

Abstract
A passively operated polydimethylsiloxane (PDMS) microfluidic device was designed for sampling of hormone secretions from eight individual murine pancreatic islets in parallel. Flow control was achieved using a single hand-held syringe and by exploiting inherent fluidic resistances of the microchannels (Rsampling = 700 ± 20 kPa s mm−3 at 37 °C). Basal (3 mM) or stimulatory (11 mM) glucose levels were applied to islets, with stimulation timing (tstim) minimized to 15 ± 2 s using modified reservoirs. Using enzyme-linked immunosorbent assays (ELISA) for postsampling analyses, we measured statistically equal levels of 1 h insulin secretion (1.26 ± 0.26 and 6.55± 1.00 pg islet−1 min−1, basal and stimulated; 62 islets) compared to standard, bulk sampling methods (1.01± 0.224 and 6.04 ± 1.53 pg islet−1 min−1, basal and stimulated; 200 islets). Importantly, the microfluidic platform revealed novel information on single-islet variability. Islet volume measurements with confocal reflectance microscopy revealed that insulin secretion had only limited correlation to islet volume, suggesting a more significant role for cellular architecture and paracrine signaling within the tissue. Compared to other methods using syringe pumps or electroosmotic flow control, this approach provides significant advantages in ease-of-use and device disposability, easing the burden on nonexperts.
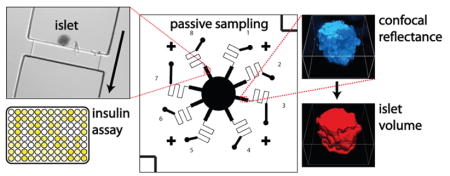
Graphical Abstract
The islets of Langerhans are considered “micro-organs,” composed of five different cell types (α, β, δ, PP, and ε), each of which is responsible for the secretion of different hormones.1–4 These small clusters of tissue can vary in diameter from tens to hundreds of micrometers and are dispersed throughout the pancreas, each with their own blood supply via microcapillaries.5,6 Impaired or lost function of the islet can lead to diabetic disease states and is linked to obesity and metabolic syndrome.7 Islet transplantation is a promising treatment that has been shown to reverse the effects of islet loss in type 1 diabetes, yet further research is needed to enhance islet survival and to address the immune reaction of transplant recipients.8,9 Most research to date has focused on the insulin-secreting β-cells within islets, which helps to ensure blood glucose homeostasis. However, recent work has highlighted the importance of paracrine communication among the multiple cell types within the islet,10,11 indicating a need for more refined methods in studying the multicellular architecture of islets and its relation to function.
A promising approach to aid in the understanding of fundamental islet function is the study of single islets rather than the ensemble response of many islets. In this respect, the microfluidic platform has proven ideal for minimizing dilution, allowing sampling of single islet secretions.3,12–14 The Kennedy group has pioneered microfluidic sampling of single islets, utilizing a downstream competitive immunoassay with microchip electrophoresis and fluorescence detection.12,13 Their recent device is capable of sampling from 15 islets with 10 s temporal resolution for insulin quantitation.13,14 The sampling and measurement system requires flow control with external pumps, microchip electrophoresis and electroosmotic flow control using computer-controlled high-voltage power supplies, and an optical system for fluorescence detection. This instrumental complexity will likely hinder the use of these devices in routine biological experiments or in a clinical setting, for example, in monitoring islet viability prior to transplantation. The Eddington group has developed a microfluidic device for sampling and monitoring of multiple islets in a single large reservoir.15,16 Although possibly more amenable to use in a clinical setting, this approach is not capable of acquiring single-islet information and also requires attachment to external instruments. There is a clear need for simple-to-use microfluidic approaches for single islet sampling and measurement.
We address this need with a passively operated microfluidic system capable of sampling secretions in parallel from eight single pancreatic islets. One major advantage associated with our system is its ease of operation, because no electrical components or external instrumentation are required. The only equipment needed to operate our device is an 8-channel pressure manifold and hand-held syringe. Building upon previous work,3,17 the microchannels’ fluidic resistances are used for passive flow control, such that no electrical power is needed for secretion sampling.3 Following sampling over 1 h periods, commercially available enzyme-linked immunosorbent assays (ELISA) were used to quantify insulin secretion from each individual islet. Thus, single-islet information was obtained in higher throughput than our previous work,3 allowing measurements on many islets from multiple mice to sample the inherent biological variability. A 1 h secretion sampling period allowed straightforward comparison to multi-islet, static methods, where the microfluidic approach provided statistically equal results. Each islet was also scanned by confocal microscopy to determine its volume, allowing the first measurements of single-islet insulin secretion versus islet volume. With this system, it should be possible to further interrogate the consequences of single-islet variability, particularly in relation to paracrine signaling among the multiple cell types. With considerable improvement in ease-of-use, this type of microfluidic system should also be generally applicable to sampling from small quantities of various tissues and multicellular systems.
EXPERIMENTAL SECTION
Reagents and Materials
Polydimethylsiloxane (PDMS) precursors, Sylgard 184 elastomer base and curing agent, were obtained from Dow Corning. NaCl, KCl, CaCl2, MgCl2, HEPES, bovine serum albumin (BSA), RPMI medium, phosphate buffered saline (PBS), fetal bovine serum, and Hank’s buffer were purchased from VWR. D-Glucose was purchased from Sigma Aldrich. Fluo-4 acetoxymethyl (AM) ester and monolabeled fluorescein isothiocyanate (FITC)-insulin were obtained from Invitrogen. Unless otherwise noted, “imaging medium” consisted of 125 mM NaCl, 5.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, 3 mM glucose, and 0.1% BSA at a pH of 7.4.
Microfluidic Device Fabrication
All devices (Figure 1) were fabricated using PDMS as the channeled substrate and No. 1 glass coverslips as the floor substrate using standard methods.3,18 Insulin adsorption to the microchannel walls was assayed using monolabeled fluorescent insulin standards (“FITC-Ins” ; Invitrogen). Further details on microchip and manifold fabrication are included in Supporting Information.
Figure 1.

Passively operated microfluidic device for islet secretion sampling and imaging. (A) Device layout, including an image of a trapped islet. (B) Hand-held apparatus for secretion sampling with no electrical components. (C) Insulin adsorption was prevented by dynamic coating with BSA in the microchannel (white bars) as well as in the reservoir (gray bars), by including 0.1% BSA in the media. Error bars represent standard deviations.
Statistical Analyses
In comparing population means of insulin ELISA measurements, the two-tailed Student’s t test with equal variances was calculated using Microsoft Excel. Values for the probability, p, that the means are equal are listed in the text and figures. Data sets with p ≤ 0.05 were considered statistically different.
Pancreatic Islet Extraction and Sampling
Islets were isolated as described previously22,23 from live C57BL/6 mice (male) and maintained in RPMI medium containing 10% fetal bovine serum and 11mMglucose at 37 °C under humidified 5%CO2 for 24 h before imaging. For standard bulk secretion assays, 10 islets with a wide distribution of diameters were loaded into 0.5 mL tubes that contained 200 μL imaging medium with basal glucose (3 mM) and maintained at 37 °C for 20 min. Glucose was increased to 11 mM, and tubes were incubated at 37 °C for 1 h. Aliquots (180 μL) of media were removed from each tube and stored at −20 °C for subsequent ELISA measurements. Control groups of 10 islets were held in imaging medium containing basal glucose (3 mM) over the entire study. Secretions were sampled from 40 islets per mouse, with 20 islets each at basal and stimulatory glucose. Five male C57BL/6 mice contributed islets to this study, totaling 200 islets.
For microfluidic perfusion and secretion sampling, eight single isolated islets with a wide distribution of diameters were first transferred from the RPMI culture medium into imaging medium (at 37 °C) with gentle rocking.3 After pretreatment for 1 h at basal glucose (3 mM), all eight islets were loaded onto the microchip on the stage-top incubator (37 °C). A constant vacuum was applied using an air-filled 100 mL syringe, with initial and final volumes defined by wooden spacers fabricated in-house (Figure 1B). Once the islets were loaded onto the microdevice, they were imaged with an inverted fluorescence microscope (Nikon Ti-E) in DIC mode to confirm successful loading. After 10 min at basal glucose, the central reservoir was emptied, and imaging medium containing stimulatory glucose levels (11 mM) was added (3 mM for controls). Secretions were sampled for an additional 50 min. Vacuum was then released; outlet tubes were removed, and islet perfusates were collected into microcentrifuge tubes and stored at −20 °C for subsequent ELISA measurements. Islets were left on the chip, and confocal images were acquired for volume measurements.
RESULTS AND DISCUSSION
8-Channel Perfusion and Sampling Device
In this work, a passively operated 8-channel microfluidic device (Figure 1A) was used for sampling secretions from multiple individual islets in parallel. The two-depth PDMS device contained deep (156 μm) and shallow (14.8 μm) channeled regions. Deep channels were designed for islet trapping, and shallow channels were used to passively control the flow rate of nutrients to and hormone secretions away from trapped islets.3 Ease of operation is a key advantage of the system. With flow rates controlled passively by fluidic resistors (Rsampling), secretion sampling was accomplished using only a hand-held syringe and 8-tube manifold (Figure 1B), precluding the need for electrical components or syringe pumps and making the devices disposable after a single use. Islets were trapped at the junction between deep and shallow channels (Figure 1A). Eight sampling channels directed flow outward in a circularly symmetric pattern from a central supply reservoir into plastic tubing (Tygon; SmallParts, Inc.) for collection of secretions. Outlets of these 8 tubes were connected to a PDMS/glass manifold (Figure 1B), fabricated in-house, which reduced to a single tube and connected to a 100 mL glass syringe (SGE Analytical Science). The syringe was used to apply and maintain a vacuum over the 1 h sampling experiments. As previously shown by others,24 it was confirmed that manual application of pressure using this hand-held syringe could be accomplished with ~2% relative standard deviation (Figure S-2, Supporting Information).
With high surface area-to-volume ratios in microfluidic devices, it was important to evaluate the likelihood of insulin adsorption to the channel walls. There have been significant efforts by others to understand and prevent protein adsorption to microchannel walls,18,25–28 and one approach that has found success due to its simplicity is the dynamic coating of channel walls by bovine serum albumin (BSA) by including BSA in the media.27,29 Since our imaging medium already contains 0.1% BSA, this was an attractive option. By flowing 100 nM monolabeled fluorescent insulin (“FITC-Ins”) through the device, it was possible to monitor insulin adsorption directly. As shown in Figure 1C, the presence of 0.1% BSA was important for preventing FITC-Ins adsorption to the microchannel walls. When 100 nM FITC-Ins solution was passed through the channels for 30 min in the absence of BSA, only 66 ± 16% of the FITC-Ins was recovered at the outlet (white bars). In the presence of 0.1% BSA, 106± 10% of the FITC-Ins was recovered at the outlet after 30 min. A similar effect was observed when 100 nM FITC-Ins was simply allowed to sit in the large reservoir (gray bars) of the device for 30 min, with 69 ± 15% and 104 ± 10% recovery without and with BSA, respectively. These tests were carried out at 37 °C under humidity control, using 2 batches (separate PDMS mixing and curing), 2 chips per batch, and 3 channels per chip to ensure representative sampling. Although the value of 100% recovery fell within the window of experimental error, mean recoveries were slightly larger than 100%, a discrepancy that is attributed to slight evaporation of the 200 μL volume in the reservoir during the 30 min experiments. The results shown in Figure 1C indicated that a pretreatment and continuous dynamic coating with 0.1% BSA in imaging medium is sufficient for preventing insulin adsorption to the microchannel walls; thus, all subsequent secretion sampling experiments followed this protocol.
Passive Flow Control
One of the key advantages of the presented system is the simplicity of use afforded by passive control over flow rates. First, to ensure nutrient supply to the islets in such a small-volume microchannel, a minimum flow rate should be established. From in vivo imaging of blood flow through islets,5 it was estimated that the blood flow rate through an average islet is ~2 μL h−1. During surgical removal, collagenase digestion of surrounding tissue collapses microcapillary vasculature within islets.3 Consequently, a minimum flow rate of ~20 μL h−1 was estimated for islet survival, consistent with previous reports.12,13 On the contrary, higher flow rates would dilute hormones to undetectable levels and increase shear stress on cells. With these restrictions, a flow rate of ~40 μL h−1 (0.011 mm3 s−1) was determined as adequate to ensure islet survival and minimize dilution.
This volumetric flow rate guided the design of shallow microfluidic channels (fluidic resistors or Rsampling in Figure 1A). Drawing analogies to electrical current flow through resistive elements, the fluidic resistance of rectangular microfluidic channels can be estimated.17,24,30 The final channel dimensions (length = 10.8 mm, depth = 14.8 μm, width = 60 μm; Figure 1A) give a Rsampling of 710 kPa s mm−3 with a working fluid of distilled water. Using the Ohm’s law analogy with Q representing the volumetric flow rate (ΔP = Q Rsampling), the theoretical pressure differential (ΔP) required to maintain the target flow rate would be −7.8 kPa. To determine the ΔP required in practice (using imaging medium), various vacuum pressures (negative ΔP) were applied to the outlets of the 8-channel device, and the fluid’s linear velocity was measured by simple meniscus tracking in the outlet tubes.25,26 At 37 °C, a vacuum was applied, and the final position of the meniscus in the outlet tubing was measured after 15 min. Multiple channels from three different chips were tested in this manner. Volumetric flow rates of the fluid were determined at each ΔP using the cross-sectional area of the tubing. As expected, a linear dependence of Q versus ΔP was observed (R2 = 0.9973), and Rsampling was determined to be 700 ± 20 kPa s mm−3 using the reciprocal of the slope (Figure S-3, Supporting Information). The agreement with the calculated value of 710 kPa s mm−3 is attributed to the decreased viscosity at 37 °C being offset by the increased viscosity of 0.1% BSA in the imaging medium. With Rsampling = 700 ± 20 kPa s mm−3 at 37 °C, a vacuum of −7.7 kPa would be required to achieve the target flow rate of 0.011 mm3 s−1 (40 μL h−1). This vacuum level was used in all secretion sampling experiments.
Stimulation Timing
To ensure ease of operation of the device, it was important to determine the timing for newly introduced stimulants to reach the islet tissue, such as increased glucose concentration.31 Rapid increase in glucose at the islet is preferred following a change of perfusate in the reservoir. Here, we define stimulation timing (tstim) as the difference between the time of solution replacement in the large reservoir (t = 0 in Figure 2B) and the time at which this new solution reached the islet trapping region. A preliminary design of the microdevice with individually punched reservoirs (0.8–1.0 cm depths; ≥ 6.3 μL dead volume) requires ~10 min to fill the dead volume upon a change in perfusate at 40 μL h−1, much too long for the 1 h measurements (~17% of total time). Gradient formation and tstim was unpredictable. To reduce tstim and improve precision, the interface to the channels was redesigned to limit dead volume. As shown by the device cross-section in Figure 2A, the final interface design included a large reservoir positioned above the channels, with a 1 mm thick PDMS layer defining access to the channels. Cross-sectioning is described in Supporting Information, (Figure S-4). The large reservoir was patterned during PDMS curing on the silicon/photoresist master, using a custom-made 11 mm diameter insert (refer to Supporting Information). Individual access reservoirs to each of the 8 channels were 1 mm in diameter and 1 mm in depth (“small reservoirs” in Figure 2A, dotted blue line). These smaller individual ports allowed for ease of loading the islets into their channels, faster delivery of solution changes to the islet, elimination of cross-talk between islets, and minimal dead volume within the large channel.
Figure 2.

Stimulation timing (tstim) was minimized by improving the input reservoir design of the PDMS device. (A) A cross-section of the device. A large input reservoir transitions to small reservoirs (blue dotted lines) that interface with the microfluidic channels. (B) Representative data from tstim measurement and analysis. The average measured tstim of 15 ± 2 s was essentially negligible on the 1 h scale of sampling experiments. Three batches, four different chips, and two channels per chip were tested at least in triplicate.
With these design modifications incorporated, any change in solution conditions, such as an increase in glucose concentration, was accomplished by simply removing the solution from the large reservoir, adding new solution, and then pipetting in and out five times rapidly. To sample variability in solution replacement and reservoir punching, tstim was characterized with multiple PDMS chips and multiple channels. A simple voltage divider was employed during solution conductivity changes, a method modified from previous reports.19–21 With microfluidic channels dominating both the fluidic and electrical resistances of the system, the voltage drop across a 20 MΩ resistor in series with a channel was used to determine tstim (apparatus and details in Supporting Information, Figure S-1). Representative voltage traces after increased or decreased conductivity are shown in Figure 2B. Voltage changes were normalized to start at zero and increase to 1.0; thus, a typical measured decrease is represented as an increase in this figure. The total microchannel volume for a single channel (deep and shallow) was 0.36 μL, and the volume of the channel leading up to the islet trapping region was 0.14 μL, a fraction of the total channel volume, f = 0.39. tstim was determined by analysis of data such as that shown in Figure 2B, where
| (1) |
With time-derivative traces of the data (Figure 2B, inset), tstart and tend were defined as the starting and ending times of the peak in the derivative using chromatographic peak analysis software.32 The average tstim was determined to be 15 ± 2 s. Batch to batch variation (separate PDMS mixing and curing) was sampled in these tests, where three batches, four different chips, and two channels per chip were tested at least in triplicate. Since secretion sampling experiments were performed over periods of 1 h, this value of tstim could then be approximated as instantaneous (0.4% of total time).
Validation of Single-Islet Microfluidic Secretion Sampling
With passive flow control and tstim optimized, we first used fluorescence microscopy to confirm that trapped islets were functioning correctly within our microfluidic device. It is well documented that calcium influx into β-cells of the islet is altered upon stimulation by high concentrations of glucose.3,4,14,33–36 Glucose transport and metabolism result in depolarization of the cells, which stimulates oscillatory calcium influx, which in turn stimulates insulin secretion. Importantly, intercellular communication can be observed in healthy islets by measuring calcium levels, since these levels in essentially all of the β-cells will oscillate in synchrony. Using the calcium indicator Fluo-4, relative calcium levels in stained islets were imaged by excitation at 470 ± 20 nm and emission measurement at 525 ± 25 nm using fluorescence microscopy. As shown in Figure 3A, synchronized calcium oscillations were indeed observed within islets trapped in our passively operated microfluidic device. As expected, we observed an increase in calcium after changing from 3.0 to 11.0 mM glucose (around 5 min), followed by intra islet synchronization of β-cell calcium oscillations beginning ~12.5 min. The break in the curve near the 5 min time point is due to interference with microscopy during switching of the solution in the reservoir. The trace in Figure 3A represents the integrated intensity of Fluo-4 emission from the entire islet (ImageJ processing), indicating that our extraction, isolation, and microfluidic trapping process does not alter this important intercellular communication within the islet.
Figure 3.

Validation of passive microfluidic method for islet secretion sampling. (A) Synchronized calcium oscillations were observed in islets trapped in the microfluidic system after exposure to stimulatory glucose (11mMG), confirming that intercellular communication was intact. (B) Microfluidic secretion sampling was validated. Average insulin secretion from islets in the device (gray bars; 62 islets, 5 mice) was statistically equal to that measured by multi-islet, static methods (“Bulk” ; white bars; 200 islets, 5 mice). Black bars show microchip analysis when excluding undetectable secretion from 11 islets (less comparable to bulk methods). Error bars represent standard error of means of ELISA measurements for each islet or tube.
Next, the passively operated devices shown in Figure 1 were validated for single islet secretion sampling by comparison to the conventional, tube-based method. With standard bulk secretion sampling approaches, groups of islets were placed into a centrifuge tube and exposed to high glucose for ~1 h, and then, the supernatant was removed and stored at −20 °C for subsequent ELISA measurements. For this study, 10 islets with a wide distribution of diameters were loaded into each 0.5 mL tube with 200 μL of imaging medium. After treatment for 1 h, 180 μL aliquots of media were removed from each tube and stored at −20 °C for subsequent ELISA measurements. In total, 40 islets per mouse were sampled from five different wild-type C57BL/6J mice. Twenty islets from each mouse were held at basal glucose while 20 were stimulated with 11 mM glucose. Thus, a total of 200 islets were sampled in the conventional, or “Bulk,” manner. As shown by the white bars in Figure 3B, insulin secretion at basal glucose was found to be 1.01 ± 0.224 pg islet−1 min−1 (100 islets, 5 mice), while secretion at 11 mM glucose was found to be 6.04 ± 1.53 pg islet−1 min−1 (100 islets, 5 mice). These means were statistically different with 99.8% confidence, or p < 0.002. Error bars depict standard errors of the means of duplicate ELISA measurements conducted on supernatants from each tube. These results followed the expected trend of increased insulin secretion at higher glucose concentration.
For microfluidic sampling experiments, flow was first initiated with the syringe, and single islets with a wide distribution of diameters were individually loaded into the trapping channels of the microfluidic device. Loading of each islet required ~30 s. After the 1 h sampling period, the vacuum source (hand-held syringe) was disconnected, and the ~40 μL volume samples in tubing were eluted into microcentrifuge tubes and stored at −20 °C for subsequent insulin ELISA measurements. From 5 different wild-type C57BL/6J mice, 27 individual islets were perfused with basal (3 mM) glucose and sampled, while 35 islets were perfused with stimulatory (11 mM) glucose and sampled; a total of 62 individual islets were sampled, a number significantly larger than has been achieved in previously reported single-islet measurements.3,12,13,31 Due to the limited dynamic range of the insulin ELISA assay, several islets secreted insulin at levels above or below the assay detection range. Samples from islets secreting above the assay detection range were simply diluted and remeasured. However, samples from 11 islets did not show detectable insulin, suggesting these islets either secreted insulin at levels lower than the assay range or did not secrete insulin. Considering that 9 of these 11 islets were from the basal glucose (3 mM) group, it appears more likely that the islets were simply secreting at levels too low to detect with ELISA. Nonetheless, these islets were not excluded from the results and were assigned an insulin secretion of zero. As shown in Figure 3B (gray bars), the mean value of the single-islet insulin secretion at basal glucose (3 mM) was found to be 1.26 ± 0.26 pg islet−1 min−1 (27 islets, 5 mice), statistically equal to the insulin secreted at basal treatment using multi-islet, static methods (p > 0.65). Single-islet insulin secretion at stimulatory glucose (11 mM) was found to be 6.55± 1.00 pg islet−1 min−1 (35 islets, 5 mice), also statistically equal to the stimulatory treatment using conventional methods (p > 0.80). Since the magnitudes of the means of single-islet insulin secretions (gray bars) were statistically equal to those obtained with conventional multi-islet, static methods (“Bulk” ; white bars), the microfluidic secretion sampling approach was considered to be validated for use in insulin secretion sampling of single islets. This validation experiment, which was not conducted in previous reports of islet secretion sampling,3,12,13,31 was possible due to the ease-of-use and higher throughput capabilities of our passively operated, multichannel microfluidic device. These results also provide additional support to our earlier findings that insulin adsorption can be eliminated using dynamic coating with BSA.
Since 9 of the 11 islets with undetectable levels of secretions were from the basal glucose (3 mM) group, it is likely that they were simply secreting at levels too low to detect with ELISA. On the basis of the volume of sampling from the microfluidic device, other “ultra-sensitive” ELISA kits would not suffice for these measurements, since they require a larger volume than is available in our system. In the future, it would be preferable to develop new, low-volume assays for insulin to solve this problem.37 Nonetheless, the fact that a significant fraction of tested islets (33% of basal treated, 6% of stimulated) secreted insulin at undetectable levels (or possibly did not secrete) highlights the importance of our single-islet measurements in understanding islets as individuals. If these islets are left out of the analysis, the mean values increase somewhat (Figure 3B, black bars), although this data is less representative of the population. More importantly, with the ease of use of this passive microfluidic sampling device, it was possible to make observations that could not be made by conventional methods on groups of islets. It is reasonable to assume that a similar percentage of islets in the bulk treatment were also secreting very low levels of insulin.
Insulin Secretion versus Islet Volume
With the ability to sample secretions from single islets and image each islet using microscopy, it is feasible to make a variety of measurements then correlate these results with hormone secretion levels. In this work, islet volume was chosen as the additional measured parameter, since the dependence of insulin secretion on islet volume is unknown. Confocal reflectance microscopy was used to image islets in successive Z-slices to assemble three-dimensional representations of the islet-occupied volume. As shown in Figure 4A, confocal reflectance (upper rightmost images; blue) highlights inner and outer membranes within cells and is insensitive to cell type within the islet. These images were converted into binary images using automated thresholding (lower right red images in Figure 4A). The three-dimensional representations (leftmost images in Figure 4A) of the islets were assembled after collecting Z-slices of 2 μm thickness in reflectance mode (blue), converting to binary mode (red), and then interpolating and reconstructing the islet volume using Nis Elements (Nikon). Scale bars are 25 μm. Further details on this analysis are included in Supporting Information.
Figure 4.

Dependence of insulin secretion on islet volume. (A) Confocal reflectance and 3-D reconstructions of volumes of each islet. Individual Z-slices are shown at right, with 3-D reconstructions at the left (reflectance in blue; binary representation in red). Scale bars are 25 μm. (B) Insulin secretion from single islets had surprisingly little correlation with islet volume at 11 mM glucose (gray triangles; R2 = 0.0678) or at 3 mM glucose (black circles; R2 = 0.0906), with high levels of secretion from some of the smallest islets. Islets secreting undetectable levels of insulin were not excluded and are shown as empty data points. Inset shows the log–log plot of the same data.
In this way, islet volume (in nL) was measured, postsampling, for each islet. Figure 4B shows the dependence of insulin secretion on islet volume for islets treated with basal (3 mM G; black circles) and stimulatory levels of glucose (11 mM G; gray triangles). Islets secreting insulin below the detection range of ELISA are included as zero insulin secretion and highlighted as empty black circles (3 mM G) or empty gray triangles (11 mM G). Islets in basal glucose (3 mM G) showed very little correlation (R2 = 0.0906, Pearson coefficient =0.301) with islet volume, as expected. Interestingly, at stimulatory levels of glucose (11mMG), there was also very little correlation of insulin secretion to islet volume (R2 = 0.0678, Pearson coefficient = 0.260). Removal of the zero points, defined by empty data points, actually decreased correlations for both data sets. One would assume that larger islets, since they contain more β-cells, would secrete more insulin in the presence of high glucose. However, as shown in Figure 4B, several of the largest levels of insulin secretion (10 to 25 pg islet−1 min−1) were sampled from islets of approximately 1 nL volume, with some of these smaller islets secreting at least double the levels of insulin of islets 5- and 10- fold larger in volume. In fact, this finding is supported by recent reports in which multiple islets were separated by size and assayed for function, and small islets were shown to secrete as much as double the insulin of larger islets.38,39 Our results with individual islets further show that signaling between islets is not necessary for this effect. This finding could be important in fundamental islet studies, where often the larger islets are preferentially selected for analysis. The results could have even more impact in islet preparation for transplantation. One possibility is that large islets more quickly lose viability, with cells in the center of the islet receiving insufficient nutrient levels and waste removal. Another explanation is that islet architecture plays a larger role in the level of secreted insulin through paracrine signaling. On the basis of data from Figure 4 and on recent reports highlighting intercellular communication in islets,11,40 we hypothesize that the level of secreted insulin is more strongly influenced by the percentage of different cells types (i.e., architecture) than the total number of β-cells. Our device, coupled with immunostaining or gene expression analysis, provides a platform to test this hypothesis. The ease-of-use benefit should make these future studies straightforward.
CONCLUSIONS
We have presented and validated a passively operated microfluidic device for secretion sampling and volume measurements of single pancreatic islets. A key advantage offered by this device is ease-of-use, since only a hand-held syringe is required for secretion sampling. Postsampling volume measurements with confocal reflectance microscopy revealed surprisingly little correlation of insulin secretion to islet volume, even at high glucose. This finding could be important for islet transplantation studies and could improve understanding of diabetes, obesity, and metabolic syndrome.
These results help to emphasize that the microfluidic platform, particularly when designed for passive operation,3,17 can enable facile collection of novel information on single islet variability. For presented results and preliminary work, >50 microdevices were fabricated, used, and disposed of. This type of single-use device with simplified operation could be valuable to nonexperts who are interested in making single islet hormone secretion measurements and/or interfacing with various methods in microscopy.
Unlike multiwell plate formats, our device allows facile interfacing with confocal microscopy, and the platform is now amenable to integration with droplet fluidics in the future to improve temporal resolution.3 A final point is that our device design could feasibly be scaled up by approximately 5-fold (~40 sampling channels) before islet loading times become prohibitive, making the device even more useful in islet viability testing for transplantation.
Supplementary Material
Acknowledgments
Support for this work was provided by the Auburn University Department of Chemistry and Biochemistry and the College of Science and Mathematics. Thanks go to Charles Ellis in Electrical Engineering for clean room use and Tony Overfelt in Materials Engineering for use of their confocal microscope.
Footnotes
Supporting Information. Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Göpel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. J Physiol. 1999;521:717–728. doi: 10.1111/j.1469-7793.1999.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rocheleau JV, Remedi MS, Granada B, Head WS, Koster JC, Nichols CG, Piston DW. PLoS Biol. 2006;4:e26. doi: 10.1371/journal.pbio.0040026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Easley CJ, Rocheleau JV, Head SW, Piston DW. Anal Chem. 2009;81:9086–9095. doi: 10.1021/ac9017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benninger RKP, Zhang M, Head WS, Satin LS, Piston DW. Biophys J. 2008;95:5048–5061. doi: 10.1529/biophysj.108.140863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nyman LR, Wells KS, Head WS, McCaughey M, Ford E, Brissova M, Piston DW, Powers AC. J Clin Invest. 2008;118:3790–3797. doi: 10.1172/JCI36209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim A, Miller K, Jo J, Kilimnik G, Wojcik P, Hara M. Islet. 2009;1(2):129–136. doi: 10.4161/isl.1.2.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butler AE, Janson J, Soeller WC, Butler PC. Diabetes. 2003;52:2304–2314. doi: 10.2337/diabetes.52.9.2304. [DOI] [PubMed] [Google Scholar]
- 8.Guanawardana SC, Benninger RK, Piston DW. Am J Physiol Endocrinol Metab. 2009;296:E323–E332. doi: 10.1152/ajpendo.90544.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuehn B. J Am Med Assoc. 2009;301:1521–1525. [Google Scholar]
- 10.Cabrera O, Berman DM, Ricordi C, Berggen P, Caicedo A. Proc Natl Acad Sci USA. 2006;103:2334–2339. doi: 10.1073/pnas.0510790103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LeMarchand SJ, Piston DW. J Biol Chem. 2010;285:14389–14398. doi: 10.1074/jbc.M109.069195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roper MG, Shackman JG, Dahlgren GM, Kennedy RT. Anal Chem. 2003;75:4711–4717. doi: 10.1021/ac0346813. [DOI] [PubMed] [Google Scholar]
- 13.Dishinger JF, Reid KR, Kennedy RT. Anal Chem. 2009;81:3119–3127. doi: 10.1021/ac900109t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nunemaker CS, Dishinger JF, Dula SB, Wu R, Merrins MJ, Reid KR, Sherman A, Kennedy RT, Satin CS. PLoS One. 2009;4(e84281):1–11. doi: 10.1371/journal.pone.0008428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohammed JS, Wang Y, Harvat TA, Oberholzer J, Eddington DT. Lab Chip. 2009;69:97–106. doi: 10.1039/b809590f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adewola AF, Dongyoung L, Harvat T, Mohammed J, Eddington DT, Oberholzer J, Wang Y. Biomed Microdevices. 2010;12:409–417. doi: 10.1007/s10544-010-9398-1. [DOI] [PubMed] [Google Scholar]
- 17.Lesile DC, Easley CJ, Seker E, Karlinsey JM, Utz M, Begley MR, Landers JP. Nature Phys. 2009;5:231–235. [Google Scholar]
- 18.Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. Anal Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Gordon MJ, Zare RN. Anal Chem. 1988;60:1837–1838. [Google Scholar]
- 20.Locascio LE, Perso CE, Lee CS. J Chromatogr, A. 1999;857:257–284. doi: 10.1016/s0021-9673(99)00774-8. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Fanguy JC, Bledsoe JM, Henry CS. Anal Chem. 2000;72:5939–5944. doi: 10.1021/ac000932l. [DOI] [PubMed] [Google Scholar]
- 22.Stefan Y, Meda P, Neufeld M, Orci L. J Clin Invest. 1987;80:175–183. doi: 10.1172/JCI113045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scharp DW, Kemp CB, Knight MJ, Ballinger WF, Lacy PE. Transplantation. 1973;16:686–689. doi: 10.1097/00007890-197312000-00028. [DOI] [PubMed] [Google Scholar]
- 24.Lesile DC, Melnikoff BA, Marchiarullo DJ, Cush DR, Ferrance JP, Landers JP. Lab Chip. 2010;10:1960–1966. doi: 10.1039/c003244a. [DOI] [PubMed] [Google Scholar]
- 25.Unger MA, Chou H, Thorsen T, Scherer A, Quake SR. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 26.Thorsen T, Maerkl SJ, Quake SR. Science. 2002;298:580–584. doi: 10.1126/science.1076996. [DOI] [PubMed] [Google Scholar]
- 27.Gómez-Sjöberg R, Leyrat AA, Pirone DM, Chen CS, Quake SR. Anal Chem. 2007;79:8557–8563. doi: 10.1021/ac071311w. [DOI] [PubMed] [Google Scholar]
- 28.Vickers JA, Caulum MM, Henry CS. Anal Chem. 2006;78:7446–7452. doi: 10.1021/ac0609632. [DOI] [PubMed] [Google Scholar]
- 29.Chen D, Du W, Liu Y, Liu W, Kuznetsov A, Mendez FE, Philipson LH, Ismagilov RF. Proc Natl Acad Sci USA. 2008;105:16843–16848. doi: 10.1073/pnas.0807916105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Attiya S, Jemere AB, Tang, Fitzpatrick G, Seiler K, Chiem N, Harrison DJ. Electrophoresis. 2001;22:318–327. doi: 10.1002/1522-2683(200101)22:2<318::AID-ELPS318>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 31.Shackman JG, Dahlgren GM, Peters JL, Kennedy RT. Lab Chip. 2005;5:56–63. doi: 10.1039/b404974h. [DOI] [PubMed] [Google Scholar]
- 32.Shackman JG, Watson CJ, Kennedy RT. J Chromatogr, A. 2004;1040:273–282. doi: 10.1016/j.chroma.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Newsholme P, Gaudel C, McClenaghan NH. In: The Islets of Langerhans: Advances in Experimental Medicine and Biology. Islam S, editor. Springer; New York: 2010. pp. 91–113. [DOI] [PubMed] [Google Scholar]
- 34.Jonkers FC, Henquin JC. Diabetes. 2001;50:540–550. doi: 10.2337/diabetes.50.3.540. [DOI] [PubMed] [Google Scholar]
- 35.Jo J, Choi MY, Koh DS. Biophys J. 2007;93:2655–2666. doi: 10.1529/biophysj.107.104125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Roper MG. Anal Chem. 2009;81:1163–1168. [Google Scholar]
- 37.Kim J, Hu J, Sollie RS, Easley CJ. Anal Chem. 2010;82:6976–6982. doi: 10.1021/ac101762m. [DOI] [PubMed] [Google Scholar]
- 38.Lehmann R, Zuellig RA, Kugelmeier P, Baenninger PB, Mortiz W, Perren A, Clavien PA, Weber M, Spinas GA. Diabetes. 2007;56:594–603. doi: 10.2337/db06-0779. [DOI] [PubMed] [Google Scholar]
- 39.Nam KH, Yong W, Harvat T, Adewola A, Wang S, Oberholzer J, Eddington DT. Biomed Microdevices. 2010;12:865–874. doi: 10.1007/s10544-010-9441-2. [DOI] [PubMed] [Google Scholar]
- 40.Bosco D, Armanet M, Morel P, Nadja N, Sgroi A, Muller YD, Giovannoni L, Parnaud G, Berney T. Diabetes. 2010;59:1202–1210. doi: 10.2337/db09-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.