

Abstract
The A-kinase anchoring protein (AKAP) smAKAP has three extraordinary features; it is very small, it is anchored directly to membranes by acyl motifs, and it interacts almost exclusively with the type I regulatory subunits (RI) of cAMP-dependent kinase (PKA). Here, we determined the crystal structure of smAKAP’s A-kinase binding domain (smAKAP-AKB) in complex with the dimerization/docking (D/D) domain of RIα which reveals an extended hydrophobic interface with unique interaction pockets that drive smAKAP’s high specificity for RI subunits. We also identify a conserved PKA phosphorylation site at Ser66 in the AKB domain which we predict would cause steric clashes and disrupt binding. This correlates with in vivo colocalization and fluorescence polarization studies, where Ser66 AKB phosphorylation ablates RI binding. Hydrogen/deuterium exchange studies confirm that the AKB helix is accessible and dynamic. Furthermore, full-length smAKAP as well as the unbound AKB is predicted to contain a break at the phosphorylation site, and circular dichroism measurements confirm that the AKB domain loses its helicity following phosphorylation. As the active site of PKA’s catalytic subunit does not accommodate α-helices, we predict that the inherent flexibility of the AKB domain enables its phosphorylation by PKA. This represents a novel mechanism, whereby activation of anchored PKA can terminate its binding to smAKAP affecting the regulation of localized cAMP signaling events.
Keywords: AKAP, inhibition, phosphorylation, PKA, structure
Introduction
Many important cellular functions are regulated by signaling through parallel cAMP-dependent pathways [1]. Among other targets, intracellular cAMP activates cAMP-dependent protein kinase (PKA) [2]. PKA is a holoenzyme consisting of a homo-dimerized regulatory subunit (PKA-R) with a catalytic subunit (PKA-C) bound to each PKA-R. Four genetically distinct PKA-R isoforms are expressed in mammalian systems, designated as PKA-RIα, PKA-RIβ, PKA-RIIα, and PKA-RIIβ. Upon binding of cAMP to PKA-R, PKA-C is released and becomes active to subsequently phosphorylate Ser/Thr residues on protein substrates [3]. Due to PKA’s importance and involvement in many pathways, it needs additional levels of regulation in space and time. It achieves this through interaction with the protein scaffolding family of A-kinase anchoring proteins (AKAPs), which localize PKA, often together with many other signaling proteins (phosphatases, phosphodiesterases, other kinases, etc.) to a specific location in the cell [4,5]. At present, more than 30 mammalian AKAPs have been reported and experimentally verified [4,5]. Thus far, mainly AKAPs binding to the PKA-RII isoforms have been identified, as well as a handful of dual-specific AKAPs, binding to both PKA-RI and PKA-RII (dAKAP1 [6], dAKAP2 [7], and Opa1 [8]).
Initially, PKA-RI was proposed to be cytosolic and not anchored; however recently, we reported the chemical proteomics-based discovery of two novel AKAPs with exclusive specificity for PKA-RI, small membrane AKAP (smAKAP) [9], and sphingosine kinase-interacting protein (SKIP, SPHKAP) [10,11]. smAKAP has an unprecedented affinity for PKA-RIα (Kd = 6.7 nM) and PKA-RIβ (Kd = 6.9 nM) and localizes only these two isoforms to the plasma membrane using myristoylation and palmitoylation at Gly2 and Cys3, respectively. Most AKAPs are large protein scaffolds (300–500 kDa) and hold next to their typical PKA-binding domain many more domains, often of vague or unknown function. The small size of smAKAP (only 11 kDa) makes it a convenient model system to investigate the specific determinants of PKA-RI’s interactions.
Previous structural characterizations have indicated that AKAPs bind to the N-terminal dimerization and docking (D/D) domain of the PKA-R dimer via their A-kinase-binding (AKB) domain [12]. This AKB domain is an amphipathic helix of 3–4 turns long and its hydrophobic edge interacts with the hydrophobic face of the D/D domain. The drivers of PKA-R/AKAP specificity are only partially understood, although it is clear that both sides of the interaction have adopted specific structural determinants. The D/D domain structures of both PKA-RIα and PKA-RIIα demonstrated a tightly packed X-type bundle consisting of four helices [13,14]. The monomers are positioned antiparallel to one another and in the core, there is a large hydrophobic plateau to accommodate AKAP binding. The N-terminal helix (α0) shows isoform-specific differences. Specifically, in RIα it contains cysteines, which lead to two identical interchain disulfide bridges between Cys16 and Cys37. This connection reinforces the monomer–monomer interaction along the boundary and the antiparallel alignment [15]. Also, the RIα D/D module presents a deeper cleft, allowing the presence of larger hydrophobic amino acids in the AKB domain [13]. This was later confirmed by peptide screening [16–18] and the presence of a phenylalanine residue in the AKB peptide of SPHKAP [10] (Fig. 1A).
Fig. 1.
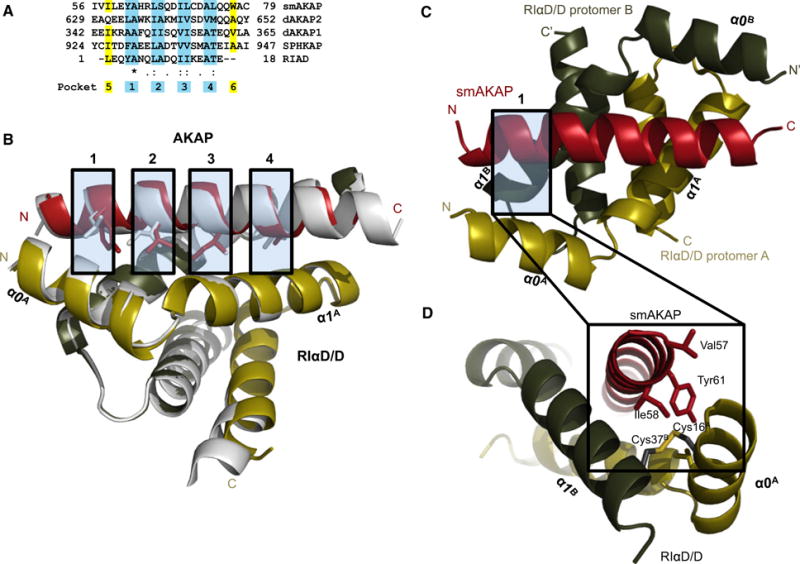
Crystal structure of the smAKAP-AKB/PKA-RIαD/D complex. (A) A sequence alignment of four AKAPs whose AKB domains each bind to PKA-RI with the established four binding pockets highlighted in blue. (B) A side view displaying the interaction surface between smAKAP-AKB (red) and PKA-RIαD/D (gold (A)/green (B)) with an overlay of dAKAP2:PKA-RIαD/D (silver). (C) A top view of smAKAP overlaying PKA-RIαD/D. (D) The N terminus of the AKB domain (red) establishes the fifth pocket. Tyr61C and Ile58C (highlighted in yellow in the sequence alignment of A) bury the disulfide bond Cys16A-Cys37B into a hydrophobic pocket. Leu13A of α0-helix (gold) and Ile33B of α1-helix (green) also close up the hydrophobic pocket. The atomic coordinates have been deposited in the Protein Data Bank (accession code 5HVZ).
More recently, crystal structures revealing the interactions of PKA-RI and PKA-RII with the dual-specific dAKAP2 (gene name AKAP10) provided a first glimpse into isoform-specific interactions [19,20]. Firstly, the PKA-RII–dAKAP2 interaction is indeed characterized by a smaller interaction surface than the PKA-RI– dAKAP2 interaction. The former utilizes only two hydrophobic pockets of the AKB domain to interact, while in the latter, four hydrophobic contact sites were observed. The larger interaction surface is largely due to the above-mentioned disulfide bridges, which create a more stable domain at the end of the D/D helices [13]. As the PKA-RI-specific AKAPs have only recently been discovered [9,10], no detailed information on their structural determinants has been elucidated yet.
Protein kinase is targeted to its substrates in the cell via AKAPs. PKA recognizes its substrates via a rather promiscuous phosphorylation motif: [R/K]-[R/K]-X-[S/T][noP] [21, 22]. PKA has been found to phosphorylate AKAPs, although only to a limited extent, suggesting that a regulatory mechanism controls AKAP complex composition, activity, or perhaps localization. A few illustrative examples include PKA phosphorylation of AKAP-Lbc on Ser1565 to allow 14-3-3β/ɛ/ξ. Binding at this site inhibits the Rho-GEF activity [23]. Similarly, AKAP79 has a number of PKA phosphorylation sites, which allow binding to phosphatidylinositol 4,5-bisphosphate [24].
Here, we report that smAKAP contains a PKA phosphorylation site within its AKB domain, which drastically affects the PKA-RI–smAKAP interaction. To investigate the structural basis for this observation, first we solved the crystal structure of the PKA-RIαD/D:smAKAP-AKB complex. This not only provided novel insights into what makes smAKAP specific toward PKA-RIα/β but also led us to define the role of smAKAP phosphorylation in the AKB domain. Using a set of complementary cell imaging, biophysical, bioinformatics, and structural biological techniques, we uncovered, and structurally characterized, a novel auto-inhibitory mechanism of PKA-RI anchoring. This further extends the repertoire of nature’s tools to regulate localized cAMP signaling in mammalian systems.
Results
Structural analysis of the PKA-RIα–smAKAP interaction
To understand the selectivity of smAKAP toward PKA-RI better, we set out to determine the crystal structure of the complex consisting of the D/D domain of PKA-RIα and the AKB domain of smAKAP. Although the crystals, solved at 2 Å resolution, showed an overall architecture that is similar to previously reported PKA-R D/D:AKB structures [13,17,19,20,25], there are novel features associated with smAKAP that may account for its higher affinity and selectivity. The complex is crystallized with one RIα-D/D dimer; the monomer whose N terminus is next to the N terminus of smAKAP is named ‘A’ (AA12-58), whereas the other monomer is named ‘B’ (AA12-61) bound to one smAKAP-AKB domain (AA 56-79), which is named ‘C’. The majority of residues in the smAKAP peptide had clear electron density; however, Trp77C and Cys79C did not, and therefore the side chains of Trp77C and Cys79C have been omitted from the model. Similarly, most of the residues of both PKA-RIαD/D monomers are accounted for, except Ser12B, Leu13B, Lys57A, and Gly58A, of which the side chains have been omitted, and Glu59A, Ala60A, and Lys61A which have been omitted entirely.
The structure of the PKA-RIαD/D dimer consists of an antiparallel, four-helix bundle. The smAKAP-AKB peptide lies diagonally across the hydrophobic interaction surface of the PKA-RIαD/D domain (Fig. 1B). The root-mean-square deviation (RMSD) between the PKA-RIαD/D of the dAKAP2 [20] structure and the smAKAP complex presented here is 0.39 Å (Cα carbons), indicating that binding of AKB peptides with different PKA-R specificity has little influence on the conformation of PKA-RI. The RMSD between the AKB domains is larger, 0.58 Å (Cα carbons), as expected due to the presence of several differences between the AKB domains of dual-specific and PKA-RI-specific AKAPs. There are four binding pockets in the D/D domain, each allowing two hydrophobic residues of the amphipathic helix to dock (Fig. 1A). The smAKAP-AKB domain has the hydrophobic amino acids fitting snugly in each binding pocket. Structure comparison of the D/D domains of RIα and RIIα shows that there is a more extended hydrophobic surface at the N terminus of RIα that includes a third helix (α0-Helix) instead of a short strand that is a conserved feature of RII subunits. Embedded within these RI-specific pockets are the disulfide bonds that are also a unique feature of RI D/D domains. Cys 16A in the α0-Helix forms a disulfide bond with Cys36B. The disulfide bond stabilizes the N terminus of the RI subunits potentially allowing them to serve as redox sensors. In contrast, the RII subunits have a short flexible strand that contains two conserved Ile residues, Ile3 and Ile5, that contribute to AKAP binding; RII subunits have no cysteines in their D/D domains. What makes the smAKAP interaction different is that at each end of the domain, there are additional hydrophobic interactions assisting in anchoring to the RIαD/D domain by burying the disulfide bonds into hydrophobic pockets. This creates a possible fifth and sixth pocket. In the fifth pocket, the disulfide bridge between Cys16A and Cys36B is surrounded by Ile33B and Val34B of helix αI, Leu13A of helix α0 and Ile58C and Tyr61C (of the amphipathic helix), creating an enhanced hydrophobic pocket (Fig. 1C). In addition, there are hydrogen bonds between side chains of both proteins, Gln76C and Lys30A in helix α1 and Asp72C and Gln26A in helix α1, which also help to stabilize the interaction due to many polar residues of PKA-RIα protruding from the edges of the interaction surface. All these factors lead to a very strong and specific interaction of smAKAP with PKA-RIα. This is further illustrated by the conservation of Ile58C throughout species and in SPHKAP [10] (Ile926), RIAD [18] (Leu1), and dAKAP1 [6] (Ile344) but not in dAKAP2 [7] (Gln631) (Fig. 1A). Tyr61C is also conserved in RIAD, whereas in SPHKAP, another bulky hydrophobic amino acid resides at this site (Phe929). The bulky amino acid at this position has been proposed earlier as a PKA-RI specifier, as it would only fit in the deeper groove of the PKA-RI D/D domains [16, 20]. It is likely that at the other end of the AKB domain, where the second disulfide bridge resides (Cys16B and Cys36A), a putative sixth pocket is formed in a similar way with Leu74C and Trp77C. Although the side chain of Trp77C is not well resolved in the structure, one could assume a function similar to the Ile58C and Tyr61C couple at the other end of the helix due to the internal symmetry present. However, the putative sixth pocket is much less defined and neither Leu74C nor Trp77C seem conserved in the other PKA-RI-specific AKB domains of SPHKAP and RIAD (Fig. 1A) [10, 18]. We hypothesize that the extra hydrophobic residues at the N and C termini of the smAKAP amphipathic helix will enhance AKAP binding to the RIα D/D domain by shielding the RI-specific disulfide bonds. In the absence of the AKAP, these pockets will be solvent exposed allowing the disulfide bonds to be broken.
The AKB domain of smAKAP is phosphorylated by PKA
BLAST analysis and sequence alignment of smAKAP and its homologs revealed several conserved serines and threonines [9]. One of them, Ser66, lies in the middle of the AKB domain and displays the phosphorylation motif H-R-L-S (Fig. 2A), a PKA substrate site proposed from peptide screens [22,26]. This PKA motif in smAKAP is conserved from zebrafish to human with the positively charged residue (arginine or histidine) occupying either the −2 or −3 positions with respect to the serine (Fig. 2A). Even though serine to alanine mutations are very common in evolution, Ser66 in smAKAP remains stable across species. Interestingly, Ser66 seems to reside on the hydrophobic side of the amphipathic AKB domain helix (Fig. 2B), which could possibly induce steric and electrostatic hindrance for the hydrophobic-based binding of smAKAP with PKA-RI.
Fig. 2.
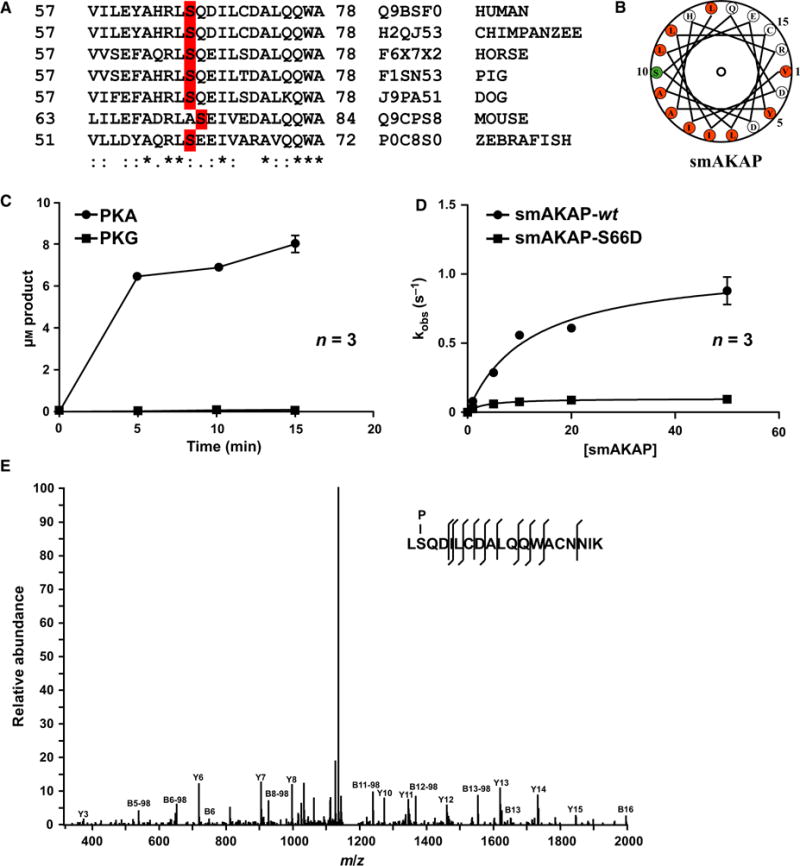
PKA phosphorylates smAKAP at Ser66 in the A-kinase anchoring domain. (A) Sequence alignment of human smAKAP (AA 57-78) with various orthologs in other vertebrate species. Identity (*), similarity (:), and the serines highlighted in red. (B) Helical wheel alignment of the smAKAP’s AKB domain reveals an amphipathic helix with a hydrophobic surface on one side (orange) which contains Ser66 (green). (C) 32P phosphorylation assay using PKA-Cα and PKG Iα as kinase and recombinant His6-smAKAP as substrate (arithmetic means ± SD and n = 3 independent experiments). (D) 32P phosphorylation assay using PKA-Cα on recombinant smAKAP-wt and smAKAP-S66D substrate (arithmetic means ± SD and n = 3 independent experiments). (E) Annotated MS/MS spectrum of the peptide LpSQDILCDALQQWACNNIK with a phosphorylated serine at position 2, corresponding to Ser66 in smAKAP.
An in vitro assay with purified His6-smAKAP and the catalytic subunit of PKA and as a control, its homolog PKG Iα revealed that only PKA phosphorylated smA-KAP (Fig. 2C). Phosphorylation of smAKAP-wt had a Vmax of 1.07 ± 0.08 μM·s−1 and a Km of 11.91 ± 2.42 μM. The filter binding assay was repeated with Ser66 mutated to an aspartic acid (smAKAP-S66D), which showed no detectable levels of phosphorylation (Fig. 2D). To confirm further that Ser66 is indeed the substrate site of PKA, a digest of in vitro phosphorylated smAKAP was analyzed by LC-MS/MS analysis. The tandem MS spectrum of the only observed phosphopeptide LpSQDILCDALQQWACNNIK (Mascot score 89) unambiguously identifies Ser66 as the phosphorylation target site of PKA (Fig. 2E).
Serine66 phosphorylation effectively abrogates binding of PKA-RI
We showed previously using a fluorescence polarization assay that smAKAP has a strong affinity (Kd ≈ 7 nM) for PKA-RIα and RIβ [9]. Executing this assay with the AKB peptide carrying a phosphorylated serine (IVILEYAHRLpSQDILCDALQQWAC), we revealed a dramatic decrease in the binding affinity (Kd > 500 nM; Table 1).
Table 1.
Fluorescence anisotropy measurements to determine the binding affinity (n = 3, Kd ± SD in nM) of the phosphorylated smAKAP-AKB domain peptide, tagged with 5-TAMRA (excitation at 535 nm and emission at 580 nm) with the full-length regulatory subunit dimers: PKA-RIα and PKA-RIβ. The smAKAP-wt, dAKAP2, and AKAP79 results were previously published [9].
| smAKAP-wt (nM) | smAKAP-P (nM) | dAKAP2 (nM) | AKAP79 (nM) | |
|---|---|---|---|---|
| RIα | 6.7 ± 0.7 | > 1000 | 50 | nd |
| RIβ | 6.9 ± 0.6 | > 1000 | – | nd |
HeLa cells transfected with smAKAP-wt-GFP and PKA-RIα-mCherry [9] or PKA-RIβ-mKO2 (Fig. 3A) show strong colocalization along the plasma membrane. In order to deduce the impact of phosphorylated smAKAP in the cellular context, phosphomimetic mutants were created: smAKAP-S66E-GFP and smAKAP-S66D-GFP. Notably, when the HeLa cells were transfected with these mutants along with PKA-RIβ-mKO2 (Fig. 3B,C), or PKA-RIα-mKO2 (Fig. 3D), smAKAP still localized at the plasma membrane, while both PKA-RI isoforms now predominantly resided in the cytoplasm, evidence that phosphorylation of Ser66 inhibits PKA-RI binding to smAKAP. In order to deduce if the phosphorylation of Ser66 by PKA occurs in a cellular context, HeLa cells were subjected to a triple transfection (smAKAP-wt-GFP, PKA-RIβ-mKO2, and PKA-C-HA). After addition of 8-CPT-cAMP and calyculin A, disruption of the association between smAKAP-wt-GFP and PKA-RIβ-mKO2 was observed by live cell microscopy (Fig. 3E). Upon phosphorylation of smAKAP, most of the PKA-RIβ cannot bind to smAKAP anymore and relocates to the cytosol. To further confirm this finding, a Förster resonance energy transfer (FRET)-based kinetic study was performed. Eight cells were transfected with smAKAP-wt-GFP, PKA-RIβ-mKO2, and PKA-C-HA. After stimulation with forskolin, there is a delayed but clear decrease of FRET signal: − 2.3 ± 0.4% (Fig. 3F). The decrease in FRET signal together with the confocal studies and binding assays suggest that the interaction between smAKAP and PKA-RI is disturbed upon phosphorylation of smAKAP.
Fig. 3.
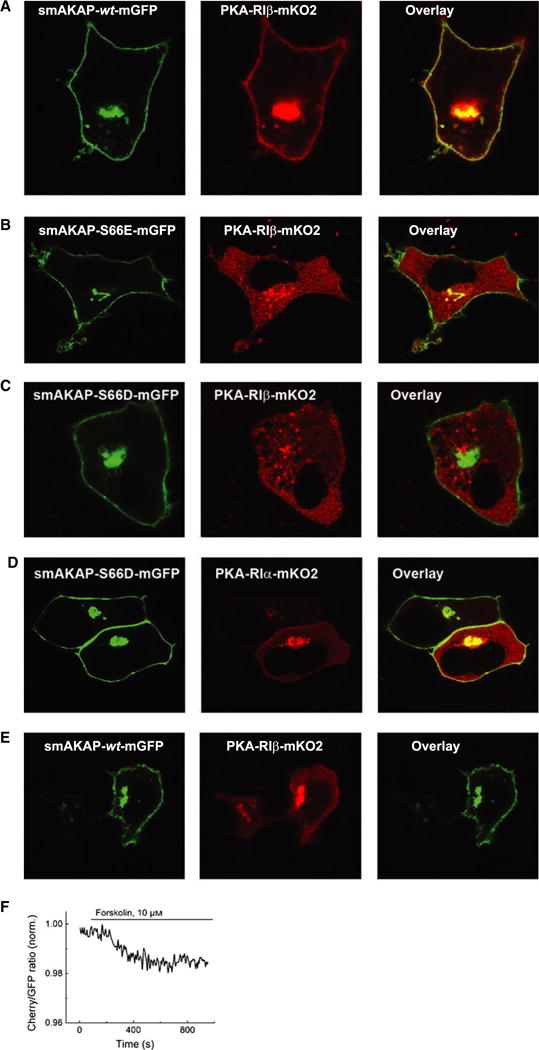
Phosphorylated smAKAP no longer localizes PKA-mRI at the plasma membrane. (A) Transfection of HeLa cells with smAKAP-wt-GFP and PKA-RIβ-mKO2 show colocalization at the plasma membrane. (B) In contrast, in the HeLa cells transfected with the phosphomimetic smAKAP-S66E-mGFP and PKA-RIβ-mKO2, no colocalization occurs. (C) After adding 8-CPT-cAMP and calyculin A, smAKAP-wt-mGFP is partially phosphorylated upon Ser66 by PKA-C, thus releasing PKA-RIβ-mKO2 into the cytosol. (D) A FRET-based kinetic assay was employed upon eight cells transfected with smAKAP-S66-mGFP, PKA-RIβ-mKO2, and PKA-C-HA. Upon stimulation with forskolin, there is a − 2.3 ± 0.4% reduction in FRET.
Hydrogen–deuterium exchange mass spectrometry
To investigate further the interaction dynamics between smAKAP and PKA-RIα, we employed hydrogen/deuterium exchange (HDX) mass spectrometry using both full-length proteins. First, smAKAP, in the presence and absence of an excess of PKA-RIα, was incubated in D2O for various time points between 10 s and 4 h. The reverse experiments, using an excess of smAKAP, were also conducted. For PKA-RI RIα with excess smAKAP, as expected, we observed a distinct difference in deuterium incorporation in specific regions. For instance, peptide AA248-255 of PKA-RIα displayed no difference in deuterium uptake (Fig. 4A), whereas peptides in the N-terminal region such as AA19-37 revealed a substantial reduction in deuterium uptake (Fig. 4B and Table S1). To a minor extent, the peptides between AA38 and AA67 also revealed differential deuterium uptake (Table S1). Although we were able to cover large parts of the PKA-RIα sequence (77%), no other region displayed uptake differences. Similarly, focusing on smAKAP, we observed between the presence and absence of PKA-RI that only the peptides covering the region AA61-78, which is the AKB domain (Fig. 1A), displayed a substantial decrease in deuterium uptake, whereas all other peptides, covering nearly the whole sequence of smAKAP (96%) displayed no substantial difference in deuterium uptake (Fig. 4C,D and Table S2). Focusing on the kinetics of deuterium uptake in the protected regions, we see PKA-RI RIα behaves very different than smA-KAP. Figure 2B illustrates that the uptake of deuterium in the D/D domain of PKA-RIα is very slow with only detectable levels of incorporation after 60 min, indicating that the hydrophobic groove is very protected in the absence of excess smAKAP. In contrast, kinetics on smAKAP are very fast (Fig. 2D). In the presence of excess PKA-RI RIα, smAKAP is initially withheld from incorporating deuterium, but already after 1 min, the incorporation is initiated and rises steep to reach full incorporation after 60 min. This suggests that smAKAP is able to dislodge from PKA-RIα, making its Ser66 residue available for phosphorylation by a nearby PKA-C.
Fig. 4.
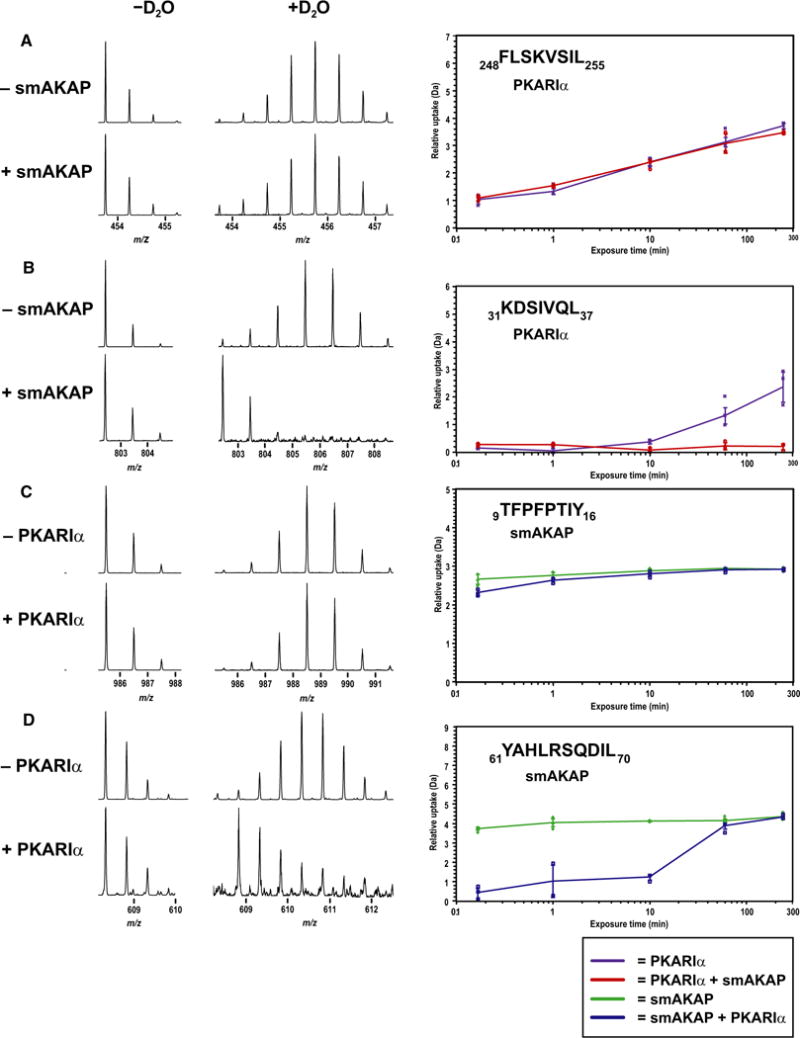
H/D exchange profiling of the interaction surface of smAKAP and PKA-RIα. (A) No difference in deuterium uptake was observed between PKA-RIα (purple) and PKA-RIα + smAKAP (red) for AA248-255 substrate (arithmetic means ± SD and n = 3 independent experiments) (B) for AA31-37, there was an increasing difference in deuterium uptake starting after 10 min (arithmetic means ± SD and n = 3 independent experiments) (C) For AA9-16 of smAKAP, there was no difference in deuterium incorporation between smAKAP (green) and smAKAP + PKA-RIα (blue) substrate (arithmetic means ± SD and n = 3 independent experiments) (D) However, a large initial difference in uptake was noted for AA61-70 substrate (arithmetic means ± SD and n = 3 independent experiments).
As the structural interaction between smAKAP and PKA-RIα was determined by both crystallography and hydrogen/deuterium exchange studies, we were able to further explore the detailed molecular mechanism of the binding abrogation induced by Ser66-phosphorylation. Initially, we hypothesized that electronegativity and steric clashing could inhibit binding of the phosphorylated AKB domain peptide (AKB-Phospho) to the hydrophobic PKA-RIαD/D. However, molecular modeling using HADDOCK [27, 28], of smAKAP AKB-wt, AKB-S66D, AKB-S66E, and AKB-pSer66 with PKA-RIαD/D suggested that the phosphorylated wild-type and the phosphomimetic mutants could bind at least as well to the D/D domain of PKA-RIα as the nonphosphorylated equivalent. These data indicated that a phosphorylated Ser66 can still fit within the hydrophobic groove and is able to extend its side chain with the electronegative moiety out of the hydrophobic groove and create hydrogen bond interactions with Lys30B and Gln26B of PKA-RIα (Fig. 5). Clearly, this is only a model, as experimentally we observed complete disruption of binding upon phosphorylation, prompting us to investigate other putative modes of binding disruption.
Fig. 5.
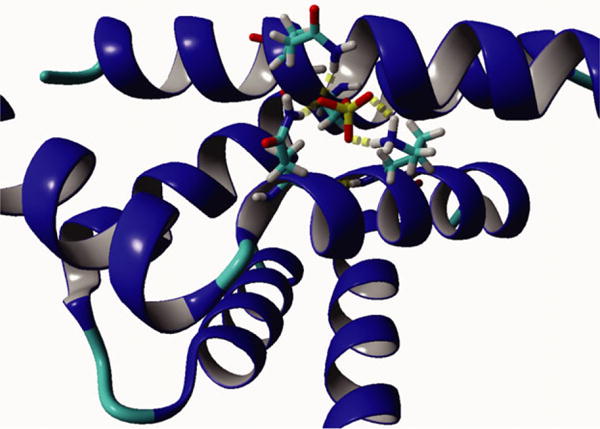
Molecular model of the smAKAP-AKB-Phospho:PKA-RIαD/D interaction. Using the HADDOCK molecular modeling program, we obtained a best model wherein the phosphoserine is sticking out of the hydrophobic domain, which in turn allows hydrogen bonding with Gln26 and Lys 30 of PKA-RIα and Gln67 of smAKAP.
Molecular mechanism of the disruption of PKA: AKAP interaction
In molecular dynamic (MD) simulations on the AKB helices of AKB-wt, AKB-S66D, AKB-S66E, and AKB-pSer66, all peptides partially or even fully lost their secondary structure in 20 ns simulations. The AKB-wt helix started to lose its helicity relatively slowly at both termini of the peptide after ~ 1 ns. In contrast, the other three peptides quickly lost helicity next to the mutated serine after ~ 1 ns (Fig. 6A). This suggests that the phosphorylation causes a severe distortion in the helix, which could possibly lead to the entire amphipathic helix being lost. Using NetSurfP 1.1, the helical propensity of the AKB-wt, AKB-S66D, and AKB-S66E peptides was determined (Fig. 6B). Mutation to an aspartic acid caused a severe drop in helical propensity which led us to investigate this further using circular dichroism (CD). The four peptides used in the MD simulations (AKB-wt, AKB-S66D, AKB-S66E, and AKB-pSer66) were synthesized and measured by CD. As suggested by the MD simulations, only the AKB-wt peptide displayed a strong α-helix propensity (~ 45%), whereas the phosphoserine and phosphomimetic mutants showed no secondary structure (Fig. 6C). Furthermore, upon further analysis of the HDX data but specifically looking at deuterium incorporation of smAKAP itself with respect to time, it is clearly shown that there is secondary structure present at the AKB domain, as previously revealed by the computational secondary structure prediction tool and CD. The largest difference of incorporation, based on the theoretical uptake of the individual peptides, was shown to be the AKB domain (Table S3). This confirms the findings of the MD simulations and suggests that the formation of an amphipathic helix, required for PKA binding, is seriously hampered when Ser66 of smAKAP is phosphorylated.
Fig. 6.
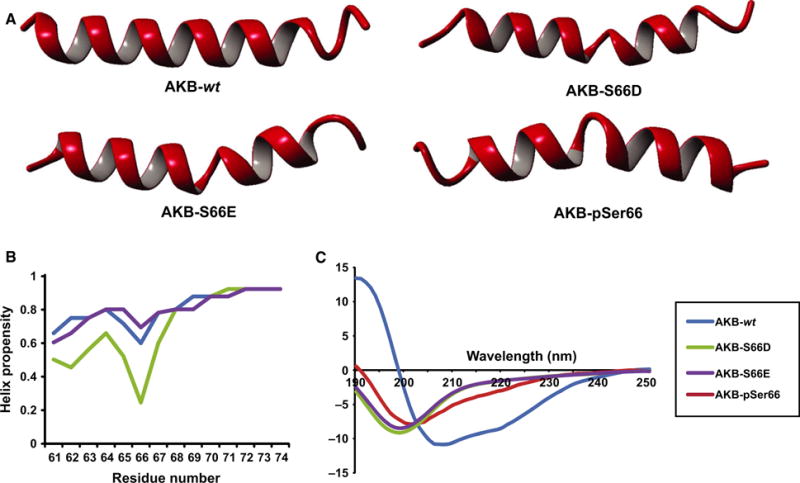
Loss of helical structure due to Ser66 phosphorylation abolishes PKA-R–smAKAP interaction. (A) Molecular dynamics simulations of the AKB-wt peptide placed in water were run for 20 ns. The peptide started to lose its helix from the N and C termini. In contrast, the AKB-S66D, AKB-S66E, and AKB-pSer66 peptides already lost their secondary structure in the middle near the serine after 1 ns. (B) Helix propensity prediction of AKB-wt, AKB-S66D, and AKB-S66E were measured via NetSurfP 1.1. Upon the mutation of serine to aspartic acid, there is a severe drop in helix propensity. NetSurfP 1.1 is unable to use phosphorylated serines. (C) CD measurements revealed a helix propensity of ~ 45% for the AKB-wt peptide. The AKB-S66D, AKB-S66E, and AKB-pSer66 peptides showed no secondary structure.
Discussion
Thus far, molecular understanding of the PKA-RI: AKAP interface has been limited to studying its interaction with the dual-specific dAKAP2 [20] and the creation of PKA-RI-specific peptide libraries [18]. Here, we took advantage of the recent discovery of two PKA-RI-specific AKAPs, SPHKAP [10], and smA-KAP [9], to extend our understanding of these specific interactions.
Comparison of dAKAP2 binding to either PKA-RI or PKA-RII revealed a remarkable difference in interaction surface [19,20]. The PKA-RIα:dAKAP2 interaction makes contact through four hydrophobic pockets on dAKAP2’s amphipathic helix (Fig. 1A), whereas PKA-RII utilizes only two hydrophobic binding pockets, resulting in a much larger interaction surface for PKA-RIα:dAKAP2 [20]. In the structure of the PKA-RIαD/D:smAKAP-AKB complex solved here, we clearly see similar contacts via four binding pockets (Fig. 1A). Another interesting difference between the D/D domains of PKA-RIα and RIIα is the presence of two identical disulfide bridges between Cys16 of one PKA-RIα monomer and Cys37 of the second PKA-RIα monomer [13,25]. These reside at the edges of the AKAP interaction surface of the PKA-RIα-D/D domain [13]. It was suggested that these disulfide bridges could contribute to the interaction with dAKAP2, although in the crystal structure of Sarma et al. [20], no direct interaction with any of dAKAP2’s residues was observed. Interestingly, smAKAP did show these interactions as Ile58C and Tyr61C at the N-terminal end (Fig. 1C) and Leu74C (possibly in conjunction with Trp77) seal both disulfide bridges into the hydrophobic pockets. An additional interaction between Ile58C and Leu13A of PKA-RIα (helix α0) also assists. In dAKAP2, the locations of Gln631 and Met647 are identical to those of smAKAP’s Ile58C and Leu74C, respectively; however these are not able to accomplish the same task with respect to binding [20].
These additional hydrophobic interactions could account for the much stronger binding affinity that PKA-RIα displays for smAKAP compared to dAKAP2 [9,16]. Three other PKA-RI-binding sequences have an isoleucine or a leucine at the equivalent Ile58 position of smAKAP (Fig. 1A): dAKAP1 (dual-specific, Ile344), SPHKAP [10] (RI-specific, Ile926), and RIAD [18] (synthetic, Leu1). dAKAP2 does not have a hydrophobic residue at the Ile58 equivalent position but a glutamine (Gln631, Fig. 1A). A peptide substitution array performed on the AKB domain of dAKAP2 showed that phenylalanine, leucine, isoleucine, or valine substitution of Gln631 increased the PKA-RIα binding affinity of dAKAP2 by sixfold [16]. These findings support the importance of creating an additional, fifth, hydrophobic pocket around Ile58C and the Cys16A:Cys37B disulfide bridge to drive high-affinity PKA-RIα binding. Our structure also suggests that Tyr61C, located in pocket 1, is part of this extended hydrophobic interaction. This residue is likely not only important for affinity but also for PKA-RI specificity. For instance, if the first amino acid in pocket 1 of dAKAP2 (Leu634) is mutated to an aromatic residue, the PKA-RII binding affinity decreases approximately 40-fold. As well, when RIAD was generated [18], it was observed that PKA-RI tolerates a bulky (aromatic) amino acid at that position (Tyr4 of RIAD), while PKA-RII did not, in line with the observation of Banky et al. [13] that PKA-RIα contains a much deeper hydrophobic groove in the D/D domain to accommodate this large side chain. This was confirmed by the establishment of SPHKAP as the first PKA-RI-specific AKAP (Phe929) [10].
In further support of the proposed sixth pocket at the C-terminal end of smAKAP’s AKB domain, Burns-Hamuro et al. [16] also created a dAKAP2 AKB domain peptide in which both Gln631 and Met647 (equivalent to Ile58 and Leu74 of smAKAP) were substituted for phenylalanines. This resulted in an affinity increase for PKA-RIα of 24-fold, instead of sixfold when substituting only Gln631 for phenylalanine. PKA-RII affinity was not affected, indicating that these interactions do not drive specificity, but rather affinity. The conservation of a hydrophobic amino acid at Trp77 (smAKAP, Fig. 1A) in the other PKA-RI-binding AKAPs suggests that this could further assist binding in this region; however, due to disorder of Trp77 in our crystal structure, verification of this hypothesis awaits.
A-kinase anchoring proteins accommodate spatial/temporal signaling of PKA. As such, AKAPs anchor PKA close to its substrates in a specific cellular compartment allowing it to respond instantly to local rises in cAMP levels. Phosphorylation of substrates may continue until halted via several feedback loops. The best described mechanism proceeds through nearby phosphodiesterases (PDEs), which degrade cAMP to AMP, thereby terminating PKA activity at the second messenger level [30]. Often, specific PDEs and PKA are sequestered by the same AKAP [5]. An example of such a negative feedback mechanism is membrane-bound PDE3A, which is activated by PKA phosphorylation [30]. Dropping concentrations lead to dissociation of cAMP from PKA-R and subsequent reformation of the inactive PKA holoenzyme on the AKAP. Dephosphorylation of PDE3 by a nearby, or even AKAP-anchored, phosphatase then resets the system to respond to rises in cAMP again.
Direct disruption of the PKA:AKAP interaction, as suggested here, could be another potential mechanism to control localized cAMP signaling; however, thus far this has only been shown pharmacologically. For instance, the Ht31 peptide [31] and more recently the isoform-specific peptides, super-AKAP-IS [17] (PKA-RII) and RIAD [18] (PKA-RI), have been proven to be useful tools in disrupting the PKA:AKAP interaction. FMP-API-1, a small molecule, was also shown to inhibit the binding of PKA to AKAPs, through an allosteric effect away from the D/D domain [32]. Here, we show an alternative negative feedback loop in which PKA inhibits its own anchoring to smAKAP by phosphorylating Ser66 in the AKB domain. Based on these findings, we propose the following model (Fig. 7): At low cAMP concentrations, smAKAP is bound to the PKA holoenzyme. Once the cAMP concentration increases, it will bind to PKA-RI, causing PKA-C to be released. The released PKA-C, which is still in close vicinity of smAKAP, will phosphorylate smAKAP when the regulatory subunit dimer dissociates under equilibrium conditions (Fig. 4D). This phosphorylation results in the amphipathic helix AKB domain of smAKAP to (partially) unfold. As it unfolds, PKA-RI gains more freedom to operate as the dimer cannot rebind to smAKAP. If the phosphorylation is removed, by a nearby phosphatase, the random coil AKB domain can re-form its α-helix, allowing PKA-RI to bind to smAKAP again. That this may be a bona fide mechanism used by other AKAPs as well is illustrated by the presence of Ser, Thr, and Tyr residues in at least six other human AKB domains (Fig. 8), of which several (AKAP1 and AKAP4) are already annotated as sites of phosphorylation in online phosphoproteomics databases. Not all of these bear the typical PKA phosphorylation motif in the AKB domain, although AKAP4 and AKAP18 do. This could indicate that the mechanism revealed here is a more common inhibitory mechanism for the regulation of localized cAMP signaling.
Fig. 7.
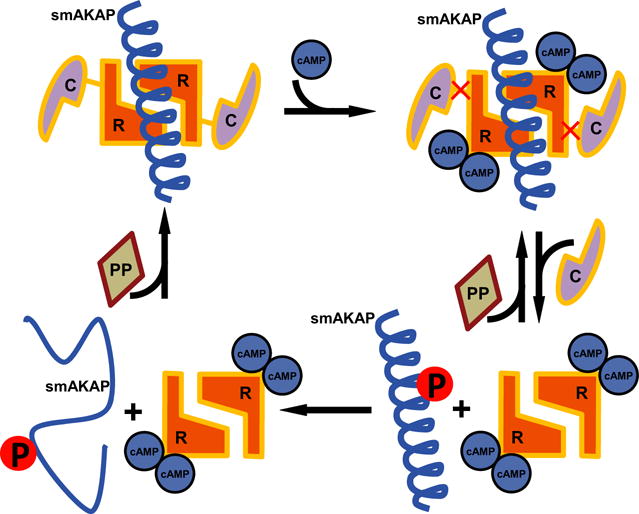
Proposed model for how PKA-Ser66 phosphorylation disrupts smAKAP binding. The PKA holoenzyme is tethered to smAKAP (signified by the amphipathic helical AKB domain as the remainder of the protein is unstructured). To each of the PKA-R in the dimer, a PKA-C is noncovalently bound. Upon binding of two cAMP to each PKA-R, the catalytic subunits are released. Upon release, PKA-C is able to phosphorylate Ser66 of smAKAP. Due to the phosphoserine present in the amphipathic helix, the secondary structure is lost and the energy barrier too high to form an α-helix again. A protein phosphatase could potentially dephosphorylate Ser66, in turn lowering the energy barrier to form an α-helix and allowing smAKAP to bind to PKA-RIα again.
Fig. 8.

Alignment of the AKB domain from smAKAP and six other human AKAPs. AKAP proteins whose AKB domains contain serines (red), threonines (green), and/or tyrosines (blue) are shown.
What is the physiological role of PKA-RI release from smAKAP? This is a question we cannot answer at this time, although it is tempting to speculate that release of the PKA-RI subunit induces a (temporary) signal termination at the smAKAP location. However, one could also imagine that PKA-RI release may assist in more efficient capturing of the catalytic subunit when cAMP concentrations are dropping or PKA phosphorylation of smAKAP could play a role in the dynamics of degradation of this particular PKA signaling node. This should be particularly important in the light of the very strong interaction between smAKAP and PKA-RI.
In conclusion, we present here the first structure of PKA-RI with one of its specific AKAPs, of which only two have been identified thus far (smAKAP and SPHKAP). The structure revealed several interesting points: (a) the interaction between PKA-RIα and smA-KAP utilizes six hydrophobic contact sites, and extends over a much larger portion of the amphipathic helix, whereas PKA-RII interactions typically use fewer hydrophobic pockets to achieve high-affinity binding; (b) the extended interaction surface provides a hydrophobic Cα for the disulfide bond thus shielding it from solvent; and (c) phosphorylation of smAKAP’s AKB domain by PKA causes destabilization of the amphipathic helix which reduces smAKAP’s affinity for PKA-RIα at least 1000-fold. This makes it tempting to speculate that PKA-RIα interacting with its specific AKAPs occurs at much higher affinities. This may actually require the observed phosphorylation event to prevent the interaction under physiological conditions. Additionally, other AKAPs have been shown to be phosphorylated in their AKB domains, suggesting this to be a more general mechanism.
Experimental procedures
Protein purification
Bovine full-length PKA-RIα [33] (P00514) and PKA-RIαD/D (AA 12-61) [20] were purified as previously described. His-smAKAP (and mutant) were expressed in Escherichia coli (BL21 (DE3) [20]. At OD600, the cell culture was induced with isopropyl β-D-thiogalactopyranoside and grown for another 6 h at 37 °C prior to harvesting at 6000 rpm. The cells were resuspended in lysis buffer (50 mM KH2PO4, 300 mM NaCl, 10 mM imidazole, 5% glycerol, 5 mM β-mercaptoethanol, pH 8.0) after which they were lysed using a Microfluidizer (Microfluidics, Westwood, MA, USA) at 18 000 p.s.i. The cells were then spun down for 75 min at 4 °C in a Beckman JA20 rotor. The supernatant was then run on a Profinia (Bio-Rad, Hercules, CA, USA) according to the native IMAC protocol. The eluent was dialyzed in 20 mM Tris, 100 mM NaCl, 5 mM DTT, and at pH 8.3. The resulting protein was purified on an S75 column and concentrated to 30 mg·mL−1. The smAKAP-AKB peptide was dissolved into 0.5 M Tris at pH 8.5 to a concentration of 5 mg·mL−1. The complex formed, consisting of PKA-RIαD/D and smAKAP-AKB peptide, had a final concentration of, respectively, 10 and 2.52 mg·mL−1, and was used for crystallization.
Phosphorylation analyses
Human His-tagged smAKAP (wt and S66D) (10 μM) [9] was taken up in a reaction buffer containing MgCl2 (10 mM), ATP (200 μM), [32P]ATP (500–1000·cmp·ρmol−1), and MOPS (50 mM) (pH 7.5). Addition of recombinant PKA-Cα (20 nM) and/or PKG Iα (20 nM). About 10 lL of reaction volume was quenched with 90 μL of 30% acetic acid at the following time points: 0, 5, 10, and 15 min. The phosphorylation assays were also analyzed by LC-MS/MS to identify the exact phosphorylation site. An in-solution digestion of His-smAKAP using trypsin was performed as described previously [10]. The sample was desalted with an HBL Oasis system (Waters, Milford, MA, USA). The desalted sample was dried down and reconstituted in 10% formic acid. A vented-column setup was used for analyzing protein digests through an Agilent 1200-Series LC system coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Schwerte, Germany) with previously described conditions [9].
Crystallography
Crystals of PKA-RIαD/D:smAKAP-AKB complex appeared approximately 2 months after being set with an Oryx8 protein crystallization robot at room temperature at a 2 : 3 ratio of protein solution:crystallizing (crystallizing solution: 0.1 M citric acid, pH 3.5, 28% w/v polyethylene glycol 8000). Data were collected on the synchrotron beamline 8.2.2 of the Advanced Light Source, Lawrence Berkeley National Labs (Berkeley, CA, USA). The data were processed and scaled via HKL2000 [34]. For phasing, the PKA-RIα D/D:dAKAP2-AKB structure (3IM4) was used as molecular replacement with CCP4 using Phaser [20,35]. This model was further refined in COOT and Refmac 5.2 [36,37]. The refinement statistics are shown in Table 2. The atomic coordinates and structure factor have been deposited in the Protein Data Bank (accession code 5HVZ).
Table 2.
Collection and refinement statistics of the crystallographic data.
| PKA-RIαD/D:smAKAP AKB | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions (Å) | |
| a | 37.5 |
| b | 55.7 |
| c | 57.3 |
| No. of molecule per asymmetrical unit | 1 |
| Resolution (Å) | 2.0 |
| Rmerge (%) | 7.6 (46) |
| Completeness (%) | 98.0 (100.0) |
| I/sigma | 26.9 (4.0) |
| No. of reflections | 8480 |
| Refinement | |
| Resolution (Å) | 40.0–2.0 |
| Rwork/Rfree (%) | 21.0/24.1 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.024 |
| Bond angles (°) | 1.9 |
| Ramachandran angles (%) | |
| Most favored | 90.9 |
| Disallowed | None |
Values in parentheses are for the highest resolution shell: (2.00–2.05 Å).
Structure analysis
The final crystallography model was evaluated using MolProbity [38].
Hydrogen–deuterium exchange mass spectrometry
A 30-fold dilution with either 100% H2O, pH 7 for the nondeuterated experiments or deuterium oxide (Sigma-Aldrich, Darmstadt, Germany), pD 7, for deuterated experiments was carried out for the following complexes: unbound smAKAP (60 pmol), smAKAP-excess PKA-RIα (60 pmol of smAKAP and 75 pmol of PKA-RIα), unbound PKA-RIα (60 pmol), and PKA-RIα-excess smA-KAP (60 pmol of PKA-RIα and 75 pmol of smAKAP). Diluted samples were incubated at room temperature for time intervals of 0 min for the nondeuterated experiments and 10 s, 1 min, 10 min, 60 min, and 240 min for the deuterated experiments. The deuteration reaction was quenched by pH reduction to 2.5 with a 1 : 1 dilution using ice-cold 4 M guanidine hydrochloride adjusted to pH 1.85. Quenched samples were immediately injected into a 50 lL injection loop on a nano-ACQUITY UPLC system with HDX technology (Waters). Online digestion was performed using an in-house built immobilized pepsin column for 2 min in 0.05% formic acid in H2O (flow rate of 25 μL min−1), held at a temperature of 15 °C. Peptides were trapped and desalted online using an ACQUITY UPLC BEH C18 1.7 μm VanGuard Pre-column (Waters) at 0 °C, with subsequent elution onto an ACQUITY UPLC BEH C18 1.7 μm, 1 mm × 100 mm column (Waters) held at 0 °C. Peptide separation was achieved using a 7-min linear acetonitrile gradient (5–85%) in 0.1% formic acid (flow rate of 40 μL min−1). The eluent was directed into a Xevo G2 instrument (Waters) with electrospray ionization and lock-mass correction (using Glufibrinogen peptide). Mass spectra were acquired in MSE mode over the m/z range 50–2000. Two blank injections were performed between each sample injection to prevent sample carryover. Peptides were identified prior to deuteration using PROTEINLYNX GLOBAL SERVER 2.5 software (Waters).
Calculation of exchange data
Deuterium uptake was calculated and compared to the nondeuterated control samples using DYNAMX 1.0.0 software (Waters). Experiments were carried out in triplicate at each time point. Absolute deuterium incorporation at a given time point was determined by comparison with t = 0 in the nondeuterated sample. The deuterium incorporation at a given time point corresponded to the centroid value across the backbone amide population. Results were averaged across repeat analyses and a SD derived. To examine the differences in a comparable way, the percentage difference uptake (Da) between the excess and unbound sets was calculated per peptide using Eqn (1).
| (1) |
Cell culture
HeLa and HEK293 cells were cultured and transfected with the plasmids (smAKAP-wt-GFP, PKA-RIα-mKO2, PKA-RIα-mKO2, PKA-C-HA, and smAKAP-S66E-mGFP) as described previously [9] for the confocal imaging. For the FRET experiment, HEK293A cells were grown in Dulbecco’s modified Eagle’s medium media (supplemented with 10% FCS, L-glutamine, and antibiotics) and plated on 24 mm round coverslides. Twenty-four hours after plating, cells were transfected with the smAKAP-GFP, PKA-RIα-mCherry, and PKA-C-HA plasmids using calcium phosphate precipitation.
Fluorescence imaging
Cells were washed once with ice-cold PBS, fixed in 4% ice-cold formaldehyde in PBS and afterwards washed twice with PBS. Upon addition of calyculin A and 8-CPT-cAMP, live imaging was employed. The confocal images were obtained with a Zeiss LSM700 confocal system with a 63× oil objective lens.
FRET measurements
About 24–36 h after transfection, coverslides were transferred into a measuring chamber and rinsed once with a buffer containing 148 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, pH 7.3. The measuring chamber was then transferred onto a ZEISS AxioObserver A1 epifluorescence microscope equipped with an oil immersion Plan Apochromat 63× objective, polychrome V light source (TILL Photonics, Gräfelfing, Germany), DV2 QuadlView (Photometrics, Tucson, AZ, USA), and a Cool-SNAP-HQ CCD-camera (Visitron Systems, Puchheim, Germany). Cells were then treated with 10 μM forskolin added in the same buffer (all chemicals were purchased from Sigma-Aldrich). To monitor FRET between GFP and mCherry, cells were excited at 490 nm, the emission light was split into two channels using the DV2 QuadView (565dcxr dichroic mirror) and detected at 515 ± 15 (GFP) and 590 ± 20 (mCherry) nm. FRET changes were monitored using VISIVIEW software (Visitron Systems) as the emission ratio of mCherry over GFP and analyzed as previously described [39].
Mutagenesis
The single-site mutations (S66D and S66E) were introduced in the previously described smAKAP-GFP plasmid [9] and pLICHIS His6-smAKAP plasmid via the QuikChange mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA).
Circular dichroism and fluorescence anisotropy
Four peptides (a) IVILEYAHRLSQDILCDALQQWAC, (b) IVILEYAHRLpSQDILCDALQQWAC, (c) IVILEYAHRLDQDILCDALQQWAC, and (d) IVILEYAHRLEQDILCDALQQWAC (mutations in bold) were synthesized and purified (NKI, Amsterdam, the Netherlands). These were dissolved in a 0.1 M Tris buffer at pH 8.5. CD measurements were carried out on a Jasco J-810 spectropolarimeter and analyzed via the Jasco Spectra Manager. For fluorescence anisotropy measurements on the interaction between PKA-RIα and smAKAP, peptides 1 and 2 were N-terminally tagged with 5-TAMRA as described previously [9]. Measurements were also performed as described previously [9].
Molecular dynamics
Molecular dynamics was performed with the program YASARA version 8.12.26 in an AMBER03 force field [40]. The simulations were run as described previously [40,41] with the exception of the following. In an aqueous continuous phase, the smAKAP-AKB peptide from structure solved in this article was placed in a 50 Å × 50 Å × 50 Å box. Simulations were run for 25 ns, and coordinates were saved every 7.5 ps. The results were then analyzed using analysis programs written in our laboratory. The AKB-Phospho peptide was constructed using the FoldX package.
Statistical methods
Statistical analyses used the Michaelis–Menten model, as implemented in PRISM (version 5.0a).
Supplementary Material
Table S1. Identified PKA-RIα peptides of the hydrogen/deuterium exchange experiment displaying their individual deuterium incorporation over time.
Table S2. The entire list of smAKAP peptides of the hydrogen/deuterium exchange displaying the large amount of deuterium incorporation in the AKB domain after 10 s.
Table S3. The smAKAP peptides of the hydrogen/deuterium exchange displaying the percentage of deuterium incorporation in the AKB domain throughout 240 min.
Acknowledgments
This work was supported in part by the PRIME-XS project, Grant Agreement Number 262067, funded by the European Union 7th Framework Programme. The Netherlands Proteomics Centre and the Netherlands Organization for Scientific Research (NWO), through funding the roadmap program Proteins@Work (project 184.032.201) are acknowledged for support. We thank A. M. J. J. Bonvin (Utrecht University, the Netherlands) for his assistance with HADDOCK modeling and Dr. Alexander Fish for his assistance with the fluorescence anisotropy measurements. S. S. T acknowledges support by the NIH (grant NIH GM034921).
Abbreviations
- 5-TAMRA
5-carboxytetramethylrhodamine
- AKAP
A-kinase anchoring protein
- AKB
A-kinase-binding domain
- C
catalytic subunit
- CD
circular dichroism
- D/D
dimerization and docking domain
- dAKAP
dual-specific AKAP
- FRET
Förster resonance energy transfer
- HDX
hydrogen/deuterium exchange mass spectrometry
- MD
molecular dynamics
- Opa1
optic atrophy 1
- PDE
phosphodiesterase
- PKA
protein kinase A
- PKG
protein kinase G
- RI/RII
regulatory subunit I/II
- RMSD
root-mean-square deviation
- smAKAP
small membrane AKAP
- SPHKAP
sphingosine kinase type 1-interaction protein
Footnotes
Database
Structural data are available in the PDB under accession number 5HVZ.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article:
Author contributions
PB and AS designed the study. PB, RJR, JB, VON, and MK performed the experiments; PB, JB, BJCJ, and MK analyzed the data. PB, SST, AJRH, and AS wrote the manuscript. SST, AJRH, and AS provided supervision.
Figures
Structure images were generated by using PYMOL Version 1.5.0 (http://www.pymol.org/) and YASARA 8.12.26. Figures were created by using Adobe Photoshop and Illustrator.
References
- 1.Mao T, Shao M, Qiu Y, Huang J, Zhang Y, Song B, Wang Q, Jiang L, Liu Y, Han JD, et al. PKA phosphorylation couples hepatic inositol-requiring enzyme 1alpha to glucagon signaling in glucose metabolism. Proc Natl Acad Sci USA. 2011;108:15852–15857. doi: 10.1073/pnas.1107394108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- 3.Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G. PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259–269. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 4.Scholten A, Aye TT, Heck AJ. A multi-angular mass spectrometric view at cyclic nucleotide dependent protein kinases: in vivo characterization and structure/function relationships. Mass Spectrom Rev. 2008;27:331–353. doi: 10.1002/mas.20166. [DOI] [PubMed] [Google Scholar]
- 5.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 6.Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem. 1997;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- 7.Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. D-AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc Natl Acad Sci USA. 1997;94:11184–11189. doi: 10.1073/pnas.94.21.11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pidoux G, Witczak O, Jarnaess E, Myrvold L, Urlaub H, Stokka AJ, Kuntziger T, Tasken K. Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J. 2011;30:4371–4386. doi: 10.1038/emboj.2011.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgers PP, Ma Y, Margarucci L, Mackey M, van der Heyden MA, Ellisman M, Scholten A, Taylor SS, Heck AJ. A small novel A-kinase anchoring protein (AKAP) that localizes specifically protein kinase A-regulatory subunit I (PKA-RI) to the plasma membrane. J Biol Chem. 2012;287:43789–43797. doi: 10.1074/jbc.M112.395970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovanich D, van der Heyden MA, Aye TT, van Veen TA, Heck AJ, Scholten A. Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type I cAMP-dependent protein kinase. ChemBioChem. 2010;11:963–971. doi: 10.1002/cbic.201000058. [DOI] [PubMed] [Google Scholar]
- 11.Means CK, Lygren B, Langeberg LK, Jain A, Dixon RE, Vega AL, Gold MG, Petrosyan S, Taylor SS, Murphy AN, et al. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc Natl Acad Sci USA. 2011;108:E1227–E1235. doi: 10.1073/pnas.1107182108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carr DW, Stofko-Hahn RE, Fraser ID, Bishop SM, Acott TS, Brennan RG, Scott JD. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem. 1991;266:14188–14192. [PubMed] [Google Scholar]
- 13.Banky P, Roy M, Newlon MG, Morikis D, Haste NM, Taylor SS, Jennings PA. Related protein-protein interaction modules present drastically different surface topographies despite a conserved helical platform. J Mol Biol. 2003;330:1117–1129. doi: 10.1016/s0022-2836(03)00552-7. [DOI] [PubMed] [Google Scholar]
- 14.Newlon MG, Roy M, Morikis D, Hausken ZE, Coghlan V, Scott JD, Jennings PA. The molecular basis for protein kinase A anchoring revealed by solution NMR. Nat Struct Biol. 1999;6:222–227. doi: 10.1038/6663. [DOI] [PubMed] [Google Scholar]
- 15.Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schroder E, Wait R, Begum S, Kentish JC, Eaton P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem. 2006;281:21827–21836. doi: 10.1074/jbc.M603952200. [DOI] [PubMed] [Google Scholar]
- 16.Burns-Hamuro LL, Ma Y, Kammerer S, Reineke U, Self C, Cook C, Olson GL, Cantor CR, Braun A, Taylor SS. Designing isoform-specific peptide disruptors of protein kinase A localization. Proc Natl Acad Sci USA. 2003;100:4072–4077. doi: 10.1073/pnas.2628038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gold MG, Lygren B, Dokurno P, Hoshi N, McConnachie G, Tasken K, Carlson CR, Scott JD, Barford D. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell. 2006;24:383–395. doi: 10.1016/j.molcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Carlson CR, Lygren B, Berge T, Hoshi N, Wong W, Tasken K, Scott JD. Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. J Biol Chem. 2006;281:21535–21545. doi: 10.1074/jbc.M603223200. [DOI] [PubMed] [Google Scholar]
- 19.Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarma GN, Kinderman FS, Kim C, von Daake S, Chen L, Wang BC, Taylor SS. Structure of D-AKAP2:PKA RI complex: insights into AKAP specificity and selectivity. Structure. 2010;18:155–166. doi: 10.1016/j.str.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neuberger G, Schneider G, Eisenhaber F. pkaPS: prediction of protein kinase A phosphorylation sites with the simplified kinase-substrate binding model. Biol Direct. 2007;2:1. doi: 10.1186/1745-6150-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tegge W, Frank R, Hofmann F, Dostmann WR. Determination of cyclic nucleotide-dependent protein kinase substrate specificity by the use of peptide libraries on cellulose paper. Biochemistry. 1995;34:10569–10577. doi: 10.1021/bi00033a032. [DOI] [PubMed] [Google Scholar]
- 23.Diviani D, Abuin L, Cotecchia S, Pansier L. Anchoring of both PKA and 14-3-3 inhibits the Rho-GEF activity of the AKAP-Lbc signaling complex. EMBO J. 2004;23:2811–2820. doi: 10.1038/sj.emboj.7600287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dell’Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5-bisphosphate. EMBO J. 1998;17:2246–2260. doi: 10.1093/emboj/17.8.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 2001;20:1651–1662. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hennrich ML, Marino F, Groenewold V, Kops GJ, Mohammed S, Heck AJ. Universal quantitative kinase assay based on diagonal SCX chromatography and stable isotope dimethyl labeling provides high-definition kinase consensus motifs for PKA and human Mps1. J Proteome Res. 2013;12:2214–2224. doi: 10.1021/pr400074f. [DOI] [PubMed] [Google Scholar]
- 27.de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AM. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- 28.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 29.Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct Biol. 2009;9:51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maurice DH, Palmer D, Tilley DG, Dunkerley HA, Netherton SJ, Raymond DR, Elbatarny HS, Jimmo SL. Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol. 2003;64:533–546. doi: 10.1124/mol.64.3.533. [DOI] [PubMed] [Google Scholar]
- 31.Klussmann E, Edemir B, Pepperle B, Tamma G, Henn V, Klauschenz E, Hundsrucker C, Maric K, Rosenthal W. Ht31: the first protein kinase A anchoring protein to integrate protein kinase A and Rho signaling. FEBS Lett. 2001;507:264–268. doi: 10.1016/s0014-5793(01)02995-7. [DOI] [PubMed] [Google Scholar]
- 32.Christian F, Szaszak M, Friedl S, Drewianka S, Lorenz D, Goncalves A, Furkert J, Vargas C, Schmieder P, Gotz F, et al. Small molecule AKAP-protein kinase A (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J Biol Chem. 2011;286:9079–9096. doi: 10.1074/jbc.M110.160614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J, Jones JM, Nguyen-Huu X, Ten Eyck LF, Taylor SS. Crystal structures of RIalpha subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry. 2004;43:6620–6629. doi: 10.1021/bi0302503. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 38.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borner S, Schwede F, Schlipp A, Berisha F, Calebiro D, Lohse MJ, Nikolaev VO. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat Protoc. 2011;6:427–438. doi: 10.1038/nprot.2010.198. [DOI] [PubMed] [Google Scholar]
- 40.Krieger E, Darden T, Nabuurs SB, Finkelstein A, Vriend G. Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins. 2004;57:678–683. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- 41.van Hell AJ, Klymchenko A, Burgers PP, Moret EE, Jiskoot W, Hennink WE, Crommelin DJ, Mastrobattista E. Conformation and intermolecular interactions of SA2 peptides self-assembled into vesicles. J Phys Chem B. 2010;114:11046–11052. doi: 10.1021/jp103440d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Identified PKA-RIα peptides of the hydrogen/deuterium exchange experiment displaying their individual deuterium incorporation over time.
Table S2. The entire list of smAKAP peptides of the hydrogen/deuterium exchange displaying the large amount of deuterium incorporation in the AKB domain after 10 s.
Table S3. The smAKAP peptides of the hydrogen/deuterium exchange displaying the percentage of deuterium incorporation in the AKB domain throughout 240 min.