbonyl carbon atom, the nature of alkyl substituents in the proximity of the carbonyl group, the existence of other functional groups in the aromatic ring, and the alkalinity of the reaction mixture. Generally, hydroxybenzaldehydes are more reactive in the Dakin oxidation than hydroxyacetophenones. This is due to the fact, that the carbonyl carbon atom of ketones is less electrophilic than the carbonyl carbon atom of an aldehyde. Under weakly basic conditions, о-hydroxybenzaldehydes and о-hydroxyacetophenones are oxidized more rapidly than p-hydroxybenzaldehydes and p-hydroxyacetophenones, whereas m-hydroxybenzaldehydes and m-hydroxyacetophenones are unreactive [387]. Electron-donating substituents in the ortho and para positions of the aromatic ring enhance the electron density on the migrating carbon atom thus promoting the [1,2]-aryl migration and accelerating the oxidation. Electron-donating substituents in the meta position have little effect on the electron density on the migrating carbon atom. Electron-withdrawing substituents in the ortho and para positions of the aromatic ring reduce the electron density on the migrating carbon atom, interfering with the [1,2]-aryl migration. The hydroperoxide anion is a more reactive nucleophile than neutral hydrogen peroxide. The reaction rate of the oxidation of hydroxyphenylaldehydes or ketones increases with increasing pH value, however, at pH higher than 13.5 the oxidation does not take place [387].
The efficient oxidation of hydroxylated aldehydes and ketones to hydroquinones and catechols was performed using a complex of urea with hydrogen peroxide as an oxidant [388]. The main advantage of this method is, that the reaction is performed under solvent-free conditions and provides the products in high yields.
A solvent-free Dakin reaction of aromatic aldehydes 356 with m-CPBA resulted in corresponding phenols 357 with high yields within a few minutes (Scheme 103) [389].
Scheme 103.

A solvent-free Dakin reaction of aromatic aldehydes 356.
The phenols 359 were prepared from electron-rich arylaldehydes 358 by a flavin-catalyzed Dakin oxidation under the action of H2O2 and sodium bicarbonate with high yields (Scheme 104) [390].
Scheme 104.

The organocatalytic Dakin oxidation of electron-rich arylaldehydes 358.
The flavin-catalyzed Dakin oxidation provides a more selective formation of phenols in comparison with the base-catalyzed rearrangement. The Dakin oxidation of arylaldehydes 361 is performed in the presence of molecular oxygen as the oxidant, a flavin organocatalyst and a Hantzsch ester. The oxidation products, catechols and electron-rich phenols 362, were prepared with 0.1–10 mol % of catalyst, 1 equiv of the Hantzsch ester, and O2 or air in a stoichiometric amount (Scheme 105) [391].
Scheme 105.

The Dakin oxidation of electron-rich arylaldehydes 361.
Dakin reactions of benzaldehydes 358 with H2O2 were successfully performed in natural feedstock extract ‘Water Extract of Banana’ (WEB) at room temperature under aerobic conditions in short reaction times. Under these conditions, phenols 359 could be obtained with 90–98% yields (Scheme 106) [392]. The WEB was prepared by extraction of banana ash with distilled water. The authors suggested that the potassium carbonate and sodium carbonate present in the extract serve as the internal base to promote the Dakin oxidation.
Scheme 106.

The Dakin oxidation of arylaldehydes 358 in water extract of banana (WEB).
The Dakin oxidation was applied for the synthesis of indolo[2,1-b]quinazolines 364 from indole-3-carbaldehydes 363. In the first step, the oxidation of indole-3-carbaldehydes 363 with further cyclization leads to isatoic anhydrides 365. Then, the anhydrides 365 react with indole-3-carbaldehydes 363 to produce the target indolo[2,1-b]quinazolines 364 (Scheme 107) [393].
Scheme 107.

A one-pot approach towards indolo[2,1-b]quinazolines 364 from indole-3-carbaldehydes 363 through the Dakin oxidation.
The Dakin oxidation is widely used for the synthesis of benzenediols and alkoxyphenols. For example, catechol generated from o-alkoxybenzaldehydes is employed as the starting reagent in the synthesis of catecholamine derivatives [394]. Catechols, for example are substrates in the manufacture of synthetic adhesives and coatings. Their multifaceted reactions with both, organic and inorganic reagents, make catechols widely applied compounds for surface modifications [395].
Vanillin was oxidized under Dakin conditions under formation of 2-methoxyhydroquinone with 97% yield. This vanillin-derivative was used as a building block in the synthesis of bio-based compounds applicable in polymer field [396].
The Dakin oxidation of mixtures of lignin depolymerization products is an important process for increasing the number of hydroxy groups in arene cycles. Then, these byproducts are glycidylated with mixtures of epoxy monomers. The obtained products are interesting compounds for the synthesis of bio-based epoxy thermosets with outstanding thermomechanical indexes [397].
Acid-catalyzed Dakin oxidation: The mechanism of Dakin oxidation under mild acidic conditions is similar to the base-catalyzed mechanism. A 30–35% aqueous H2O2/acid system can be employed as the oxidizing agent to synthesize phenols 367a–c from benzaldehydes 366a–c. The oxidation of 366a using traditional peracids produces a mixture of aryl formate 368 and epoxides 369 and 370 (Scheme 108) and cannot be applied to substrates containing peracid-labile functional groups [398].
Scheme 108.

The synthesis of phenols 367a–c from benzaldehydes 366a-c via acid-catalyzed Dakin oxidation.
The addition of boric acid to the H2O2/acid system leads to an increase in the yield of phenols 372a–f even in the case of benzaldehydes 371a–c or acetophenones 371d–f containing electron-donating groups in the meta position or electron-withdrawing groups in the ortho or para positions with respect to the carbonyl group (Table 16) [399].
Table 16.
Acid-catalyzed Dakin oxidation of benzaldehydes 371a–c and acetophenones 371d–f by H2O2/H3BO3 in THF.
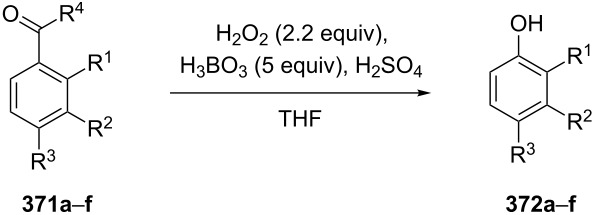 | |||
| Compound | Carbonyl compound | Reaction time, h | Yield, % 372a–f |
| 371a | R1 = R2 = R3 = R4 = H | 12 | 74 |
| 371b | R1 = OH, R2 = R3 = R4 = H | 7 | 80 |
| 371c | R1 = R2 = R4 = H, R3 = OH | 24 | 90 |
| 371d | R1 = OH, R2 = R3 = H, R4 = Me | 36 | 90 |
| 371e | R1 = R2 = R3 = H, R4 = Me | 24 | 63 |
| 371f | R1 = R2 = H, R3 = NO2, R4 = Me | 48 | 60 |
Presumably, the coordination of the H2O2–aldehyde adduct 373 by the highly polarized boric acid is responsible for the increased yields of phenols 372. The adduct 373 easily eliminates a borate ion with concerted migration of the aryl group giving phenols 372. The migrating rate of the aryl group is higher in comparison with hydride migration and formation of 374 (Scheme 109).
Scheme 109.

Possible transformation paths of the highly polarized boric acid coordinated H2O2–aldehyde adduct 373.
1.6. Elbs persulfate oxidation of phenols
The Elbs oxidation is the oxidation of phenols 375 with potassium persulfate in the presence of alkali hydroxides to form p-hydroquinones 376 (Scheme 110) [400–401].
Scheme 110.

The Elbs oxidation of phenols 375 to hydroquinones.
The Elbs oxidation is a multistep process, which commences by the formation of the phenolate anion 377. This is followed by the nucleophilic substitution of peroxide oxygen in the peroxydisulfate ion 378 [402] and the resulting sulfoxy group positioned in the para position (compound 379) is hydrolyzed the with formation of p-hydroquinone 376 (Scheme 111).
Scheme 111.

The mechanism of the Elbs persulfate oxidation of phenols 375 affording p-hydroquinones 376.
The oxidation of phenols containing electron-donating substituents to dihydroxybenzenes gives products in higher yields compared with phenols containing electron-withdrawing substituents (Table 17) [403–405].
Table 17.
Oxidation of phenols 375a–f with potassium persulfate in the presence of alkali.
| Phenol | Product | Yield, % | Phenol | Product | Yield, % |
 375a |
 376a |
47 |
 375d |
 376d |
69 |
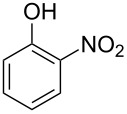 375b |
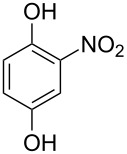 376b |
35 |
 375e |
 376e |
42 |
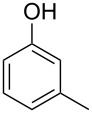 375c |
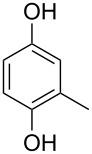 376c |
66 |
 375f |
 376f |
49 |
The main drawback of the persulfate-mediated Elbs oxidation of phenols, are the normally observed moderate conversions and yields. Remarkably, under the above Elbs oxidation conditions 5-hydroxy-2-pyridones 381 were prepared from pyridines 380 with good yields (Scheme 112) [406].
Scheme 112.

Oxidation of 2-pyridones 380 under Elbs persulfate oxidation conditions.
Later, the synthesis of 3-hydroxy-4-pyridone (384) via the Elbs oxidation of 4-pyridone (382) and isolation of 4-pyridone-3-sulfate (383) was described (Scheme 113) [407]. The synthesis of 5-hydroxy-6-bromo-2-pyridone was described under similar conditions [408].
Scheme 113.

Synthesis of 3-hydroxy-4-pyridone (384) via an Elbs oxidation of 4-pyridone (382).
1.7. Schenck and Smith rearrangements
In 1958, Schenck observed that the storage of 5α-hydroperoxide 385 in chloroform for 3 days results in the shift of the OOH group from the 5α to 7α position and a double-bond migration with formation of 386. This reaction is nowadays known as the Schenck rearrangement (Scheme 114) [409–411].
Scheme 114.

The Schenck rearrangement.
In 1973, Smith discovered another type of rearrangement of allylic hydroperoxides [412]. The 7α-hydroperoxide 386 underwent a 20–30% isomerization to the 7β-hydroperoxide 387 if a solution of 386 in ethyl acetate was kept at 40 °C for 48 h (Scheme 115). This process is called the Smith rearrangement.
Scheme 115.

The Smith rearrangement.
The mechanisms of these, at first glance simple, reactions were systematically investigated 40 years after their discovery.
Three main pathways for the Schenck rearrangement have been proposed (Scheme 116). Path A involves the cyclization resulting in the formation of a carbon-centered radical. Path B comprises the formation of a transition state with the electron density distributed over a cyclic system. Path C proceeds through a dissociation to form an allylic radical and triplet oxygen (Scheme 116) [186,413].
Scheme 116.

Three main pathways of the Schenck rearrangement.
Path A, the initially considered most favorable pathway, was excluded because the isomerization of hydroperoxides 388 and 389 following this route would lead to a β-scission ring opening of 390 (Scheme 117).
Scheme 117.

The isomerization of hydroperoxides 388 and 389.
However, this process was not observed and none of the possible carbon-centered radicals 390 was trapped by molecular oxygen [414]. Meanwhile, it is known that the dioxacyclopentyl radical 392 formed from 391 is trapped by oxygen to form hydroperoxide 393 (Scheme 118) [415].
Scheme 118.

Trapping of dioxacyclopentyl radical 392 by oxygen.
It was hypothesized that the Schenck rearrangement of peroxide 394 proceeds through a cyclic structure 395 according to the pathway shown in Scheme 119 [414].
Scheme 119.

The hypothetical mechanism of the Schenck rearrangement of peroxide 394.
However, this hypothesis was also rejected because the ESR spectra recorded after the photolysis of 5α- and 7α-hydroperoxides 385 and 386 showed that the tertiary allylperoxyl radical and secondary allylperoxyl radical are separate and distinct species, and that they do not have the common cyclic structure 395 [416].
In a study using labeled isotope 18O2 it was found that the two hydroperoxides 398 and 399 derived from autoxidation of oleic acid (397) underwent the Schenck rearrangement without incorporating dioxygen from the atmosphere (Scheme 120) [417–418]. Later on, Beckwith and Davies confirmed this fact for cholesterol hydroperoxide [416] and the hydroperoxide generated from valencene [419].
Scheme 120.

The autoxidation of oleic acid (397) with the use of labeled isotope 18O2.
Based on these results, no formation of triplet oxygen occurs in the reaction, thus excluding path C in Scheme 116. Instead, a cyclic transition state (path B, Scheme 116) became more likely, which was confirmed by the stereoselective rearrangement of optically pure olefinic hydroperoxides [420].
However, the study on the rearrangement of hydroperoxides 398, 399 obtained from oleic acid (397) using stereochemical, oxygen-isotopic labeling and solvent viscosity analyses demonstrated that, in hexane, a small amount of atmospheric oxygen is incorporated into the product. The replacement of the solvent by more viscous dodecane and then by octadecane led to a decreased content of atmospheric oxygen in the final product [421–422]. These results provided evidence that the Schenck rearrangement proceeds also through path C in Scheme 116.
Besides, path C was also confirmed by the rearrangement of 18O-labeled hydroperoxide 400 under an atmosphere of 16O2 with formation of isotopomers 401–403 (Scheme 121) [423].
Scheme 121.

The rearrangement of 18O-labeled hydroperoxide 400 under an atmosphere of 16O2.
Examples of the Schenck rearrangement are given in Table 18.
Table 18.
Examples of the Schenck rearrangement.
| Entry | Allylic isomer A | Allylic isomer B | Ref. |
| 1 |
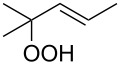 404a |
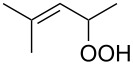 404b |
[424] |
| At 40 °C in non-polar solvents, an approximately equimolar mixture of A and B is formed | |||
| 2 |
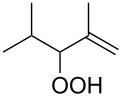 405a |
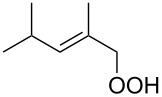 405b |
[414] |
| In hexane, A is rearranged to an equilibrium mixture of ~80% A and ~20% B | |||
| 3 |
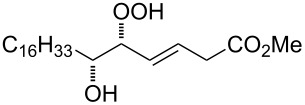 406a |
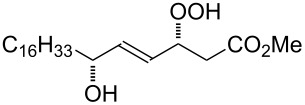 406b |
[425] |
| At 60–70 °C in C6H6 or MeCN in the presence of TBHN or AIBN within 16–22 h, a 50:50 A:B mixture is formed | |||
| 4 |
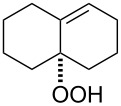 407a |
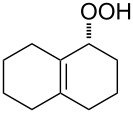 407b |
[426] |
| In CCl4 at 40 °C for 141 h, the rearrangement proceeds by 80% | |||
| 5 |
 408a |
 408b |
[427] |
| In CDCl3, the rearrangement of A into B is completed in 24 h | |||
| 6 |
 409a |
 409b |
[428] |
| In CDCl3, the rearrangement is completed in 72 h | |||
| 7 |
 410a |
 410b |
[429] |
| In C6H6 in presence of 10 equiv TBHP and 20 mol % DTBN at 40 °C for 16 h, isomers A and B are formed in equal amounts | |||
| 8 |
 411a |
 411b |
[430] |
| In CDCl3 the rearrangement is completed in 48 h | |||
| 9 |
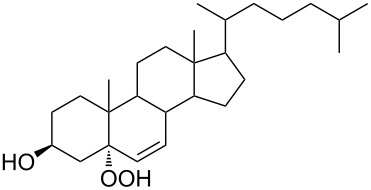 412a |
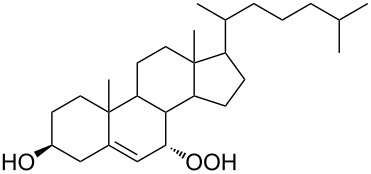 412b |
[431] |
| In CHCl3 for 5 d at room temperature, only partial conversion | |||
| 10 |
 413a |
 413b |
[432] |
| In CDCl3 the rearrangement is completed after 3–4 weeks; R: CO2H, CO2Me, CH2OH, CH3 | |||
| 11 |
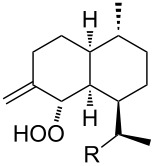 414a |
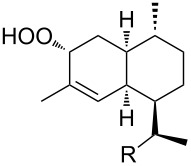 414b |
|
| In CDCl3 the rearrangement is completed after 2–4 weeks; R: CO2H, CO2Me, CH2OH, CH3 | |||
| 12 |
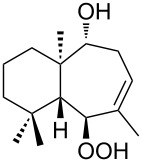 415a |
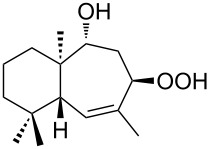 415b |
[433] |
| In CDCl3 the rearrangement is completed after 2 d | |||
| 13 |
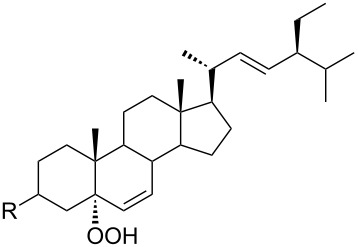 416a |
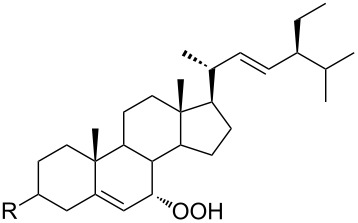 416b |
[434] |
| In pyridine for 24 h, R: OH,CH3COO, F, Cl, conversion 12–58% | |||
| 14 |
 417a |
 417b |
[435] |
| In a 5 M solution of LiClO4 in Et2O the rearrangement is completed in 24 h | |||
| 15 |
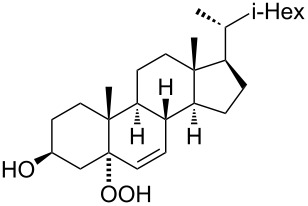 418a |
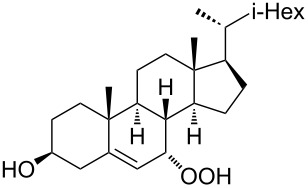 418b |
[436] |
| In CDCl3/D2O, lyophilized PBS buffer at pH 7 for 20 h, the conversion is 14% | |||
| 16 |
 419a |
 419b |
[136] |
| In CH2Cl2 at −78 °C with BF3·OEt2 (1 mol %) | |||
| 17 |
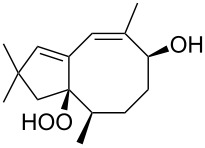 420a |
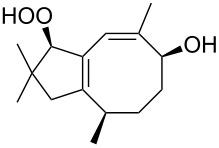 420b |
[437] |
| In MeCN/H2O, only partial conversion. | |||
The Schenck rearrangement takes also place with allylic hydroperoxides derived from lipids. The rearrangement of the oleate-derived allylic hydroperoxides (S)-421, and (R)-425 involved free radicals includes the oxygen-centered radicals 422, 423, 426, 427. The E-oleate hydroperoxide (S)-421 transforms into the corresponding (R)-E-product 424 at room temperature with a high (S) → (R) stereoselectivity of more than 97%. A decreased selectivity (~90%) was observed for product 428 obtained from the Z-hydroperoxide (R)-425. In this case, the configurational direction of the reaction was (R) → (R) (Scheme 122) [438].
Scheme 122.

The rearrangement of the oleate-derived allylic hydroperoxides (S)-421 and (R)-425.
The Smith rearrangement is a free-radical chain reaction in which atmospheric oxygen may play a greater role than in the Schenck rearrangement. Apparently, the Smith rearrangement proceeds through a dissociation to the allylic radical and 3O2. Presumably, the distance between these active species is large enough to allow an exchange with atmospheric oxygen (Scheme 123). The Schenck and Smith rearrangements are both a consequence of the reversibility of the reaction of allyl radicals with triplet dioxygen and differ mechanistically in the degree of separation of these two components [186]. There are only a few examples of the Smith rearrangement known and some of them are collected in Table 19.
Scheme 123.

Mechanisms of Schenck and Smith rearrangements.
Table 19.
Examples of the Smith rearrangement.
| Entry | Allylic isomer B | Allylic isomer C | Comments | Ref. |
| 1 |
 429a |
 429b |
In CDCl3 within 259 h, approximately 5% of B was transformed into C | [427] |
| 2 |
 430a |
 430b |
In CHCl3 at room temperature within 150 h, the B:C ratio reached 1.8:1 | [439] |
| 3 |
 431a |
 431b |
In CDCl3 at 40 °C within 3.5 h, B is transformed into C by 20%. In EtOAc at 40 °C, the yield of C was 25–30% | [416] |
| In CHCl3 after 5 d at room temperature, only partial conversion | [431] | |||
| 4 |
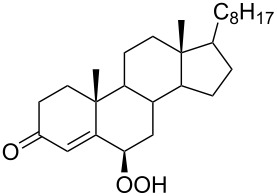 432a |
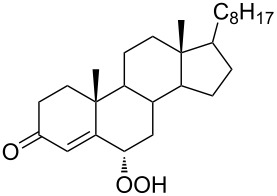 432b |
In CDCl3, the B:C ratio reached 1:1.5 | [439] |
In diene or triene-containing systems (433), both the rearrangement and cyclization of allylic peroxyl radicals can take place with formation of 434–436 (Scheme 124) [440].
Scheme 124.

The rearrangement and cyclization of 433.
1.8. Wieland rearrangement
In 1911 Wieland performed the decomposition of bis(triphenylmethyl)peroxide (437) under an atmosphere of CO2 in boiling xylene for 10 min and obtained the crystalline product 438 in 70% yield (Scheme 125) [441].
Scheme 125.

The Wieland rearrangement.
The mechanism of the Wieland rearrangement involves the following three steps: Initial formation of O-centered radical A, the rearrangement of radical A into diphenylphenoxymethyl radical C, and the dimerization of radical C [442–443].
Radical 1,2-aryl migrations from silicon or germanium to oxygen is similar to the Wieland rearrangement. The thermal decomposition of either bis(triphenylsilyl) 439 or bis(triphenylgermyl) 441 peroxides leads to the rearranged products 440, 442 in high yields (Scheme 126) [444–445].
Scheme 126.

The rearrangement of bis(triphenylsilyl) 439 or bis(triphenylgermyl) 441 peroxides.
2. Unnamed rearrangements of organic peroxides and related processes
2.1. Protic acid-catalyzed rearrangements of organic peroxides and related processes
The oxidative transformation of cyclic ketones 58d and 443a–d in the reaction with hydrogen peroxide in alcohols in the presence of sulfuric acid proceeds through the formation of geminal dihydroperoxides 444a–e. The latter compounds are oxidized to dicarboxylic acids 445a–e followed by their transformation into the corresponding dicarboxylates 446a–e, rather than formation of lactones via the Baeyer–Villiger reaction (Scheme 127) [446].
Scheme 127.

The oxidative transformation of cyclic ketones.
This transformation requires the following key conditions to proceed: a reaction temperature higher than 80 °C, the H2SO4 concentration in the range of 0.2–1.0 mol/L, and a molar ratio of hydrogen peroxide/ketone in the range of 5–10. The corresponding dibutyl esters were prepared in 53−70% yields by oxidation in butanol, which keeps the temperature in the range of 98−106 °C (Table 20).
Table 20.
Examples of oxidation of ketones 58d, 443a–d in butanol to diesters 446a–e.
| Ketone | Diester | Yield of diester, % | |
| aqueous H2O2 solution | ethereal solution of H2O2 | ||
 443a |
 446a |
59 | 64 |
 443b |
 446b |
57 | 63 |
 58d |
 446c |
62 | 67 |
 443c |
 446d |
61 | 65 |
 443d |
 446e |
64 | 70 |
In a study on the hydroxylation of compounds containing a double bond to the corresponding α-glycols, the tungstic acid-catalyzed reaction of cyclohexene (447) with 90% hydrogen peroxide in methanol, ethanol, or isopropanol afforded the corresponding 2-alkoxycyclohexanols 448a–c in 70, 41, and 21% yields, respectively, as well as the trans-1,2-cyclohexanediols 449a–d (Scheme 128) [447].
Scheme 128.

The hydroxylation of cyclohexene (447) in the presence of tungstic acid.
A detailed study on the hydroxylation of cyclohexene (447) in tert-butanol using 30% hydrogen peroxide showed that in this reaction the formation and rearrangements of 2-hydroperoxyalkanols 451 is involved. The treatment of 2-hydroperoxycyclohexanol (451) with acetone afforded the cyclic peroxide 452. The acid-catalyzed rearrangement of the peroxide 452 gave dialdehyde 453, which further transformed into aldehyde 454. The isolation and characterization of the latter compound was crucial to an understanding of the oxidation of olefins to aldehydes under the action of hydrogen peroxide (Scheme 129).
Scheme 129.

The oxidation of cyclohexene (447) under the action of hydrogen peroxide.
The study of the reactions of various unsaturated molecules with hydrogen peroxide demonstrated that the reaction of butenylacetylacetone 455 with H2O2 at pH 5–6 at 38–40 °C produces 2-methyl-3-hexenoic acid (457). Other possible products 456 resulting from a double-bond oxidation reaction were not observed. Apparently, the formation of carbanion A is the driving force of this reaction. Carbanion A transforms into the symmetrical dihydroxyperoxide B, which subsequently rearranges through a deacetoxylation to finally afford 2-methyl-3-hexenoic acid (457) (Scheme 130) [448].
Scheme 130.

The reaction of butenylacetylacetone 455 with hydrogen peroxide.
The oxidation of bridged 1,2,4,5-tetraoxanes 458 upon heating in an acidic medium in the presence of H2O2 is leading to esters 459 (Scheme 131) [449].
Scheme 131.

The oxidation of bridged 1,2,4,5-tetraoxanes.
It is assumed that the reaction of tetraoxanes 458a–f proceeds as an acid-catalyzed oxidative transformation, similar to the Baeyer–Villiger and Hock rearrangements, to yield intermediate A. This is further transformed into esters 459a–f through the oxidation of the CH group and esterification (Scheme 132).
Scheme 132.

The proposed mechanism for the oxidation of bridged 1,2,4,5-tetraoxanes.
In another study [450], the rearrangement of isomeric ozonides was described. Here, the ozonides 460a,b were interconverted and rearranged into the tricyclic monoperoxide 461 under the action of phosphomolybdic acid (PMA). This result is attributable to the protic acid nature of PMA as well as its ability to form peroxo compounds containing M–O–O groups that influence the direction of the reaction (Scheme 133).
Scheme 133.

The rearrangement of ozonides.
The observed interconversion of ozonides may be useful for the interpretation of the data on the ozonolysis of unsymmetrical unsaturated compounds.
Carboxylic acids 464 were prepared through a camphorsulfonic acid-catalyzed oxidative rearrangement of a 1,2-dioxolane intermediate 463 prepared from malondialdehydes 462 and H2O2 (Scheme 134) [451].
Scheme 134.

The acid-catalyzed oxidative rearrangement of malondialdehydes 462 under the action of H2O2.
2.2. Lewis acid-catalyzed cleavage of peroxides
The Lewis acid-catalyzed cleavage of peroxides follows mainly two pathways: the O–O-bond heterolysis to form an oxycarbenium ion 467 accompanied by the migration of the adjacent substituent, and the acid-catalyzed ionization of the C–O bond to yield carbenium ion 468. The reaction pathway is mainly determined by the nature of the starting compound and the C–O ionization pathway is promoted by the stabilization of the final carbocation, whereas the O–O-bond heterolysis is facilitated by a high migratory ability of the adjacent groups. The fragmentation of dialkyl peroxides 465 and ozonides 466 mainly depends on the nature of the applied Lewis acid. In this way, SnCl4 and BF3·Et2O facilitate the O–O-bond heterolysis (A), whereas TiCl4 promotes the C–O ionization (SN1 mechanism) in tertiary peroxides (B). The formation of ketones 469, 471 and ester 470 is the result of the Lewis acid-catalyzed decomposition of ozonides through the ionization of peroxide, ionization of alkoxide, or oxygen–oxygen heterolysis (C) (Scheme 135) [452].
Scheme 135.

Pathways of the Lewis acid-catalyzed cleavage of dialkyl peroxides 465 and ozonides 466.
The TiCl4-promoted rearrangement of (tert-butyldioxy)cyclohexanedienones 472a–d, which are generated by the ruthenium-catalyzed oxidation of phenols with tert-butyl hydroperoxide, provides an efficient route to 2-substituted quinones 473a–d (Table 21) [453–454]. The mechanism of this transformation is depicted in Scheme 136.
Table 21.
TiCl4-promoted rearrangement of (tert-butyldioxy)cyclohexanedienones 472a–d.
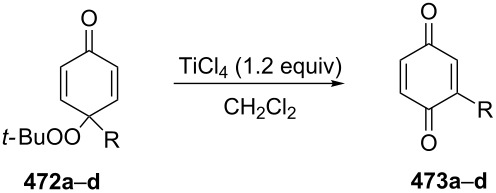 | |||
| Peroxide | Reaction conditions | Quinone | Yield, % |
 472a |
25 °C, 1 h |
 473a |
92 |
 472b |
−15 °C, 4 h |
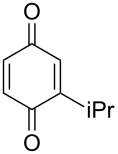 473b |
98 |
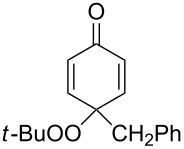 472c |
−78 °C, 0.5 h |
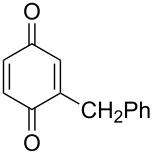 473c |
93 |
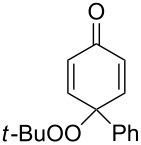 472d |
−78 °C, 0.5 h |
 473d |
91 |
Scheme 136.

The mechanism of the transformation of (tert-butyldioxy)cyclohexanedienones 472.
In the first step, the coordination of dienone 472 to the Lewis acid gives rise to cation 474. The second step involves a 1,2-alkyl migration to form cation 475. The subsequent deprotonation of the latter affords aromatic intermediate 476. In the final step, trichloro-tert-butoxytitanium is eliminated from intermediate 476 to produce 2-alkylquinones 473.
The transformation of 4-methyl-4-tert-butyldioxycyclohexadienone 472a into 2-methylbenzoquinone (473a) can be used also for the regioselective synthesis of vitamin K3 477 (Scheme 137) [455–456].
Scheme 137.

The synthesis of Vitamin K3 from 472a.
The use of SnCl4 or TMSOTf as the catalyst made it possible to prepare trimethylsilyl-substituted cyclic peroxides 479a–d and 480a,b in a cis configuration starting from allyltrimethylsilane and bicyclic [2.2.n]endoperoxides 478a–d (Table 22) [457].
Table 22.
Conditions of the synthesis of trimethylsilyl-substituted cyclic peroxides (1,2-dioxanes) 479a–d and 480a,b.
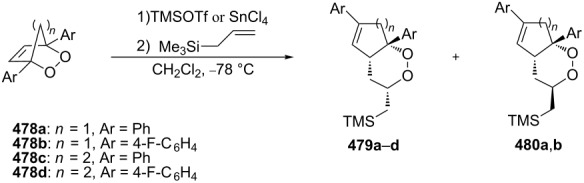 | ||||
| Endoperoxide | Equivalents of TMSOTf (or SnCl4) | Reaction time, min | Product/ratio of diastereomers | Total yield, % |
| 478a | 0.033 | 15 | 479a/480a, 1:0 | 54 |
| 478a | 1.0 SnCl4 | 30 | 479a/480a, 1:1 | 53 |
| 478b | 1.1 | 15 | 479b/480b, 1:0.8 | 60 |
| 478c | 1.1 | 40 | 479c, 1:0 | 10 |
| 478d | 1.1 | 40 | 479d, 1:0 | 48 |
The mechanism of this reaction implies that TMSOTf or SnCl4 promote the heterolytic cleavage of the C–O bond in 478d to form carbocation 481d, which is then attacked by allyltrimethylsilane through a chair-like transition state 482d. The subsequent cyclization of 482d through the stabilized carbocation 483d affords silyl-substituted peroxide, 1,2-dioxane 479d, containing the substituent (–CH2SiMe3) in the equatorial position (Scheme 138).
Scheme 138.

Proposed mechanism for the transformation of 478d into silylated endoperoxide 479d.
The employment of BF3·Et2O as the catalyst for the rearrangement of hydroperoxide 485, which is generated by the oxidation of steroid 484, enables the opening of the D ring between C-14 and C-16 to form diketone 486 (Scheme 139) [458].
Scheme 139.

The rearrangement of hydroperoxide 485 to form diketone 486.
2.3. Rearrangements and related processes of organic peroxides in the presence of bases
The base-catalyzed rearrangement of cyclic peroxides 488a–g, which are prepared by the manganese-catalyzed oxidation of 1- and 1,2-disubstituted cyclopropanols 487a–g, provides a convenient approach to the synthesis of aliphatic and arylaliphatic α,β-epoxy ketones 489a–g. The latter compounds are attractive substrates for the synthesis of for example natural compounds (Scheme 140) [459].
Scheme 140.

The base-catalyzed rearrangement of cyclic peroxides 488a–g.
Peroxy hemiketals 491 are the starting reagents in the synthesis of epoxides 492 and aldols 493. Scheme 141 shows the synthesis of epoxides and aldols from inexpensive and readily available α,β-unsubstituted ketones 490 through the intermediate formation of peroxy hemiketal 491 in the presence of a chiral catalyst [460].
Scheme 141.

Synthesis of chiral epoxides and aldols from peroxy hemiketals 491.
A 1:1 mixture of the diastereomeric hydroperoxides 495a–e was synthesized by ozonolysis of (R)-carvone (494) and in situ trapping with primary alcohols ROH (R = Me, Et, Bu, Pent, Oct). Further cyclization of these hydroperoxides 495a–e using the sodium methanolate/MeOH system results in endoperoxides 496a–e exhibiting antimalarial activity (Scheme 142) [461].
Scheme 142.

The multistep transformation of (R)-carvone (494) to endoperoxides 496a–e.
The intramolecular rearrangement of 1,2-dioxetanes 497 containing an aromatic electron-donating substituent is accompanied by emission of light. This process is of special interest for the application in clinical and biological analytical methods, and the synthesis of carbonyl-containing compounds 498 (Table 23) [462–475].
Table 23.
Base-catalyzed intramolecular rearrangement of 1,2-dioxetanes.
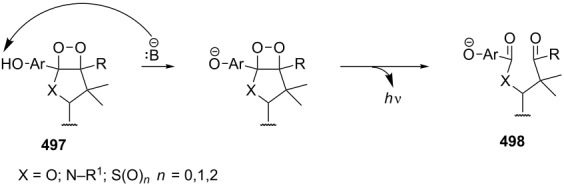 | |||||
| Entry | X | R | Ar-OH | Reaction conditions | Ref. |
| 1 | O | t-Bu |
 R1, R2 = H, OMe, CO2Me, CO2H, CH2OH |
TBAF in DMSO at 25 °C for 1 h | [463] |
| 2 | O | t-Bu |
 R = H, OMe; Y = O, S |
NaOH in СH3CN/H2O at 45 °C | [464] |
| 3 | O | t-Bu |  |
TBAF in DMSO (NMP or DMF) at 45–100 °C | [465] |
| 4 | O | Me, Et, iPr, iBu | in NMP at 50–100 °C or in TBAF/NMP at 35–60 °C | [466] | |
| 5 | O | t-Bu |  |
TBAF in CH3CN at 45 °C | [467] |
| 6 | NBoc | t-Bu | 3-OH-C6H4 3-OMe-C6H4 6-OH-C10H6 |
TBAF in DMSO at 25 °C | [468–469,473] |
| 7 | S, SO, S(O)2 | t-Bu | 3-OH-C6H4 3-OMe-C6H4 3-OAc-C6H4 |
TBAF in DMSO at 25 °C | [472] |
| 8 | O | t-Bu | HO-phenanthrenyl | TBAF in CH3CN at 45 °C | [474] |
| 9 | O | t-Bu |
 R = H, Me, Ph |
TBAF in CH3CN or NaOH in H2O at 45 °C | [475] |
| 10 | 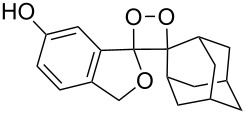 |
TBAF in DMSO at 25 °C | [470] | ||
| 11 |  |
TBAF in THF/DMSO (1:1) at 25 °C | [476] | ||
| 12 |  |
DBU in CH3CN at 25 °C | [477] | ||
| 13 |  |
TBAF in DMSO/PBS buffer | [471] | ||
Catalytic amounts of a sodium bicarbonate are sufficient to induce the decomposition of anthracene endoperoxide 499 to anthraquinone (500) (Scheme 143) [478].
Scheme 143.

The decomposition of anthracene endoperoxide 499.
An intramolecular rearrangement of α-azidoperoxides 502 promoted by DBU provides esters 503. The reaction takes place with alkyl, aryl and heteroaryl α-azidoperoxides generated from the corresponding aldehydes 501 (Scheme 144) [479].
Scheme 144.

Synthesis of esters 503 from aldehydes 501 via rearrangement of peroxides 502.
There could be two possible paths for base-promoted decomposition of α-azidoperoxides 502 (Scheme 145). The abstraction of the α-hydrogen in the azidoperoxide leads to the direct decomposition of the peroxide bond, which provides acylazide 504 and alkoxide ion 505 (path A). Further, the exchange of the azide moiety in the acylazide with an alkoxide ion generates esters 503. On the other hand, an abstraction of the α-hydrogen in the azidoperoxide leads to a resonance-stabilized intermediate I (path B). Then, an intramolecular 1,2-alkoxy migration of I, via scission of the peroxide bond, followed by cleavage of the C–N bond (intermediate IV) affords the desired ester 503. On basis of control experiments, the reaction is probably following the latter path.
Scheme 145.

Two possible paths for the base-promoted decomposition of α-azidoperoxides 502.
2.4. Thermal and photochemical transformations of organic peroxides
Story and co-workers discovered that the thermal and photochemical decomposition of cyclic ketone peroxides 506 produces cycloalkanes 507 and cyclic lactones 508 (Scheme 146 and Scheme 147) [480–483]. This transformation is a general method for the synthesis of macrocyclic compounds from readily available starting materials.
Scheme 146.

The Story decomposition of cyclic diperoxide 506a.
Scheme 147.

The Story decomposition of cyclic triperoxide 506b.
Examples of the thermal decomposition and photolysis of diperoxide 506a and triperoxide 506b are given in Table 24.
Table 24.
Products of thermal and photochemical rearrangement of diperoxide 506a and triperoxide 506b.
| Peroxide | Conditions | Yields, % | ||
| Cycloalkane (507) |
Macrolactone (508) |
Ketone (45b) |
||
| 506a | 150 °C, 30 min | 44 | 23 | 21 |
| hv, MeOH, 3 h | 14 | 10 | 20 | |
| 506b | 150 °C, 30 min | 16 | <1 | 15 |
| hv, MeOH, 3 h | 15 | 25 | 20 | |
Unsaturated endoperoxides are convenient starting compounds for thermal and photochemical rearrangements. The thermal rearrangement of endoperoxides A into diepoxides B (Scheme 148) is one of the commonly used transformations [353,484–485].
Scheme 148.
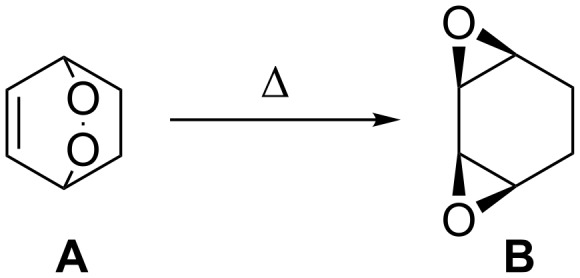
The thermal rearrangement of endoperoxides A into diepoxides B.
The transformation of peroxide 510 is a key step in the synthesis of the cytotoxic agent stemolide (511) from methyl dehydroabietate (509) (Scheme 149) [486].
Scheme 149.

The transformation of peroxide 510 in the synthesis of stemolide (511).
It was shown that thermal and photochemical transformations of endoperoxides 261g, 263, and 512a–с afford, in addition to diepoxides 513a–e, keto epoxides 514a–e [487–489]. Examples of the thermal decomposition and photolysis of endoperoxides 261g, 263, and 512a–c are given in Table 25.
Table 25.
Products of thermal decomposition and photolysis of endoperoxides 261g, 263, 512a-c.
| Endoperoxide | Diepoxide, 513 | Keto epoxides, 514 | Ratio of products, 513:514 |
 261g |
 a |
 a |
Δ 36:65 hν 28:72 |
 263 |
 b |
 b |
Δ 90:10 hν 33:67 |
 512a |
 c |
 c |
Δ – hν 35:65 |
 512b |
 d |
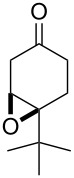 d |
Δ 65:35 hν 53:37 |
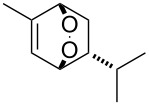 512c |
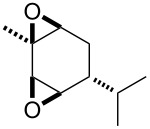 e |
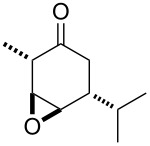 e |
Δ 58:42 hν 24:76 |
The possible mechanism of the rearrangement of endoperoxide 261g is shown in Scheme 150. It is supposed that diepoxide 513a and keto epoxide 514a are generated from diradical 516 via cyclization of the diradical or a 1,2-hydride shift, respectively. Since 1,4-cyclohexanedione is not generated from endoperoxide, it can be concluded that the first cyclization of 515 to 1,3-biradical 516 occurs rapidly and the formation of epoxide rings takes place successively rather than simultaneously.
Scheme 150.

The possible mechanism of the rearrangement of endoperoxide 261g.
The photooxidation of indene 517 without a sensitizer provides dioxetane 518, in the presence of Rose Bengal, the diepoxyendoperoxide 521 is obtained. Product 521 originates probably from a [2 + 4] addition of singlet oxygen to give 519, followed by rearrangement to diepoxydiene 520, which is capable of adding a second mole of oxygen. The use of meso-tetraphenylporphyrin instead of Rose Bengal leads to the formation of diendiperoxide 522 (Scheme 151) [484].
Scheme 151.

The photooxidation of indene 517.
Ascaridole (523) was slowly isomerized into isoascaridole (524) under irradiation with visible light (Scheme 152) [490]. Thermal and photochemical isomerization of related endoperoxides have been applied to the syntheses of other ascaridole analogs [491].
Scheme 152.
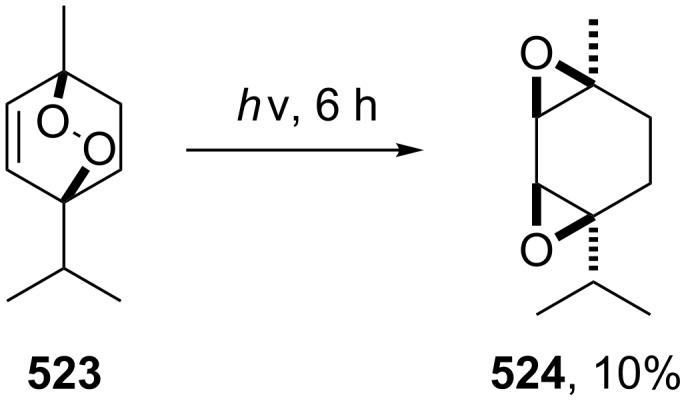
The isomerization of ascaridole (523).
The diepoxide 526 was obtained in 67% yield by photolysis of 525 with a medium-pressure Hg vapor lamp (Scheme 153) [492].
Scheme 153.

The isomerization of peroxide 525.
The thermal transformation of endoperoxides produces mainly bis-epoxides, but can also provide unexpected products such as epoxy ketals. The heating of endoperoxide 236 to 160 °C in toluene affords epoxy ketal 528 in 53% yield through the formation of biradical 527 (Scheme 154) [347].
Scheme 154.

The thermal transformation of endoperoxide 355.
The photooxidation of cyclopentadiene (529) in an alcohol solution in the presence of polymerization inhibitors at a temperature higher than 0 °C gave cis-4,5-epoxy-2-pentenal (531) in 58% yield, cis-1,2,3,4-diepoxycyclopentane (532) as a byproduct (in 7% yield), and polymers instead of the expected peroxide 530 (Scheme 155) [344].
Scheme 155.

The photooxidation of cyclopentadiene (529) at a temperature higher than 0 °C.
The extensive development of methods for the synthesis of cyclopentenones lies in the fact that this structural unit is present in some natural compounds, such as dihydrojasmone, prostaglandins, and rethrolones. The mechanism of thermal decomposition of saturated fulvene endoperoxides 533a–d involves the formation of one of the three intermediates A, B, C, which are precursors to cyclopentenones 534a–d (Table 26) [493].
Table 26.
Synthesis of cyclopentenones 534a–d from saturated fulvene endoperoxides 533a–d.
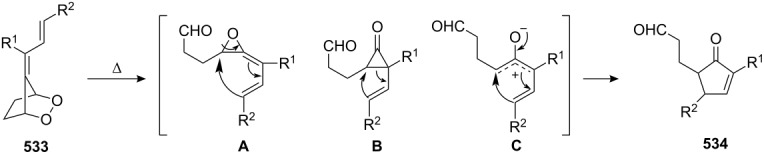 | ||
| Endoperoxide | Product | Yield (trans:cis) |
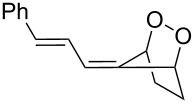 533a |
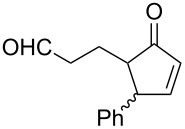 534a |
85% (8:1) |
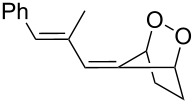 533b |
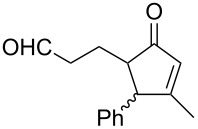 534b |
83% (2:1) |
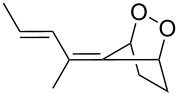 533c |
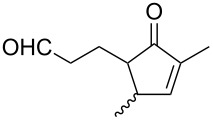 534c |
90% (6:1) |
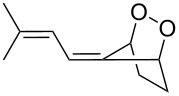 533d |
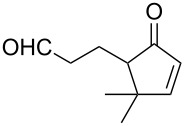 534d |
68% |
The replacement of the vinyl group at the exocyclic double bond in the fulvene precursor by a 3-butenyl group and the thermal decomposition of the resulting endoperoxides 535 at 80 °C lead to a [3,4]-sigmatropic shift of the 3-butenyl group and formation of the 5-oxo-6-heptenal derivatives 536. The mechanism of this process involves the formation of epoxide A, which undergoes a [3,4]-shift through the intermediate B [494] (Table 27).
Table 27.
The mechanism and results of the thermal rearrangement of saturated fulvene endoperoxides 535a–d.
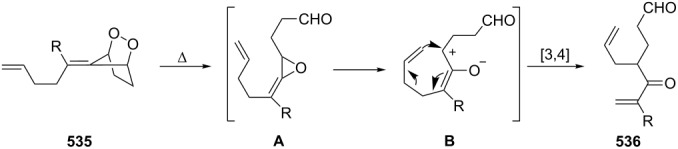 | ||
| Endoperoxide | Product | Yield, % |
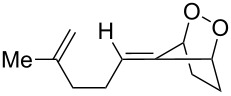 535a |
 536a |
45 |
 535b |
 536b |
83 |
 535c |
 536c |
85 |
 535d |
 536d |
79 |
The thermal rearrangement of endoperoxides 538a,b, which are generated by the photooxidation of furanosyl furans 537a,b, selectively affords glycosides 539a,b (Scheme 156) [495].
Scheme 156.

The thermal rearrangement of endoperoxides 538a,b.
The methylene blue-sensitized photooxidation of arabinofuranosyl furan 537a as an 1:6 α,β-anomeric mixture at −20 °C followed by warming of the reaction mixture to room temperature produced furanoside 539a as an anomeric mixture in the same molar ratio. The photooxidation of pure β-arabinofuranosyl furan 537a produced exclusively β-furanoside 539a. Based on these data, the intermediate endoperoxide 538a originates from the cycloaddition of 1O2 to the furanosyl furan. The selective thermal rearrangement of endoperoxide 538a, which is similar to the Baeyer–Villiger rearrangement with the retention of the configuration, results in the corresponding O-derivatives.
The thermally unstable endoperoxides 541a–d generated from 2-alkoxyfurans 540a–d rearrange through several pathways depending upon the nature of the substituent at the carbon atom C5 in 541 with formation of 542 or 543 (Table 28) [496].
Table 28.
Results of the rearrangement of endoperoxides 541a–d.
 | |||||
| Compound | R1 | R2 | Ar | Yield of 542, % | Yield of 543,% |
| a | Et | Et | Ph | 88 | traces |
| b | Et | Et | 4-Br-C6H4 | 92 | traces |
| c | Ph | Ph | 4-Br-C6H4 | 93 | traces |
| d | Me | H | Ph | 0 | 87 |
The rearrangement of endoperoxides 541a–c containing a substituent with a tertiary hydroxy group in the 5 position results in the formation of hydroperoxyoxetanes 542a–c and trace amounts of Z-ketoesters 543a–c. Under the same conditions, the rearrangement of endoperoxide 541d containing a substituent with a secondary hydroxy group in the 5 position produces exclusively the Z-keto ester 543d [497].
This difference is apparently attributable to the following two factors: (1) the lower nucleophilicity of the secondary hydroxy group compared to the tertiary hydroxy group; (2) the conformer, which would be suitably orientated towards the nucleophilic attack, is sterically unfavored in the case of R2 = H. At −20 °C, the transformation of 541d into a conformational isomer occurs more slowly than the thermal decomposition giving 543d (Scheme 157). Thermal rearrangements of strained cyclic peroxides 544a–d and 546a–e provide a versatile tool for the synthesis of carbonyl compounds 545a–d and 547a–e and heterocyclic systems 548 and 549 (Scheme 158) [498–499].
Scheme 157.

The transformation of peroxides 541.
Scheme 158.

The thermal rearrangements of strained cyclic peroxides.
The thermal rearrangement of diacyl peroxide 551 was carried out in the synthesis of the C4-epi-lomaiviticin B core 553. Diacyl peroxide 551 was prepared from p-nitroperbenzoic acid (p-NPBA) and the acid chloride of carboxylic acid 550. An ionic Criegee-like rearrangement of peroxide 551 upon heating resulted in the corresponding acyl carbonate species. The reaction of MeOH with this acyl carbonate intermediate provided a single diastereomer of secondary carbinol 552 in 38% yield (Scheme 159) [500].
Scheme 159.

The thermal rearrangement of diacyl peroxide 551 in the synthesis of C4-epi-lomaiviticin B core 553.
Two diastereoisomeric dioxindolylalanines 556 were identified after the 1O2 oxidation of tryptophan (554). Mechanistic investigations supported the dioxindolylalanine formation through a dioxetane intermediate 555 (Scheme 160) [501].
Scheme 160.

The 1O2 oxidation of tryptophan (554) and rearrangement of dioxetane intermediate 555.
2.5. Metal-catalyzed transformations of peroxides
This section focuses on transformations of peroxides under the action of the most representative metals used for these types of reactions: Fe(II), Co(II), Ru(II), and Pd(II).
The Fe(II)-promoted activation of peroxides is believed to be involved in the antimalarial activity of a number of peroxides, including the natural product artemisinin. The understanding of the underlying mechanism of the Fe(II)-promoted cleavage of bicyclic peroxides is critical to the design and preparation of more efficient antimalarial peroxides. From this perspective, metal-catalyzed transformations of peroxides are of special interest. It was shown [502] that the reaction of fluorinated cyclic peroxide 557a with FeBr2 in THF proceeds through an intermediate O-centered radical to form epoxy ketone 558a and 1,4-diol 559a. The reaction of 557b with FeCl2(PPh3)2 in CH2Cl2 proceeds in a different manner through an intermediate O-centered radical to yield diepoxide 560b (Scheme 161) [503].
Scheme 161.

The Fe(II)-promoted cleavage of aryl-substituted bicyclic peroxides.
In a related study investigating the reaction of 557а–с with FeBr2, bis-epoxides 560а–с and epoxy ketones 561а–с were obtained as the major products (Table 29) [504] and the proposed mechanism of the rearrangement of 557a–c is presented in Scheme 162.
Table 29.
Transformation of endoperoxides 557а–с under the action of FeBr2.
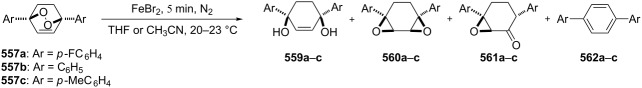 | ||||||
| Substrate | Solvent | Conversion, % | Yield, % | |||
| 559 | 560 | 561 | 562 | |||
| 557a | THF | 100 | <1 | 52 | 26 | <1 |
| 557b | THF | 100 | <1 | 52 | 20 | 3 |
| 557c | THF | 100 | 20 | 0 | 65 | 3 |
| 557a | CH3CN | 95 | <1 | 69 | <1 | 2 |
| 557b | CH3CN | 100 | <1 | 73 | <1 | 2 |
| 557c | CH3CN | 91 | 0 | 50 | 22 | 2 |
Scheme 162.

The proposed mechanism of the Fe(II)-promoted rearrangement of 557a–c.
Both in THF and CH3CN, the intermediate O-centered radical A is generated via an electron transfer from Fe(II) to 557a–c. The transformation of intermediate A can proceed through two different pathways. The first involves the intramolecular addition of an O-centered radical to the double bond in radical A to form C-centered radical B (path A). The second pathway involves an electron transfer from Fe(II) to radical A to give intermediate G (minor path B). The intramolecular electron transfer in intermediate B results in the formation of carbocation C followed by the formation of diepoxide 560a–c and concomitant elimination of Fe(II). The generation of epoxy ketone 561a–c from 560a–c can occur through paths C and D. Paths E and F apparently give rise to 1,4-diol 559a–c and diarylbenzene 562a–c, respectively, from intermediate G.
The reaction of dioxolane 563 with Fe(II) sulfate produces an O-centered radical, and the β-scission of the latter gives a C-centered radical, the oxidation and further cyclization of which yields 564 (Scheme 163) [505].
Scheme 163.

The reaction of dioxolane 563 with Fe(II) sulfate.
The monocyclic 1,2-dioxane 565, as opposed to related dioxolane 563, decomposes under the action of Fe(II) with exclusive formation of a 1:1 mixture of products 566 and 567. This is attributed to the fact that the reaction proceeds through 1,5-hydrogen transfer, while β-scission does not occur (Scheme 164) [505].
Scheme 164.

Fe(II)-promoted rearrangement of 1,2-dioxane 565.
The reaction of Fe(II) cysteinate with dioxolane 568 produced compounds 569 and 570, which were isolated from the reaction mixture. The formation of methyl acetate 571 was confirmed by GC analysis of the reaction mixture before work-up (Scheme 165) [506].
Scheme 165.

Fe(II) cysteinate-promoted rearrangement of 1,2-dioxolane 568.
The reaction of 1,2-dioxanes 572a–c with FeCl2 is accompanied by the formation of lactones 573a,b, which were isolated in the individual state (Scheme 166) [507].
Scheme 166.

The transformation of 1,2-dioxanes 572a–c under the action of FeCl2.
The reaction of synthetic tetraoxane 574 with Fe(II) cysteinate affords a complex mixture of products. Only one product, 575, could be isolated from the mixture and identified. This was the first work, where the Fe(II)-promoted cleavage of 1,2,4,5-tetraoxane was investigated (Scheme 167) [508].
Scheme 167.

Fe(II) cysteinate-promoted transformation of tetraoxane 574.
The hypothesis that this difference in the structure of the reaction products is associated with the rearrangement of intermediate endoperoxides gave impetus to research on the reaction of endoperoxides with transition metal derivatives. It was found that the catalytic rearrangement of endoperoxides using cobalt meso-tetraphenylporphyrin occurs in high yield. Therefore, this is an efficient approach to the synthesis of syn-1,2:3,4-diepoxides from 1,3-dienes under mild conditions. Table 30 summarizes the results of the cobalt(II) tetraphenylporpyrin-catalyzed rearrangement of endoperoxides 576 [509], 578 [510], 580 [511], 582 [512], 584 [353], 586 [513], 588 [514], 590, 592 [515], 594 [516], 596 [517], and 598 [518] which afforded products structurally similar to the diepoxides prepared by thermal rearrangement of endoperoxides (Table 25). All rearrangements were stereospecific and yielded only the syn-diepoxides.
Table 30.
CoTPP-catalyzed rearrangement of endoperoxides.
| Substrate | Product | Yield, % |
 576 |
 577 |
50 |
 578 |
 579 |
84 |
 580 |
 581 |
80 |
 582 |
 583 or 
|
75 |
 584 |
 585 |
45 |
 586 |
 587 |
61 |
 588 |
 589 |
96 |
 R = COOCH3 590 |
 591 |
83 |
 R = COOCH3 592 |
 593 |
55 |
 594 |
 595 |
90 |
 596 |
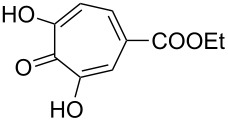 597 |
60 |
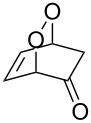 598 |
 599 |
50 |
The study of the CoTPP-catalyzed transformation of bicyclic endoperoxides containing non-strained diene moieties demonstrated that the formation of epoxides can be accomplished in yields up to 90–100%, while the side reaction giving epoxy ketones is suppressed. A detailed study on the CoTPP-catalyzed reaction of 600a showed that this reaction affords, in addition to the expected diepoxide 601a, two isomeric epoxy aldehydes 602a and 603a. The reaction of bicyclic endoperoxides 600b,c gives, instead of the expected epoxides 601b,c, exclusively epoxy aldehydes 602b, 603b and the reaction of endoperoxide 600d produces solely the diepoxide 601d (Scheme 168) [519].
Scheme 168.

The CoTPP-catalyzed transformation of bicyclic endoperoxides 600a–d.
The reaction of epoxy-1,2-dioxanes 604a–d and 606 with Co(II) complexes affords 4-hydroxy-2,3-epoxy ketones 605a–d and 607 in good yields (Scheme 169) [364]. Possibly the selectivity towards the hydroxyketones formation is provided by means of cobalt ions interaction. The obtained compounds are useful synthons in organic synthesis.
Scheme 169.

The CoTPP-catalyzed transformation of epoxy-1,2-dioxanes.
The Ru(II)-catalyzed reactions of 1,4-endoperoxide 261g involves the formation of intermediate radicals 608a,b, the structures of which differ from that of the radicals generated by photolysis or thermal decomposition. Ruthenium ions have a considerable effect on the stability and reactivity of radicals, resulting in the selective transformation of peroxides under mild conditions. The reactivity also substantially depends on steric factors (Scheme 170) [503,520–521].
Scheme 170.

The Ru(II)-catalyzed reactions of 1,4-endoperoxide 261g.
The Ru(II)-catalyzed transformation of 1,4-endoperoxide 609 is used as a key step in the synthesis of the natural compound, elyiapyrone A (610) (Scheme 171) [522–523].
Scheme 171.

The Ru(II)-catalyzed transformation as a key step in the synthesis of elyiapyrone A (610) from 1,4-endoperoxide.
Transformations of endoperoxides catalyzed by variable-valence metals are well studied for metals such as Cu(II), Fe(II), or Co(II), which can initiate the reaction through a one-electron oxidation–reduction mechanism. The decomposition of endoperoxides catalyzed by Ru(II)phosphine complexes also belongs to this type of reaction and the decomposition produces diepoxides as the major products.
The reactions of endoperoxides with Pd(0) proceed through different pathways. Thus, bicyclic 2,3-saturated 1,4-endoperoxides 611a–d are transformed into the corresponding 4-hydroxyketones and 1,4-diols. Bicyclic 2,3-unsaturated 1,4-endoperoxides 530, 261g, 263 produce 4-hydroxyenones, 1,4-diols, and diepoxides. Monocyclic endoperoxides 611e–g are transformed into enones, 1,4-diols, 1,4-diketones, or furan derivatives (Table 31) [524–525].
Table 31.
Pd(PPh3)4-catalyzed transformation of 1,4-endoperoxides.
| Endoperoxide | Temperature, °C (time, h) | Products, % | |||
 611a |
17 (3) |
 612a, 41 |
 613a, 29 |
 614a, 20 |
|
 611b |
60 (5) |
 612b, 49 |
 613b, 3 |
 614b, 37 |
|
 611c |
60 (10) |
 612c, 62 |
 613c, 13 |
 614c, 25 |
|
 611d |
65 (15) |
 612d, 73 |
 614d, 23 |
||
 530 |
4 (20) |
 612e, 54 |
 614e, 16 |
||
 261g |
50–60 (5) |
 262g, 42 |
 614f, 32 |
 513a, 9 |
|
 263 |
60 (29) |
 612f, 45 |
 613d, 10 |
 614g, 17 |
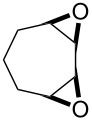 513b,28 |
 611e |
60 (39) |
 612g, 34 |
 614h, 59 |
 615, 5 |
|
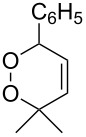 611f |
100 (15) |
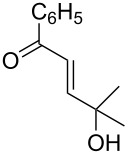 612h, 66 |
|||
 611g |
70 (12) |
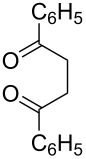 612i, 74 |
|||
The reactivity of bicyclic substrates depends on the carbon-ring size. Strained 1,4-endoperoxide derivatives are readily decomposed under the action of Pd(PPh3)4 at room or elevated temperatures, whereas substrates containing larger rings require more severe conditions. Monocyclic substrates are less reactive than bicyclic endoperoxides and require even more harsh conditions.
3. Rearrangements and related processes of important natural and synthetic peroxides
3.1. Antimalarial, antiparasitic, and antitumor peroxides
The extensive development of the chemistry of organic peroxides has been stimulated largely by the isolation of the antimalarial agent artemisinin from leaves of the annual wormwood Artemisia annua in 1972. The structural identification showed that artemisinin contains a cyclic endoperoxide moiety (1,2,4-trioxane ring), which plays a key role in its antimalarial activity [526–527]. The highly reactive and unusual chemical structure, in addition to low yields isolated from natural sources gave impetus to the development of total synthesis methods of artemisinin. Several routes towards the total synthesis of this compound were elaborated and several semisynthetic derivatives were prepared [12,16,528–533]. The high costs of these products stimulated the search for alternative peroxides, which are synthetically easier accessible and less expensive compared with the natural and semisynthetic structures. It was shown that 1,2-dioxolanes [35], 1,2-dioxanes [40], 1,2,4-trioxolanes [534–536], 1,2,4-trioxanes [44], and 1,2,4,5-tetraoxanes [537] exhibit antimalarial activity, which was sometimes higher than that of the parent artemisinin (Scheme 172). As a milestone of this research, arterolane, a fully synthetic 1,2,4-trioxolane was discovered and in 2012, the arterolane-based drug synriam was approved to the market.
Scheme 172.

Peroxides with antimalarial activity.
Although artemisinin has been used in medicine for about three decades, the mechanism of its action remains unclear [538–539]. Two main theories of its antiparasitic action are assumed. In accordance with one theory, the endoperoxide bond is reduced by means of iron ions leading to the formation of oxygen-centered radicals, which are responsible for the initiation of oxidative stress in infected erythrocytes. An alternative theory proposes that specific parasites’ proteins or heme are alkylated by carbon-centered radicals derived from the peroxide [540–541]. In infected human erythrocytes, malaria parasites digest more than 70% of the hemoglobin with formation of globin and heme. After the hydrolysis of globin, the resulting amino acids are used by the parasites for protein synthesis. Malaria parasites detoxify the toxic heme via a heme polymerization process with preparation of hemozoin, which exists in the crystalline form. Parasite metalloproteins, superoxide dismutase and ferredoxin, use a small part of the host’s iron for their construction. In such a manner parasite cells always contain heme iron and non-heme iron, allows for the interaction with artemisinin or other peroxides [542].
Numerous studies on the interaction of iron ions with artemisinin (616) demonstrated that Fe(II) promotes the O–O-bond cleavage via two paths. Thus, Fe(II) may bind to either O1 or O2 in artemisinin (Scheme 173) [542–551]. The interaction of Fe(II) with O1 gives rise to an intermediate oxy radical 617a, which undergoes β-scission to form the primary C-centered radical 617b. The subsequent elimination of Fe(II) is accompanied by the formation of compound 618 containing a tetrahydrofuran ring. The pathway involving the interaction of Fe(II) with O2 affords the O-centered radical 619a. A subsequent [1,5]-H shift results in the formation of the secondary C-centered radical 619b, and the β-scission of the latter produces vinyl ester 620, which can be epoxidated by the resulting high-valent iron-oxo species. Epoxide 621 is finally cyclized to hydroxydeoxoartemisinin 622. The formation of 618 and 622 is evidence in favor of the proposed two pathways of the Fe(II)-promoted transformation of artemisinin. The highly reactive intermediates 617 and 619 apparently lead to the damage of some parasite biomolecules [552].
Scheme 173.

The interaction of iron ions with artemisinin (616).
The 1,2-dioxanes 623 and 624 exhibiting antimalarial activity were isolated from the Caribbean sponge Plakortis simplex and their reactions with Fe(II) result in compounds 625a,b and 626a,b, respectively (Scheme 174) [553].
Scheme 174.

The interaction of FeCl2 with 1,2-dioxanes 623, 624.
Scheme 175 shows the mechanism including the formation of oxygen radicals 627, 629 from cyclic peroxides 623 and 624. The 1,5-rearrangement of the latter produces the alkyl-side chain carbon-centered radicals 628, 630. The reaction of these toxic intermediates with parasite biomolecules determines the biological effect observed for 1,2-dioxanes 623 and 624 (Scheme 175).
Scheme 175.

The mechanism of reaction 623 and 624 with Fe(II)Cl2.
Depending on the nature of the substituents in close vicinity of the peroxide group, the bicyclic natural endoperoxides G3-factors 631–633 which are involved in plant defense and extracted from the leaves of Eucalyptus grandis, react with Fe(II) to form different types of products. For instance, treatment of the 631 with Fe(II)SO4, gives rise to 634 in 82% yield. On the other hand the reaction of 632 under the same reaction conditions affords three products 635, 636, and 637 in a 1:1:1 ratio. The fluorinated endoperoxide 633 gives exclusively 638 under these conditions (Scheme 176) [554–555].
Scheme 176.

The reaction of bicyclic natural endoperoxides G3-factors 631–633 with FeSO4.
In the reaction with Fe(II), the natural antimalarial terpene cardamom peroxide 639 isolated from Amomum krervanh Pierre (Siam cardamom) is transformed into acids 640, 641, and 642 (Scheme 177) [164].
Scheme 177.

The transformation of terpene cardamom peroxide 639.
However, the cleavage of tetraoxane 643 gives two major products, namely 644 and 645, in yields of 44% and 51%, respectively. The reaction mechanism based on the results of this study is shown in Scheme 178 [556].
Scheme 178.

The different ways of the cleavage of tetraoxane 643.
Presumably, in accordance with the direction from Fe(II) to O2, tetraoxane 646 interacts with iron(II) heme 647. Starting heme 647 reacts within 30 min with formation of three products. The LC–MS study proved the formation of the covalent coupling product 648 formed from heme (mass 616) and the tetroxane-derived secondary C-centered radical. The molecular ion [M]+ of coupling product 648 was observed at m/z 782.3, which is consistent with the prediction (Scheme 179) [537,556].
Scheme 179.

The LC–MS analysis of interaction of tetraoxane 646 with iron(II)heme 647.
Under similar conditions, the same alkylated heme adduct was obtained with trioxolanes [557]. Four peaks at m/z 782.3 were detected which were assigned to the four possible regioisomers of alkylated heme adduct 648 as reported for heme–artemisinin adducts [558]. Later, in an initial study dealing with monoclonal antibodies that recognize the alkylation signature (sum of heme and protein alkylation) of synthetic peroxides it was shown that the artemisinins alkylate proteins in P. falciparum [559].
All the above-mentioned transformations involve the homolytic O–O-bond cleavage resulting in the formation of an O-centered radical, which is followed by the rearrangement into a C-centered radical, as a key step. The subsequent transformation of the C-centered radical determines the structure of the final product.
The peroxide, 3,6-epidioxy-1,10-bisaboladiene (EDBD, 649), isolated from wild plants, Cacalia delphiniifolia and Cacalia hastata, possesses cytotoxicity against the human promyelocytic leukemia cell line HL60. It was shown that the mechanism of biological activity of EDBD involves a rearrangement with formation of an unstable C-centered radical intermediate 650, followed by its transformation into product 651 (Scheme 180) [560].
Scheme 180.

The rearrangement of 3,6-epidioxy-1,10-bisaboladiene (EDBD, 649).
3.2. Rearrangement of lipid peroxides
Lipids contained in cell membranes maintain the structure and control of the vital functions of cells. Lipids are the targets of the reactions with reactive oxygen species (ROS) such as various oxygen-centered radicals, which play a key role in several pathological states [561]. Compounds containing double bonds, polyunsaturated fatty acids and esters, cholesterol and its derivatives easily undergo oxidation by action of oxygen-centered radicals (Scheme 181) [562–563].
Scheme 181.

Easily oxidized substrates.
Rearrangements of organic peroxides play an important role in such biological processes as the synthesis of prostaglandins from fatty acids. Prostaglandins are physiologically active substances produced by the reaction of arachidonic acid (652) with cyclooxygenase (COX) isoenzymes. Prostaglandin G2 (PGG2, 653) containing an endoperoxide fragment undergoes transformations mediated by a series of specific isomerases and synthases with production of PGE2, PGI2, PGD2, PGF2, and TXA2 (Scheme 182) [564–567].
Scheme 182.

Biopathway of synthesis of prostaglandins.
The formation of the metabolites isoprostanes, neuroprostanes, phytoprostanes, and isofurans 655–657 from fatty acids under autoxidative conditions in vivo involves both the reduction of peroxides and their rearrangements (Scheme 183). These compounds proved to be widespread in nature. Compounds 655–657 display significant biological activities, and the isoprostanes are currently the most reliable indicators of oxidative stress [568–570].
Scheme 183.

The reduction and rearrangements of isoprostanes.
One of the essential fatty acids, linoleic acid, contains a homoconjugated diene fragment, which is responsible for a specific peroxidation mechanism without the formation of cyclic peroxides. In addition to linoleic acid, its esters are present in the human circulating low-density lipoprotein (LDL). For this reason, the oxidation of linoleic acid esters is of special biomedical interest [566]. A mechanism for linoleate (658) oxidation, which involves hydroperoxyoctadecadienoates (HPODE, 660–662) preparation, is presented in Scheme 184. The first step of the oxidation process is the formation of the carbon-centered pentadienyl radical 659. The reaction of 659 with O2 produces three peroxyl radicals, one of them having a nonconjugated diene part with the oxygen at C-11 position. The two other radicals have Z,E- and E,E-conjugated diene parts with oxygen substituents at the C-9 and C-13 positions. These peroxyl radical intermediates after abstracting hydrogen atoms transform to the hydroperoxyoctadecadienoates (HPODE, 660–662) [570].
Scheme 184.

The partial mechanism for linoleate 658 oxidation.
The Hock cleavage mechanism is a possible route to transform lipid hydroperoxide 663 into smaller carbonyl compounds 664–666, although this transformation seems to occur only in the presence of photosensitizers (Scheme 185) [571].
Scheme 185.

The transformation of lipid hydroperoxide.
In mammalian tissues and cells, cholesterol is found to a large extent. One of the main cholesterol functions represents to maintaining the stability of plasma membranes. The oxidation of cholesterol by means of free radical particles is responsible for the initiation of a range of pathological conditions [572–573]. Many processes including the rearrangement of intermediately formed peroxides accompany the oxidation of cholesterol. The major product of 1O2 oxidation of cholesterol (667), cholesterol 5α-hydroperoxide (668), readily forms 5,6-secosterol ketoaldehyde 669 and the product of its intramolecular aldolization 670 through an acid-catalyzed (Hock) cleavage of the C5–C6 bond in 668 (Scheme 186) [67].
Scheme 186.

The acid-catalyzed cleavage of the product from free-radical oxidation of cholesterol (667).
3.3. Rearrangement of dioxygenase enzyme–substrate systems
A useful chemical property of most soil bacteria concludes in their capability to oxidize aromatic compounds. This multistep process depends on the structure of dioxygenase enzymes, which utilize molecular oxygen for oxidation [574]. This oxidation has attracted much attention as a green chemistry approach for the conversion of aromatic compounds to water-soluble products and for degradation of lignin [575–576]. The ring cleavage of 1,2-dihydroxybenzene (catechol) is likely the most thoroughly studied reaction which is catalyzed by iron-dependent catechol dioxygenase enzymes [577–579]. The oxidation of catechols 671 and 673 by two types of enzymes – intradiol dioxygenase and extradiol dioxygenase – affords 3-carboxyhexa-2,4-dienedioic acid (672) and 2-hydroxy-6-ketonona-2,4-dienoic acid (674) (Scheme 187) [580–581].
Scheme 187.

Two pathways of catechols oxidation.
A key step in the cleavage of the aromatic ring is the oxygen-atom insertion into the C–C-double bond as the result of a Criegee-like or Hock-like intermediate rearrangement [582–583]. It was demonstrated that, despite the different mechanisms of the initial step of the substrate/molecular oxygen activation, both reactions produce hydroperoxide 675 as the intermediate. This hydroperoxide undergoes Criegee-like or Hock-like rearrangement through different pathways. Intradiol dioxygenase catalyzes the 1,2-acyl migration (path B) and the formation of an intermediate anhydride 677. On the other hand, extradiol dioxygenase catalyzes the 1,2-migration of the alkenyl moiety (path A) through the intermediate formation of lactone 676 (Scheme 188) [584].
Scheme 188.

Criegee-like or Hock-like rearrangement of the intermediate hydroperoxide 675 in dioxygenase enzyme–catechol system.
Therefore, the catalyst for the O–O-bond cleavage in the Criegee-like intermediate determines the regioselectivity of the catechol oxidation.
A similar rearrangement of the Criegee intermediate with the cleavage of the С–С bond occurs in oxidative cleavage of natural organic pigments, carotinoides 679 by carotenoid cleavage dioxygenases (Scheme 189) [585–586].
Scheme 189.

Carotinoides 679 cleavage by carotenoid cleavage dioxygenases.
In this section, we considered rearrangements of the most important natural and synthetic peroxides, which proceed or can take place in biological systems. Apparently, there are is a much larger number of biological processes, involving rearrangements of peroxides, which has to be discovered and studied in the future.
Conclusion
The rearrangements of organic peroxides and related processes are covered in the literature in hundreds of publications and several specialized reviews. However, these reviews are limited in scope, narrow in their approach, and do not provide an overall picture of this field of chemistry. The present review is the first to offer a complex analysis of the available data on rearrangements of peroxides published in the last 15−20 years with an excursion to the history of the discovery of particular reactions and transformations. The rearrangements and related processes are classified according to the type of the catalysts used: acid- and base-catalyzed processes, reactions catalyzed by variable-valence metals, photochemical and thermal action. Special emphasis is drawn to current trends in the performance and application of rearrangements of organic peroxides, such as asymmetric synthesis, organocatalysis, and the use of transition metal-peroxo complexes for the preparation of compounds interesting for pharmacological applications. The published data summarized in the review provide, for the first time, an insight into the common and different features of the reaction mechanisms and allow predicting experimental and structural requirements for performing rearrangements with specified results. An analysis of the published data shows that there are numerous new and unnamed processes related to name reactions. The development and investigation of these processes are apparently the future of peroxide chemistry.
Rearrangements of organic peroxides are the key steps in processes such as the Baeyer−Villiger, the Criegee and Hock reactions, the Kornblum−DeLaMare rearrangement, and Dakin and Elbs oxidation reactions. These reactions are widely used in chemistry: The Baeyer−Villiger oxidation is widely used for the synthesis of esters and lactones. The Criegee reaction is employed to transform tertiary alcohols into ketones and aldehydes. The Kornblum−DeLaMare rearrangement is an important tool in the synthesis of γ-hydroxy enones. The Dakin oxidation is applied in the synthesis of phenols from arylaldehydes or aryl ketones and the Elbs persulfate oxidation is used to prepare hydroxyphenols from phenols.
The comprehensive analysis of the published data makes the knotty term "peroxide rearrangement” more exact. Two types of processes are actually included under the term "peroxide rearrangement”: processes that fall under the definition of a classical rearrangement, resulting in the formation of a compound of isomeric structure, and processes, in which the O–O-bond cleavage is followed by the rearrangement of one of the resulting fragments.
The pathways of peroxide rearrangements mainly depend on the type of the catalysts used, the reaction conditions, and the structure of the starting peroxide. Rearrangements can be accompanied by a homolytic or heterolytic O–O-bond cleavage, through the formation of a carbocation (e.g., the Criegee rearrangement), a carbanion (e.g., the Kornblum−DeLaMare rearrangement), or an O-centered radical (e.g., the Wieland rearrangement or rearrangements promoted by variable-valence metals).
In recent years, there has been a growing interest in organic peroxides as a base for the design of antiparasitic and antitumor agents, which led to an extensive search for new classes of peroxides. New compounds and new structural classes play a key role in the development of the chemistry of rearrangements and the performance of related useful transformations of peroxides.
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (Grant no 16-29-10678).
References
- 1.Ando W. Organic Peroxides. Chichester: Wiley; 1992. [Google Scholar]
- 2.Reetz I, Yagci Y, Mishra M K. Handbook of Vinyl Polymers. Boca Raton: CRC Press; 2008. Initiation of Vinyl Polymerization by Organic Molecules and Nonmetal Initiators; pp. 27–48. [Google Scholar]
- 3.Flory P J. Principles of Polymer Chemistry. Ithaca, New York: Cornell University Press; 1953. [Google Scholar]
- 4.Denisov E T, Denisova T G, Pokidova T S. Handbook of free radical initiators. New York: John Wiley & Sons; 2005. [Google Scholar]
- 5.Adam W. Four-membered ring peroxides: 1,2-dioxetanes and α-peroxylactones. In: Patai S, editor. Peroxides. Chichester, U.K.: John Wiley & Sons, Ltd.; 1983. pp. 829–920. [DOI] [Google Scholar]
- 6.Bouillon G, Lick C, Schank K. Diacyl peroxides, peroxycarboxylic acids and peroxy esters. In: Patai S, editor. Peroxides. Chichester, U.K.: John Wiley & Sons, Ltd.; 1983. pp. 279–309. [DOI] [Google Scholar]
- 7.Saito I, Nittala S S. Endoperoxides. In: Patai S, editor. Peroxides. Chichester, U.K.: John Wiley & Sons, Ltd.; 1983. pp. 311–374. [DOI] [Google Scholar]
- 8.Sheldon R A. Synthesis and uses of alkyl hydroperoxides and dialkyl peroxides. In: Patai S, editor. Peroxides. Chichester, U.K.: John Wiley & Sons, Ltd.; 1983. pp. 161–200. [DOI] [Google Scholar]
- 9.Adam W. Peroxide Chemistry: Mechanistic and Preparative Aspects of Oxygen Transfer. Weinheim: Wiley-VCH; 2000. [DOI] [Google Scholar]
- 10.World Health Organization [Jul 4;2016 ];World Health Organization. World malaria report 2015; WHO Press, 2005. Available from: http://www.who.int/malaria/publications/world-malaria-report-2015/en/
- 11.Jefford C W. Curr Top Med Chem. 2012;12:373–399. doi: 10.2174/156802612799362940. [DOI] [PubMed] [Google Scholar]
- 12.Slack R D, Jacobine A M, Posner G H. Med Chem Commun. 2012;3:281–297. doi: 10.1039/c2md00277a. [DOI] [Google Scholar]
- 13.Muregi F W, Ishih A. Drug Dev Res. 2010;71:20–32. doi: 10.1002/ddr.20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernández I, Robert A. Org Biomol Chem. 2011;9:4098–4107. doi: 10.1039/c1ob05088e. [DOI] [PubMed] [Google Scholar]
- 15.Chadwick J, Amewu R K, Marti F, Bousejra-El Garah F, Sharma R, Berry N G, Stocks P A, Burrell-Saward H, Wittlin S, Rottmann M, et al. ChemMedChem. 2011;6:1357–1361. doi: 10.1002/cmdc.201100196. [DOI] [PubMed] [Google Scholar]
- 16.Opsenica D M, Šolaja B A. J Serb Chem Soc. 2009;74:1155–1193. doi: 10.2298/JSC0911155O. [DOI] [Google Scholar]
- 17.Ghorai P, Dussault P H, Hu C. Org Lett. 2008;10:2401–2404. doi: 10.1021/ol800657m. [DOI] [PubMed] [Google Scholar]
- 18.Dong Y. Mini-Rev Med Chem. 2002;2:113–123. doi: 10.2174/1389557024605537. [DOI] [PubMed] [Google Scholar]
- 19.Vennerstrom J L, Fu H N, Ellis W Y, Ager A L, Jr, Wood J K, Andersen S L, Gerena L, Milhous W K. J Med Chem. 1992;35:3023–3027. doi: 10.1021/jm00094a015. [DOI] [PubMed] [Google Scholar]
- 20.Yadav N, Sharma C, Awasthi S K. RSC Adv. 2014;4:5469–5498. doi: 10.1039/c3ra42513d. [DOI] [Google Scholar]
- 21.Terent’ev A O, Borisov D A, Yaremenko I A. Chem Heterocycl Compd. 2012;48:55–58. doi: 10.1007/s10593-012-0969-3. [DOI] [Google Scholar]
- 22.Hao H-D, Wittlin S, Wu Y. Chem – Eur J. 2013;19:7605–7619. doi: 10.1002/chem.201300076. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Hao H-D, Wittlin S, Wu Y. Chem – Asian J. 2012;7:1881–1886. doi: 10.1002/asia.201200166. [DOI] [PubMed] [Google Scholar]
- 24.Boissier J, Portela J, Pradines V, Coslédan F, Robert A, Meunier B. C R Chim. 2012;15:75–78. doi: 10.1016/j.crci.2011.11.008. [DOI] [Google Scholar]
- 25.Keiser J, Veneziano V, Rinaldi L, Mezzino L, Duthaler U, Cringoli G. Res Vet Sci. 2010;88:107–110. doi: 10.1016/j.rvsc.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Keiser J, Ingram K, Vargas M, Chollet J, Wang X, Dong Y, Vennerstrom J L. Antimicrob Agents Chemother. 2012;56:1090–1092. doi: 10.1128/AAC.05371-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shuhua X, Tanner M, N'Goran E K, Utzinger J, Chollet J, Bergquist R, Minggang C, Jiang Z. Acta Trop. 2002;82:175–181. doi: 10.1016/S0001-706X(02)00009-8. [DOI] [PubMed] [Google Scholar]
- 28.Ingram K, Yaremenko I A, Krylov I B, Hofer L, Terent’ev A O, Keiser J. J Med Chem. 2012;55:8700–8711. doi: 10.1021/jm3009184. [DOI] [PubMed] [Google Scholar]
- 29.Dembitsky V M. Eur J Med Chem. 2008;43:223–251. doi: 10.1016/j.ejmech.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 30.Dembitsky V M, Gloriozova T A, Poroikov V V. Mini-Rev Med Chem. 2007;7:571–589. doi: 10.2174/138955707780859396. [DOI] [PubMed] [Google Scholar]
- 31.del Sol Jimenez M, Garzón S P, Rodriguez A D. J Nat Prod. 2003;66:655–661. doi: 10.1021/np030021h. [DOI] [PubMed] [Google Scholar]
- 32.Posner G H, D’Angelo J, MO’Neill P, Mercer A. Expert Opin Ther Pat. 2006;16:1665–1672. doi: 10.1517/13543776.16.12.1665. [DOI] [Google Scholar]
- 33.Lai H C, Singh N P, Sasaki T. Invest New Drugs. 2013;31:230–246. doi: 10.1007/s10637-012-9873-z. [DOI] [PubMed] [Google Scholar]
- 34.Dwivedi A, Mazumder A, du Plessis L, du Preez J L, Haynes R K, du Plessis J. Nanomedicine. 2015;11:2041–2050. doi: 10.1016/j.nano.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 35.Martyn D C, Ramirez A P, Berattie M J, Cortese J F, Patel V, Rush M A, Woerpel K A, Clardy J. Bioorg Med Chem Lett. 2008;18:6521–6524. doi: 10.1016/j.bmcl.2008.10.083. [DOI] [PubMed] [Google Scholar]
- 36.Schiaffo C E, Rottman M, Wittlin S, Dussault P H. ACS Med Chem Lett. 2011;2:316–319. doi: 10.1021/ml100308d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ingram K, Schiaffo C E, Sittiwong W, Benner E, Dussault P H, Keiser J. J Antimicrob Chemother. 2012;67:1979–1986. doi: 10.1093/jac/dks141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X, Dong Y, Wittlin S, Charman S A, Chiu F C K, Chollet J, Katneni K, Mannila J, Morizzi J, Ryan E, et al. J Med Chem. 2013;56:2547–2555. doi: 10.1021/jm400004u. [DOI] [PubMed] [Google Scholar]
- 39.Chaudhary S, Sharma V, Jaiswal P K, Gaikwad A N, Sinha S K, Puri S K, Sharon A, Maulik P R, Chaturvedi V. Org Lett. 2015;17:4948–4951. doi: 10.1021/acs.orglett.5b02296. [DOI] [PubMed] [Google Scholar]
- 40.O'Neill P M, Stocks P A, Pugh M D, Araujo N C, Korshin E E, Bickley J F, Ward S A, Bray P G, Pasini E, Davies J, et al. Angew Chem, Int Ed. 2004;43:4193–4197. doi: 10.1002/anie.200453859. [DOI] [PubMed] [Google Scholar]
- 41.Holla H, Labaied M, Pham N, Jenkins I D, Stuart K, Quinn R J. Bioorg Med Chem Lett. 2011;21:4793–4797. doi: 10.1016/j.bmcl.2011.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lombardo M, Sonawane D P, Quintavalla A, Trombini C, Dhavale D D, Taramelli D, Corbett Y, Rondinelli F, Fattorusso C, Persico M, et al. Eur J Org Chem. 2014:1607–1614. doi: 10.1002/ejoc.201301394. [DOI] [Google Scholar]
- 43.Givelet C, Bernat V, Danel M, André-Barrès C, Vial H. Eur J Org Chem. 2007:3095–3101. doi: 10.1002/ejoc.200700086. [DOI] [Google Scholar]
- 44.Amewu R, Gibbons P, Mukhtar A, Stachulski A V, Ward S A, Hall C, Rimmer K, Davies J, Vivas L, Bacsa J, et al. Org Biomol Chem. 2010;8:2068–2077. doi: 10.1039/b924319d. [DOI] [PubMed] [Google Scholar]
- 45.Reiter C, Fröhlich T, Zeino M, Marschall M, Bahsi H, Leidenberger M, Friedrich O, Kappes B, Hampel F, Efferth T, et al. Eur J Med Chem. 2015;97:164–172. doi: 10.1016/j.ejmech.2015.04.053. [DOI] [PubMed] [Google Scholar]
- 46. [Jul 4;2016 ];World Health Organization; Management of severe malaria – A practical handbook, 3rd ed.; WHO Press: 2013. Available from: http://www.who.int/malaria/publications/atoz/9789241548526/en/
- 47. [Jul 4;2016 ];World Health Organization; Guidelines for the treatment of malaria, 3rd ed.; WHO Press, 2015. Available from: http://www.who.int/malaria/publications/atoz/9789241549127/en/ [PubMed]
- 48.Johnson R A, Sharpless K B. Addition Reactions with Formation of Carbon–Oxygen Bonds: (ii) Asymmetric Methods of Epoxidation. In: Fleming B M T, editor. Comprehensive Organic Synthesis. Oxford: Pergamon Press; 1991. pp. 389–436. [DOI] [Google Scholar]
- 49.Baer H, Bergamo M, Forlin A, Pottenger L H, Lindner J. Ullmann's Encyclopedia of Industrial Chemistry. Weinheim, Germany: Wiley-VCH; 2012. Propylene Oxide. [DOI] [Google Scholar]
- 50.Sienel G, Rieth R, Rowbottom K T. Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH; 2000. Epoxides. [DOI] [Google Scholar]
- 51.Wang Z. Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons, Inc.; 2010. Prilezhaev Reaction; pp. 2270–2274. [DOI] [Google Scholar]
- 52.Weber M, Weber M, Kleine-Boymann M. Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH; 2000. Phenol. [DOI] [Google Scholar]
- 53.Schmidt R J. Appl Catal, A. 2005;280:89–103. doi: 10.1016/j.apcata.2004.08.030. [DOI] [Google Scholar]
- 54.Weissermel K, Arpe H-J. Industrial Organic Chemistry. Wiley-VCH; 2008. Benzene Derivatives; pp. 337–385. [DOI] [Google Scholar]
- 55.Ritz J, Fuchs H, Kieczka H, Moran W C. Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH; 2000. Caprolactam. [DOI] [Google Scholar]
- 56.Vollhardt K P C, Schore N E. Organic Chemistry: Structure and Function. 7th ed. W. H. Freeman and Company; 2014. [Google Scholar]
- 57.Poth U. Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH; 2000. Drying Oils and Related Products. [DOI] [Google Scholar]
- 58.Mallégol J, Gardette J-L, Lemaire J. J Am Oil Chem Soc. 2000;77:249–255. doi: 10.1007/s11746-000-0041-5. [DOI] [Google Scholar]
- 59.van Gorkum R, Bouwman E. Coord Chem Rev. 2005;249:1709–1728. doi: 10.1016/j.ccr.2005.02.002. [DOI] [Google Scholar]
- 60.Juita, Dlugogorski B Z, Kennedy E M, Mackie J C. Fire Sci Rev. 2012;1:No. 3. doi: 10.1186/2193-0414-1-3. [DOI] [Google Scholar]
- 61.Oakley L H, Casadio F, Shull K R, Broadbelt L J. Appl Phys A. 2015;121:869–878. doi: 10.1007/s00339-015-9363-1. [DOI] [Google Scholar]
- 62.Roda A. Chemiluminescence and bioluminescence: past, present and future. Cambridge: RSC Press; 2010. [DOI] [Google Scholar]
- 63.Dodeigne C, Thunus L, Lejeune R. Talanta. 2000;51:415–439. doi: 10.1016/S0039-9140(99)00294-5. [DOI] [PubMed] [Google Scholar]
- 64.Ciardelli F, Ruggeri G, Pucci A. Chem Soc Rev. 2013;42:857–870. doi: 10.1039/C2CS35414D. [DOI] [PubMed] [Google Scholar]
- 65.Roda A, Mirasoli M, Michelini E, Di Fusco M, Zangheri M, Cevenini L, Roda B, Simoni P. Biosens Bioelectron. 2016;76:164–179. doi: 10.1016/j.bios.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 66.Takeuchi C, Galvé R, Nieva J, Witter D P, Wentworth A D, Troseth R P, Lerner R A, Wentworth P., Jr Biochemistry. 2006;45:7162–7170. doi: 10.1021/bi0604330. [DOI] [PubMed] [Google Scholar]
- 67.Brinkhorst J, Nara S J, Pratt D A. J Am Chem Soc. 2008;130:12224–12225. doi: 10.1021/ja804162d. [DOI] [PubMed] [Google Scholar]
- 68.Murphy R C, Johnson K M. J Biol Chem. 2008;283:15521–15525. doi: 10.1074/jbc.R700049200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Terao J, Minami Y, Bando N. J Clin Biochem Nutr. 2011;48:57–62. doi: 10.3164/jcbn.11-008FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Terao J. Cholesterol Hydroperoxides and Their Degradation Mechanism. In: Kato Y, editor. Lipid Hydroperoxide-Derived Modification of Biomolecules. Vol. 77. Netherlands: Springer; 2014. pp. 83–91. [DOI] [PubMed] [Google Scholar]
- 71.Havaux M. Plant J. 2014;79:597–606. doi: 10.1111/tpj.12386. [DOI] [PubMed] [Google Scholar]
- 72.Milas N A, Mageli O L, Golubovic A, Arndt R W, Ho J C J. J Am Chem Soc. 1963;85:222–226. doi: 10.1021/ja00885a022. [DOI] [Google Scholar]
- 73.Rieche A, Bischoff C. Chem Ber. 1962;95:77–82. doi: 10.1002/cber.19620950115. [DOI] [Google Scholar]
- 74.Criegee R. Organic Peroxides. New York, London: Wiley-Interscience; 1972. [Google Scholar]
- 75.Cocker W, Grayson D H. J Chem Soc, Perkin Trans 1. 1975:1347–1352. doi: 10.1039/P19750001347. [DOI] [Google Scholar]
- 76.Vinogradova L P, Zav’yalov S I. Izv Akad Nauk SSSR, Ser Khim. 1961:2050–2054. Chem. Abstr.1962,57, 12344c. [Google Scholar]
- 77.Vinogradova L P, Zav’yalov S I. Izv Akad Nauk SSSR, Ser Khim. 1961:1482–1486. Chem. Abstr.1962,56, 338b. [Google Scholar]
- 78.Vinogradova L P, Rudenko B A, Zav’yalov S I. Izv Akad Nauk SSSR, Ser Khim. 1962:1436–1441. Chem. Abstr.1963,58, 2378g. [Google Scholar]
- 79.Rieche A, Bischoff C, Prescher D. Chem Ber. 1964;97:3071–3075. doi: 10.1002/cber.19640971116. [DOI] [Google Scholar]
- 80.Rieche A, Seyfarth H E, Brand F. Justus Liebigs Ann Chem. 1969;725:93–98. doi: 10.1002/jlac.19697250112. [DOI] [Google Scholar]
- 81.Wolffenstein R. Ber Dtsch Chem Ges. 1895;28:2265–2269. doi: 10.1002/cber.189502802208. [DOI] [Google Scholar]
- 82.Criegee R, Schnorrenberg W, Becke J. Justus Liebigs Ann Chem. 1949;565:7–21. doi: 10.1002/jlac.19495650103. [DOI] [Google Scholar]
- 83.Dickey F H, Rust F F, Vaughan W E. J Am Chem Soc. 1949;71:1432–1434. doi: 10.1021/ja01172a081. [DOI] [Google Scholar]
- 84.Criegee R, Metz K. Chem Ber. 1956;89:1714–1718. doi: 10.1002/cber.19560890720. [DOI] [Google Scholar]
- 85.Hawkins E G E. J Chem Soc C. 1969:2663–2670. doi: 10.1039/j39690002663. [DOI] [Google Scholar]
- 86.Hawkins E G E. J Chem Soc C. 1969:2671–2677. doi: 10.1039/j39690002671. [DOI] [Google Scholar]
- 87.Hawkins E G E. J Chem Soc C. 1969:2678–2681. doi: 10.1039/j39690002678. [DOI] [Google Scholar]
- 88.Hawkins E G E. J Chem Soc C. 1969:2682–2686. doi: 10.1039/j39690002682. [DOI] [Google Scholar]
- 89.Hawkins E G E. J Chem Soc C. 1969:2686–2691. doi: 10.1039/j39690002686. [DOI] [Google Scholar]
- 90.Hawkins E G E. J Chem Soc C. 1969:2691–2697. doi: 10.1039/j39690002691. [DOI] [Google Scholar]
- 91.Milas N A, Golubović A. J Am Chem Soc. 1959;81:6461–6462. doi: 10.1021/ja01533a033. [DOI] [Google Scholar]
- 92.Rieche A. Angew Chem. 1958;70:251–266. doi: 10.1002/ange.19580700902. [DOI] [Google Scholar]
- 93.Edwards J O, Sauer M C V. J Phys Chem. 1971;75:3004–3011. doi: 10.1021/j100688a023. [DOI] [Google Scholar]
- 94.Hine J, Redding R W. J Org Chem. 1970;35:2769–2772. doi: 10.1021/jo00833a065. [DOI] [Google Scholar]
- 95.Jefford C W, Jaggi D, Kohmoto S, Boukouvalas J, Bernardinelli G. Helv Chim Acta. 1984;67:2254–2260. doi: 10.1002/hlca.19840670831. [DOI] [Google Scholar]
- 96.Singh C. Tetrahedron Lett. 1990;31:6901–6902. doi: 10.1016/S0040-4039(00)97202-2. [DOI] [Google Scholar]
- 97.Jefford C W, Li Y, Jaber A, Boukouvalas J. Synth Commun. 1990;20:2589–2596. doi: 10.1080/00397919008051466. [DOI] [Google Scholar]
- 98.Antonovskii V L, Fedorova E V, Shtivel N E, Emelin Y D. Zh Org Khim. 1991;27:820–823. [Google Scholar]
- 99.Avery M A, Gao F, Chong W K M, Hendrickson T F, Inman W D, Crews P. Tetrahedron. 1994;50:957–972. doi: 10.1016/S0040-4020(01)80810-3. [DOI] [Google Scholar]
- 100.Rieche A, Bischoff C, Dietrich P. Chem Ber. 1961;94:2932–2936. doi: 10.1002/cber.19610941115. [DOI] [Google Scholar]
- 101.Shreibert A I, Chardin A P, Pil’dus I E, Ermarchenko V I. Zh Org Khim. 1971;7:967–971. [Google Scholar]
- 102.Razuvaev G A, Dodonov V A, Zaburdyaeva S N. Zh Org Khim. 1968;4:1302–1303. J. Org. Chem. USSR (Eng. Trans.)1968,4, 1256–1257. [Google Scholar]
- 103.Dussault P H, Lee I Q. J Am Chem Soc. 1993;115:6458–6459. doi: 10.1021/ja00067a090. [DOI] [Google Scholar]
- 104.Dussault P H, Eary C T. J Am Chem Soc. 1998;120:7133–7134. doi: 10.1021/ja9808503. [DOI] [Google Scholar]
- 105.Dussault P H, Lee H-J, Niu Q J. J Org Chem. 1995;60:784–785. doi: 10.1021/jo00109a001. [DOI] [Google Scholar]
- 106.Kuznetsov M L, Rocha B G M, Pombeiro A J L, Shul’pin G B. ACS Catal. 2015;5:3823–3835. doi: 10.1021/acscatal.5b00077. [DOI] [Google Scholar]
- 107.Wang C, Yamamoto H. J Am Chem Soc. 2014;136:1222–1225. doi: 10.1021/ja411379e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bunge A, Hamann H-J, Dietz D, Liebscher J. Tetrahedron. 2013;69:2446–2450. doi: 10.1016/j.tet.2013.01.032. [DOI] [Google Scholar]
- 109.Jadhav V H, Bande O P, Puranik V G, Dhavale D D. Tetrahedron: Asymmetry. 2010;21:163–170. doi: 10.1016/j.tetasy.2010.01.007. [DOI] [Google Scholar]
- 110.Cussó O, Garcia-Bosch I, Ribas X, Lloret-Fillol J, Costas M. J Am Chem Soc. 2013;135:14871–14878. doi: 10.1021/ja4078446. [DOI] [PubMed] [Google Scholar]
- 111.Nieto N, Munslow I J, Fernández-Pérez H, Vidal-Ferran A. Synlett. 2008:2856–2858. doi: 10.1055/s-0028-1083545. [DOI] [Google Scholar]
- 112.Burke C P, Shi Y. J Org Chem. 2007;72:4093–4097. doi: 10.1021/jo070205r. [DOI] [PubMed] [Google Scholar]
- 113.Lifchits O, Mahlau M, Reisinger C M, Lee A, Farès C, Polyak I, Gopakumar G, Thiel W, List B. J Am Chem Soc. 2013;135:6677–6693. doi: 10.1021/ja402058v. [DOI] [PubMed] [Google Scholar]
- 114.Wang L, Sha W, Dai Q, Feng X, Wu W, Peng H, Chen B, Cheng J. Org Lett. 2014;16:2088–2091. doi: 10.1021/ol500277u. [DOI] [PubMed] [Google Scholar]
- 115.Dragan A, Kubczyk T M, Rowley J H, Sproules S, Tomkinson N C O. Org Lett. 2015;17:2618–2621. doi: 10.1021/acs.orglett.5b00953. [DOI] [PubMed] [Google Scholar]
- 116.Chen H-H, Wang G-Z, Han J, Xu M-Y, Zhao Y-Q, Xu H-J. Tetrahedron. 2014;70:212–217. doi: 10.1016/j.tet.2013.11.085. [DOI] [Google Scholar]
- 117.Gogoi A, Modi A, Guin S, Rout S K, Das D, Patel B K. Chem Commun. 2014;50:10445–10447. doi: 10.1039/C4CC04407J. [DOI] [PubMed] [Google Scholar]
- 118.Wu X-F, Gong J-L, Qi X. Org Biomol Chem. 2014;12:5807–5817. doi: 10.1039/C4OB00276H. [DOI] [PubMed] [Google Scholar]
- 119.Zhao J, Fang H, Han J, Pan Y, Li G. Adv Synth Catal. 2014;356:2719–2724. doi: 10.1002/adsc.201400032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dai Q, Jiang Y, Yu J-T, Cheng J. Synthesis. 2015;48:329–339. doi: 10.1055/s-0035-1560536. [DOI] [Google Scholar]
- 121.Terent’ev A O, Borisov D A, Chernyshev V V, Nikishin G I. J Org Chem. 2009;74:3335–3340. doi: 10.1021/jo900226b. [DOI] [PubMed] [Google Scholar]
- 122.Li Y, Hao H-D, Wu Y. Org Lett. 2009;11:2691–2694. doi: 10.1021/ol900811m. [DOI] [PubMed] [Google Scholar]
- 123.Opsenica D M, Pocsfalvi G, Milhous W K, Šolaja B A. J Serb Chem Soc. 2002;67:465–471. doi: 10.2298/JSC0207465O. [DOI] [Google Scholar]
- 124.Terent'ev A O, Kutkin A V, Platonov M M, Ogibin Y N, Nikishin G I. Tetrahedron Lett. 2003;44:7359–7363. doi: 10.1016/S0040-4039(03)01844-6. [DOI] [Google Scholar]
- 125.Žmitek K, Zupan M, Stavber S, Iskra J. J Org Chem. 2007;72:6534–6540. doi: 10.1021/jo0708745. [DOI] [PubMed] [Google Scholar]
- 126.Ghorai P, Dussault P H. Org Lett. 2008;10:4577–4579. doi: 10.1021/ol801859c. [DOI] [PubMed] [Google Scholar]
- 127.Amewu R, Stachulski A V, Ward S A, Berry N G, Bray P G, Davies J, Labat G, Vivas L, O'Neill P M. Org Biomol Chem. 2006;4:4431–4436. doi: 10.1039/b613565j. [DOI] [PubMed] [Google Scholar]
- 128.Landenberger K B, Bolton O, Matzger A J. Angew Chem, Int Ed. 2013;52:6468–6471. doi: 10.1002/anie.201302814. [DOI] [PubMed] [Google Scholar]
- 129.Climent E, Gröninger D, Hecht M, Walter M A, Martinez-Máñez R, Weller M G, Sancenón F, Amorós P, Rurack K. Chem – Eur J. 2013;19:4117–4122. doi: 10.1002/chem.201300031. [DOI] [PubMed] [Google Scholar]
- 130.Terent'ev A O, Borisov D A, Yaremenko I A, Chernyshev V V, Nikishin G I. J Org Chem. 2010;75:5065–5071. doi: 10.1021/jo100793j. [DOI] [PubMed] [Google Scholar]
- 131.Terent'ev A O, Borisov D A, Semenov V V, Chernyshev V V, Dembitsky V M, Nikishin G I. Synthesis. 2011:2091–2100. doi: 10.1055/s-0030-1260027. [DOI] [Google Scholar]
- 132.Singh C, Srivastav N C, Puri S K. Bioorg Med Chem. 2004;12:5745–5752. doi: 10.1016/j.bmc.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 133.Griesbeck A G, El-Idreesy T T, Höinck L-O, Lex J, Brun R. Bioorg Med Chem Lett. 2005;15:595–597. doi: 10.1016/j.bmcl.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 134.Singh C, Pandey S, Saxena G, Srivastava N, Sharma M. J Org Chem. 2006;71:9057–9061. doi: 10.1021/jo061414z. [DOI] [PubMed] [Google Scholar]
- 135.Liu Y-H, Deng J, Gao J-W, Zhang Z-H. Adv Synth Catal. 2012;354:441–447. doi: 10.1002/adsc.201100561. [DOI] [Google Scholar]
- 136.Griesbeck A G, Schlundt V. Synlett. 2011:2430–2432. doi: 10.1055/s-0030-1261225. [DOI] [Google Scholar]
- 137.Kazakov D V, Kazakova O B, Ishmuratov G Yu, Terent'ev A O, Nikishin G I, Tolstikov G A. Dokl Chem. 2011;436:34–38. doi: 10.1134/S0012500811010022. [DOI] [Google Scholar]
- 138.Sashidhara K V, Avula S R, Singh L R, Palnati G R. Tetrahedron Lett. 2012;53:4880–4884. doi: 10.1016/j.tetlet.2012.07.001. [DOI] [Google Scholar]
- 139.Hall J F B, Bourne R A, Han X, Earley J H, Poliakoff M, George M W. Green Chem. 2013;15:177–180. doi: 10.1039/C2GC36711D. [DOI] [Google Scholar]
- 140.Terent'ev A O, Yaremenko I A, Vil' V A, Moiseev I K, Kon'kov S A, Dembitsky V M, Levitsky D O, Nikishin G I. Org Biomol Chem. 2013;11:2613–2623. doi: 10.1039/c3ob27239g. [DOI] [PubMed] [Google Scholar]
- 141.Terent'ev A O, Yaremenko I A, Chernyshev V V, Dembitsky V M, Nikishin G I. J Org Chem. 2012;77:1833–1842. doi: 10.1021/jo202437r. [DOI] [PubMed] [Google Scholar]
- 142.Terent'ev A O, Yaremenko I A, Vil' V A, Dembitsky V M, Nikishin G I. Synthesis. 2013;45:246–250. doi: 10.1055/s-0032-1317895. [DOI] [Google Scholar]
- 143.Nesvadba P. Encyclopedia of Radicals in Chemistry, Biology and Materials. John Wiley & Sons; 2012. Radical Polymerization in Industry. [DOI] [Google Scholar]
- 144.Moad G, Solomon D H. The chemistry of radical polymerization. 2nd ed. Elsevier; 2006. [Google Scholar]
- 145.Ebewele R O. Polymer Science and Technology. Boca Raton, New York: CRC Press; 2000. [DOI] [Google Scholar]
- 146.Mark H F. Encyclopedia of Polymer Science and Technology. 3rd ed. John Wiley & Sons; 2007. [Google Scholar]
- 147.Matyjaszewski K, Davis T P. Handbook of Radical Polymerization. John Wiley & Sons; 2003. Future Outlook and Perspectives; pp. 895–900. [DOI] [Google Scholar]
- 148.Hao H-D, Li Y, Han W-B, Wu Y. Org Lett. 2011;13:4212–4215. doi: 10.1021/ol2015434. [DOI] [PubMed] [Google Scholar]
- 149.Li Y, Hao H-D, Zhang Q, Wu Y. Org Lett. 2009;11:1615–1618. doi: 10.1021/ol900262t. [DOI] [PubMed] [Google Scholar]
- 150.Yan X, Chen J, Zhu Y-T, Qiao C. Synlett. 2011:2827–2830. doi: 10.1055/s-0031-1289864. [DOI] [Google Scholar]
- 151.Žmitek K, Zupan M, Stavber S, Iskra J. Org Lett. 2006;8:2491–2494. doi: 10.1021/ol060590r. [DOI] [PubMed] [Google Scholar]
- 152.Azarifar D, Khosravi K, Soleimanei F. Synthesis. 2009:2553–2556. doi: 10.1055/s-0029-1217394. [DOI] [Google Scholar]
- 153.Terent'ev A O, Kutkin A V, Platonov M M, Vorontsov I I, Antipin M Yu, Ogibin Yu N, Nikishin G I. Russ Chem Bull. 2004;53:681–687. doi: 10.1023/B:RUCB.0000035657.58776.cc. [DOI] [Google Scholar]
- 154.Zhang Q, Jin H-X, Liu H-H, Wu Y-K. Chin J Chem. 2006;24:1190–1195. doi: 10.1002/cjoc.200690222. [DOI] [Google Scholar]
- 155.Zhang Q, Li Y, Wu Y-K. Chin J Chem. 2007;25:1304–1308. doi: 10.1002/cjoc.200790242. [DOI] [Google Scholar]
- 156.Terent'ev A O, Kutkin A V, Platonov M M, Starikova Z A, Ogibin Yu N, Nikishin G I. Russ Chem Bull. 2005;54:1214–1218. doi: 10.1007/s11172-005-0383-4. [DOI] [Google Scholar]
- 157.Terent'ev A O, Kutkin A V, Starikova Z A, Antipin M Yu, Ogibin Yu N, Nikishin G I. Synthesis. 2004:2356–2366. doi: 10.1055/s-2004-831171. [DOI] [Google Scholar]
- 158.Hamann H-J, Hecht M, Bunge A, Gogol M, Liebscher J. Tetrahedron Lett. 2011;52:107–111. doi: 10.1016/j.tetlet.2010.10.151. [DOI] [Google Scholar]
- 159.Terent'ev A O, Platonov M M, Sonneveld E J, Peschar R, Chernyshev V V, Starikova Z A, Nikishin G I. J Org Chem. 2007;72:7237–7243. doi: 10.1021/jo071072c. [DOI] [PubMed] [Google Scholar]
- 160.Terent'ev A O, Platonov M M, Krylov I B, Chernyshev V V, Nikishin G I. Org Biomol Chem. 2008;6:4435–4441. doi: 10.1039/b809661a. [DOI] [PubMed] [Google Scholar]
- 161.Ito T, Tokuyasu T, Masuyama A, Nojima M, McCullough K J. Tetrahedron. 2003;59:525–536. doi: 10.1016/S0040-4020(02)01556-9. [DOI] [Google Scholar]
- 162.Terent'ev A O, Borisov A M, Platonov M M, Starikova Z A, Chernyshev V V, Nikishin G I. Synthesis. 2009:4159–4166. doi: 10.1055/s-0029-1217062. [DOI] [Google Scholar]
- 163.Eliasen A M, Thedford R P, Claussen K R, Yuan C, Siegel D. Org Lett. 2014;16:3628–3631. doi: 10.1021/ol501497y. [DOI] [PubMed] [Google Scholar]
- 164.Hu X, Maimone T J. J Am Chem Soc. 2014;136:5287–5290. doi: 10.1021/ja502208z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gamage N-D H, Stiasny B, Stierstorfer J, Martin P D, Klapötke T M, Winter C H. Chem Commun. 2015;51:13298–13300. doi: 10.1039/C5CC05015D. [DOI] [PubMed] [Google Scholar]
- 166.Starkl Renar K, Pečar S, Iskra J. Org Biomol Chem. 2015;13:9369–9372. doi: 10.1039/C5OB01503K. [DOI] [PubMed] [Google Scholar]
- 167.Rozhko E, Solmi S, Cavani F, Albini A, Righi P, Ravelli D. J Org Chem. 2015;80:6425–6431. doi: 10.1021/acs.joc.5b00861. [DOI] [PubMed] [Google Scholar]
- 168.Hilf J A, Witthoft L W, Woerpel K A. J Org Chem. 2015;80:8262–8267. doi: 10.1021/acs.joc.5b01326. [DOI] [PubMed] [Google Scholar]
- 169.Lu X, Deng L. Org Lett. 2014;16:2358–2361. doi: 10.1021/ol500677v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fisher T J, Mattson A E. Org Lett. 2014;16:5316–5319. doi: 10.1021/ol502494h. [DOI] [PubMed] [Google Scholar]
- 171.Wang X, Pan Y, Huang K-W, Lai Z. Org Lett. 2015;17:5630–5633. doi: 10.1021/acs.orglett.5b02881. [DOI] [PubMed] [Google Scholar]
- 172.Yu H, Shen J. Org Lett. 2014;16:3204–3207. doi: 10.1021/ol5012168. [DOI] [PubMed] [Google Scholar]
- 173.Carney J M, Hammer R J, Hulce M, Lomas C M, Miyashiro D. Synthesis. 2012;44:2560–2566. doi: 10.1055/s-0031-1289764. [DOI] [Google Scholar]
- 174.Yan X, Qiao C, Guo Z. Synlett. 2013;24:502–506. doi: 10.1055/s-0032-1318213. [DOI] [Google Scholar]
- 175.Hurlocker B, Miner M R, Woerpel K A. Org Lett. 2014;16:4280–4283. doi: 10.1021/ol5020015. [DOI] [PubMed] [Google Scholar]
- 176.Liu Q, Wang W, Liu Z, Wang T, Wu L, Ge M. RSC Adv. 2014;4:19716–19724. doi: 10.1039/C4RA02486A. [DOI] [Google Scholar]
- 177.Parrish J D, Ischay M A, Lu Z, Guo S, Peters N R, Yoon T P. Org Lett. 2012;14:1640–1643. doi: 10.1021/ol300428q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wei W, Zhang C, Xu Y, Wan X. Chem Commun. 2011;47:10827–10829. doi: 10.1039/c1cc14602e. [DOI] [PubMed] [Google Scholar]
- 179.Daeppen C, Kaiser M, Neuburger M, Gademann K. Org Lett. 2015;17:5420–5423. doi: 10.1021/acs.orglett.5b02773. [DOI] [PubMed] [Google Scholar]
- 180.Kazakova O B, Kazakov D V, Yamansarov E Yu, Medvedeva N I, Tolstikov G A, Suponitsky K Yu, Arkhipov D E. Tetrahedron Lett. 2011;52:976–979. doi: 10.1016/j.tetlet.2010.12.047. [DOI] [Google Scholar]
- 181.Lau S-H, Galván A, Merchant R R, Battilocchio C, Souto J A, Berry M B, Ley S V. Org Lett. 2015;17:3218–3221. doi: 10.1021/acs.orglett.5b01307. [DOI] [PubMed] [Google Scholar]
- 182.Feng Y, Holte D, Zoller J, Umemiya S, Simke L R, Baran P S. J Am Chem Soc. 2015;137:10160–10163. doi: 10.1021/jacs.5b07154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yablokov V A. Russ Chem Rev. 1980;49:833–842. doi: 10.1070/RC1980v049n09ABEH002509. Usp. Khim. 1980,49, 1711–1729. [DOI] [Google Scholar]
- 184.Dussault P. Reactions of Hydroperoxides and Peroxides. In: Foote C S, Valentine J, Greenberg A, et al., editors. Active Oxygen in Chemistry. Vol. 2. Netherlands: Springer; 1995. pp. 141–203. [DOI] [Google Scholar]
- 185.Renz M, Meunier B. Eur J Org Chem. 1999:737–750. doi: 10.1002/(SICI)1099-0690(199904)1999:4<737::AID-EJOC737>3.0.CO;2-B. [DOI] [Google Scholar]
- 186.Davies A G. J Chem Res. 2009:533–544. doi: 10.3184/030823409X12491375725131. [DOI] [Google Scholar]
- 187.Syrkin Ya K, Moiseev I I. Russ Chem Rev. 1960;29:193–214. doi: 10.1070/RC1960v029n04ABEH001227. Usp. Khim. 1960,29, 193. [DOI] [Google Scholar]
- 188.Ishmuratov G Yu, Legostaeva Yu V, Botsman L P, Tolstikov G A. Russ J Org Chem. 2010;46:1593–1621. doi: 10.1134/S1070428010110011. [DOI] [Google Scholar]
- 189.Smith M B. March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. 7th ed. John Wiley & Sons, Inc.; 2013. p. 2080. [Google Scholar]
- 190.Wang Z. Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons; 2010. Baeyer–Villiger Oxidation; pp. 150–155. [DOI] [Google Scholar]
- 191.Hassall C H. The Baeyer-Villiger Oxidation of Aldehydes and Ketones. In: Adams R, editor. Organic Reactions. New York: John Wiley & Sons, Inc.; 2011. [DOI] [Google Scholar]
- 192.Baeyer A, Villiger V. Ber Dtsch Chem Ges. 1899;32:3625–3633. doi: 10.1002/cber.189903203151. [DOI] [Google Scholar]
- 193.Baeyer A, Villiger V. Ber Dtsch Chem Ges. 1900;33:858–864. doi: 10.1002/cber.190003301153. [DOI] [Google Scholar]
- 194.Belov V N, Heyfitz L A, Virezub S I. Reaczii I Metodi issledovaniya organicheskich soedineniy. Vol. 10. Moscow: Goshimizdat; 1961. Okislenie karbonil'nykh soedineniy perekis'yu vodoroda i nadkislotami (reaktsiya Bayera–Villigera) pp. 7–208. [Google Scholar]
- 195.Krow G R. The Baeyer–Villiger Oxidation of Ketones and Aldehydes. In: Paquette L A, editor. Organic Reactions. Vol. 43. John Wiley & Sons, Inc.; 1993. pp. 251–798. [DOI] [Google Scholar]
- 196.Strukul G. Angew Chem, Int Ed. 1998;37:1198–1209. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1198::AID-ANIE1198>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 197.Chida N, Tobe T, Ogawa S. Tetrahedron Lett. 1994;35:7249–7252. doi: 10.1016/0040-4039(94)85373-8. [DOI] [Google Scholar]
- 198.Butkus E, Stončius S. J Chem Soc, Perkin Trans 1. 2001:1885–1888. doi: 10.1039/b103564a. [DOI] [Google Scholar]
- 199.Doering W v E, Speers L. J Am Chem Soc. 1950;72:5515–5518. doi: 10.1021/ja01168a041. [DOI] [Google Scholar]
- 200.ten Brink G-J, Arends I W C E, Sheldon R A. Chem Rev. 2004;104:4105–4124. doi: 10.1021/cr030011l. [DOI] [PubMed] [Google Scholar]
- 201.Lambert A, Macquarrie D J, Carr G, Clark J H. New J Chem. 2000;24:485–488. doi: 10.1039/b003161p. [DOI] [Google Scholar]
- 202.Chang C D, Hellring S D, inventors. Shape-selective oxidation of cyclic ketones. US4,870,192. U.S. Patent. 1989 Sep 26;
- 203.Fischer J, Hölderich W F. Appl Catal, A. 1999;180:435–443. doi: 10.1016/S0926-860X(98)00378-0. [DOI] [Google Scholar]
- 204.Taylor R T, Flood L A. J Org Chem. 1983;48:5160–5164. doi: 10.1021/jo00174a003. [DOI] [Google Scholar]
- 205.Jacobson S E, Mares F, Zambri P M. J Am Chem Soc. 1979;101:6938–6946. doi: 10.1021/ja00517a025. [DOI] [Google Scholar]
- 206.Sandaroos R, Goldani M T, Damavandi S, Mohammadi A. J Chem Sci. 2012;124:871–876. doi: 10.1007/s12039-012-0277-6. [DOI] [Google Scholar]
- 207.Hoelderich W P D, Fischer J, Schindler G P D, Arntz D D, inventors. Verfahren zur Herstellung von Lactonen durch Baeyer-Villiger Oxidation. DE19,745,442 A1. Ger. Patent. 1999 Apr 22; Chem. Abstr.1999,130, 281989u.
- 208.Gavagnin R, Cataldo M, Pinna F, Strukul G. Organometallics. 1998;17:661–667. doi: 10.1021/om970756f. [DOI] [Google Scholar]
- 209.Bolm C, Beckmann O. Chirality. 2000;12:523–525. doi: 10.1002/(SICI)1520-636X(2000)12:5/6<523::AID-CHIR39>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 210.Phillips A M F, Romão C. Eur J Org Chem. 1999:1767–1770. doi: 10.1002/(SICI)1099-0690(199908)1999:8<1767::AID-EJOC1767>3.0.CO;2-S. [DOI] [Google Scholar]
- 211.Herrmann W A, Fischer R W, Correia J D G. J Mol Catal. 1994;94:213–223. doi: 10.1016/S0304-5102(94)87043-8. [DOI] [Google Scholar]
- 212.ten Brink G-J, Vis J-M, Arends I W C E, Sheldon R A. J Org Chem. 2001;66:2429–2433. doi: 10.1021/jo0057710. [DOI] [PubMed] [Google Scholar]
- 213.Grieco P A, Yokoyama Y, Gilman S, Ohfune Y. J Chem Soc, Chem Commun. 1977:870–871. doi: 10.1039/c39770000870. [DOI] [Google Scholar]
- 214.Jacobson S E, Tang R, Mares F. J Chem Soc, Chem Commun. 1978:888–889. doi: 10.1039/c39780000888. [DOI] [Google Scholar]
- 215.Uchida T, Katsuki T, Ito K, Akashi S, Ishii A, Kuroda T. Helv Chim Acta. 2002;85:3078–3089. doi: 10.1002/1522-2675(200210)85:10<3078::AID-HLCA3078>3.0.CO;2-1. [DOI] [Google Scholar]
- 216.Berkessel A, Andreae M R M. Tetrahedron Lett. 2001;42:2293–2295. doi: 10.1016/S0040-4039(01)00141-1. [DOI] [Google Scholar]
- 217.McClure J D, Williams P H. J Org Chem. 1962;27:24–26. doi: 10.1021/jo01048a005. [DOI] [Google Scholar]
- 218.Macias-Alonso M, Morzycki J W, Iglesias-Arteaga M A. Steroids. 2011;76:317–323. doi: 10.1016/j.steroids.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 219.Carlqvist P, Eklund R, Hult K, Brinck T. J Mol Model. 2003;9:164–171. doi: 10.1007/s00894-003-0128-y. [DOI] [PubMed] [Google Scholar]
- 220.Bach R D. J Org Chem. 2012;77:6801–6815. doi: 10.1021/jo300727w. [DOI] [PubMed] [Google Scholar]
- 221.Hitomi Y, Yoshida H, Tanaka T, Funabiki T. J Mol Catal A: Chem. 2006;251:239–245. doi: 10.1016/j.molcata.2006.02.002. [DOI] [Google Scholar]
- 222.Yamabe S, Yamazaki S. J Org Chem. 2007;72:3031–3041. doi: 10.1021/jo0626562. [DOI] [PubMed] [Google Scholar]
- 223.Hamann H-J, Bunge A, Liebscher J. Chem – Eur J. 2008;14:6849–6851. doi: 10.1002/chem.200800932. [DOI] [PubMed] [Google Scholar]
- 224.Gordon N J, Evans S A., Jr J Org Chem. 1993;58:4516–4519. doi: 10.1021/jo00069a004. [DOI] [Google Scholar]
- 225.Enders D, Geibel G, Osborne S. Chem – Eur J. 2000;6:1302–1309. doi: 10.1002/(SICI)1521-3765(20000417)6:8<1302::AID-CHEM1302>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 226.Iglesias-Arteaga M A, Velazquez-Huerta G A, Mendez-Stivalet J M, Galano A, Alvarez-Idaboy J R. ARKIVOC. 2006;(vi):109–126. [Google Scholar]
- 227.Zhou G, Corey E J. J Am Chem Soc. 2005;127:11958–11959. doi: 10.1021/ja054503m. [DOI] [PubMed] [Google Scholar]
- 228.Sharrna R, Nikas S P, Guo J J, Mallipeddi S, Wood J T, Makriyannis A. ACS Med Chem Lett. 2014;5:400–404. doi: 10.1021/ml4005304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Peris G, Miller S J. Org Lett. 2008;10:3049–3052. doi: 10.1021/ol8010248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Patil N D, Yao S G, Meier M S, Mobley J K, Crocker M. Org Biomol Chem. 2015;13:3243–3254. doi: 10.1039/C4OB01771D. [DOI] [PubMed] [Google Scholar]
- 231.Poladura B, Martínez-Castañeda Á, Rodríguez-Solla H, Llavona R, Concellón C, del Amo V. Org Lett. 2013;15:2810–2813. doi: 10.1021/ol401143q. [DOI] [PubMed] [Google Scholar]
- 232.Goodman M A, Detty M R. Synlett. 2006:1100–1104. doi: 10.1055/s-2006-939692. [DOI] [Google Scholar]
- 233.Yu L, Wu Y, Cao H, Zhang X, Shi X, Luan J, Chen T, Pan Y, Xu Q. Green Chem. 2014;16:287–293. doi: 10.1039/C3GC41562G. [DOI] [Google Scholar]
- 234.ten Brink G-J, Vis J M, Arends I W C E, Sheldon R A. Tetrahedron. 2002;58:3977–3983. doi: 10.1016/S0040-4020(02)00248-X. [DOI] [Google Scholar]
- 235.Ichikawa H, Usami Y, Arimoto M. Tetrahedron Lett. 2005;46:8665–8668. doi: 10.1016/j.tetlet.2005.10.055. [DOI] [Google Scholar]
- 236.Zhang X, Ye J, Yu L, Shi X, Zhang M, Xu Q, Lautens M. Adv Synth Catal. 2015;357:955–960. doi: 10.1002/adsc.201400957. [DOI] [Google Scholar]
- 237.Yu L, Bai Z, Zhang X, Zhang X, Ding Y, Xu Q. Catal Sci Technol. 2016;6:1804–1809. doi: 10.1039/C5CY01395J. [DOI] [Google Scholar]
- 238.Del Todesco Frisone M, Giovanetti R, Pinna F, Strukul G. Stud Surf Sci Catal. 1991;66:405–410. doi: 10.1016/S0167-2991(08)62858-X. [DOI] [Google Scholar]
- 239.Bolm C, Schlingloff G, Weickhardt K. Angew Chem, Int Ed Engl. 1994;33:1848–1849. doi: 10.1002/anie.199418481. [DOI] [Google Scholar]
- 240.Bolm C, Schlingloff G, Bienewald F. J Mol Catal A: Chem. 1997;117:347–350. doi: 10.1016/S1381-1169(96)00359-7. [DOI] [Google Scholar]
- 241.Gusso A, Baccin C, Pinna F, Strukul G. Organometallics. 1994;13:3442–3451. doi: 10.1021/om00021a019. [DOI] [Google Scholar]
- 242.Strukul G, Varagnolo A, Pinna F. J Mol Catal A: Chem. 1997;117:413–423. doi: 10.1016/S1381-1169(96)00246-4. [DOI] [Google Scholar]
- 243.Lopp M, Paju A, Kanger T, Pehk T. Tetrahedron Lett. 1996;37:7583–7586. doi: 10.1016/0040-4039(96)01666-8. [DOI] [Google Scholar]
- 244.Ito K, Ishii A, Kuroda T, Katsuki T. Synlett. 2003:643–646. doi: 10.1055/s-2003-38377. [DOI] [Google Scholar]
- 245.Petersen K S, Stoltz B M. Tetrahedron. 2011;67:4352–4357. doi: 10.1016/j.tet.2011.04.046. [DOI] [Google Scholar]
- 246.Michelin R A, Sgarbossa P, Scarso A, Strukul G. Coord Chem Rev. 2010;254:646–660. doi: 10.1016/j.ccr.2009.09.014. [DOI] [Google Scholar]
- 247.Bernini R, Coratti A, Provenzano G, Fabrizi G, Tofani D. Tetrahedron. 2005;61:1821–1825. doi: 10.1016/j.tet.2004.12.025. [DOI] [Google Scholar]
- 248.Wang Y, Yokoi T, Otomo R, Kondo J N, Tatsumi T. Appl Catal, A. 2015;490:93–100. doi: 10.1016/j.apcata.2014.11.011. [DOI] [Google Scholar]
- 249.Luo H Y, Bui L, Gunther W R, Min E, Román-Leshkov Y. ACS Catal. 2012;2:2695–2699. doi: 10.1021/cs300543z. [DOI] [Google Scholar]
- 250.Kang Z, Zhang X, Liu H, Qiu J, Yeung K L. Chem Eng J. 2013;218:425–432. doi: 10.1016/j.cej.2012.12.019. [DOI] [Google Scholar]
- 251.Paul M, Pal N, Mondal J, Sasidharan M, Bhaumik A. Chem Eng Sci. 2012;71:564–572. doi: 10.1016/j.ces.2011.11.038. [DOI] [Google Scholar]
- 252.Nabae Y, Rokubuichi H, Mikuni M, Kuang Y, Hayakawa T, Kakimoto M-a. ACS Catal. 2013;3:230–236. doi: 10.1021/cs3007928. [DOI] [Google Scholar]
- 253.Uyanik M, Nakashima D, Ishihara K. Angew Chem. 2012;124:9227–9230. doi: 10.1002/ange.201204286. [DOI] [PubMed] [Google Scholar]
- 254.Uyanik M, Ishihara K. ACS Catal. 2013;3:513–520. doi: 10.1021/cs300821u. [DOI] [Google Scholar]
- 255.Nicolaou K C, Yu R, Shi L, Cai Q, Lu M, Heretsch P. Org Lett. 2013;15:1994–1997. doi: 10.1021/ol4006689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hu Y-L, Li D-J, Li D-S. RSC Adv. 2015;5:24936–24943. doi: 10.1039/C5RA02234G. [DOI] [Google Scholar]
- 257.House H O, Wasson R L. J Org Chem. 1957;22:1157–1160. doi: 10.1021/jo01361a005. [DOI] [Google Scholar]
- 258.Kaneda K, Ueno S, Imanaka T. J Mol Catal A: Chem. 1995;102:135–138. doi: 10.1016/1381-1169(95)00055-0. [DOI] [Google Scholar]
- 259.Kaneda K, Yamashita T. Tetrahedron Lett. 1996;37:4555–4558. doi: 10.1016/0040-4039(96)00902-1. [DOI] [Google Scholar]
- 260.Pillai U R, Sahle-Demessie E. J Mol Catal A: Chem. 2003;191:93–100. doi: 10.1016/S1381-1169(02)00347-3. [DOI] [Google Scholar]
- 261.Bradley T D, Dragan A, Tomkinson N C O. Tetrahedron. 2015;71:8155–8161. doi: 10.1016/j.tet.2015.08.037. [DOI] [Google Scholar]
- 262.Ito K. Asymmetric Baeyer–Villiger Oxidation. In: Carreira E M, Yamamoto H, editors. Comprehensive Chirality. Amsterdam: Elsevier; 2012. pp. 1–35. [DOI] [Google Scholar]
- 263.Bryliakov K P. Sustainable Asymmetric Oxidations. In: Poeppelmeier J R, editor. Comprehensive Inorganic Chemistry. 2nd ed. Amsterdam: Elsevier; 2013. pp. 625–664. [DOI] [Google Scholar]
- 264.Leisch H, Morley K, Lau P C K. Chem Rev. 2011;111:4165–4222. doi: 10.1021/cr1003437. [DOI] [PubMed] [Google Scholar]
- 265.Mihovilovic M D, Müller B, Stanetty P. Eur J Org Chem. 2002:3711–3730. doi: 10.1002/1099-0690(200211)2002:22<3711::AID-EJOC3711>3.0.CO;2-5. [DOI] [Google Scholar]
- 266.Mihovilovic M D, Rudroff F, Winninger A, Schneider T, Schulz F, Reetz M T. Org Lett. 2006;8:1221–1224. doi: 10.1021/ol0601040. [DOI] [PubMed] [Google Scholar]
- 267.Reetz M T, Brunner B, Schneider T, Schulz F, Clouthier C M, Kayser M M. Angew Chem, Int Ed. 2004;43:4075–4078. doi: 10.1002/anie.200460272. [DOI] [PubMed] [Google Scholar]
- 268.Baldwin C V F, Wohlgemuth R, Woodley J M. Org Process Res Dev. 2008;12:660–665. doi: 10.1021/op800046t. [DOI] [Google Scholar]
- 269.Schallmey A, Domínguez de María P, Bracco P. Biocatalytic Asymmetric Oxidations in Stereoselective Synthesis. In: Andrushko V, Andrushko N, editors. Stereoselective Synthesis of Drugs and Natural Products. Vol. 2. Hoboken, New Jersey: John Wiley & Sons, Inc.; 2013. [DOI] [Google Scholar]
- 270.Reile I, Paju A, Müürisepp A-M, Pehk T, Lopp M. Tetrahedron. 2011;67:5942–5948. doi: 10.1016/j.tet.2011.06.036. [DOI] [Google Scholar]
- 271.Jõgi A, Paju A, Pehk T, Kailas T, Müürisepp A-M, Kanger T, Lopp M. Synthesis. 2006:3031–3036. doi: 10.1055/s-2006-950193. [DOI] [Google Scholar]
- 272.Paju A, Laos M, Jõgi A, Päri M, Jäälaid R, Pehk T, Kanger T, Lopp M. Tetrahedron Lett. 2006;47:4491–4493. doi: 10.1016/j.tetlet.2006.04.013. [DOI] [Google Scholar]
- 273.Crudden C M, Chen A C, Calhoun L A. Angew Chem, Int Ed. 2000;39:2851–2855. doi: 10.1002/1521-3773(20000818)39:16<2851::AID-ANIE2851>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 274.Xu S, Wang Z, Zhang X, Zhang X, Ding K. Angew Chem, Int Ed. 2008;47:2840–2843. doi: 10.1002/anie.200705932. [DOI] [PubMed] [Google Scholar]
- 275.Xu S, Wang Z, Li Y, Zhang X, Wang H, Ding K. Chem – Eur J. 2010;16:3021–3035. doi: 10.1002/chem.200902698. [DOI] [PubMed] [Google Scholar]
- 276.Xu S, Wang Z, Zhang X, Ding K. Chin J Chem. 2010;28:1731–1735. doi: 10.1002/cjoc.201090292. [DOI] [Google Scholar]
- 277.Xu S, Wang Z, Zhang X, Ding K. Eur J Org Chem. 2011:110–116. doi: 10.1002/ejoc.201001130. [DOI] [Google Scholar]
- 278.Mazzini C, Lebreton J, Furstoss R. J Org Chem. 1996;61:8–9. doi: 10.1021/jo951905b. [DOI] [Google Scholar]
- 279.Murahashi S-I, Ono S, Imada Y. Angew Chem, Int Ed. 2002;41:2366–2368. doi: 10.1002/1521-3773(20020703)41:13<2366::AID-ANIE2366>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 280.van Berkel W J H, Kamerbeek N M, Fraaije M W. J Biotechnol. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 281.Černuchová P, Mihovilovic M D. Org Biomol Chem. 2007;5:1715–1719. doi: 10.1039/B703175K. [DOI] [PubMed] [Google Scholar]
- 282.Schulz F, Leca F, Hollmann F, Reetz M T. Beilstein J Org Chem. 2005;1:No. 10. doi: 10.1186/1860-5397-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Zhou L, Liu X, Ji J, Zhang Y, Hu X, Lin L, Feng X. J Am Chem Soc. 2012;134:17023–17026. doi: 10.1021/ja309262f. [DOI] [PubMed] [Google Scholar]
- 284.Zhou L, Liu X, Ji J, Zhang Y, Wu W, Liu Y, Lin L, Feng X. Org Lett. 2014;16:3938–3941. doi: 10.1021/ol501737a. [DOI] [PubMed] [Google Scholar]
- 285.Romney D K, Colvin S M, Miller S J. J Am Chem Soc. 2014;136:14019–14022. doi: 10.1021/ja508757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Terent'ev A O, Chodykin S V. Cent Eur J Chem. 2005;3:417–431. doi: 10.2478/BF02479272. [DOI] [Google Scholar]
- 287.Putic A, Stecher L, Prinz H, Müller K. Eur J Med Chem. 2010;45:3299–3310. doi: 10.1016/j.ejmech.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 288.An G-i, Kim M, Kim J Y, Rhee H. Tetrahedron Lett. 2003;44:2183–2186. doi: 10.1016/S0040-4039(03)00156-4. [DOI] [Google Scholar]
- 289.Kao J P Y, Muralidharan S, Zavalij P Y, Fletcher S, Xue F, Rosen G M. Tetrahedron Lett. 2014;55:3111–3113. doi: 10.1016/j.tetlet.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wang Z. Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons; 2010. Criegee Rearrangement; pp. 770–774. [DOI] [Google Scholar]
- 291.Criegee R. Ber Dtsch Chem Ges A. 1945;77:722–726. doi: 10.1002/cber.19450770912. [DOI] [Google Scholar]
- 292.Hedaya E, Winstein S. Tetrahedron Lett. 1962;3:563–567. doi: 10.1016/S0040-4039(00)76930-9. [DOI] [Google Scholar]
- 293.Hedaya E, Winstein S. J Am Chem Soc. 1967;89:1661–1672. doi: 10.1021/ja00983a023. [DOI] [Google Scholar]
- 294.Wistuba E, Rüchardt C. Tetrahedron Lett. 1981;22:3389–3392. doi: 10.1016/S0040-4039(01)81913-4. [DOI] [Google Scholar]
- 295.Schreiber S L, Liew W-F. Tetrahedron Lett. 1983;24:2363–2366. doi: 10.1016/S0040-4039(00)81926-7. [DOI] [Google Scholar]
- 296.Goodman R M, Kishi Y. J Org Chem. 1994;59:5125–5127. doi: 10.1021/jo00097a006. [DOI] [Google Scholar]
- 297.Ogibin Y N, Terent'ev A O, Kutkin A V, Nikishin G I. Tetrahedron Lett. 2002;43:1321–1324. doi: 10.1016/S0040-4039(01)02368-1. [DOI] [Google Scholar]
- 298.Baumstark A L, Vasquez P C, Chen Y-X. J Org Chem. 1994;59:6692–6696. doi: 10.1021/jo00101a030. [DOI] [Google Scholar]
- 299.Krasutsky P A, Kolomitsyn I V, Krasutsky S G, Kiprof P. Org Lett. 2004;6:2539–2542. doi: 10.1021/ol049171p. [DOI] [PubMed] [Google Scholar]
- 300.Krasutsky P A, Kolomitsyn I V. ARKIVOC. 2005;(iv):151–171. [Google Scholar]
- 301.Srikrishna A, Reddy T J. Tetrahedron. 1998;54:11517–11524. doi: 10.1016/S0040-4020(98)00672-3. [DOI] [Google Scholar]
- 302.Srikrishna A, Kumar P P, Reddy T J. Tetrahedron Lett. 1998;39:5815–5818. doi: 10.1016/S0040-4039(98)01148-4. [DOI] [Google Scholar]
- 303.Srikrishna A, Anebouselvy K. J Org Chem. 2001;66:7102–7106. doi: 10.1021/jo0105484. [DOI] [PubMed] [Google Scholar]
- 304.Srikrishna A, Gharpure S J, Kumar P P. Tetrahedron Lett. 2000;41:3177–3180. doi: 10.1016/S0040-4039(00)00325-7. [DOI] [Google Scholar]
- 305.Srikrishna A, Anebouselvy K, Reddy T J. Tetrahedron Lett. 2000;41:6643–6647. doi: 10.1016/S0040-4039(00)01107-2. [DOI] [Google Scholar]
- 306.Srikrishna A, Anebouselvy K. Tetrahedron Lett. 2002;43:2769–2771. doi: 10.1016/S0040-4039(02)00382-9. [DOI] [Google Scholar]
- 307.Srikrishna A, Mahesh K. Synlett. 2011:2537–2540. doi: 10.1055/s-0030-1260326. [DOI] [Google Scholar]
- 308.Gándara Z, Rivadulla M L, Pérez M, Gómez G, Fall Y. Eur J Org Chem. 2013:5678–5682. doi: 10.1002/ejoc.201300528. [DOI] [Google Scholar]
- 309.Taatjes C A, Shallcross D E, Percival C J. Phys Chem Chem Phys. 2014;16:1704–1718. doi: 10.1039/c3cp52842a. [DOI] [PubMed] [Google Scholar]
- 310.Chao W, Hsieh J-T, Chang C-H, Lin J J-M. Science. 2015;347:751–754. doi: 10.1126/science.1261549. [DOI] [PubMed] [Google Scholar]
- 311.Liu F, Beames J M, Petit A S, McCoy A B, Lester M I. Science. 2014;345:1596–1598. doi: 10.1126/science.1257158. [DOI] [PubMed] [Google Scholar]
- 312.Welz O, Savee J D, Osborn D L, Vasu S S, Percival C J, Shallcross D E, Taatjes C A. Science. 2012;335:204–207. doi: 10.1126/science.1213229. [DOI] [PubMed] [Google Scholar]
- 313.Wang Z. Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons; 2010. Hock Rearrangement; pp. 1438–1442. [DOI] [Google Scholar]
- 314.Udris R J, Sergeyev P G, Kruzhalov B D, inventors. Sposob polucheniya gidroperekisejj alkilirovannykh-proizvodnykh benzola ili alicikloaromaticheskikh uglevodorodov. 106,666. USSR Patent. 1947 Jan 7;
- 315.Sergeyev P G, Udris R J, Kruzhalov B D, Nyemtsov B D, inventors. Sposob odnovremennogo polucheniya fenola i acetona. 106,992. USSR Patent. 1947 Jan 7;
- 316.Hock H, Lang S. Ber Dtsch Chem Ges A. 1944;77:257–264. doi: 10.1002/cber.19440770321. [DOI] [Google Scholar]
- 317.Hock H, Kropf H. Angew Chem. 1957;69:313–321. doi: 10.1002/ange.19570691002. [DOI] [Google Scholar]
- 318.Deno N C, Billups W E, Kramer K E, Lastomirsky R R. J Org Chem. 1970;35:3080–3082. doi: 10.1021/jo00834a046. [DOI] [Google Scholar]
- 319.Olah G A, Parker D G, Yoneda N. Angew Chem, Int Ed Engl. 1978;17:909–931. doi: 10.1002/anie.197809091. [DOI] [Google Scholar]
- 320.Lange J-P, Breed A J M. Catal Commun. 2002;3:25–28. doi: 10.1016/S1566-7367(01)00071-1. [DOI] [Google Scholar]
- 321.Lillie T S, Ronald R C. J Org Chem. 1985;50:5084–5088. doi: 10.1021/jo00225a018. [DOI] [Google Scholar]
- 322.Fisher T J, Dussault P H. Tetrahedron Lett. 2010;51:5615–5617. doi: 10.1016/j.tetlet.2010.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Anderson G H, Smith J G. Can J Chem. 1968;46:1561–1570. doi: 10.1139/v68-256. [DOI] [Google Scholar]
- 324.Hamann H-J, Liebscher J. J Org Chem. 2000;65:1873–1876. doi: 10.1021/jo991457y. [DOI] [PubMed] [Google Scholar]
- 325.Hamann H-J, Liebscher J. Synlett. 2001:96–98. doi: 10.1055/s-2001-9704. [DOI] [Google Scholar]
- 326.Chan Y-Y, Zhu C, Leung H-K. J Am Chem Soc. 1985;107:5274–5275. doi: 10.1021/ja00304a041. [DOI] [Google Scholar]
- 327.Chan Y-Y, Li X, Zhu C, Liu X, Zhang Y, Leung H-K. J Org Chem. 1990;55:5497–5504. doi: 10.1021/jo00307a022. [DOI] [Google Scholar]
- 328.Kabalka G W, Reddy N K, Narayana C. Tetrahedron Lett. 1993;34:7667–7668. doi: 10.1016/S0040-4039(00)61534-4. [DOI] [Google Scholar]
- 329.Boger D L, Coleman R S. J Org Chem. 1986;51:5436–5439. doi: 10.1021/jo00376a079. [DOI] [Google Scholar]
- 330.Zheng X, Lu S, Li Z. Org Lett. 2013;15:5432–5435. doi: 10.1021/ol402509u. [DOI] [PubMed] [Google Scholar]
- 331.Zheng X, Lv L, Lu S, Wang W, Li Z. Org Lett. 2014;16:5156–5159. doi: 10.1021/ol5025053. [DOI] [PubMed] [Google Scholar]
- 332.Louillat-Habermeyer M-L, Jin R, Patureau F W. Angew Chem, Int Ed. 2015;54:4102–4104. doi: 10.1002/anie.201500089. [DOI] [PubMed] [Google Scholar]
- 333.Boger D L, Coleman R S. Tetrahedron Lett. 1987;28:1027–1030. doi: 10.1016/S0040-4039(00)95902-1. [DOI] [Google Scholar]
- 334.Amara Z, Bellamy J F B, Horvath R, Miller S J, Beeby A, Burgard A, Rossen K, Poliakoff M, George M W. Nat Chem. 2015;7:489–495. doi: 10.1038/nchem.2261. [DOI] [PubMed] [Google Scholar]
- 335.Wang Z. Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons, Inc.; 2010. Kornblum–Delamare Rearrangement; pp. 1675–1678. [DOI] [Google Scholar]
- 336.Kornblum N, DeLaMare H E. J Am Chem Soc. 1951;73:880–881. doi: 10.1021/ja01146a542. [DOI] [Google Scholar]
- 337.Mete E, Altundaş R, Seçen H, Balci M. Turk J Chem. 2003;27:145–153. [Google Scholar]
- 338.Akbulut N, Balci M. J Org Chem. 1988;53:3338–3342. doi: 10.1021/jo00249a039. [DOI] [Google Scholar]
- 339.Kelly D R, Bansal H, Morgan J J G. Tetrahedron Lett. 2002;43:9331–9333. doi: 10.1016/S0040-4039(02)02374-2. [DOI] [Google Scholar]
- 340.Avery T D, Taylor D K, Tiekink E R T. J Org Chem. 2000;65:5531–5546. doi: 10.1021/jo0002240. [DOI] [PubMed] [Google Scholar]
- 341.Greatrex B W, Kimber M C, Taylor D K, Tiekink E R T. J Org Chem. 2003;68:4239–4246. doi: 10.1021/jo020700h. [DOI] [PubMed] [Google Scholar]
- 342.Greatrex B W, Taylor D K, Tiekink E R T. J Org Chem. 2004;69:2580–2583. doi: 10.1021/jo0303315. [DOI] [PubMed] [Google Scholar]
- 343.Zhang X, Khan S I, Foote C S. J Org Chem. 1993;58:7839–7847. doi: 10.1021/jo00079a032. [DOI] [Google Scholar]
- 344.Schulte-Elte K H, Willhalm B, Ohloff G. Angew Chem. 1969;81:1045. doi: 10.1002/ange.19690812406. [DOI] [Google Scholar]
- 345.Staben S T, Linghu X, Toste F D. J Am Chem Soc. 2006;128:12658–12659. doi: 10.1021/ja065464x. [DOI] [PubMed] [Google Scholar]
- 346.Oda M, Kitahara Y. Tetrahedron Lett. 1969;10:3295–3296. doi: 10.1016/S0040-4039(01)88413-6. [DOI] [Google Scholar]
- 347.Daştan A, Balci M. Tetrahedron. 2006;62:4003–4010. doi: 10.1016/j.tet.2006.02.026. [DOI] [Google Scholar]
- 348.Koc F, Cadirci E, Albayrak A, Halici Z, Hacimuftuoglu A, Suleyman H. Med Chem Res. 2010;19:84–93. doi: 10.1007/s00044-009-9174-z. [DOI] [Google Scholar]
- 349.Morita Y, Matsumura E, Okabe T, Shibata M, Sugiura M, Ohe T, Tsujibo H, Ishida N, Inamori Y. Biol Pharm Bull. 2003;26:1487–1490. doi: 10.1248/bpb.26.1487. [DOI] [PubMed] [Google Scholar]
- 350.Balci M, Atasoy B. Tetrahedron Lett. 1984;25:4033–4036. doi: 10.1016/0040-4039(84)80059-3. [DOI] [Google Scholar]
- 351.Atasoy B, Balci M. Tetrahedron. 1986;42:1461–1468. doi: 10.1016/S0040-4020(01)87365-8. [DOI] [Google Scholar]
- 352.Sengül M E, Ceylan Z, Balci M. Tetrahedron. 1997;53:10401–10408. doi: 10.1016/S0040-4020(97)00630-3. [DOI] [Google Scholar]
- 353.Güney M, Ceylan Z C, Daştan A, Balci M. Can J Chem. 2005;83:227–235. doi: 10.1139/v05-046. [DOI] [Google Scholar]
- 354.Gu X, Zhang W, Salomon R G. J Org Chem. 2012;77:1554–1559. doi: 10.1021/jo201910g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Kawasumi M, Kanoh N, Iwabuchi Y. Org Lett. 2011;13:3620–3623. doi: 10.1021/ol201273b. [DOI] [PubMed] [Google Scholar]
- 356.Paddock V L, Phipps R J, Conde-Angulo A, Blanco-Martin A, Giró-Mañas C, Martin L J, White A J P, Spivey A C. J Org Chem. 2011;76:1483–1486. doi: 10.1021/jo102314w. [DOI] [PubMed] [Google Scholar]
- 357.Fujishima T, Kitoh F, Yano T, Irie R. Synlett. 2010:2279–2282. doi: 10.1055/s-0030-1258028. [DOI] [Google Scholar]
- 358.Erden I, Öcal N, Song J, Gleason C, Gärtner C. Tetrahedron. 2006;62:10676–10682. doi: 10.1016/j.tet.2006.07.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Wasserman H H, Terao S. Tetrahedron Lett. 1975;16:1735–1738. doi: 10.1016/S0040-4039(00)72246-5. [DOI] [Google Scholar]
- 360.Frimer A A. Chem Rev. 1979;79:359–387. doi: 10.1021/cr60321a001. [DOI] [Google Scholar]
- 361.Hewton C E, Kimber M C, Taylor D K. Tetrahedron Lett. 2002;43:3199–3201. doi: 10.1016/S0040-4039(02)00503-8. [DOI] [Google Scholar]
- 362.Rössle M, Werner T, Baro A, Frey W, Christoffers J. Angew Chem, Int Ed. 2004;43:6547–6549. doi: 10.1002/anie.200461406. [DOI] [PubMed] [Google Scholar]
- 363.Zhang F, Du P, Chen J, Wang H, Luo Q, Wan X. Org Lett. 2014;16:1932–1935. doi: 10.1021/ol5004687. [DOI] [PubMed] [Google Scholar]
- 364.Greatrex B W, Jenkins N F, Taylor D K, Tiekink E R T. J Org Chem. 2003;68:5205–5210. doi: 10.1021/jo0300845. [DOI] [PubMed] [Google Scholar]
- 365.Zagorski M G, Salomon R G. J Am Chem Soc. 1980;102:2501–2503. doi: 10.1021/ja00527a081. [DOI] [Google Scholar]
- 366.Zagorski M G, Salomon R G. J Am Chem Soc. 1984;106:1750–1759. doi: 10.1021/ja00318a034. [DOI] [Google Scholar]
- 367.Nicolaou K C, Totokotsopoulos S, Giguère D, Sun Y-P, Sarlah D. J Am Chem Soc. 2011;133:8150–8153. doi: 10.1021/ja2032635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Buchanan G S, Cole K P, Tang Y, Hsung R P. J Org Chem. 2011;76:7027–7039. doi: 10.1021/jo200936r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Kumar R A, Maheswari C U, Ghantasala S, Jyothi C, Reddy K R. Adv Synth Catal. 2011;353:401–410. doi: 10.1002/adsc.201000580. [DOI] [Google Scholar]
- 370.Tan H, Chen X, Liu Z, Wang D Z. Tetrahedron. 2012;68:3952–3955. doi: 10.1016/j.tet.2012.03.076. [DOI] [Google Scholar]
- 371.Gesinski M R, Brenzovich W E, Jr, Staben S T, Srinilta D J, Toste F D. Tetrahedron Lett. 2015;56:3643–3646. doi: 10.1016/j.tetlet.2015.02.067. [DOI] [Google Scholar]
- 372.Wiegand C, Herdtweck E, Bach T. Chem Commun. 2012;48:10195–10197. doi: 10.1039/c2cc35621j. [DOI] [PubMed] [Google Scholar]
- 373.Palframan M J, Kociok-Köhn G, Lewis S E. Chem – Eur J. 2012;18:4766–4774. doi: 10.1002/chem.201104035. [DOI] [PubMed] [Google Scholar]
- 374.Du P, Li H, Wang Y, Cheng J, Wan X. Org Lett. 2014;16:6350–6353. doi: 10.1021/ol503128j. [DOI] [PubMed] [Google Scholar]
- 375.Liu C, Shi E, Xu F, Luo Q, Wang H, Chen J, Wan X. Chem Commun. 2015;51:1214–1217. doi: 10.1039/C4CC07833K. [DOI] [PubMed] [Google Scholar]
- 376.Jiang J, Liu J, Yang L, Shao Y, Cheng J, Bao X, Wan X. Chem Commun. 2015;51:14728–14731. doi: 10.1039/C5CC05183E. [DOI] [PubMed] [Google Scholar]
- 377.Chen I T, Baitinger I, Schreyer L, Trauner D. Org Lett. 2014;16:166–169. doi: 10.1021/ol403156r. [DOI] [PubMed] [Google Scholar]
- 378.Zhang M, Liu N, Tang W. J Am Chem Soc. 2013;135:12434–12438. doi: 10.1021/ja406255j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Priest J, Longland M R, Elsegood M R J, Kimber M C. J Org Chem. 2013;78:3476–3481. doi: 10.1021/jo400177j. [DOI] [PubMed] [Google Scholar]
- 380.Hugelshofer C L, Magauer T. J Am Chem Soc. 2015;137:3807–3810. doi: 10.1021/jacs.5b02021. [DOI] [PubMed] [Google Scholar]
- 381.Kalaitzakis D, Triantafyllakis M, Alexopoulou I, Sofiadis M, Vassilikogiannakis G. Angew Chem, Int Ed. 2014;53:13201–13205. doi: 10.1002/anie.201407477. [DOI] [PubMed] [Google Scholar]
- 382.Vassilikogiannakis G, Stratakis M. Angew Chem, Int Ed. 2003;42:5465–5468. doi: 10.1002/anie.200352180. [DOI] [PubMed] [Google Scholar]
- 383.Vassilikogiannakis G, Margaros I, Montagnon T. Org Lett. 2004;6:2039–2042. doi: 10.1021/ol0493610. [DOI] [PubMed] [Google Scholar]
- 384.Vassilikogiannakis G, Margaros I, Montagnon T, Stratakis M. Chem – Eur J. 2005;11:5899–5907. doi: 10.1002/chem.200401311. [DOI] [PubMed] [Google Scholar]
- 385.Dakin H D. Am Chem J. 1909;42:477–498. [Google Scholar]
- 386.Wang Z. Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons; 2010. Dakin Reaction; pp. 829–831. [DOI] [Google Scholar]
- 387.Hocking M B, Bhandari K, Shell B, Smyth T A. J Org Chem. 1982;47:4208–4215. doi: 10.1021/jo00143a007. [DOI] [Google Scholar]
- 388.Varma R S, Naicker K P. Org Lett. 1999;1:189–192. doi: 10.1021/ol990522n. [DOI] [Google Scholar]
- 389.da Silva E T, Câmara C A, Antunes O A C, Barreiro E J, Fraga C A M. Synth Commun. 2008;38:784–788. doi: 10.1080/00397910701820673. [DOI] [Google Scholar]
- 390.Chen S, Hossain M S, Foss F W., Jr Org Lett. 2012;14:2806–2809. doi: 10.1021/ol3010326. [DOI] [PubMed] [Google Scholar]
- 391.Chen S, Foss F W., Jr Org Lett. 2012;14:5150–5153. doi: 10.1021/ol302479b. [DOI] [PubMed] [Google Scholar]
- 392.Saikia B, Borah P, Barua N C. Green Chem. 2015;17:4533–4536. doi: 10.1039/C5GC01404B. [DOI] [Google Scholar]
- 393.Abe T, Itoh T, Choshi T, Hibino S, Ishikura M. Tetrahedron Lett. 2014;55:5268–5270. doi: 10.1016/j.tetlet.2014.07.113. [DOI] [Google Scholar]
- 394.Jung M E, Lazarova T I. J Org Chem. 1997;62:1553–1555. doi: 10.1021/jo962099r. [DOI] [Google Scholar]
- 395.Faure E, Falentin-Daudré C, Jérôme C, Lyskawa J, Fournier D, Woisel P, Detrembleur C. Prog Polym Sci. 2013;38:236–270. doi: 10.1016/j.progpolymsci.2012.06.004. [DOI] [Google Scholar]
- 396.Fache M, Darroman E, Besse V, Auvergne R, Caillol S, Boutevin B. Green Chem. 2014;16:1987–1998. doi: 10.1039/c3gc42613k. [DOI] [Google Scholar]
- 397.Fache M, Boutevin B, Caillol S. Green Chem. 2016;18:712–725. doi: 10.1039/C5GC01070E. [DOI] [Google Scholar]
- 398.Matsumoto M, Kobayashi H, Hotta Y. J Org Chem. 1984;49:4740–4741. doi: 10.1021/jo00198a037. [DOI] [Google Scholar]
- 399.Roy A, Reddy K R, Mohanta P K, Ila H, Junjappat H. Synth Commun. 1999;29:3781–3791. doi: 10.1080/00397919908086017. [DOI] [Google Scholar]
- 400.Elbs K. J Prakt Chem. 1893;48:179–185. doi: 10.1002/prac.18930480123. [DOI] [Google Scholar]
- 401.Wang Z. Comprehensive Organic Name Reactions and Reagents. John Wiley & Sons; 2010. Elbs Persulfate Oxidation; pp. 977–981. [DOI] [Google Scholar]
- 402.Behrman E J. Beilstein J Org Chem. 2006;2:No. 22. doi: 10.1186/1860-5397-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Behrman E C, Chen S, Behrman E J. Tetrahedron Lett. 2002;43:3221–3224. doi: 10.1016/S0040-4039(02)00495-1. [DOI] [Google Scholar]
- 404.Capdevielle P, Maumy M. Tetrahedron Lett. 1982;23:1573–1576. doi: 10.1016/S0040-4039(00)87161-0. [DOI] [Google Scholar]
- 405.Sethna S M. Chem Rev. 1951;49:91–101. doi: 10.1021/cr60152a002. [DOI] [Google Scholar]
- 406.Behrman E J. Synth Commun. 2008;38:1168–1175. doi: 10.1080/00397910701865819. [DOI] [Google Scholar]
- 407.Behrman E J. Chem Cent J. 2009;3:No. 1. doi: 10.1186/1752-153X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Behrman E J. J Chem Res. 2014;38:121–122. doi: 10.3184/174751914X13896383516701. [DOI] [Google Scholar]
- 409.Schenck G O, Neumüller O-A, Eisfeld W. Justus Liebigs Ann Chem. 1958;618:202–210. doi: 10.1002/jlac.19586180123. [DOI] [Google Scholar]
- 410.Schenck G O, Neumüller O-A, Eisfeld W. Angew Chem. 1958;70:595. doi: 10.1002/ange.19580701906. [DOI] [Google Scholar]
- 411.Eistert B, Bock G. Angew Chem. 1958;70:595. doi: 10.1002/ange.19580701905. [DOI] [Google Scholar]
- 412.Teng J I, Kulig M J, Smith L L, Kan G, Van Lier J E. J Org Chem. 1973;38:119–123. doi: 10.1021/jo00941a024. [DOI] [PubMed] [Google Scholar]
- 413.Porter N A, Dussault P H. Rearrangements of optically pure hydroperoxides. In: Minisci F, editor. Free Radicals in Synthesis and Biology. Kluwer Academic Publishers; 1989. pp. 407–421. [DOI] [Google Scholar]
- 414.Brill W F. J Chem Soc, Perkin Trans 2. 1984:621–627. doi: 10.1039/p29840000621. [DOI] [Google Scholar]
- 415.Porter N, Zuraw P. J Chem Soc, Chem Commun. 1985:1472–1473. doi: 10.1039/c39850001472. [DOI] [Google Scholar]
- 416.Beckwith A L J, Davies A G, Davison I G E, Maccoll A, Mruzek M H. J Chem Soc, Perkin Trans 2. 1989:815–824. doi: 10.1039/P29890000815. [DOI] [Google Scholar]
- 417.Porter N A, Wujek J S. J Org Chem. 1987;52:5085–5089. doi: 10.1021/jo00232a004. [DOI] [Google Scholar]
- 418.Beckwith A L J, Davies A G, Davison I G E, Maccoll A, Mruzek M H. J Chem Soc, Chem Commun. 1988:475–476. doi: 10.1039/c39880000475. [DOI] [Google Scholar]
- 419.Davies A G, Davison I G E. J Chem Soc, Perkin Trans 2. 1989:825–830. doi: 10.1039/p29890000825. [DOI] [Google Scholar]
- 420.Porter N A, Kaplan J K, Dussault P H. J Am Chem Soc. 1990;112:1266–1267. doi: 10.1021/ja00159a068. [DOI] [Google Scholar]
- 421.Porter N A, Mills K A, Caldwell S E, Dubay G R. J Am Chem Soc. 1994;116:6697–6705. doi: 10.1021/ja00094a027. [DOI] [Google Scholar]
- 422.Mills K A, Caldwell S E, Dubay G R, Porter N A. J Am Chem Soc. 1992;114:9689–9691. doi: 10.1021/ja00050a076. [DOI] [Google Scholar]
- 423.Lowe J R, Porter N A. J Am Chem Soc. 1997;119:11534–11535. doi: 10.1021/ja9723038. [DOI] [Google Scholar]
- 424.Brill W F. J Am Chem Soc. 1965;87:3286–3287. doi: 10.1021/ja01092a077. [DOI] [Google Scholar]
- 425.Dussault P H, Eary C T, Woller K R. J Org Chem. 1999;64:1789–1797. doi: 10.1021/jo981128q. [DOI] [PubMed] [Google Scholar]
- 426.Dang H S, Davies A G, Davison I G E, Schiesser C H. J Org Chem. 1990;55:1432–1438. doi: 10.1021/jo00292a012. [DOI] [Google Scholar]
- 427.Avila D V, Davies A G, Davison I G E. J Chem Soc, Perkin Trans 2. 1988:1847–1852. doi: 10.1039/p29880001847. [DOI] [Google Scholar]
- 428.Dang H-S, Davies A G. J Chem Soc, Perkin Trans 2. 1992:1095–1101. doi: 10.1039/P29920001095. [DOI] [Google Scholar]
- 429.Dussault P H, Woller K R. J Am Chem Soc. 1997;119:3824–3825. doi: 10.1021/ja970174p. [DOI] [Google Scholar]
- 430.Nonami Y, Baran J, Sosnicki J, Mayr H, Masuyama A, Nojima M. J Org Chem. 1999;64:4060–4063. doi: 10.1021/jo990127a. [DOI] [Google Scholar]
- 431.Bortolomeazzi R, De Zan M, Pizzale L, Conte L S. J Agric Food Chem. 1999;47:3069–3074. doi: 10.1021/jf9812580. [DOI] [PubMed] [Google Scholar]
- 432.Sy L-K, Ngo K-S, Brown G D. Tetrahedron. 1999;55:15127–15140. doi: 10.1016/S0040-4020(99)00987-4. [DOI] [Google Scholar]
- 433.Ngo K-S, Brown G D. Tetrahedron. 1999;55:14623–14634. doi: 10.1016/S0040-4020(99)00938-2. [DOI] [Google Scholar]
- 434.Ponce M A, Ramirez J A, Galagovsky L R, Gros E G, Erra-Balsells R. J Chem Soc, Perkin Trans 2. 2000:2351–2358. doi: 10.1039/b000144i. [DOI] [Google Scholar]
- 435.Kinart W J, Kinart A, Sendecki M. Phys Chem Liq. 2008;46:627–630. doi: 10.1080/00319100802072656. [DOI] [Google Scholar]
- 436.Wentworth A D, Song B-D, Nieva J, Shafton A, Tripurenani S, Wentworth P., Jr Chem Commun. 2009:3098–3100. doi: 10.1039/B821584G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Chen D, Chen W, Liu D, van Ofwegen L, Proksch P, Lin W. J Nat Prod. 2013;76:1753–1763. doi: 10.1021/np400480p. [DOI] [PubMed] [Google Scholar]
- 438.Pratt D A, Tallman K A, Porter N A. Acc Chem Res. 2011;44:458–467. doi: 10.1021/ar200024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Dang H-S, Davies A G, Schiesser C H. J Chem Soc, Perkin Trans 1. 1990;3:789–794. doi: 10.1039/p19900000789. [DOI] [Google Scholar]
- 440.Courtneidge J L. J Chem Soc, Chem Commun. 1992:1270–1272. doi: 10.1039/c39920001270. [DOI] [Google Scholar]
- 441.Wieland H. Ber Dtsch Chem Ges. 1911;44:2550–2556. doi: 10.1002/cber.19110440380. [DOI] [Google Scholar]
- 442.Ingold K U, Smeu M, DiLabio G A. J Org Chem. 2006;71:9906–9908. doi: 10.1021/jo061898z. [DOI] [PubMed] [Google Scholar]
- 443.DiLabio G A, Ingold K U, Lin S, Litwinienko G, Mozenson O, Mulder P, Tidwell T T. Angew Chem, Int Ed. 2010;49:5982–5985. doi: 10.1002/anie.201001008. [DOI] [PubMed] [Google Scholar]
- 444.Dannley R L, Farrant G C. J Org Chem. 1969;34:2432–2437. doi: 10.1021/jo01260a037. [DOI] [Google Scholar]
- 445.Dannley R L, Shubber A K. J Org Chem. 1971;36:3784–3787. doi: 10.1021/jo00823a027. [DOI] [Google Scholar]
- 446.Terent'ev A O, Platonov M M, Kashin A S, Nikishin G I. Tetrahedron. 2008;64:7944–7948. doi: 10.1016/j.tet.2008.06.027. [DOI] [Google Scholar]
- 447.Payne G B, Smith C W. J Org Chem. 1957;22:1682–1685. doi: 10.1021/jo01363a042. [DOI] [Google Scholar]
- 448.Payne G. J Org Chem. 1959;24:1830–1832. doi: 10.1021/jo01093a633. [DOI] [Google Scholar]
- 449.Terent'ev A O, Borisov D A, Yaremenko I A, Ogibin Y N, Nikishin G I. Synthesis. 2010:1145–1149. doi: 10.1055/s-0029-1219225. [DOI] [Google Scholar]
- 450.Yaremenko I A, Terent'ev A O, Vil' V A, Novikov R A, Chernyshev V V, Tafeenko V A, Levitsky D O, Fleury F, Nikishin G I. Chem – Eur J. 2014;20:10160–10169. doi: 10.1002/chem.201402594. [DOI] [PubMed] [Google Scholar]
- 451.Yu X, Liu Z, Xia Z, Shen Z, Pan X, Zhang H, Xie W. RSC Adv. 2014;4:53397–53401. doi: 10.1039/C4RA11237G. [DOI] [Google Scholar]
- 452.Dussault P H, Lee H-J, Liu X. J Chem Soc, Perkin Trans 1. 2000:3006–3013. doi: 10.1039/b001391i. [DOI] [Google Scholar]
- 453.Murahashi S-I, Naota T, Miyaguchi N, Noda S. J Am Chem Soc. 1996;118:2509–2510. doi: 10.1021/ja954009q. [DOI] [Google Scholar]
- 454.Murahashi S-I, Miyaguchi N, Noda S, Naota T, Fujii A, Inubushi Y, Komiya N. Eur J Org Chem. 2011:5355–5365. doi: 10.1002/ejoc.201100740. [DOI] [Google Scholar]
- 455.Mal D, Ray S, Sharma I. J Org Chem. 2007;72:4981–4984. doi: 10.1021/jo062271j. [DOI] [PubMed] [Google Scholar]
- 456.Murahashi S-I, Fujii A, Inubushi Y, Komiya N. Tetrahedron Lett. 2010;51:2339–2341. doi: 10.1016/j.tetlet.2010.02.134. [DOI] [Google Scholar]
- 457.O'Neill P M, Rawe S L, Storr R C, Ward S A, Posner G H. Tetrahedron Lett. 2005;46:3029–3032. doi: 10.1016/j.tetlet.2005.03.022. [DOI] [Google Scholar]
- 458.Afonso A. Can J Chem. 1970;48:691–693. doi: 10.1139/v70-113. [DOI] [Google Scholar]
- 459.Kulinkovich O G, Astashko D A, Tyvorskii V I, Ilyina N A. Synthesis. 2001:1453–1455. doi: 10.1055/s-2001-16089. [DOI] [Google Scholar]
- 460.Reisinger C M, Wang X, List B. Angew Chem, Int Ed. 2008;47:8112–8115. doi: 10.1002/anie.200803238. [DOI] [PubMed] [Google Scholar]
- 461.O'Neill P M, Searle N L, Raynes K J, Maggs J L, Ward S A, Storr R C, Park B K, Posner G H. Tetrahedron Lett. 1998;39:6065–6068. doi: 10.1016/S0040-4039(98)01248-9. [DOI] [Google Scholar]
- 462.Bernat V, André C, André-Barrès C. Org Biomol Chem. 2008;6:454–457. doi: 10.1039/b715491g. [DOI] [PubMed] [Google Scholar]
- 463.Hoshiya N, Watanabe N, Ijuin H K, Matsumoto M. Tetrahedron. 2006;62:12424–12437. doi: 10.1016/j.tet.2006.09.108. [DOI] [Google Scholar]
- 464.Matsumoto M, Akimoto T, Matsumoto Y, Watanabe N. Tetrahedron Lett. 2005;46:6075–6078. doi: 10.1016/j.tetlet.2005.07.008. [DOI] [Google Scholar]
- 465.Matsumoto M, Tanimura M, Akimoto T, Watanabe N, Ijuin H K. Tetrahedron Lett. 2008;49:4170–4173. doi: 10.1016/j.tetlet.2008.04.110. [DOI] [Google Scholar]
- 466.Tanimura M, Watanabe N, Ijuin H K, Matsumoto M. J Org Chem. 2011;76:902–908. doi: 10.1021/jo1021822. [DOI] [PubMed] [Google Scholar]
- 467.Tanimura M, Watanabe N, Ijuin H K, Matsumoto M. J Org Chem. 2012;77:4725–4731. doi: 10.1021/jo300417e. [DOI] [PubMed] [Google Scholar]
- 468.Matsumoto M, Suzuki H, Sano Y, Watanabe N, Ijuin H K. Tetrahedron Lett. 2008;49:5372–5375. doi: 10.1016/j.tetlet.2008.06.103. [DOI] [Google Scholar]
- 469.Matsumoto M, Suzuki H, Watanabe N, Ijuin H K, Tanaka J, Tanaka C. J Org Chem. 2011;76:5006–5017. doi: 10.1021/jo2006945. [DOI] [PubMed] [Google Scholar]
- 470.Matsumoto M, Takamido Y, Nomura K, Shiono T, Watanabe N, Ijuin H K. Tetrahedron Lett. 2008;49:6145–6147. doi: 10.1016/j.tetlet.2008.08.024. [DOI] [Google Scholar]
- 471.Turan I S, Akkaya E U. Org Lett. 2014;16:1680–1683. doi: 10.1021/ol5003412. [DOI] [PubMed] [Google Scholar]
- 472.Watanabe N, Kikuchi M, Maniwa Y, Ijuin H K, Matsumoto M. J Org Chem. 2010;75:879–884. doi: 10.1021/jo902477n. [DOI] [PubMed] [Google Scholar]
- 473.Watanabe N, Sano Y, Suzuki H, Tanimura M, Ijuin H K, Matsumoto M. J Org Chem. 2010;75:5920–5926. doi: 10.1021/jo101114b. [DOI] [PubMed] [Google Scholar]
- 474.Koyama Y, Watanabe N, Ijuin H K, Matsumoto M. Heterocycles. 2015;90:462–481. doi: 10.3987/COM-14-S(K)43. [DOI] [Google Scholar]
- 475.Hagiwara H, Watanabe N, Ijuin H K, Yamada M, Matsumoto M. Heterocycles. 2013;87:65–78. doi: 10.3987/COM-12-12602. [DOI] [Google Scholar]
- 476.Bastos E L, da Silva S M, Baader W J. J Org Chem. 2013;78:4432–4439. doi: 10.1021/jo400426y. [DOI] [PubMed] [Google Scholar]
- 477.Ciscato L F M L, Bartoloni F H, Colavite A S, Weiss D, Beckert R, Schramm S. Photochem Photobiol Sci. 2014;13:32–37. doi: 10.1039/C3PP50345C. [DOI] [PubMed] [Google Scholar]
- 478.Klaper M, Wessig P, Linker T. Chem Commun. 2016;52:1210–1213. doi: 10.1039/C5CC08606J. [DOI] [PubMed] [Google Scholar]
- 479.Pramanik S, Reddy R R, Ghorai P. Org Lett. 2015;17:1393–1396. doi: 10.1021/acs.orglett.5b00190. [DOI] [PubMed] [Google Scholar]
- 480.Story P R, Denson D D, Bishop C E, Clark B C, Jr, Farine J-C. J Am Chem Soc. 1968;90:817–818. doi: 10.1021/ja01005a063. [DOI] [Google Scholar]
- 481.Sanderson J R, Story P R. J Org Chem. 1974;39:3463–3469. doi: 10.1021/jo00938a001. [DOI] [Google Scholar]
- 482.Sanderson J R, Story P R, Paul K. J Org Chem. 1975;40:691–695. doi: 10.1021/jo00894a006. [DOI] [Google Scholar]
- 483.Paul K, Story P R, Busch P, Sanderson J R. J Org Chem. 1976;41:1283–1285. doi: 10.1021/jo00869a054. [DOI] [Google Scholar]
- 484.Boyd J D, Foote C S. J Am Chem Soc. 1979;101:6758–6759. doi: 10.1021/ja00516a051. [DOI] [Google Scholar]
- 485.Celik M, Akbulut N, Balci M. Helv Chim Acta. 2000;83:3131–3138. doi: 10.1002/1522-2675(20001220)83:12<3131::AID-HLCA3131>3.0.CO;2-6. [DOI] [Google Scholar]
- 486.van Tamelen E E, Taylor E G. J Am Chem Soc. 1980;102:1202–1203. doi: 10.1021/ja00523a067. [DOI] [Google Scholar]
- 487.Carless H A J, Atkins R, Fekarurhobo G K. Tetrahedron Lett. 1985;26:803–806. doi: 10.1016/S0040-4039(00)89142-X. [DOI] [Google Scholar]
- 488.Maheshwari K K, de Mayo P, Wiegand D. Can J Chem. 1970;48:3265–3268. doi: 10.1139/v70-548. [DOI] [Google Scholar]
- 489.Wilson R M, Rekers J W. J Am Chem Soc. 1981;103:206–207. doi: 10.1021/ja00391a046. [DOI] [Google Scholar]
- 490.Ronzani F, Costarramone N, Blanc S, Benabbou A K, Le Bechec M, Pigot T, Oelgemöller M, Lacombe S. J Catal. 2013;303:164–174. doi: 10.1016/j.jcat.2013.04.001. [DOI] [Google Scholar]
- 491.Delort E, Jaquier A, Decorzant E, Chapuis C, Casilli A, Frérot E. Phytochemistry. 2015;109:111–124. doi: 10.1016/j.phytochem.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 492.El-Mansy M F, Flister M, Lindeman S, Kalous K, Sem D S, Donaldson W A. Chem – Eur J. 2015;21:10886–10895. doi: 10.1002/chem.201501274. [DOI] [PubMed] [Google Scholar]
- 493.Erden I, Xu F-P, Drummond J, Alstad R. J Org Chem. 1993;58:3611–3612. doi: 10.1021/jo00066a007. [DOI] [Google Scholar]
- 494.Erden I, Xu F-P, Cao W-G. Angew Chem, Int Ed Engl. 1997;36:1516–1518. doi: 10.1002/anie.199715161. [DOI] [Google Scholar]
- 495.Cermola F, Iesce M R, Buonerba G. J Org Chem. 2005;70:6503–6505. doi: 10.1021/jo0504159. [DOI] [PubMed] [Google Scholar]
- 496.Scarpati R, Iesce M R, Cermola F, Guitto A. Synlett. 1998:17–25. doi: 10.1055/s-1998-1556. [DOI] [Google Scholar]
- 497.Iesce M R, Cermola F, De Lorenzo F, Orabona I, Graziano M L. J Org Chem. 2001;66:4732–4735. doi: 10.1021/jo0102112. [DOI] [PubMed] [Google Scholar]
- 498.Matsumoto M, Murakami H, Watanabe N. Chem Commun. 1998:2319–2320. doi: 10.1039/a806823b. [DOI] [Google Scholar]
- 499.Matsumoto M, Sano Y, Watanabe N, Ijuin H K. Chem Lett. 2006;35:882–883. doi: 10.1246/cl.2006.882. [DOI] [Google Scholar]
- 500.Lee A S, Shair M D. Org Lett. 2013;15:2390–2393. doi: 10.1021/ol400832r. [DOI] [PubMed] [Google Scholar]
- 501.Ronsein G E, de Oliveira M C B, de Medeiros M H G, Di Mascio P. Photochem Photobiol Sci. 2011;10:1727–1730. doi: 10.1039/c1pp05181d. [DOI] [PubMed] [Google Scholar]
- 502.Posner G H, Tao X, Cumming J N, Klinedinst D, Shapiro T A. Tetrahedron Lett. 1996;37:7225–7228. doi: 10.1016/0040-4039(96)01625-5. [DOI] [Google Scholar]
- 503.Suzuki M, Ohtake H, Kameya Y, Hamanaka N, Noyori R. J Org Chem. 1989;54:5292–5302. doi: 10.1021/jo00283a023. [DOI] [Google Scholar]
- 504.Kamata M, Satoh C, Kim H-S, Wataya Y. Tetrahedron Lett. 2002;43:8313–8317. doi: 10.1016/S0040-4039(02)02024-5. [DOI] [Google Scholar]
- 505.Bloodworth A J, Shah A. Tetrahedron Lett. 1995;36:7551–7554. doi: 10.1016/0040-4039(95)01530-2. [DOI] [Google Scholar]
- 506.Liu H-H, Jin H-X, Wu Y-K. Chin J Chem. 2004;22:1029–1033. doi: 10.1002/cjoc.20040220930. [DOI] [Google Scholar]
- 507.Persico M, Quintavalla A, Rondinelli F, Trombini C, Lombardo M, Fattorusso C, Azzarito V, Taramelli D, Parapini S, Corbett Y, et al. J Med Chem. 2011;54:8526–8540. doi: 10.1021/jm201056j. [DOI] [PubMed] [Google Scholar]
- 508.Liu H-H, Wu Y-K, Shen X. Chin J Chem. 2003;21:875–877. doi: 10.1002/cjoc.20030210731. [DOI] [Google Scholar]
- 509.Boyd J D, Foote C S, Imagawa D K. J Am Chem Soc. 1980;102:3641–3642. doi: 10.1021/ja00530a063. [DOI] [Google Scholar]
- 510.Baran A, Bekarlar M, Aydin G, Nebioglu M, Sahin E, Balci M. J Org Chem. 2012;77:1244–1250. doi: 10.1021/jo202494v. [DOI] [PubMed] [Google Scholar]
- 511.Baran A, Cambul S, Nebioglu M, Balci M. J Org Chem. 2012;77:5086–5097. doi: 10.1021/jo300655p. [DOI] [PubMed] [Google Scholar]
- 512.Özen R, Kormali F, Balci M, Atasoy B. Tetrahedron. 2001;57:7529–7535. doi: 10.1016/S0040-4020(01)00702-5. [DOI] [Google Scholar]
- 513.Coşkun A, Güney M, Daştan A, Balci M. Tetrahedron. 2007;63:4944–4950. doi: 10.1016/j.tet.2007.03.145. [DOI] [Google Scholar]
- 514.Griffen J A, Kenwright S J, Abou-Shehada S, Wharry S, Moody T S, Lewis S E. Org Chem Front. 2014;1:79–90. doi: 10.1039/c3qo00057e. [DOI] [Google Scholar]
- 515.Özer G, Saraçoglu N, Balci M. J Org Chem. 2003;68:7009–7015. doi: 10.1021/jo0345300. [DOI] [PubMed] [Google Scholar]
- 516.Kiliç H, Balci M. Tetrahedron. 2001;57:9889–9897. doi: 10.1016/S0040-4020(01)01008-0. [DOI] [Google Scholar]
- 517.Dastan A, Saracoglu N, Balci M. Eur J Org Chem. 2001:3519–3522. doi: 10.1002/1099-0690(200109)2001:18<3519::AID-EJOC3519>3.0.CO;2-2. [DOI] [Google Scholar]
- 518.Adam W, Balci M, Kiliç H. J Org Chem. 2000;65:5926–5931. doi: 10.1021/jo000120p. [DOI] [PubMed] [Google Scholar]
- 519.Sutbeyaz Y, Seçen H, Balci M. J Org Chem. 1988;53:2312–2317. doi: 10.1021/jo00245a034. [DOI] [PubMed] [Google Scholar]
- 520.Durie A J, Slawin A M Z, Lebl T, Kirsch P, O'Hagan D. Chem Commun. 2011;47:8265–8267. doi: 10.1039/c1cc13016a. [DOI] [PubMed] [Google Scholar]
- 521.Sar A, Lindeman S, Donaldson W A. Org Biomol Chem. 2010;8:3908–3917. doi: 10.1039/c004730a. [DOI] [PubMed] [Google Scholar]
- 522.Barbarow J E, Miller A K, Trauner D. Org Lett. 2005;7:2901–2903. doi: 10.1021/ol050831f. [DOI] [PubMed] [Google Scholar]
- 523.Miller A K, Trauner D. Angew Chem, Int Ed. 2005;44:4602–4606. doi: 10.1002/anie.200500488. [DOI] [PubMed] [Google Scholar]
- 524.Suzuki M, Oda Y, Hamanaka N, Noyori R. Heterocycles. 1990;30:517–535. doi: 10.3987/COM-89-S45. [DOI] [Google Scholar]
- 525.Suzuki M, Oda Y, Noyori R. Tetrahedron Lett. 1981;22:4413–4416. doi: 10.1016/S0040-4039(01)82971-3. [DOI] [Google Scholar]
- 526.The Research Group of Artemisinin Structure Chin Sci Bull. 1977;22:142. [Google Scholar]
- 527.The Qinghaosu Antimalarial Coordinating Research Group Chin Med J (Beijing, China, Engl Ed) 1979;92:811–816. [Google Scholar]
- 528.Chaturvedi D, Goswami A, Saikia P P, Barua N C, Rao P G. Chem Soc Rev. 2010;39:435–454. doi: 10.1039/B816679J. [DOI] [PubMed] [Google Scholar]
- 529.Chen H-J, Han W-B, Hao H-D, Wu Y. Tetrahedron. 2013;69:1112–1114. doi: 10.1016/j.tet.2012.11.056. [DOI] [Google Scholar]
- 530.Opsenica D M, Solaja B A. Maced J Chem Chem Eng. 2012;31:137–182. [Google Scholar]
- 531.Schmid G, Hofheinz W. J Am Chem Soc. 1983;105:624–625. doi: 10.1021/ja00341a054. [DOI] [Google Scholar]
- 532.Yadav J S, Thirupathaiah B, Srihari P. Tetrahedron. 2010;66:2005–2009. doi: 10.1016/j.tet.2010.01.051. [DOI] [Google Scholar]
- 533.Zhu C, Cook S P. J Am Chem Soc. 2012;134:13577–13579. doi: 10.1021/ja3061479. [DOI] [PubMed] [Google Scholar]
- 534.Dong Y, Wittlin S, Sriraghavan K, Chollet J, Charman S A, Charman W N, Scheurer C, Urwyler H, Santo Tomas J, Snyder C, et al. J Med Chem. 2010;53:481–491. doi: 10.1021/jm901473s. [DOI] [PubMed] [Google Scholar]
- 535.Uhlemann A-C, Wittlin S, Matile H, Bustamante L Y, Krishna S. Antimicrob Agents Chemother. 2007;51:667–672. doi: 10.1128/AAC.01064-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Vennerstrom J L, Arbe-Barnes S, Brun R, Charman S A, Chiu F C K, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, et al. Nature. 2004;430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 537.O'Neill P M, Amewu R K, Nixon G L, ElGarah F B, Mungthin M, Chadwick J, Shone A E, Vivas L, Lander H, Barton V, et al. Angew Chem, Int Ed. 2010;49:5693–5697. doi: 10.1002/anie.201001026. [DOI] [PubMed] [Google Scholar]
- 538.Edikpo N, Ghasi S, Elias A, Oguanobi N. Mol Cell Pharmacol. 2013;5:75–89. [Google Scholar]
- 539.Klonis N, Creek D J, Tilley L. Curr Opin Microbiol. 2013;16:722–727. doi: 10.1016/j.mib.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 540.Li J, Zhou B. Molecules. 2010;15:1378–1397. doi: 10.3390/molecules15031378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Robert A, Bonduelle C, Laurent S A-L, Meunier B. J Phys Org Chem. 2006;19:562–569. doi: 10.1002/poc.1059. [DOI] [Google Scholar]
- 542.Pereira M S C, Kiralj R, Ferreira M M C. J Chem Inf Model. 2008;48:85–98. doi: 10.1021/ci700011f. [DOI] [PubMed] [Google Scholar]
- 543.Creek D J, Chiu F C K, Prankerd R J, Charman S A, Charman W N. J Pharm Sci. 2005;94:1820–1829. doi: 10.1002/jps.20400. [DOI] [PubMed] [Google Scholar]
- 544.Bousejra-El Garah F, Meunier B, Robert A. Eur J Inorg Chem. 2008:2133–2135. doi: 10.1002/ejic.200800129. [DOI] [Google Scholar]
- 545.Haynes R K, Chan W C, Lung C-M, Uhlemann A-C, Eckstein U, Taramelli D, Parapini S, Monti D, Krishna S. ChemMedChem. 2007;2:1480–1497. doi: 10.1002/cmdc.200700108. [DOI] [PubMed] [Google Scholar]
- 546.Moles P, Oliva M, Safont V S. J Phys Chem B. 2011;115:333–346. doi: 10.1021/jp1064903. [DOI] [PubMed] [Google Scholar]
- 547.Navacchia M L, Capobianco M L, D'Angelantonio M, Marconi G. Tetrahedron Lett. 2012;53:1296–1299. doi: 10.1016/j.tetlet.2012.01.007. [DOI] [Google Scholar]
- 548.Nosoongnoen W, Pratuangdejkul J, Sathirakul K, Jacob A, Conti M, Loric S, Launay J-M, Manivet P. Phys Chem Chem Phys. 2008;10:5083–5093. doi: 10.1039/b804516j. [DOI] [PubMed] [Google Scholar]
- 549.O'Neill P M, Barton V E, Ward S A. Molecules. 2010;15:1705–1721. doi: 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Robert A, Benoit-Vical F, Meunier B. Coord Chem Rev. 2005;249:1927–1936. doi: 10.1016/j.ccr.2004.12.022. [DOI] [Google Scholar]
- 551.Tang Y, Dong Y, Wang X, Sriraghavan K, Wood J K, Vennerstrom J L. J Org Chem. 2005;70:5103–5110. doi: 10.1021/jo050385+. [DOI] [PubMed] [Google Scholar]
- 552.Posner G H, Cumming J N, Ploypradith P, Oh C H. J Am Chem Soc. 1995;117:5885–5886. doi: 10.1021/ja00126a042. [DOI] [Google Scholar]
- 553.Taglialatela-Scafati O, Fattorusso E, Romano A, Scala F, Barone V, Cimino P, Stendardo E, Catalanotti B, Persico M, Fattorusso C. Org Biomol Chem. 2010;8:846–856. doi: 10.1039/B918600J. [DOI] [PubMed] [Google Scholar]
- 554.André-Barrès C, Najjar F, Bottalla A-L, Massou S, Zedde C, Baltas M, Gorrichon L. J Org Chem. 2005;70:6921–6924. doi: 10.1021/jo050439f. [DOI] [PubMed] [Google Scholar]
- 555.Najjar F, Gorrichon L, Baltas M, André-Barrès C, Vial H. Org Biomol Chem. 2005;3:1612–1614. doi: 10.1039/b503402g. [DOI] [PubMed] [Google Scholar]
- 556.Bousejra-El Garah F, Wong M H-L, Amewu R K, Muangnoicharoen S, Maggs J L, Stigliani J-L, Park B K, Chadwick J, Ward S A, O'Neill P M. J Med Chem. 2011;54:6443–6455. doi: 10.1021/jm200768h. [DOI] [PubMed] [Google Scholar]
- 557.Creek D J, Charman W N, Chiu F C K, Prankerd R J, Dong Y, Vennerstrom J L, Charman S A. Antimicrob Agents Chemother. 2008;52:1291–1296. doi: 10.1128/AAC.01033-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Robert A, Coppel Y, Meunier B. Inorg Chim Acta. 2002;339:488–496. doi: 10.1016/S0020-1693(02)00940-4. [DOI] [Google Scholar]
- 559.Jourdan J, Matile H, Reift E, Biehlmaier O, Dong Y, Wang X, Mäser P, Vennerstrom J L, Wittlin S. ACS Infect Dis. 2016;2:54–61. doi: 10.1021/acsinfecdis.5b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Kimura K-i, Sakamoto Y, Fujisawa N, Uesugi S, Aburai N, Kawada M, Ohba S-i, Yamori T, Tsuchiya E, Koshino H. Bioorg Med Chem. 2012;20:3887–3897. doi: 10.1016/j.bmc.2012.04.025. [DOI] [PubMed] [Google Scholar]
- 561.Davies K J A. IUBMB Life. 2000;50:279–289. doi: 10.1080/713803728. [DOI] [PubMed] [Google Scholar]
- 562.Arai H. Oxidative Modification of Lipoproteins. In: Kato Y, editor. Lipid Hydroperoxide-Derived Modification of Biomolecules. Vol. 77. Netherlands: Springer; 2014. pp. 103–114. [DOI] [Google Scholar]
- 563.Mohd Fauzi N, Spickett C. Lipid Oxidation. In: Roberts S M, Kehrer J P, Klotz L-O, editors. Studies on Experimental Toxicology and Pharmacology. Springer International Publishing; 2015. pp. 43–79. [DOI] [Google Scholar]
- 564.Cadet J, Di Mascio P. PATAI'S Chemistry of Functional Groups. John Wiley & Sons, Ltd.; 2009. Peroxides in Biological Systems. [DOI] [Google Scholar]
- 565.Jahn U, Galano J-M, Durand T. Angew Chem, Int Ed. 2008;47:5894–5955. doi: 10.1002/anie.200705122. [DOI] [PubMed] [Google Scholar]
- 566.Porter N A. J Org Chem. 2013;78:3511–3524. doi: 10.1021/jo4001433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Ricciotti E, FitzGerald G A. Arterioscler, Thromb, Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Roberts L J, Fessel J P. Chem Phys Lipids. 2004;128:173–186. doi: 10.1016/j.chemphyslip.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 569.Yin H, Havrilla C M, Morrow J D, Porter N A. J Am Chem Soc. 2002;124:7745–7754. doi: 10.1021/ja0201092. [DOI] [PubMed] [Google Scholar]
- 570.Yin H, Xu L, Porter N A. Chem Rev. 2011;111:5944–5972. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- 571.Itri R, Junqueira H C, Mertins O, Baptista M S. Biophys Rev. 2014;6:47–61. doi: 10.1007/s12551-013-0128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Xu L, Porter N A. Free Radical Res. 2015;49:835–849. doi: 10.3109/10715762.2014.985219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Miyoshi N, Iuliano L, Tomono S, Ohshima H. Biochem Biophys Res Commun. 2014;446:702–708. doi: 10.1016/j.bbrc.2013.12.107. [DOI] [PubMed] [Google Scholar]
- 574.Costas M, Mehn M P, Jensen M P, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 575.Sainsbury P D, Mineyeva Y, Mycroft Z, Bugg T D H. Bioorg Chem. 2015;60:102–109. doi: 10.1016/j.bioorg.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 576.Guzik U, Hupert-Kocurek K, Wojcieszyńska D. Intradiol Dioxygenases – The Key Enzymes in Xenobiotics Degradation. In: Chamy R, Rosenkranz F, editors. Biodegradation of Hazardous and Special Products. InTech; 2013. [DOI] [Google Scholar]
- 577.Komatsuzaki H, Shiota A, Hazawa S, Itoh M, Miyamura N, Miki N, Takano Y, Nakazawa J, Inagaki A, Akita M, et al. Chem – Asian J. 2013;8:1115–1119. doi: 10.1002/asia.201300029. [DOI] [PubMed] [Google Scholar]
- 578.Arnold A, Metzinger R, Limberg C. Chem – Eur J. 2015;21:1198–1207. doi: 10.1002/chem.201405155. [DOI] [PubMed] [Google Scholar]
- 579.Poureskandari M, Safaei E, Sajjadi S M, Karimpour T, Jaglicic Z, Lee Y-I. J Mol Struct. 2015;1094:130–136. doi: 10.1016/j.molstruc.2015.04.008. [DOI] [Google Scholar]
- 580.Borowski T, Siegbahn P E M. J Am Chem Soc. 2006;128:12941–12953. doi: 10.1021/ja0641251. [DOI] [PubMed] [Google Scholar]
- 581.Mendel S, Arndt A, Bugg T D H. Biochemistry. 2004;43:13390–13396. doi: 10.1021/bi048518t. [DOI] [PubMed] [Google Scholar]
- 582.Kovaleva E G, Rogers M S, Lipscomb J D. Biochemistry. 2015;54:5329–5339. doi: 10.1021/acs.biochem.5b00709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Váradi T, Pap J S, Giorgi M, Párkányi L, Csay T, Speier G, Kaizer J. Inorg Chem. 2013;52:1559–1569. doi: 10.1021/ic302378r. [DOI] [PubMed] [Google Scholar]
- 584.Xin M, Bugg T D H. J Am Chem Soc. 2008;130:10422–10430. doi: 10.1021/ja8029569. [DOI] [PubMed] [Google Scholar]
- 585.Harrison P J, Bugg T D H. Arch Biochem Biophys. 2014;544:105–111. doi: 10.1016/j.abb.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 586.Harrison P J, Newgas S A, Descombes F, Shepherd S A, Thompson A J, Bugg T D H. FEBS J. 2015;282:3986–4000. doi: 10.1111/febs.13400. [DOI] [PubMed] [Google Scholar]