

Abstract
Basal-like breast cancer (BLBC) accounts for the most aggressive types of breast cancer, marked by high rates of relapse and poor prognoses and with no effective clinical therapy yet. Therefore, investigation of new targets and treatment strategies is more than necessary. Here, we identified a receptor that can be targeted in BLBC for efficient and specific siRNA mediated gene knockdown of therapeutically relevant genes such as the histone demethylase GASC1, which is involved in multiple signaling pathways leading to tumorigenesis. Breast cancer and healthy breast cell lines were compared regarding transferrin receptor (TfR) expression via flow cytometry and transferrin binding assays. Nanobioconjugates made of low molecular weight polyethylenimine (LMW-PEI) and transferrin (Tf) were synthesized to contain a bioreducible disulfide bond. siRNA complexation was characterized by condensation assays and dynamic light scattering. Cytotoxicity, transfection efficiency, and the targeting specificity of the conjugates were investigated in TfR positive and negative healthy breast and breast cancer cell lines by flow cytometry, confocal microscopy, RT-PCR, and Western blot. Breast cancer cell lines revealed a significantly higher TfR expression than healthy breast cells. The conjugates efficiently condensed siRNA into particles with 45 nm size at low polymer concentrations, showed no apparent toxicity on different breast cancer cell lines, and had significantly greater transfection and gene knockdown activity on mRNA and protein levels than PEI/siRNA leading to targeted and therapeutic growth inhibition post GASC1 knockdown. The synthesized nanobioconjugates improved the efficiency of gene transfer and targeting specificity in transferrin receptor positive cells but not in cells with basal receptor expression. Therefore, these materials in combination with our newly identified siRNA sequences are promising candidates for therapeutic targeting of hard-to-treat BLBC and are currently further investigated regarding in vivo targeting efficacy and biocompatibility.
Keywords: nanoparticle, siRNA, polyethylenimine (PEI), transferrin, transferrin receptor, GASC1, artificial virus, targeting, breast cancer, oncogene, targeted therapy
Graphical Abstract
INTRODUCTION
Basal-like breast cancer (BLBC) accounts for the most aggressive types of breast cancer and currently cannot be treated specifically. Therefore, investigation of new targets and treatment strategies is more than necessary. One possible new group of targets are “epigenetic enzymes” (e.g., histone modifying and DNA methylating enzymes) as the causative links between genetics and epigenetics in cancer development are well documented.1,2 There is also an increasing appreciation that epigenetic aberrations that are often directly caused by genetic defects resulting in loss or gain of function of epigenetic regulators contribute significantly to cancer onset, severity, and progression.3 Specifically, a genetic alteration in the gene encoding an “epigenetic enzyme” can lead to changes within the histone code,4 which is involved in both tumorigenesis and cancer stem cell generated hierarchies in multiple tumor types.4
The possibility of reversing epigenetic codes may provide new targets for therapeutic intervention. In the present work, we focused on therapeutically downregulating a recently discovered gene which was first identified and cloned as a novel cancer gene by Yang et al.5 The gene amplified in squamous cell carcinoma 1 (GASC1, also referred to as JMJD2C/KDM4C) encodes a nuclear protein that catalyzes lysine demethylation of histones5 and is involved in several cancers, including breast cancer.6,7 Thus, downregulation of GASC1 expression represents an excellent therapeutic approach but is currently not possible in a cell specific manner. Moreover, recent findings clearly established that the GASC1 gene is amplified in approximately 15% of breast cancers,8,9 and its overexpression is more prevalent in basal-like breast cancers (BLBC).10 Besides, GASC1 overexpression is correlated with a poor prognosis and induces transformed phenotypes.9 Previous studies have demonstrated that therapeutic interference with GASC1 using an shRNA-based approach inhibits proliferation of breast cancer cells in vitro and in vivo.9,11 However, in these studies, viral vectors were used to express shRNA for GASC1 knockdown. Besides the immunologic challenges and safety issues viral vectors bear, viruses do not transduce cells in a specific manner. However, targeting can be achieved with chemically engineered artificial viruses.12
Human breast carcinomas represent a heterogeneous group of tumors that are diverse in their behavior, outcome, and response to therapy. Breast cancer is generally classified in four main groups based on their expression of estrogen receptor (ER), progesterone receptor (PR), and Her2/neu receptor (HER2): two of the main groups are ER+ and include luminal A (ER+, PR+, and HER2−) and luminal B (ER+, PR+, and HER2+) subtypes, and two groups are ER−, HER2 overexpressing (ER2−, PR−, HER+) and basal-like (ER−/low, PR−/low, HER2−/low).13
Among the different types of breast cancer, basal-like breast cancers (BLBC) are particularly aggressive and are associated with high histological grade, prevalence of younger patient age, and poor prognoses, and they often relapse rapidly.13 The treatment for BLBC consists of standard chemotherapy regimens as no effective molecularly targeted therapies have been developed.14
Failure in the current conventional therapies for BLBC patients urges the development of novel strategies to achieve favorable outcomes.
Combining molecular genetic information to derive new cancer-specific therapeutics with nanotechnologies applied to medicine via nanoparticle drug delivery will lead to the design of more specifically tailored therapies.15 RNA interference holds promise for molecular therapy of targets so far believed to be “undruggable”, including epigenetic targets in breast cancer.16 To be able to downregulate the newly identified oncogenic histone modifying enzyme GASC1, artificial viruses, i.e., nanoparticles, were chemically engineered to deliver therapeutic siRNA. Nanoparticles offer an unprecedented opportunity in rational drug and gene delivery by enhancing therapeutic efficacy and reducing side effects through overcoming major obstacles such as nonspecific biodistribution with consequent dose-limiting toxicity and multidrug resistance (MDR).17 In order to further improve the tumor targeting capability of the nanoparticle system described here, the targeting ligand transferrin (Tf) was conjugated after investigating TfR as a potential target in BLBC. Upregulation of TfR expression in a wide variety of cancers and its function and highly efficient recycling pathway provide a gate for intracellular gene and drug delivery to BLBC in a tumor-specific manner.18 Therefore, we designed here two specifically targeted nanobioconjugates to improve the delivery of therapeutic siRNA for efficient downregulation of the overexpressed GASC1 in BLBC. We identified a new siRNA sequence that efficiently reduces GASC1 expression if delivered intracellularly. To the best of our knowledge, this is the first report of developing a BLBC-targeted delivery system with a newly identified siRNA for therapeutic GASC1 gene knockdown. Therefore, our approach is promising for therapeutic targeting of hard-to-treat BLBC and could result in new receptor mediated therapy options for basal-like breast cancer.
MATERIALS AND METHODS
Materials
Polyethylenimine (PEI, MW 5 kDa) was obtained from BASF (Lupasol G100, Ludwigshafen, Germany). N-Succinimidyl 3-(2-pyridyldithio)propionate (SPDP), a PEGylated SPDP (PEG4-SPDP), Lipofectamine 2000, trypsin, penicillin/streptomycin, 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES), and heparin sodium (140,000 U/g) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Anti-human CD71 (transferrin receptor) PE was acquired from eBioscience (San Diego, CA, USA). Dithiothreitol (DTT) was purchased from Alfa Aesar, MA, USA. SYBR Gold dye and CellTiter-Blue were obtained from Life Technologies (Carlsbad, CA, USA). RPMI-1640 medium (1×) with 2.05 mM L-glutamine, Ham’s F-12 medium, and trypan blue (0.4%, sterile filtered) was purchased from HyClone (GE Health Care, Pittsburgh, PA, USA). Human holo-transferrin (80 kDa) was obtained from Calbiochem (Billerica, MA, USA). Dulbecco’s phosphate buffered saline (PBS), fetal bovine serum (FBS), D-(+)-glucose, 2-mercaptoethanol, dimethyl sulfoxide (DMSO, ≥99.7%), and sodium bicarbonate (NaHCO3) were purchased from Sigma-Aldrich (St. Louis, MO, USA). DNase I reaction buffer and DNase/RNase-free water were purchased from Zymo Research (Irvine, CA, USA). Hs_GASC1 primer (forward and reverse) and sequences V4, V6, and V7 (self-designed siRNA against GASC1 using the GENtle software by Magnus Manske) were ordered from Invitrogen (Life Technologies, Carlsbad, CA, USA). Commercially available GASC1 specific siRNA was purchased from Qiagen (Valencia, CA, USA) for sequences A, B, C, and D. SMART pool and sequences E, F, G, and H (Table 1) were purchased from Dharmacon (Thermo Fisher, Waltham, MA, USA).
Table 1a.
| sequence name | sequences | |
|---|---|---|
| negative control | MISSION siRNA Universal Negative Control #1 | |
| hGAPDH | IDT sense | 5′-pGGUCGGAGUCAACGGAUUUGGUC[dG][dT] |
| IDT antisense | 3′-UmUmCCmAGmCCmUCmAGmUUmGCmCUmAAACCAGCA | |
| hGASC1 | V4 sense | 5′-ACCGAAGACAUGGACCUCU[dT][dT] |
| V4 antisense | 3′-[dT][dT]UGGCUUCUGUACCUGGAGA | |
| hGASC1 | V6 sense | 5′-CAGAAUUUAUCAGAUCAUA |
| V6 antisense | 3′-[dT][dT]GUCUUAAAUAGUCUAGUAU | |
| hGASC1 | V7 sense | 5′-GUGAAGAUUUCAAUGGAUA[dT][dT] |
| V7 antisense | 3′-[dT][dT]CACUUCUAAAGUUACCUAU | |
| hGASC1-A | Hs_KDM4C1 sense | 5′-CGAAGCUUCCAGUGUGCUA[dT][dT] |
| Hs_KDM4C1 antisense | 3′-[dG][dG]GCUUCGAAGGUCACACGAU | |
| hGASC1-B | Hs_JMJD2C3 sense | 5′-GAUAUUAAUGGGAGCAUAU[dT][dT] |
| Hs_JMJD2C3 antisense | 3′-[dG][dT]CUAUAAUUACCCUCGUAUA | |
| hGASC1-C | Hs_JMJD2C4 sense | 5′-CCAGAAGUUCGAUUCACUA[dT][dT] |
| Hs_JMJD2C4 antisense | 3′-[dA][dG]GGUCUUCAAGCUAAGUGAU | |
| hGASC1-D | Hs_JMJD2C5 sense | 5′-CAUCGGAGGGAAAGACUAA[dT][dT] |
| Hs_JMJD2C5 antisense | 3′-[dA][dT]GUAGCCUCCCUUUCUGAUU | |
| hGASC1-E | J-004293-12 sense | 5′-GAGAAGUCGUCCAAGUCAAUU |
| J-004293-12 antisense | 3′-UUCUCUUCAGCAGGUUCAGUU | |
| hGASC1-F | J-004293-11 sense | 5′-GCAUAUAUGAUGAGGGUGUUU |
| J-004293-11 antisense | 3′-UUCGUAUAUACUACUCCCACA | |
| hGASC1-G | J-004293-10 sense | 5′-ACGAAGAUUUGGAGCGCAAUU |
| J-004293-10 antisense | 3′-UUUGCUUCUAAACCUCGCGUU | |
| hGASC1-H | J-004293-9 sense | 5′-AUUACAUGCUUUCGACAUAUU |
| J-004293-9 antisense | 3′-UUUAAUGUACGAAAGCUGUAU |
Deoxyribonucleotides are presented in parentheses with the letter d, and 2′O-methylated ribonucleotides are denoted with the letter m. The letter p indicates an additional phosphate.
Cell Line Characteristic and Culture Conditions
The nontumorigenic human mammary epithelial cell line MCF10A and human breast cancer cell line HCC1954 were purchased from the American Type Culture Collection (Manassas, VA, USA). The COLO824 cell line was obtained from DSMZ (Germany), and the cell lines SUM102, SUM225, SUM52, and SUM149 were a kind gift from Dr. Stephen P. Ethier (Department of Pathology and Laboratory Medicine, Medical University of South Carolina). The COLO824 and HCC1954 cell lines were retained and grown in RPMI-1640 cell culture medium supplemented with 10% fetal bovine serum. MCF10A and SUM cell lines were cultured in a special DMEM/F12 medium, containing 1% EGF (10 ng/mL), 0.1% hydrocortisone (1 μg/mL), and 0.5% insulin (5 μg/mL). The cell line characteristics are shown in Table 2.
Table 2.
Different Breast Cell Line Models and Their Specifications
| cell line | cell source | subtype | PR | ER | Her2/neu | GASC1 |
|---|---|---|---|---|---|---|
| HCC1954 | grade III invasive ductal carcinoma | basal | − | − | + | +++ |
| COLO824 | mammary gland carcinoma | basal | − | − | − | +++ |
| MCF10A | mammary gland-immortalized | − | − | − | − | |
| SUM102 | intraductal carcinoma/microinvasion | basal | − | − | − | − |
| SUM225 | chest wall recurrence of ductal carcinoma | her2+ | − | − | + | − |
| SUM149 | invasive ductal carcinoma (inflammatory) | basal | − | − | − | + |
| SUM52 | pleural effusion | luminal | − | + | − | − |
Transferrin Receptor Expression Study
Selected breast cancer cell lines were used to evaluate transferrin receptor expression levels (CD71, TfR) by flow cytometry. Of each cell suspension, 200,000 cells were transferred into an Eppendorf tube and spun down (350g for 5 min). The cell pellet was washed with PBS containing 2 mM EDTA. Afterward, PE-labeled CD71 antibody and its corresponding isotype control were each added to assigned cell samples and incubated for 30 min at 4 °C before analysis by flow cytometry (Attune acoustic focusing cytometer Life Technologies, Carlsbad, CA). Blank cells were not stained with any antibody. The excitation wavelength of 488 nm and an emission filter at 585 nm/42 nm were used. Cell gating and data analysis in the Attune cytometric software were performed using cell morphology and mean fluorescence intensity (MFI), respectively. The mean values and standard deviation (SD) of experiments with n = 3 are shown.
Specific Binding of Transferrin to the Corresponding Receptor
To analyze the specificity of transferrin binding to TfR, Tf was fluorescently labeled with FITC (Invitrogen, life Technologies). Bovine serum albumin (BSA) was used as a control. Transferrin and BSA were dissolved in 0.1 M NaHCO3. A 10-fold molar excess of FITC (dissolved in DMSO) was incubated with transferrin and BSA overnight at 4 °C. The protein–FITC conjugates were subsequently separated from unbound FITC by gel permeation method (Sephadex G25 M, GE Healthcare PD-10 columns) using 20 mM HEPES-150 mM NaCl buffer. The BCA protein assay (Thermo Scientific) was used to measure the final concentration of transferrin and albumin after purification. The presence of FITC was found to not impact the measurements at the applied concentrations. After purification and characterization of the labeled (glyco-) proteins, HCC1954 and MCF10A cell lines (60,000 cells in 24 well plates) were incubated with different amounts of FITC–transferrin or FITC–BSA (20, 200, and 400 pmol) for 4 h at 37 °C and 5% CO2. Flow cytometry was used as described above to measure the binding of Tf–FITC or albumin–FITC to the cells with the emission filter set to 530/30 nm bandpass.
Coupling of Cell-Binding Ligand to Branched PEI as Targeting Moiety
Conjugation Synthesis and Characterization
Two different heterobifunctional cross-linking agents, namely, N-succinimidyl 3-(2-pyridyldithio)propionate (SPDP) and a PEGylated SPDP (PEG4-SPDP) containing four ethylene glycol units, were used to conjugate transferrin to a 5 kDa LMW PEI as described before.19 PEI (1 mg/mL) was reacted with SPDP or PEG4-SPDP (20 mM) in DMSO stirring at room temperature overnight (1:10 molar ratio relative to amines present). The intermediate products were purified by ultrafiltration using Amicon Ultra 4 mL filters (MWCO 3000) and Vivaspin centrifugal ultrafilters (10,000 MWCO) as described.19 The conjugates were separated from uncoupled Tf-SPDP by ion exchange FPLC via ÄKTA prime plus (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) as reported.20 The products (Tf–PEI and Tf-PEG-PEI) were preserved at 4 °C until use. The degree of SPDP coupling onto PEI was determined as previously reported by spectrophotometric detection of release of the pyridine-2-thione group.21 The characterization of the final products was performed by spectrophotometric detection of transferrin at 280 nm compared to a standard curve20 and quantification of PEI amines in using a modified TNBS method22 in the presence of the respective amount of transferrin as described by Germershaus et al.20
Preparation of siRNA-Polymer Nanoparticles
For preparing nanoparticles based on electrostatic interaction resulting in the formation of polyelectrolyte complexes, or polyplexes, the PEI/siRNA charge ratios (defined as N/P ratios) were calculated based on PEI nitrogen (N) groups per siRNA phosphate (P) group.23 Polymer (1 mg/mL) and siRNA (100 μM) stock solutions were diluted with a 5% glucose solution (pH 7.4, prepared with RNase free water). Equal volumes of PEI or modified PEI and siRNA solutions were mixed to result in the respective N/P ratio, vortexed for 30 s, and incubated for 20 min at room temperature before use.
Physicochemical Properties of siRNA/PEI Polyplexes
Analysis of Hydrodynamic Diameters and Zeta (ζ) Potentials
The hydrodynamic diameters and zeta potential of polyplexes prepared with 40 pmol siRNA and polymer at N/P 7 were measured in PBS using a Zetasizer Nano ZS (Malvern Instruments Inc., Westborough, MA, USA) and the refractive index and viscosity settings for water. Dynamic light scattering (DLS) was detected at 25 °C at an angle of 173°. Results from triplicate samples are shown as Z average in nanometers ± SD. Samples were then diluted with 700 μL of PBS buffer for ζ-potential measurements. Results of three independent measurements are given in millivolts ± SD.
Atomic Force Microscopy
Polyplex morphology was determined using atomic force microscopy (AFM). For image acquisition, a drop of polyplexes prepared at N/P 7 was incubated for 5 min on a freshly cleaved mica surface. Excess solution was rinsed off with deionized water, and the mica was dried and imaged at room temperature using a Pico LE atomic force microscope (Molecular Imaging, Agilent Technologies) with a Si3Ni4 V-shaped cantilever in contact mode as described before.23
SYBR Gold Condensation Assays
The capacity of the Tf–PEI and Tf-PEG-PEI to condense siRNA at different N/P ratios (0–20) was evaluated in SYBR Gold assays, and PEI (5 kDa) was used as control. Formulations with 50 pmol of siRNA per well were placed in a 96-well plate, vortexed, and incubated at room temperature for 20 min. Fluorescence (Ex/Em: 495/537 nm) was measured after 10 min of incubation with a 4× SYBR Gold solution using a Synergy 2 microplate reader (BioTek, Winooski, VT, USA).
Stability of siRNA/PEI Complexes: Heparin Dissociation Assays
Polyplex formulations with 50 pmol of siRNA per well prepared at N/P 7 were incubated with a fixed heparin concentration (0.2 IU) over varying periods of time (5–180 min). Fluorescence was measured with a Synergy 2 microplate reader (BioTek Instruments) after adding a 4× SYBR Gold solution (Ex/Em: 495/537 nm). The mean values of three fluorescence intensity measurements of SYBR Gold dye only (0) and of three samples of SYBR Gold intercalating with free siRNA (1) were used for normalization and calculation of the relative stability of the polyplexes. Results of triplicate samples are shown as mean values ± SD.
Quantification of Cellular siRNA Uptake by Flow Cytometry
Cells were seeded with 80,000 cells per well in 24-well plates 24 h before they were transfected with polyplexes prepared with 50 pmol of Alexa Fluor 488 labeled siRNA per well at N/P 7. Negative controls included untreated cells and cells treated with free Alexa Fluor 488 labeled siRNA. PEI (5 kDa) was used as a positive control for comparison. After 4 h of transfection with 100 μL of polyplexes, 1 mL of medium was added and cells were incubated at 37 °C and 5% CO2 for another 24 h. The cells were washed with PBS and trypsinized. Intracellular fluorescence from siRNA was detected using an Attune acoustic focusing flow cytometer (Thermo Fisher Scientific, Carlsbad, CA) and corresponding software. For the distinction between internalized and surface-bound siRNA, trypan blue (0.4%) was used to quench surface-bound fluorescence. Data were analyzed after gating for 10,000 viable cells, and median fluorescence intensity (MFI) values are given for three independent studies.
Confocal Laser Scanning Microscopy
HCC 1954 cells were seeded in 8-well chamber slides at a density of 25,000 cells per well (Permanox Slide, Nunc, Thermo Fisher Scientific) and allowed to grow for 24 h at 37 °C in 5% CO2. Polyethylenimine (PEI), Lipofectamine (LF), or Tf–PEI polyplexes made with Tye563-labeled siRNA (Ex/Em: 543/563 nm) were used to transfect cells as described earlier. Subsequently, the cells were washed with PBS and fixed using 3% paraformaldehyde (Affymetrix, Cleveland, OH, USA) for 30 min, followed by nuclei staining with DAPI (Ex/Em: 405/470 nm). Finally, the cells were embedded using Fluorsave reagent (Calbiochem, Merck, Darmstadt, Germany) and covered with a coverslip. The images were recorded with a Zeiss LSM 780 microscope (Zeiss, Oberkochen, Germany). Representative pictures are shown in Figure 9 (100× magnification).
Figure 9.
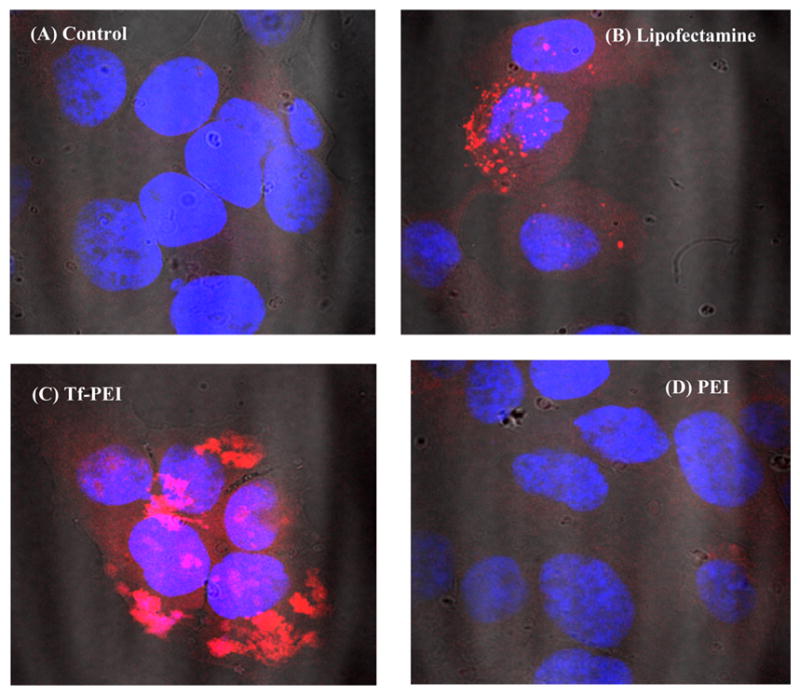
Confocal microscopy images of cellular interaction of different complexes with the HCC1954 cell line after 4 h incubation with siRNA polyplexes. HCC1954 cells were seeded with 25,000 cells per chamber and transfected with 25 pmol of siRNA (TYE 563 labeled, indicated by red color) for 4 h. Cells were fixed and the nuclei stained with DAPI (indicated by blue color). Representative pictures are shown (100× magnification): (A) blank; siRNA polyplexes with (B) Lipofectamine; (C) Tf–PEI (N/P 7); and (D) PEI (N/P 7).
Cytotoxicity of Polyplexes
Cells (HCC1954, COLO824, SUM102, and MCF10A) were seeded at a density of 10,000 cells in a 96-well plate (Thermo Scientific) and incubated for 24 h at 37 °C and 5% CO2. Polyplexes containing 50 pmol of siRNA were added to the cells, and plates were incubated for 24 h at 37 °C. Subsequently, CellTiter-Blue reagent was added directly to each well, and the plates were incubated at 37 °C for 2 h to allow the cells to convert resazurin to resorufin. The obtained fluorescence was measured at Ex/Em 560/590 nm. The percentage of cell viability and proliferation was calculated as the ratio between the fluorescence of a sample and the untreated control cells as follows: ([A] test/[A] control) × 100%. Results are shown as the mean value ± SD of triplicate samples.
Knockdown Measured by qRT-PCR and Western Blot
HCC1954 cells were seeded with 500,000 cells per well in 6-well plates (Corning Incorporated) and were incubated for 24 h before transfection at 37 °C and 5% CO2. PEI, Tf–PEI, and Tf-PEG-PEI polyplexes were prepared with 100 pmol of GASC1 siRNA at N/P 7 in a total volume of 100 μL and were added to 900 μL of cell culture medium per well. The cells were incubated with the polyplexes for 4 h before 2 mL of fresh medium was added to each well. The cells were then incubated for an additional 24 h for PCR or 72 h for Western Blot analysis. Lipofectamine (LF) (0.5 μL/10 pmol of siRNA) was used as a positive transfection control. Cells were transfected with samples in fresh medium, and subsequently cells were washed with PBS and lysed with lysis buffer (PureLink RNA mini, Ambion, Fisher Scientific, USA). Total RNA was then isolated from cells according to the manufacturer’s protocol with supplementary DNase I digestion; mRNA was mixed with qScript cDNA SuperMix kit (Quanta Biosciences, Gaithersburg, MD, USA) and then converted into cDNA through a reverse transcription reaction for real time PCR (Eppendorf Master-cycler gradient, Hauppauge, NY, USA). Primers set for GASC1 genes were ordered from Invitrogen (Life Technologies). A PUM1 primer (Qiagen, Valencia, CA, USA) was used for normalization. Semiquantitative RT-PCR was performed using the FastStart Universal SYBR Green Master (ROX) using a Bio-Rad iCycler Thermal Cycler (IQ 5 multicolor read thermal PCR). Results are given as the average of n = 3 ± SD.
For measuring protein levels after transfection of the HCC1954 cell lines with modified PEI formulations, Western blot analysis was performed after protein extraction of the transfected cells following a 7 day in vitro incubation based on previous protocol optimization. The cells were lysed in RIPA buffer containing protease inhibitor cocktail (Calbiochem, San Diego, CA, USA). Samples were size-fractionated by 7.5% SDS–PAGE (XCell SureLock Mini-Cell), and the blot was transferred to Immunobilon-PVDF membranes (Millipore, Bedford, MA, USA). The blot was then incubated in blocking buffer (5% milk, 0.1% Tween in TBS buffer) at room temperature for 1 h and subsequently incubated with rabbit anti-JMJD2C antibody (Bethyl Laboratories, Inc., Montgomery, TX, USA) in precooled blocking buffer overnight at 4 °C. After washing in TBS/0.1% Tween, The blot was incubated with horseradish peroxidase (HRP) labeled anti-rabbit secondary antibody at room temperature for 1 h. After washing with TBS/0.1% Tween, the blot was developed by enhanced chemiluminescence (Thermo Scientific SuperSignal West Pico), and visualized with a HyBlot CL film (Denville).
Therapeutic Effects of GASC1 Gene Knockdown on Cell Proliferation
Therapeutic effects on cell proliferation were measured in MTT assays after transfection with siRNA against GASC1 or a scrambled control. In 24-well plates (Corning Incorporated), HCC1954 cells were seeded at a density of 8,000 cells per well and incubated for 24 h at 37 °C and 5% CO2. Polyplexes were prepared with 50 pmol of GASC1 siRNA or a scrambled negative control siRNA, and the cells were transfected on days 1, 3, and 6 after seeding. MTT assays were performed as described above on days 2 and 10 after seeding. The results are shown as mean values ± SD of 3 independent measurements in comparison to proliferation of untreated cells.
Statistical Analysis
Results from all experiments represent at least three independent measurements and are presented as mean value ± SD as analyzed by Graph Pad Prism 6.0 software (GraphPad Software, La Jolla, CA, USA). Statistical tests were either one or two way ANOVA with a Bonferroni post hoc analysis (significance level 0.05). Significantly different results are indicated with an asterisk (*) in figures. Corresponding significance values are *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
RESULTS
Screening for TfR Expression Levels
Transferrin receptor expression levels were determined via flow cytometry in a series of breast cell lines which were characterized as GASC1 positive or negative breast cell line to select a valid cell line model and its controls for the targeted siRNA delivery.9 The receptor expression of TfR (CD71) was compared to the MCF10A cell line, an immortalized, nontransformed epithelial cell line, showing no GASC1 amplification.9 The expression was verified using a PE-labeled anti-CD71 antibody. Non-specific binding of the antibody was precluded based on experimental results using an isotype IgG (negative control) and nontreated cells (blank). PE-labeled CD71 antibody and its corresponding isotype control were each added to assigned cell samples. Figure 1 depicts that MCF-10A cells bear only basal levels of TfR whereas the expression level in HCC1954 cells was almost 9-fold higher than in MCF10A. Therefore, HCC1954 cells were selected as cell model to verify GASC1 knockdown via targeting the internalizing transferrin receptor. Screening of TfR expression levels in other breast cancer model cell lines showed that GASC1-amplified COLO824 cells present higher TfR expression levels than SUM102, SUM52 (p < 0.001), and MCF10A (p < 0.0001) but TfR levels were not significantly different between COLO824, SUM149, and SUM225 cell lines (P > 0.05). Therefore, all three of those cell lines could be used as another valid cell culture model for therapeutic GASC1 knockdown.
Figure 1.
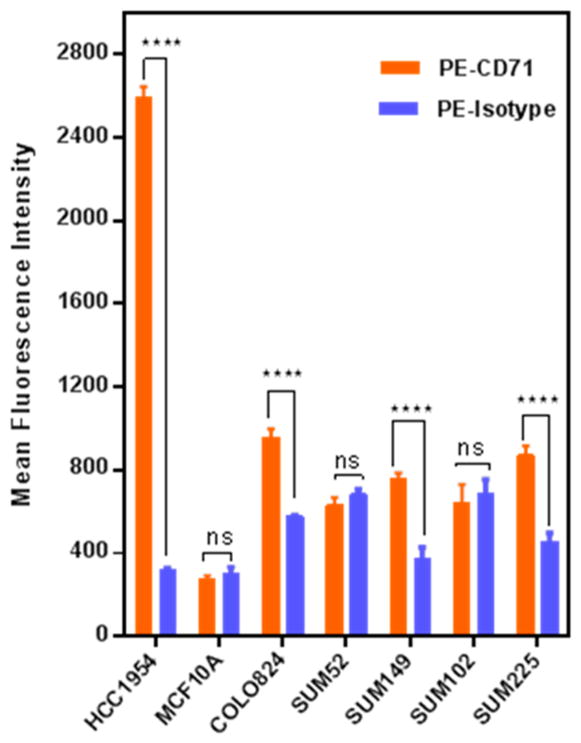
Screening of TfR expression levels in different cancer cell lines compared to MCF 10A cells. Using flow cytometry, TfR overexpression on the breast cancer cell line HCC1954 was verified, which also shows a comparably high GASC1 amplification. The transferrin receptor expression (CD71, TfR) was compared to the MCF-10A cell line, an immortalized, nontransformed epithelial cell line, showing no GASC1 amplification. The expression was verified by using a PE-labeled anti-CD71 antibody, and nonspecific binding of the antibody was precluded based on results with an isotype control. Data are expressed as the mean ± SD (n = 3); ns, nonsignificant; ****p < 0.0001.
Specific Binding of Tf to Transferrin Receptor
To confirm the endocytosis activity of Tf receptors on cell lines determined to overexpress the receptor, HCC1954 and MCF10A cell lines were incubated with FITC-labeled transferrin in comparison to FITC-labeled albumin. The same amount of transferrin (20 pmol) as used in the Tf–PEI uptake experiment was chosen, and results were compared to higher amounts of transferrin or albumin (200 and 400 pmol). Quenching of noninternalized Tf or albumin was achieved by trypan blue treatment. Figure 2 clearly shows that a 2.3-fold increase in Tf internalization is detectable in the HCC1954 cell line compared to albumin after incubation with 20 pmol of FITC-labeled proteins. Meanwhile, FITC–transferrin showed an almost 3-fold higher internalization in HCC1954 cells compared to MCF10A for the same amount of transferrin. There was no significant difference in the uptake of FITC–Tf in the MCF10A cell line compared to FITC–albumin (p > 0.05). A notable increase in MFI of the HCC1954 cell line was detected with increasing the amount of FITC–transferrin from 20 to 200 and 400 pmol (p < 0.0001), however this was not the case for FITC–albumin (p > 0.05). The MFI in MCF10A cell lines with basal levels of TfR, however, did not change with increasing the amount of transferrin from 20 to 200 pmol or 200 to 400 pmol (p > 0.05).
Figure 2.

Comparison of FITC–transferrin or FITC–albumin internalization in HCC1954 (lower left bar graph) and MCF10A (lower right bar graph) cell lines at different concentrations of transferrin and albumin via flow cytometry. Data are expressed as the mean ± SD (n = 3); ns, nonsignificant; ****p < 0.0001.
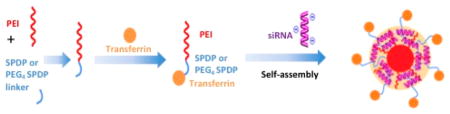
Development of Transferrin Conjugation with PEI through Bioreducible Linker
Coupling procedures for the synthesis of Tf–PEI conjugates are illustrated in Figure 3. SPDP and its PEGylated form (PEG4-SPDP) with a 25.7 Å ethylene glycol spacer are amine- and sulfhydryl-reactive heterobifunctional cross-linkers that produce disulfide-containing linkages which are stable extracellularly but can be cleaved intracellularly.24 This design was chosen to prevent premature release of the targeting ligand but will mediate release of the nanoparticle from Tf and the recycling TfR inside the cell.
Figure 3.

Schematic illustration of transferrin cross-linking with PEI by SPDP.
Physicochemical Properties of the Nanoparticles
Based on DLS and zeta potential measurements, both Tf–PEI and Tf-PEG-PEI more efficiently formed dense polyplexes with siRNA than nonmodified 5 kDa PEI (Figure 4). Tf–PEI/siRNA and Tf-PEG-PEI/siRNA complexes at N/P 7 were compactly packed with 30 and 140 nm hydrodynamic diameters, respectively, and narrow size distributions (PDI: 0.1). However, PEI/siRNA exhibited larger mean diameter of 700 nm with somehow broader distribution (PDI: 0.4). In addition, the zeta potentials of Tf–PEI (−0.6 ± 0.07 mV) and Tf-PEG-PEI (−0.01 ± 0.04 mV) complexes with siRNA were lower than those of PEI (39.9 ± 7 mV) complexes, implying that most of the positively charged portion of PEI in was shielded by Tf in the Tf-modified polyplexes. These results reveal that modified PEI produced significantly smaller and more compact structures with siRNA compared to PEI at the same N/P ratio. The same trend was observed via AFM imaging, which showed the tendency of PEI polyplexes to aggregate into particles of several hundred nanometers in size, whereas the morphology of the Tf–PEI complexes was more homogeneous, the size distribution was narrow, and the particles did not show any aggregation tendency (Figure 5). In additional experiments, the capacity of PEI and the modified PEI polymers to condense siRNA at different N/P ratios was evaluated by SYBR Gold condensation assays. In these experiments, free (unbound) siRNA is accessible to the intercalating dye SYBR Gold. The free siRNA is thus subsequently quantified on the basis of fluorescence emitted after SYBR Gold treatment. Results were compared to PEI (5 kDa) as a control (Figure 6). PEI and Tf–PEI showed better siRNA condensation at N/P ratios 1–5, but all formulations showed similar condensation efficiency at N/P ratio of ≥7.
Figure 4.
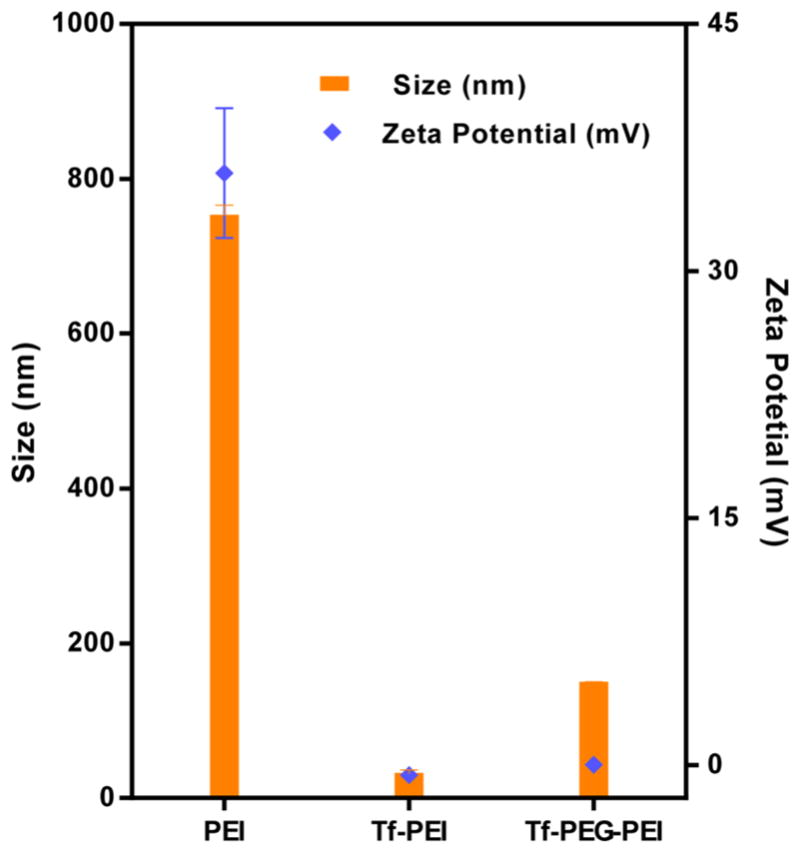
Hydrodynamic diameter (nm) and zeta potential (mV) of siRNA polyplexes made with PEI, Tf–PEI, and Tf-PEG-PEI at N/P ratio of 7 at room temperature. Data are expressed as the mean ± SD (n = 3).
Figure 5.

AFM topography of siRNA polyplexes made with PEI (left) and Tf–PEI (right) at N/P ratio of 7 and imaged after drying at room temperature.
Figure 6.

Condensation efficiency of Tf–PEI and Tf-PEG-PEI complexes with siRNA measured by SYBR Gold assay at different N/P ratios (1–20) compared to polyethylenimine (PEI 5 kDa). Results are given as the average of three measurements ± SD (n = 3).
A heparin-induced decomplexation stability test was performed to determine the stability of the polyplexes. The release profiles of siRNA from all polyplexes reached a maximum siRNA release after 30 min of incubation and then leveled off over the remaining incubation time (Figure 6). After 30 min of incubation with 0.2 IU heparin, PEI and Tf-PEG-PEI displayed around 50% and 85% of siRNA release, respectively, but Tf–PEI showed only 25% of siRNA release at that time point (Figure 7). The siRNA release from Tf–PEI polyplexes initially increased, then decreased gradually and remained unchanged (7%) until the end of the experiments (180 min) (Figure 7).
Figure 7.
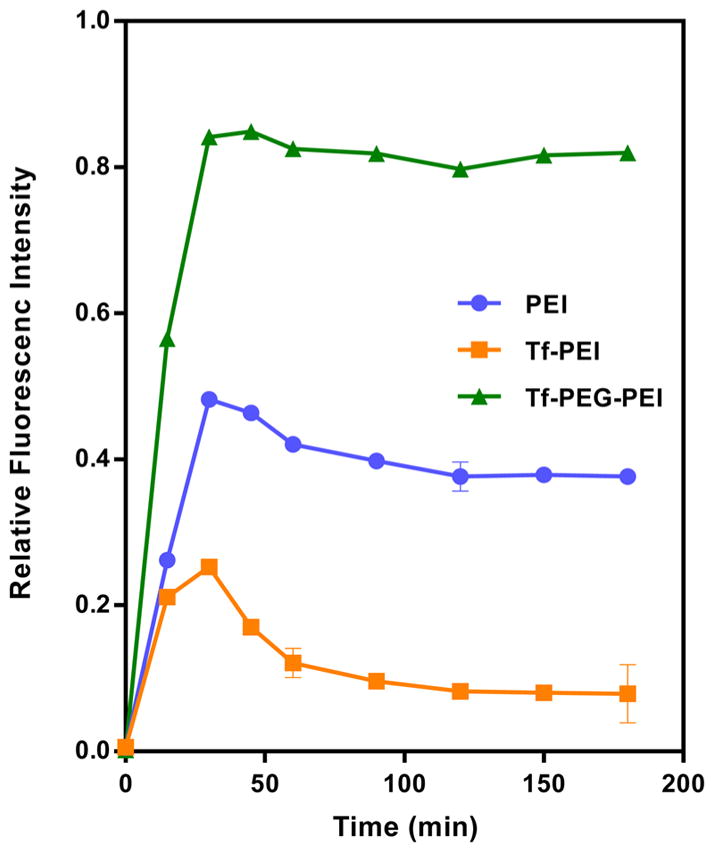
Release profile of siRNA from transferrin modified polyplexes (N/P 7; 50 pmol of siRNA/well) compared to polyethylenimine (PEI 5 kDa) as a function of time (0–185 min) with heparin concentration of 0.2 IU per well. Results are shown as relative mean fluorescence ± SD (n = 3).
Cellular Uptake of the Nanoparticles
Cellular uptake of polyplexes with 50 pmol of Alexa Fluor 488 labeled siRNA (N/P 7) was quantified in different cell lines (GASC1 positive and GASC1 negative) by flow cytometry. Trypan blue staining was employed on half of the samples to differentiate between surface bound and internalized siRNA. The results were compared to cells that were not treated with trypan blue, while cells treated with trypan blue are indicated with an asterisk in Figure 8. Overall, trypan blue treated cells showed slightly lower mean fluorescence intensities compared to cells that did not undergo quenching of surface-bound siRNA, indicating that a small fraction of the siRNA polyplexes was attached to the cell membrane and was not taken up intracellularly. However, this difference was not significant in any of the samples (p > 0.05). As can be seen in Figure 8, there is a significant difference (p < 0.0001) in uptake of PEI/siRNA in GASC1 positive cell lines (HCC1954 and COLO824) and GASC1 negative cells (MCF10A and SUM102). Interestingly, PEI alone showed better uptake in MCF10A and SUM102, which could be caused by higher endocytic activity of these two cell lines.25 However, PEI conjugated transferrin (Tf–PEI and Tf-PEG-PEI) showed very high uptake in the HCC1954 cell line, which was expected due to the higher level of TfR expression. The uptake efficiency of Tf–PEI polyplexes did not show any significant differences between HCC1954 and SUM102 cell lines (p > 0.05), although the TfR expression of HCC1954 cells is about 4 times higher than that of SUM102 cells (Figure 1). The mean fluoresce intensity in HCC1954 cells treated with Tf–PEI polyplexes was 2.5- and 10-fold higher than in MCF10A and COLO824 cells, respectively. As can be seen in Figure 8, Tf-PEG-PEI/siRNA polyplexes showed 2.6 times higher uptake in HCC1954 than in SUM102 cells. Uptake efficiency of the conjugates containing the PEG spacer was 3.6- and 6-fold higher in HCC1954 than MCF10A and COLO824 cells, respectively (Figure 8).
Figure 8.

Comparison of siRNA uptake with different formulations (Blank (untreated cells), PEI (positive control), Tf–PEI, Tf-PEG-PEI) in GACS1 positive (HCC1954 and COLO824) and GASC1 negative (MCF10A and SUM102) cell line models after fluorescence quenching of membrane bound nanoparticles with trypan blue (asterisks indicate cell samples treated with trypan blue). Data are expressed as the mean ± SD (n = 3); ns, nonsignificant.
The intracellular uptake of the nanoparticles was then analyzed by confocal microscopy (Figure 9). The images obtained revealed the presence of concentrated fluorescence after 4 h of incubation with Tf–PEI conjugates. The internalization was detected using a TYE 563 labeled siRNA (Figure 9). Higher nanoparticle binding to the cell surface and cell entry of the Tf–PEI conjugate compared to LF and polyethylenimine was visible. Interestingly, PEI delivered fluorescently labeled siRNA was not detected by microscopy, which was consistent with the flow cytometry results showing very poor uptake of PEI polyplexes in HCC1954 cells.
Cytotoxicity
In order to test one aspect of biocompatibility of the nanoparticle systems investigated here, the cytotoxicity of PEI and Tf-shielded polyplexes was analyzed. The rate of viability was assessed by CellTiter-Blue assays which are based on the ability of active mitochondria to convert a redox dye (resazurin) into a fluorescent end product (resorufin). Neither the conjugate polyplexes (Tf–PEI/siRNA and Tf-PEG-PEI/siRNA) nor PEI/siRNA at N/P 7 appeared to have any apparent cytotoxicity, and cell survival rates greater than 85% were maintained for all treatments (Figure 10). There is a significant change in cell viability observed alone in PEI treated COLO824 cells compared to Tf–PEI (p < 0.05). No significant differences in cytotoxicity among the different cells lines were observed (p > 0.05).
Figure 10.
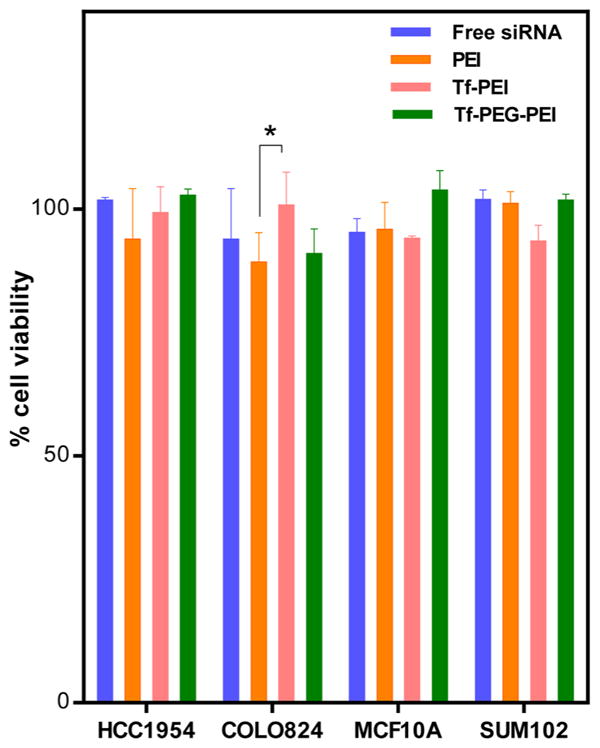
Cytotoxicity profile of siRNA polyplexes made with Tf–PEI or Tf-PEG-PEI measured by the CellTiter-Blue method compared to PEI/siRNA polyplexes. HCC1954, COLO824, SUM102, and MCF10A cell lines were treated with polyplexes for 4 h, and the viability of cells was measured after 24 h. Cell toxicity is presented as impaired cell viability as a percentage relative to control cells. Data are expressed as the mean ± SD (n = 5). *P < 0.05.
GASC1 Knockdown Optimization
To identify the amount of GASC1 knockdown on the mRNA level, several transfection parameters were optimized and determined by RT-PCR. The primary studies were performed with 9 commercially available siRNA sequences (A–D, E–H, and mixed) to screen their efficacy against GASC1 since no validated sequences are available. LF was used as standard transfection agent. Figure 11 shows the GASC1 knockdown efficiency in HCC1954 cells with the above-mentioned siRNA sequences. Among the different sequences, the strongest relative reduction in GASC1 RNA was achieved with sequence A. However, there was no significant difference between A and sequences C, D, and E (p > 0.05). Based on this result, sequence A was applied for further optimization experiments. Gene silencing efficiencies were investigated after 6, 17, 24, and 48 h post transfection (Figure 12). Clearly, the GASC1 mRNA level was reduced significantly after 24 h but was not significantly further reduced 48 h post transfection (p > 0.05). However, there was no measurable gene knockdown compared with the negative/nontreated control at the 6 h and 17 h time points. Additionally, the GASC1 mRNA levels measured at 6 and 17 h were not significantly different as well as the levels measured at 17 and 48 h post transfection with sequence A (Figure 12). The optimum transfection time point of 24 h was then used to probe the effect of different siRNA doses (100, 150, 200, 250 pmol). However, the most efficient gene knockdown was already achieved at 100 pmol, so that further optimization was not successful. As these sequences did not reach an acceptable and therapeutically relevant level of knockdown, we designed three new sequences of GASC1 siRNA (named V4, V6, and V7) using GENtle software. Figure 13A presents the results of mRNA knockdown with the newly designed GASC1 siRNA duplexes. GASC1 siRNA (V4, V6, and V7, 100 pmol) were used to transfect HCC1954 cells with LF, PEI, and the different Tf-modified nanobioconjugates. hGAPDH siRNA was used as a positive control. Among the different siRNA sequences, V7 showed the most efficient mRNA reduction with LF, which was comparable with the positive control. Additionally, the Tf conjugate (Tf–PEI) also showed promising knockdown with sequence V7. There was no significant difference between the mRNA knockdown achieved with V7 when delivered with PEI or Tf-PEG-PEI. To verify the result of the RT-PCR assay, we performed Western blot analysis to quantify the target protein levels. The analysis indicated that cells treated with the optimized Tf–PEI polyplexes with GASC1 siRNA (N/P 7) expressed less GASC1 protein compared to lysates of cells that were treated with scrambled control or nontargeted PEI/GASC1 siRNA complexes as was predicted based on the RT-PCR results (Figure 13 B).
Figure 11.

GASC1 gene knockdown optimization with different siRNA sequences (A–D sequences from Qiagen, and siRNA sequences E–H and mixed from Dharmacon). Per well, 500,000 HCC1954 cells were transfected with Lipofectamine 2000 (LF) for 24 h with 100 pmol of different siRNA sequences against GASC1. RT-PCR was performed with specific GASC1 primers; normalization was carried out against the housekeeping gene PUM1. NC = scrambled siRNA; error bars reflect SD, n = 3. *P < 0.05 and **P < 0.01.
Figure 12.

GASC1 gene knockdown optimization with siRNA sequence A in HCC1954 cells at different time points (A), or different siRNA concentration 24 h post transfection (B). Lipofectamine 2000 (LF) was used as transfection agent, and RT-PCR was performed with specific GASC1 primers; normalization was carried out against the housekeeping gene PUM1. NC = scrambled siRNA; error bars reflect SD, n = 3; ns, nonsignificant; *P < 0.05, ***P < 0.001, and ****p < 0.0001.
Figure 13.

(A) GASC1 knockdown of Tf–PEI with siRNA sequence V7 shows that the conjugate was able to knock down mRNA levels of GASC1 more efficiently than the commercially available siRNA sequences in HCC1954 cells. V4, V6, and V7 are self-designed GASC1 siRNA sequences. NC = scrambled siRNA; PC = positive control siRNA (hGAPDH), error bars reflect SD, n = 3; ns, nonsignificant; *P < 0.05, **P < 0.01, ***P < 0.001, and ****p < 0.0001. (B) GASC1 protein knockdown in HCC1954 cells with Tf–PEI compared to PEI alone was confirmed by Western blot. Alpha-tubulin was used as the loading control. Top row: A, PEI/V7GASC1 siRNA; B, PEI/NC siRNA; C, Tf–PEI/V7GASC1; D, Tf–PEI/NC siRNA.
Therapeutic Effects
To determine if this gene knockdown on RNA and protein level would translate into a therapeutic effect, which would be reflected by a sequence-dependent inhibition of cell growth, MTT assays were performed after either one or multiple transfections. Even though a small effect on cell proliferation was observed 24 h after a single transfection (2 days after seeding the cells), a more pronounced and clearly sequence-specific effect was observed after 3 transfections on day 10 after seeding the cells (Figure 14). While the proliferation after 3 transfections with the negative control siRNA seemed to be slightly inhibited, the proliferation of those cells was not significantly different from that of the untreated control cells.
Figure 14.
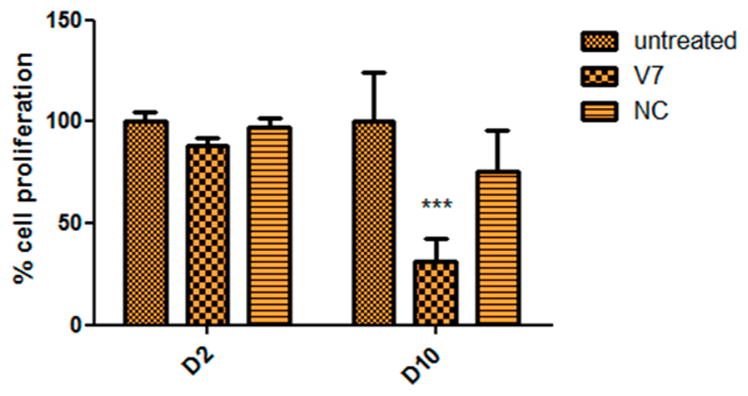
Therapeutic effects of GASC1 knockdown with Tf–PEI polyplexes on days 2 (D2) and 10 (D10) after seeding measured as inhibition of proliferation of HCC1954 cells. V7 is a self-designed GASC1 siRNA sequence; NC = scrambled siRNA; error bars reflect SD, n = 3.
DISCUSSION
Considering that epigenetics and genetics are accomplices in breast cancer tumor development, much attention has been paid to the investigation of molecular and biology insights which could lead to the design of more specifically tailored therapies (e.g., gene therapy).26 siRNA mediated RNAi has become a powerful tool for its specificity and efficiency to knock down targets that cannot be readily downregulated by conventional chemotherapy.27 However, siRNA clinical applications have been a very challenging task due to multiscale barriers, including rapid excretion of siRNA, low stability in blood serum, nonspecific accumulation in tissues, poor cellular uptake, and inefficient intracellular release.27 Several delivery strategies have been developed to address these issues.28 Here, we aimed to design a specifically targeted nanoparticle delivery system with chemical modification and ligand decoration of the complexes to improve efficient delivery of siRNA for the downregulation of GASC1 overexpression in target cells. This strategy was chosen because GASC1 overexpression correlates with poor prognosis in BLBC patients.5
Low molecular weight polyethylenimine (PEI) has been researched extensively as a safe and efficient nucleic acid delivery system based on its ability to electrostatically complex siRNA molecules into nanoscale polyplex and, hence, to protect them from nuclease degradation.29,30 However, PEI alone is not a suitable carrier for targeted gene delivery to specific cells.30 Meanwhile, TfR is a cell membrane associated glycoprotein serving as the main port of entry for iron through a clathrin/dynamin dependent endocytosis mechanism31 and can be used as a gate for delivery of PEI when conjugated to a ligand such as transferrin that selectively targets TfR on receptor expressing cells. Therefore, utilizing knowledge about the physiological function of the transferrin–transferrin receptor complex and the efficiency of its receptor mediated endocytosis provides the rationale to develop transferrin-targeted anticancer modalities. Moreover, studies have shown the elevated expression levels of TfR on cancer cells compared with normal cells, which correlates with tumor stage and poor prognosis (the Nottingham prognostic index (NPI)).32,33 Among the different ligand targeting nanoparticles, there are four transferrin-conjugated nanoparticle delivery systems currently in early clinical evaluation studies.34 Our TfR screening study confirmed distinct levels of TfR among breast cancer cell models (Figure 1), and the cell line HCC1954 (overexpressing GASC1) possesses the highest TfR expression compared to the healthy breast cell line MCF10A (GASC1 negative control). The results of the binding capacity of transferrin to actively endocytosing TfR were in accordance with the transferrin receptor expression data and led to the assumption that the fluorescently labeled Tf retained its binding capacity after FITC labeling and was successfully internalized by the TfR in HCC1954 cells as a valid model for overexpression of TfR (Figure 2).
Here, we used functionalized linkers (SPDP or PEG4-SPDP) to couple the targeting ligand transferrin to LMW-PEI (Figure 3). The effect of two different linker types and different molar stoichiometries (Tf:PEI) on nanoparticle formation and efficacy were compared. The analysis of the prepared conjugates revealed that a Tf:PEI molar ratio between 1.3 and 2 results in better condensation of siRNA in the Tf–PEI polyplex, which are considerably smaller than a ratio around 0.4 for Tf-PEG-PEI polyplex (Figure 4). The difference in transferrin coupling degrees observed in this study was a result of using different linkers, i.e., SPDP vs PEG4-SPDP. However, it is also possible that the presence of PEG in the nanobioconjugate with the lower transferrin coupling degree interfered with dense condensation of siRNA based on electrostatics. The presence of PEG in the cross-linking agent may, however, benefit the ligand–receptor interaction of those conjugates as the spacer provides longer distance of the ligand from the polyplex and better solubility. Accordingly, this space may avoid steric hindrance of ligand–receptor binding by the bulky nanoparticle. However, it needs to be kept in mind that the coupling of substances to the primary amines of PEI may reduce its charge density and the proton sponge effect as well as the interaction with siRNA. Indeed, excessive grafting influences the complexation efficacy, complex stability, and complex architecture, and thus requires detailed analysis prior to application in vivo.30
The extent of siRNA condensation with transferrin conjugates was determined by measuring the change of the fluorescence intensity of SYBR Gold as nucleic acid intercalating agent at different N/P ratios. Increasing the N/P ratios from 1 to 20 not only led to decreased amounts of free siRNA as shown in Figure 6, but also higher net positive charges of the polyplexes were obtained. This higher net charge increases the ability of polymer to electrostatically interact with negatively charged siRNA and to condense siRNA more efficiently. However, higher positive charges of the nanoparticles also correlate with nonspecific toxicity and nonspecific cell uptake. As shown by Plank et al., nanoparticles with a positive surface charge can bind nonspecifically to the cell surface and, in addition, activate the alternative complement system.35 This interaction would also lead to rapid nanoparticle uptake by the reticuloendothelial system (RES) after in vivo administration, and as a consequence, rapid clearance of the nanoparticles is expected. However, modification of the polymer surface reduces this phenomenon.36 In our experiments, the condensation efficiency for all three formulations (PEI, Tf–PEI, and Tf-PEG-PEI) reached a plateau at N/P ratio of ≥7, indicating the ability of efficiently complexing with siRNA. The results in Figure 6 showed that the condensation ability of PEI was not compromised even after modification with transferrin. Therefore, polyplexes with N/P ratio of 7 were chosen as the optimal ratio for further experiments.
Particle size and zeta potential are key factors in the biological outcomes of a drug delivery system as they govern the biodistribution profile, their efficiency to accumulate in tumors through the enhanced permeability and retention (EPR) effect, and cellular uptake.37 As a rough estimation, it is believed that the design of rigid and spherical nanoparticles ranging in size from 10 to 200 nm improves circulation half-life of nanoparticles and their uptake by tumors.38 At the aforementioned N/P ratio, Tf-modified polyplexes displayed very promising small sizes with close to neutral zeta potentials (Figure 4). The latter property is of particular interest for potential in vivo use, since a neutral surface charge reduces nonspecific charge-based interactions and undesirable clearance by the RES, such as liver and spleen macrophages, and improves the blood compatibility. Thus, small nanoparticles with close to neutral surface charge are expected to efficiently deliver anticancer drugs to tumor sites.39
The stability of polyplexes is another critical component which determines the nanoparticles’ efficiency. The presence of competing anions in the cell membrane or in serum is known to affect the stability of polyplexes, which determines their in vivo fate.40 Since conjugation of Tf to PEI might influence complex stability, this issue was addressed by incubating the complexes with the competing model polyanion heparin.41 Results confirmed the ability of modified PEI (Tf–PEI) to protect the siRNA in the presence of polyanions even more efficiently than PEI alone; however, Tf-PEG-PEI showed lower stability compared to Tf–PEI and PEI alone (Figure 7). Our results reflect the enhanced stability of Tf-shielded polyplexes (Tf–PEI), which also has been reported by others.42 Based on the zeta potential results, we hypothesize that Tf is located on the surface of the nanoparticles and shields the PEI/siRNA from competing polyanions. The PEG-containing complexes, however, exhibited compromised stability, which may be due to the less dense packaging of siRNA as hypothesized above. The peak of siRNA release that was especially prominent for the Tf–PEI and PEI formulation can be explained as an initial competition between bound siRNA and added heparin for binding with PEI. After longer incubation, however, a new equilibrium is reached, and possibly even ternary complexes of polymer, siRNA, and heparin are formed. A part of the initially released siRNA seems to be condensed again at 45 or 60 min after incubation, resulting in lower fluorescence intensities. In the case of Tf-PEG-PEI, the siRNA was released almost quantitatively upon addition of heparin and further incubation did not lead to a recomplexation of siRNA. The weak condensation of siRNA by Tf-PEG-PEI therefore precludes any in vivo administration of this formulation.
The Tf-modified PEI formulations are also expected to avoid nonspecific cytotoxicity which is often caused by excess positive charges on PEI. This was confirmed by the CellTiter-Blue assay (Figure 10). We propose that this modification of PEI could reduce the toxicity of the polymer backbone and also enhance the stability of Tf–PEI complexes as well as the circulation of polyplexes in vivo.
Superior internalization of the delivered siRNA with Tf modified PEI in GASC1 positive HCC1954 cell lines compared to PEI alone was confirmed by flow cytometry and confocal microscopy images (Figures 8 and 9). These interesting results implied that Tf–PEI/siRNA polyplexes undergo selective cellular uptake through transferrin receptor mediated endocytosis, and therefore may exhibit increased therapeutic benefits through the ability to deliver high concentrations of relevant siRNA to the tumor site.33 However, it must be taken into account that the final silencing effect depends also on the endolysosomal escape and the efficient incorporation of siRNA to the RNA-induced silencing machinery.43
While the ligand choice depends on the receptor being targeted, an additional consideration for targeting is the type of cell entry pathway that will be induced after ligand–receptor binding. Therefore, the endocytic pathway which may be used by the vector depends on the targeting ligand, cell type, and vector properties.44 It was surprising that, in COLO824 cells with relatively high levels of TfR (Figure 1), no significant transfection was detected with PEI alone or modified PEI (Figure 8). This could be related to the cell type, which may have a very low endocytic activity, but an accurate answer requires further investigations.
It has been proposed that the majority of receptor–ligand complexes (e.g., Tf/TfR) enter cells by clathrin-mediated endocytosis.45 Receptor binding typically triggers the localized accumulation of clathrin structures on the cytoplasmic surface of the plasma membrane as it begins to invaginate, forming clathrin-coated pits.45 TfR undergoes clathrin-mediated endocytosis and may enter two types of trafficking pathways.46 In the first pathway, within 10 min after uptake, the receptor undergoes rapid recycling from early endosomes to the cell surface. In the second pathway, the receptor undergoes slow recycling to the cell surface from the recycling compartment, which is slightly acidic.46 Importantly, studies showed that gene expression after delivery with nanoparticles which enter cells through clathrin-coated endocytosis is comparatively less efficient than after delivery via other pathways, suggesting that this path is linked to endosomolytic degradation of the cargo. Therefore, vectors utilizing this pathway must incorporate a mechanism of release from either organelle.31 Here, we used PEI for its important buffering feature with regard to the intracellular fate of the complexes. The so-called “proton sponge effect” postulates a proton acceptor function of PEI, leading to the buffering of low pH values in the endosomal/lysosomal system and therefore reduced nuclease activity. The influx of chloride counterions and the resulting osmotic influx of water then cause swelling and bursting of the endolysosomes, which eventually leads to release of the endolysosomal contents.47 Therefore, as stated earlier, careful formulation of vector assembly should be taken into account not to compromise this property through grafting with targeting moiety and reducing the accessible protonable amine groups.
As reported by a study for Tf-targeted nanoparticles (lipoplexes), it is likely that other yet unidentified cell surface components (nonreceptor endocytosis) also mediate complex binding and uptake. This might be the reason for the rather high transfection efficacies of Tf–PEI nanoparticles in SUM102 cells. The underlying molecular mechanisms have yet to be investigated.
It has been also demonstrated that conjugation/linker chemistry may have a significant effect on transfection efficiency and specificity in vitro.48,49 In our study, we used two structurally different conjugates (Tf–PEI and Tf-PEG-PEI) which were comparable in physicochemical properties but different in transfection, binding, and uptake efficiencies. As a result, the efficiency of PEI conjugates strongly depends on the linker chemistry. The coupling of transferrin with SPDP to LMW-PEI was most efficient. In contrast, our second approach of using PEG chains in the form of PEG4-SPDP showed that a slightly longer spacer may hinder the proper presentation of transferrin or the condensation of siRNA, and/or have an effect on proton sponge features of PEI. Introducing a cyclic RGD peptide as a targeting moiety via direct coupling or PEG spacer linking to PEI studied by Kunath et al. showed that only direct coupling of the peptide to PEI via SPDP mediated efficient and specific DNA transfection, while an RGD-PEG-PEI conjugate was inefficient. The authors concluded that the use of a PEG spacer seemed to impair targeting, possibly by shielding not only PEI but also the RGD ligand.50 In contrast, Moffatt et al. successfully utilized a PEG linker for coupling an anti-PSMA antibody to PEI/DNA polyplexes.51 Polyethylene glycol (PEG) spacers are expected to preserve protein function by moving the targeting moiety away from the surface, by providing flexibility and reducing nonspecific binding of environmental biomolecules.52 In another study, antibody–PEI conjugates were prepared with three different linkers (SPDP, SMCC, and IBFB 110001) to prepare HER2 or CD90 targeted conjugates. Using these linkers, major differences in the degrees of conjugation and in target cell specificity of the resulting conjugates were found. Interestingly, none of the conjugates prepared with SPDP as a linker showed target cell specificity. Because of significant differences in the study design, direct comparison of these results proved to be difficult, however.
Optimization of GASC1 knockdown on the mRNA level was carried out by initially screening commercially available GASC1 siRNA sequences. The results revealed that three commercially available sequences (A, C, and D obtained from Qiagen) were able to reduce the GASC1 mRNA levels to about 75% after transfection with LF (Figure 11). While the general expression levels of GASC1 cannot be compared to easy to knockdown genes like the housekeeping gene GAPDH, the reduction to 75% residual expression even with LF was not acceptable. Therefore, optimization of several parameters such as incubation time and siRNA concentration was attempted (Figure 12). Since the gene knockdown was not improved by this approach, new siRNA sequences (V4, V6, and V7) were designed and transfected with LF, PEI, and Tf-modified PEIs (Figure 13). The gene knockdown efficacy was determined via RT-PCR. Among all sequences, V7 showed very promising results after transfection of HCC1954 cells with LF and acceptable results after transfection with Tf–PEI/siRNA complexes (N/P 7, Figure 13A). This final formulation was therefore used to verify the protein knockdown level in HCC1954 cells after 7 days of incubation based on a previously optimized protocol. The Western blot analysis confirmed the RT-PCR results (Figure 13B), indicating that the combination of Tf–PEI with V7 siRNA is a promising formulation for GASC1 gene knockdown in BLBC.
The question whether GASC1 gene knockdown would result in efficient inhibition of cell proliferation was answered after optimizing the transfection approach. As shown in Figure 14, a single transfection did not yet lead to efficient inhibition of cell proliferation. This result is not surprising considering that previous approaches using shRNA6 or small molecule GASC1 inhibitors also required multiple treatments to inhibit cell proliferation.53 The results also imply that GASC1 may have a long biologic half-life. Figure 14 shows that multiple treatments with negative control siRNA can have a small but not significant impact on cell proliferation in comparison to untreated cells, whereas one single treatment does not affect the cells, which is in line with the cytotoxicity results shown in Figure 10.
CONCLUSIONS
The establishment of an efficient and cell type specific gene delivery system is a most challenging objective, which in the context of cancer therapy ideally would allow for the specific introduction of a therapeutic transgene or knockout in malignant cells. Combining the high transfection potential of PEI with the efficient and specific mechanism of receptor-mediated endocytosis through TfR improved gene transfer efficiency in cells overexpressing TfR and offers the possibility for specific targeting of gene delivery in a BLBC cell line model. Through choosing the epigenetically and therapeutically relevant target gene GASC1 which is involved in multiple signaling pathways leading to tumorigenesis, this approach of identifying a targetable receptor in basal-like breast cancer has revealed promising results for targeted siRNA therapeutics and inhibition of breast cancer cell proliferation. However, it remains to be shown whether these properties can be exploited for targeted in vivo gene delivery after systemic administration.
Future Perspective
The aggressive nature of BLBC results in failure of current conventional therapies at time of diagnosis, urging the development of novel strategies to achieve improved outcomes. The fact that the epigenome acts at the pinnacle of the hierarchy of gene control mechanisms means that the mutations probably have effects on multiple pathways relevant to the cancer phenotype and so a single mutation could cause wide scale misregulation.54 This opens the door to further drug development, since it might be possible to correct several pathways by altering or inhibiting one enzyme.54
Nanoparticles have the potential of addressing and resolving some of the most significant limitations of traditional anticancer therapeutics, namely, interactions of nanoparticles with the tumor include aiding in molecule transport to the intracellular organelles to induce the therapeutic effects. The present study designed an optimized form of targeted nanoparticles with chemically engineered transferrin that were capable of delivering siRNA specifically and efficiently. They demonstrated promising results in downregulating the newly identified oncogenic histone modifying enzyme (GASC1) in vitro. In the future, more studies will be employed to investigate the systematic gene knockdown and understand and modulate the trial.
Acknowledgments
This work was supported by the Karmanos Cancer Institute-SRIG grant and Angelika Burger Fellowship grant awarded to O.M.M. and Z.-Q.Y. and partially supported by grants from the National Institutes of Health R21CA175244-01A1 (to Z.-Q.Y.) and the Department of Defense (DoD) Prostate Cancer Program PC121737 and PC130259 (to Z.-Q.Y.). The NIH Center Grant P30CA22453 supporting the Wayne State Microscopy, Imaging and Cytometry Resources (MICR) is gratefully acknowledged. We thank Dr. Stephen P. Ethier for providing human SUM breast cancer cell lines and acknowledge Barani Govindarajan and Dr. Yuanyuan Jiang for technical support.
ABBREVIATIONS USED
- Tf
transferrin
- TfR
Tf receptor (CD71)
- PEI
polyethylenimine
- (LMW-PEI)
low molecular weight polyethylenimine
- Tf–PEI
transferrin conjugated to PEI with SPDP linker
- Tf-PEG-PEI
transferrin conjugated to PEI with PEG4-SPDP linker
- GASC1
gene amplified in squamous cell carcinoma 1
- FITC
fluorescein isothiocyanate
- BSA
bovine serum albumin
- MFI
mean fluorescent intensity
- BLBC
basal-like breast cancer
- HER2
human epidermal growth factor receptor 2
- ER
estrogen receptor
- PR
progesterone receptor
- EPR
enhanced permeability and retention
- RES
reticuloendothelial system
- SPDP
succinimidyl 3-(2-pyridyldithio)propionate
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Waldmann T, Schneider R. Targeting histone modifications–epigenetics in cancer. Curr Opin Cell Biol. 2013;25(2):184–9. doi: 10.1016/j.ceb.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 3.DeCarlo D, Hadden MK. Oncoepigenomics: Making histone lysine methylation count. Eur J Med Chem. 2012;56(0):179–194. doi: 10.1016/j.ejmech.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8(4):286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z-Q, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, Sugano S, Nakamura Y, Inazawa J. Identification of a Novel Gene, GASC1, within an Amplicon at 9p23–24 Frequently Detected in Esophageal Cancer Cell Lines. Cancer Res. 2000;60(17):4735–4739. [PubMed] [Google Scholar]
- 6.Wu J, Liu S, Liu G, Dombkowski A, Abrams J, Martin-Trevino R, Wicha MS, Ethier SP, Yang ZQ. Identification and functional analysis of 9p24 amplified genes in human breast cancer. Oncogene. 2012;31(3):333–341. doi: 10.1038/onc.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holowatyj A, Yang Z-Q. The Role of Histone Demethylase GASC1 in Cancer and its Therapeutic Potential. Curr Cancer Ther Rev. 2013;9(1):78–85. [Google Scholar]
- 8.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho Y-J, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao M-S, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP, Yang Z-Q. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene. 2009;28(50):4491–4500. doi: 10.1038/onc.2009.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han W, Jung EM, Cho J, Lee JW, Hwang KT, Yang SJ, Kang JJ, Bae JY, Jeon YK, Park IA, Nicolau M, Jeffrey SS, Noh DY. DNA copy number alterations and expression of relevant genes in triple-negative breast cancer. Genes, Chromosomes Cancer. 2008;47(6):490–9. doi: 10.1002/gcc.20550. [DOI] [PubMed] [Google Scholar]
- 11.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A. 2012;109(49):E3367–E3376. doi: 10.1073/pnas.1217394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mastrobattista E, van der Aa MAEM, Hennink WE, Crommelin DJA. Artificial viruses: a nanotechnological approach to gene delivery. Nat Rev Drug Discovery. 2006;5:115–121. doi: 10.1038/nrd1960. [DOI] [PubMed] [Google Scholar]
- 13.Pazaiti A, Fentiman IS. Basal phenotype breast cancer: implications for treatment and prognosis. Women’s Health. 2011;7(2):181–202. doi: 10.2217/whe.11.5. [DOI] [PubMed] [Google Scholar]
- 14.Shastry M, Yardley DA. Updates in the treatment of basal/triple-negative breast cancer. Curr Opin Obstet Gynecol. 2013;25(1):40–8. doi: 10.1097/GCO.0b013e32835c1633. [DOI] [PubMed] [Google Scholar]
- 15.Camp ER, Wang C, Little EC, Watson PM, Pirollo KF, Rait A, Cole DJ, Chang EH, Watson DK. Transferrin receptor targeting nanomedicine delivering wild-type p53 gene sensitizes pancreatic cancer to gemcitabine therapy. Cancer Gene Ther. 2013;20(4):222–8. doi: 10.1038/cgt.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen H, Sun T, Ferrari M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 2012;19(6):367–373. doi: 10.1038/cgt.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Essex S, Navarro G, Sabhachandani P, Chordia A, Trivedi M, Movassaghian S, Torchilin VP. Phospholipid-modified PEI-based nanocarriers for in vivo siRNA therapeutics against multidrug-resistant tumors. Gene Ther. 2015;22(3):41–50. doi: 10.1038/gt.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geninatti Crich S, Cadenazzi M, Lanzardo S, Conti L, Ruiu R, Alberti D, Cavallo F, Cutrin JC, Aime S. Targeting ferritin receptors for the selective delivery of imaging and therapeutic agents to breast cancer cells. Nanoscale. 2015;7(15):6527–6533. doi: 10.1039/c5nr00352k. [DOI] [PubMed] [Google Scholar]
- 19.Xie Y, Kim NH, Nadithe V, Schalk D, Thakur A, Kilic A, Lum LG, Bassett DJ, Merkel OM. Targeted delivery of siRNA to activated T cells via transferrin-polyethylenimine (Tf-PEI) as a potential therapy of asthma. J Controlled Release. 2016;229:120–129. doi: 10.1016/j.jconrel.2016.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Germershaus O, Neu M, Behe M, Kissel T. HER2 Targeted Polyplexes: The Effect of Polyplex Composition and Conjugation Chemistry on in Vitro and in Vivo Characteristics. Bioconjugate Chem. 2008;19(1):244–253. doi: 10.1021/bc700311n. [DOI] [PubMed] [Google Scholar]
- 21.Merkel OM, Germershaus O, Wada CK, Tarcha PJ, Merdan T, Kissel T. Integrin ανβ3 Targeted Gene Delivery Using RGD Peptidomimetic Conjugates with Copolymers of PEGylated Poly(ethylene imine) Bioconjugate Chem. 2009;20(6):1270–1280. doi: 10.1021/bc9001695. [DOI] [PubMed] [Google Scholar]
- 22.Fischer D, Bieber T, Li Y, Elsasser HP, Kissel T. A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: effect of molecular weight on transfection efficiency and cytotoxicity. Pharm Res. 1999;16(8):1273–9. doi: 10.1023/a:1014861900478. [DOI] [PubMed] [Google Scholar]
- 23.Elsayed M, Corrand V, Kolhatkar V, Xie Y, Kim NH, Kolhatkar R, Merkel OM. Influence of oligospermines architecture on their suitability for siRNA delivery. Biomacromolecules. 2014;15(4):1299–310. doi: 10.1021/bm401849d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Fong D, Navarro-Quiroga I, Ochoa I, Alvarez-Maya I, Meraz MA, Luna J, Arias-Montaño J-A. Neurotensin-SPDP-poly-l-lysine conjugate: a nonviral vector for targeted gene delivery to neural cells. Mol Brain Res. 1999;69(2):249–262. doi: 10.1016/s0169-328x(99)00114-x. [DOI] [PubMed] [Google Scholar]
- 25.Sartor CI, Dziubinski ML, Yu C-L, Jove R, Ethier SP. Role of Epidermal Growth Factor Receptor and STAT-3 Activation in Autonomous Proliferation of SUM-102PT Human Breast Cancer Cells. Cancer Res. 1997;57(5):978–987. [PubMed] [Google Scholar]
- 26.Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. Layer-by-Layer Nanoparticles for Systemic Codelivery of an Anticancer Drug and siRNA for Potential Triple-Negative Breast Cancer Treatment. ACS Nano. 2013;7(11):9571–9584. doi: 10.1021/nn4047925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shim MS, Kwon YJ. Efficient and targeted delivery of siRNA in vivo. FEBS J. 2010;277(23):4814–4827. doi: 10.1111/j.1742-4658.2010.07904.x. [DOI] [PubMed] [Google Scholar]
- 28.Ardana A, Whittaker AK, McMillan NAJ, Thurecht KJ. Polymeric siRNA delivery vectors: knocking down cancers with polymeric-based gene delivery systems. J Chem Technol Biotechnol. 2015;90(7):1196–1208. [Google Scholar]
- 29.Taranejoo S, Liu J, Verma P, Hourigan K. A review of the developments of characteristics of PEI derivatives for gene delivery applications. J Appl Polym Sci. 2015 doi: 10.1002/app.42096. [DOI] [Google Scholar]
- 30.Hobel S, Aigner A. Polyethylenimines for siRNA and miRNA delivery in vivo. Wiley Interdiscip Rev: Nanomed Nanobiotechnol. 2013;5(5):484–501. doi: 10.1002/wnan.1228. [DOI] [PubMed] [Google Scholar]
- 31.Mayle KM, Le AM, Kamei DT. The intracellular trafficking pathway of transferrin. Biochim Biophys Acta, Gen Subj. 2012;1820(3):264–281. doi: 10.1016/j.bbagen.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Habashy HO, Powe DG, Staka CM, Rakha EA, Ball G, Green AR, Aleskandarany M, Paish EC, Douglas Macmillan R, Nicholson RI, Ellis IO, Gee JM. Transferrin receptor (CD71) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen. Breast Cancer Res Treat. 2010;119(2):283–93. doi: 10.1007/s10549-009-0345-x. [DOI] [PubMed] [Google Scholar]
- 33.Tortorella S, Karagiannis T. Transferrin Receptor-Mediated Endocytosis: A Useful Target for Cancer Therapy. J Membr Biol. 2014;247(4):291–307. doi: 10.1007/s00232-014-9637-0. [DOI] [PubMed] [Google Scholar]
- 34.van der Meel R, Vehmeijer LJ, Kok RJ, Storm G, van Gaal EV. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Delivery Rev. 2013;65(10):1284–98. doi: 10.1016/j.addr.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Plank C, Mechtler K, Szoka FC, Jr, Wagner E. Activation of the complement system by synthetic DNA complexes: a potential barrier for intravenous gene delivery. Hum Gene Ther. 1996;7(12):1437–46. doi: 10.1089/hum.1996.7.12-1437. [DOI] [PubMed] [Google Scholar]
- 36.Merkel OM, Urbanics R, Bedocs P, Rozsnyay Z, Rosivall L, Toth M, Kissel T, Szebeni J. In vitro and in vivo complement activation and related anaphylactic effects associated with polyethylenimine and polyethylenimine-graft-poly(ethylene glycol) block copolymers. Biomaterials. 2011;32(21):4936–4942. doi: 10.1016/j.biomaterials.2011.03.035. [DOI] [PubMed] [Google Scholar]
- 37.Navarro G, Movassaghian S, Torchilin VP. Current Trends in the Use of Cationic Polymer Assemblies for siRNA and Plasmid DNA Delivery. Pharm Nanotechnol. 2013;1(3):165–183. [Google Scholar]
- 38.Movassaghian S, Torchilin VP. Chapter 18—Long-Circulating Therapies for Cancer Treatment. In: Singh M, Salnikova M, editors. Novel Approaches and Strategies for Biologics, Vaccines and Cancer Therapies. Academic Press; San Diego: 2015. pp. 433–462. [Google Scholar]
- 39.Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32(13):3435–3446. doi: 10.1016/j.biomaterials.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuckerman JE, Gale A, Wu P, Ma R, Davis ME. siRNA delivery to the glomerular mesangium using polycationic cyclodextrin nanoparticles containing siRNA. Nucleic Acid Ther. 2015;25(2):53–64. doi: 10.1089/nat.2014.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merdan T, Callahan J, Petersen H, Kunath K, Bakowsky U, Kopeckova P, Kissel T, Kopecek J. Pegylated polyethylenimine-Fab’ antibody fragment conjugates for targeted gene delivery to human ovarian carcinoma cells. Bioconjugate Chem. 2003;14(5):989–96. doi: 10.1021/bc0340767. [DOI] [PubMed] [Google Scholar]
- 42.Kircheis R, Wightman L, Schreiber A, Robitza B, Rossler V, Kursa M, Wagner E. Polyethylenimine/DNA complexes shielded by transferrin target gene expression to tumors after systemic application. Gene Ther. 2001;8(1):28–40. doi: 10.1038/sj.gt.3301351. [DOI] [PubMed] [Google Scholar]
- 43.Li Z, Rana TM. Molecular Mechanisms of RNA-Triggered Gene Silencing Machineries. Acc Chem Res. 2012;45(7):1122–1131. doi: 10.1021/ar200253u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medina-Kauwe LK, Xie J, Hamm-Alvarez S. Intracellular trafficking of nonviral vectors. Gene Ther. 2005;12(24):1734–51. doi: 10.1038/sj.gt.3302592. [DOI] [PubMed] [Google Scholar]
- 45.Akhtar MJ, Ahamed M, Alhadlaq HA, Alrokayan SA, Kumar S. Targeted anticancer therapy: overexpressed receptors and nanotechnology. Clin Chim Acta. 2014;436:78–92. doi: 10.1016/j.cca.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 46.Sheff DR, Daro EA, Hull M, Mellman I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J Cell Biol. 1999;145(1):123–39. doi: 10.1083/jcb.145.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Behr J-P. The Proton Sponge: a Trick to Enter Cells the Viruses Did Not Exploit. CHIMIA International Journal for Chemistry. 1997;51(1–2):34–36. [Google Scholar]
- 48.Gopal V, Xavier J, Kamal MZ, Govindarajan S, Takafuji M, Soga S, Ueno T, Ihara H, Rao NM. Synthesis and Transfection Efficiency of Cationic Oligopeptide Lipids: Role of Linker. Bioconjugate Chem. 2011;22(11):2244–2254. doi: 10.1021/bc2002874. [DOI] [PubMed] [Google Scholar]
- 49.Zhao F, Yin H, Zhang Z, Li J. Folic Acid Modified Cationic γ-Cyclodextrin-oligoethylenimine Star Polymer with Bioreducible Disulfide Linker for Efficient Targeted Gene Delivery. Biomacromolecules. 2013;14(2):476–484. doi: 10.1021/bm301718f. [DOI] [PubMed] [Google Scholar]
- 50.Kunath K, Merdan T, Hegener O, Haberlein H, Kissel T. Integrin targeting using RGD-PEI conjugates for in vitro gene transfer. J Gene Med. 2003;5(7):588–99. doi: 10.1002/jgm.382. [DOI] [PubMed] [Google Scholar]
- 51.Moffatt S, Papasakelariou C, Wiehle S, Cristiano R. Successful in vivo tumor targeting of prostate-specific membrane antigen with a highly efficient J591//PEI//DNA molecular conjugate. Gene Ther. 2006;13(9):761–772. doi: 10.1038/sj.gt.3302721. [DOI] [PubMed] [Google Scholar]
- 52.Otsuka H, Nagasaki Y, Kataoka K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv Drug Delivery Rev. 2003;55(3):403–419. doi: 10.1016/s0169-409x(02)00226-0. [DOI] [PubMed] [Google Scholar]
- 53.Ye Q, Holowatyj A, Wu J, Liu H, Zhang L, Suzuki T, Yang ZQ. Genetic alterations of KDM4 subfamily and therapeutic effect of novel demethylase inhibitor in breast cancer. Am J Cancer Res. 2015;5(4):1519–1530. [PMC free article] [PubMed] [Google Scholar]
- 54.You JS, Jones PA. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell. 2012;22(1):9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]