

Abstract
Mumps virus belongs to the family of Paramyxoviridae and has the potential to be an oncolytic agent. Mumps virus Urabe strain had been tested in the clinical setting as a treatment for human cancer four decades ago in Japan. These clinical studies demonstrated that mumps virus could be a promising cancer therapeutic agent that showed significant antitumor activity against various types of cancers. Since oncolytic virotherapy was not in the limelight until the beginning of the 21st century, the interest to pursue mumps virus for cancer treatment slowly faded away. Recent success stories of oncolytic clinical trials prompted us to resurrect the mumps virus and to explore its potential for cancer treatment. We have obtained the Urabe strain of mumps virus from Osaka University, Japan, which was used in the earlier human clinical trials. In this report we describe the development of a reverse genetics system from a major isolate of this Urabe strain mumps virus stock, and the construction and characterization of several recombinant mumps viruses with additional transgenes. We present initial data demonstrating these recombinant mumps viruses have oncolytic activity against tumor cell lines in vitro and some efficacy in preliminary pilot animal tumor models.
Introduction
Oncolytic virotherapy is a rapidly evolving field in which viruses are exploited for their targeted cell killing properties. Viruses had been utilized for cancer treatment in the 20th century,1 but they received considerable interest only at the beginning of 21st century. Oncolytic viruses specifically infect and kill tumor cells without harming healthy cells with intact interferon pathway.2 Currently, many viruses are being studied extensively for their oncolytic and immunotherapeutic properties in clinical and preclinical trials. Recent FDA approval of herpes virus underscores the importance of oncolytic viruses in the field of cancer therapeutics as an alternative therapeutic agent.3 The reports of significant responses of human cancers to oncolytic virotherapy in clinical trials kindle the interest of many researchers to explore various viruses for their usefulness in cancer treatment.
A recombinant measles virus encoding human sodium iodide symporter (MV-NIS), has shown some promising results in recent human clinical trials including a complete response of a myeloma patient in phase 1 trial at the Mayo Clinic.4 We were inspired to look for another equally competent virus in the same family. In this regard, we explored the safety and efficacy of another member of paramyxoviridae family, mumps virus (MuV). It belongs to the genus Rubulavirus and possess a single stranded negative sense RNA genome (~15 kb) which encodes at least nine viral proteins.5 Mumps virus has 12 genotypes, designated A–N (excluding E and M) based on the sequence of the SH gene.6
Mumps virus has long been used for cancer treatment as an immuno-therapeutic and antineoplastic agent.7–9 Dr. Asada, a physician from Japan demonstrated oncolytic activity of Mumps virus in cancer patients. He used a near wild-type mumps virus (Urabe strain) collected from saliva of patients with epidemic parotitis, and minimally passaged on cultured cells. For later experiments, Asada used purified mumps virus grown in tissue culture (human embryonic kidney cells), from the Department of Virology, Research Institute for Microbial Diseases, Osaka University. In this clinical trial, Asada treated 90 patients with various kinds of terminal cancers. For 37 of 90 patients treated, the tumor regressed completely or decreased to less than half of the initial size. Among which 42 patients responded moderately and their tumor showed a tendency of retreat or growth suppression. Asada also compared live mumps virus with an inactivated one and found no anticancer effect which clearly shows that live replicating virus is essential for antitumor efficacy. He also noticed that oncolytic efficacy was terminated once antimumps immunity developed. Local or intratumoral administration was more effective than systemic therapy that requires a large dose of mumps virus. Many patients were in remission for a long time after discontinuation of therapy, suggesting development of antitumor immunity. He also concluded that it is essential to start virotherapy when the immune system is intact in the early stages of cancer or before other conventional therapies.
A second clinical trial was conducted using the same Urabe strain mumps virus but after additional passages in cultured cells and with improved purity.10 In this trial, patients with various cancers, most of them at terminal stages, were treated with mumps virus intravenously (i.v.) and tumor regression were observed in 26 out of 200 patients. This trial was followed by a third one, in which patients with advanced gynecologic cancer were preimmunized with mumps virus before treatment.11 Marked clinical response was observed with patients treated locally and no response was noticed in unprimed patients or patients with large tumor mass. The above clinical trials strongly demonstrate the oncolytic and immune-therapeutic potential of Urabe strain mumps virus.
Recently, we were able to obtain Urabe strain mumps virus that was subjected to cancer clinical trials in Japan by Dr. Asada and coworkers.7 Since modern day clinical trial requires preclinical studies with detailed information on safety and efficacy of oncolytic virus derived from infectious clone, we developed a reverse genetics system for this Urabe strain of mumps virus and conducted preliminary studies on oncolytic efficacy both in in-vitro and in-vivo models with the aim of translating this virus again to the clinic.
Results
Mumps virus Urabe strain
An aliquot of the mumps virus Urabe strain (MuV-U) used in oncolytic virotherapy human clinical trials in Japan in the 1970’s and 1980’s was obtained. A representative clone, MuV-U Clone 1-C-3 (MuV-UC-WT) was isolated, characterized, and used in this study. A unique aspect of this MuV-UC virus stock was the minimal amplification in cultured cells that may have minimized the attenuation from the original patient isolate.
The development of a reverse genetics system based on MuV-U
A reverse genetics platform based on the nucleotide sequence of the MuV-UC isolate was constructed initially with an additional transcription unit containing the green flourescent protein (GFP) coding sequence flanked by unique restriction enzyme cloning sites between MuV-UC genes HN and L, in a plasmid vector pMuV-UC-GFP. Negative-strand RNA can be synthesized using T7 polymerase and the T7 transcription elements flanking the MuV-UC genome along with three MuV-UC-based helper plasmids expressing MuV-UC N, P and L proteins. Baby hamster kidney cells infected with a vaccinia virus vector encoding the T7 polymerase were then transfected with the infectious MuV-UC plasmid along with the three helper plasmids to rescue recombinant virus. After successful rescue in baby hamster kidney cells, the recombinant MuV-UC virus was further propagated in Vero cells to produce virus stocks (Figure 1a). When growth potential of recombinant mumps virus expressing GFP was compared with wild-type virus, it showed better growth rate (Figure 1b).
Figure 1.

Rescue of recombinant mumps viruses Urabe strain (rMuV-UC). (a) Rescue of rMuV-UC-GFP infectious virus in baby hamster kidney (BHK) cells and further propagation of the same virus in Vero cells visualized by fluorescent microscopy. (b) Mumps virus wild-type and rescued virus rMuV-UC-GFP were compared for their growth potential in Vero cell in multistep growth curve. Cells were infected with multiplicity of infection (MOI) of 0.1, supernatants and cell pellets were collected at different time intervals and titered in Vero cells using standard plaque assay. Left panel shows virus titer of cell free supernatants and right panel shows virus titer of cell associated virus.
To test the genomic stability and transgene expression abilities of the recombinant MuV-UC platform, we constructed several other recombinant viruses with different useful transgenes for in vivo bio-distribution and efficacy studies (Figure 2a). The GFP coding region was replaced with the luciferase gene (rMuV-UC-LUC), or the human sodium iodide symporter (NIS) gene (rMUV-UC-NIS). We also tested whether two transgenes could be inserted and expressed from the rMuV-UC platform by adding an additional transgene, mouse interferon beta (mIFNβ), in between M and F genes of rMuV-UC-GFP creating rMuV-UC-mIFNβ-GFP that should express both the mIFNβ and GFP. All the recombinant mumps viruses replicated well on Vero cells (Figure 2b). Also, as expected, the rMuV-UC-mIFNβ-GFP with two transgenes replicated to a lower titer compared with viruses with single transgenes. All transgenes delivered by the recombinant mumps viruses were well expressed (Figure 2c–e).
Figure 2.

Characterization of the replication and transgene expression of the recombinant mumps viruses. (a) Schematic diagram of various constructs of mumps virus expressing foreign genes. (b) Growth curve of various recombinant mumps viruses in Vero cells after infection at MOI = 0.1. (c) Luciferase assay demonstrating the expression of LUC transgene by the rMuV-UC-LUC in infected cells compared with only background LUC expression by the original MuV-UC virus isolate. (d) Measurement and comparison of activity of human sodium iodide symporter (NIS) produced by rMuV-UC-NIS and wild-type MuV-UC-wt. (e) Measurement of secreted mouse interferon beta (mIFN-β) in the supernatant of rMuV-UC-mIFNβ and MuV-UC infected Vero cells by enzyme-linked immunosorbent assay (ELISA).
Oncolytic activity of rMuV-UC-GFP replication in tumor cell lines
Since oncolytic virotherapy not only expects the virus to directly kill tumor cells but then cause a unique immune response to the tumor to completely cure the patient, efficacy models that can evaluate both strategies of this two-pronged therapeutic approach are the best. Therefore, while we want to know the efficacy of mumps virus for treating human tumors, human tumor xenografts in nude mice can only evaluate the virus killing therapeutic component. Since some syngeneic mouse tumors models respond similarly to their human tumor counterparts, we next investigated the infectivity and oncolytic activity of the rMuV-UC-GFP virus on a variety of human and mouse tumor cell lines. We tested the oncolytic efficacy of rMuV-UC-GFP in various human cancer cell lines (Figure 3a). The human tumor cell lines were infected with rMuV-UC-GFP at an MOI = 10, and analyzed 5 days later by fluorescence microscopy for GFP expression. This analysis demonstrated that mumps virus could infect most human tumor cell lines tested.
Figure 3.

rMuV-UC-GFP replication in tumor cell lines. (a) Human cancer cell lines were infected with mumps virus at multiplicity of infection (MOI) of 10 and images were taken 5 days of postinfection. (b) Mouse cancer cell lines were infected with mumps virus at MOI of 10 and images were taken 5 days postinfection. (c) Selected cell lines were infected with MOI of 1.0 and supernatants were collected at indicated time points and titered in Vero cells.
At the same time we found out that most of the mouse tumor cell lines are nonpermissive to robust rMuV-UC-GFP virus infection and replication, with only the N2A neuroblastoma cell line and the CT-26-LacZ colon cancer cell lines showing significant numbers of GFP positive cells (Figure 3b). Both the N2A and CT-26-LacZ tumor lines permitted some rMuV-UC-GFP replication but at significantly lower titers compared with rMuV-UC-GFP replication on human KAS6/1 tumor cells (Figure 3c). Significant cell killing was observed in most human tumor lines tested, however the extent as well as the rate of cell killing can differ substantially between individual tumor cell lines (see Supplementary Figure S1). Very little cell killing was observed in the infected mouse tumor lines with the best in the N2A and CT-26-LacZ by day 7, where the mumps virus replicated relatively well.
Since the neurovirulence studies of mumps virus has been carried out in rat models, we tested the infectivity and replication of rMuV-UC-GFP in some of the rat tumor cell lines. C6 and RG2 are two rat glioma tumor cell lines, and were infected with mumps virus at different multiplicity of infection. RG2 glioma cells were more permissive to MuV infection compared with C6 cells (Figure 4a). Mumps virus multiplies better in RG2 cells reaching its peak titer (6 x 105 pfu/ml) at 72 hours post-infection (Figure 4b). The titer produced by RG2 cells is almost two logs higher than C6 cells but more than a log lower than Vero cells. In correlation with the higher rMuV-UC-GFP replication in RG2 cells compared with C6 cells, up to 60% of the RG2 cells were killed by the mumps virus compared with just 10–20% of C6 cells as determined by MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay, confirmed the higher infectivity in RG2 cells (Figure 4c).
Figure 4.

Infectivity of rMuV-UC-GFP in rat cancer cells. (a) Rat glioma cells C6 and RG2 were infected at multiplicity of infection (MOI) 0.1 and compared with Vero cells. The fluorescent images were taken at indicated time points. (b) Vero, RG2, and C6 cells were infected with mumps virus at 0.1 MOI. Supernatants were collected at indicated time points and were titered in Vero cells (Left panel). Cell viability assay for C6 and RG2 cells infected with mumps virus (middle and right panels) GFP, green fluorescent protein.
Modulation of interferon pathway and its effect on mumps virus replication
It has been shown that mumps virus infection in nonpermissive rodent cells resulted in abortive infection.12 This may be caused by the presence of antiviral machinery or due to inefficient penetration of virus into the cell. Since interferon is one of the obstacles that could prevent MuV replication, we treated some of the less permissive cell lines with Janus kinase inhibitor, Ruxolitinib,13 and then infected the treated cells with rMuV-UC-GFP. The Ruxolitinib treatment increased the viral infection significantly in rat glioma (C6), mouse lung carcinoma (LLC) cells and also to some extent in CT-26 colon carcinoma cells, but not in plasmacytoma (MPC11) and human myeloma (MM1) cells (Figure 5). This suggests that individual cell lines not only differ in the innate immune defense but also employ more than one mechanism to restrict viral replication. So the low infectivity of MuV in mouse tumor cells may be the result of multiple cellular factors rather than single one that control different steps in the mumps virus life-cycle.
Figure 5.
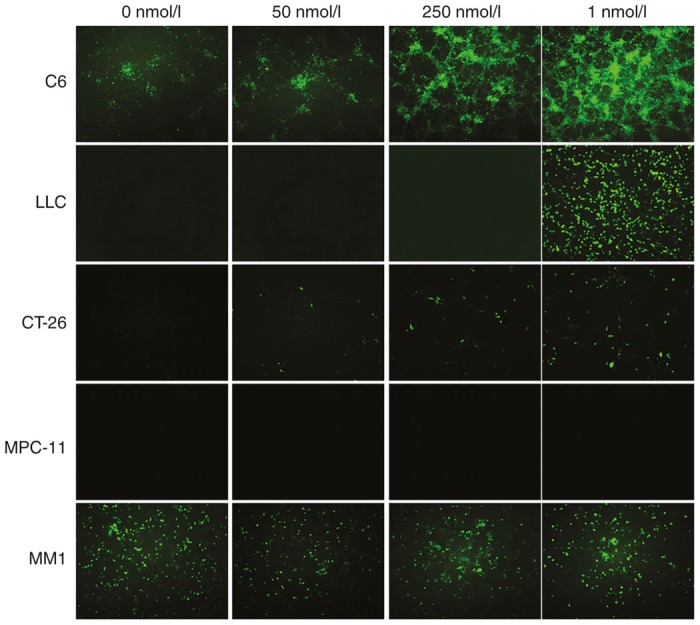
Effect of a Janus kinase inhibitor on mumps virus infectivity. Selected tumor cells lines were treated with Ruxolitinib, a Janus kinase inhibitor, at the indicated doses. After 48 hours the treated and controlled cells were infected with rMuV-UC-GFP mumps virus (multiplicity of infection (MOI,. 10). Fluorescent images were taken 5 days postinfection. GFP, green fluorescent protein.
Oncolytic efficacy of mumps viruses in immunocompetent mouse models
To test the oncolytic activity of mumps virus in immunocompetent mouse models, we conducted pilot preliminary studies using the two relatively permissive mouse cancer cell lines to mumps virus infection, colon carcinoma (CT-26-LacZ) and neuroblastoma (N2A). These tumor cells were implanted into the flanks of syngeneic mice, Balb/C, and A/J respectively. Once the tumors reached a significant size, mumps viruses were administered i.v. through the tail vein. In CT-26-LacZ model, groups of mice were treated with rMuV-UC-GFP at 106 and 107, rMuV-UC-LUC at 107, MuV-UC at 107, and a saline control. Some of the mice with CT-26-LacZ tumors treated with rMuV-UC-LUC or MuV-UC virus had delay in their tumor growth and had better overall survival with one rMuV-UC-LUC treated mouse and two MuV-UC virus treated mice having a complete response (Figure 6). However, immunohistochemical analysis of tumor tissue on day 14 does not show any mumps virus positive staining and also the luciferase imaging on day 7 and 14 yielded no positive signal (data not shown). This suggests that there may be an involvement of immune system in tumor suppression.
Figure 6.

Oncolytic efficacy of mumps viruses in the mouse CT-26-LacZ colon carcinoma tumor model. Balb/c mice (n = 5) were implanted with 5 × 106 CT-26-LacZ cells subcutaneously. Once the tumor volume reached 0.2–0.5 mm3, mice were treated with saline or mumps viruses (106–107 PFU) administered i.v. through by tail vein: (a) MuV-UC-wt (107), (b) rMuV-UC-GFP-106; (c) rMuV-UC-GFP-107; (d) rMuV-UC-Luc (107); or (e) saline. Tumor size was measured using handheld calipers three times a week. Saline versus GFP106, P = 0.6117; Saline versus GFP107, P = 0.0966; Saline versus Luc, P ≤0.0006; Saline versus WT, P≤0.0001 (f) Kaplan–Meier survival curves for the mice are shown in the survival curve. *P = 0.0072 rMuV-UC-wt versus Saline; P = 0.1316 rMuV-UC-Luc versus Saline. GFP, green fluorescent protein.
In our pilot studies, we found out that mutating a single amino acid in polymerase gene increased the replication rate of mumps virus (nt13328, aa N to H). When this virus was compared with MuV-UC-WT and rMuV-UC-GFP, no significance difference was observed in oncolytic activity in in-vitro studies (see Supplementary Figure S2). But we decided to use this virus (rMuV-UC-L13328-GFP) for the rest of the animal studies. In the N2A model, mice were treated with rMuV-UC-LUC, rMuV-UC-L13328-GFP, MuV-UC, and equal amount of saline. No significant antitumor activity was seen in the N2A tumor model possibly due to the aggressive nature of N2A tumor with most of the mice requiring sacrifice ~10 days postinfection (Figure 7). However, one mouse survived in rMuV-UC-L13328-GFP treated group.
Figure 7.

Oncolytic efficacy of mumps virus in the mouse N2A neuroblastoma tumor model. A/J mice (4 weeks old, n = 7) bearing subcutaneous N2A tumors were treated with a single dose (1 × 107) administered i.v. through by tail vein: (a) MuV-UC-wt; (b) rMuV-UV-L13328-GFP; (c) rMuV-UC-LUC; (d) or saline. Tumor size was measured using handheld calipers three times a week. Saline versus GFP-L13328, P = 0.0498 (e) Kaplan–Meier survival curves for the mice are shown in the survival curve. GFP, green fluorescent protein.
Oncolytic efficacy of mumps virus in human myeloma model
In order to initially assess the antitumor activity of the mumps viruses, in-vivo in a human myeloma model, we implanted human myeloma tumor cells (KAS6/1) in the flanks of nude mice. Once the tumor reached an appreciable size, 107 PFU of rMuV-UC-LUC, rMuV-UC-L13328-GFP, MuV-UC or saline were injected i.v. through the tail vein, and the mice was observed for 60 days (Figure 8). In this study, unfortunately there was more variability in the growth of the individual tumor xenografts than we wanted as shown with the saline treated animals, with four of five animals succumbing to tumor load by 60 days. This variability was seen in all four groups and prevented the survival results from reaching statistical significance. However, the data clearly shows the MuV-UC isolate significantly suppressed the tumor growth in all five animals, with one animal having a complete response. The results from mice treated with the recombinant viruses were promising with a complete response and possibly two tumors controlled when treated with rMuV-UC-LUC, while two animals had a complete response to treatment with rMuV-UC-L13328-GFP. To confirm virus replication, tumors were harvested from mice on day 7 and day 12 after virus administration and analyzed for mumps virus antigens. All tumors were positive for mumps viral proteins on day 7 and showed increased staining on day 12. MuV-UC treated tumors having comparatively better infectivity and spread relative to the tumors treated with the recombinant mumps viruses (see Supplementary Figure S3). Since these studies involved single administration of mumps virus, other treatment regimens could possibly improve oncolytic efficacy significantly.
Figure 8.

Oncolytic efficacy of mumps viruses’ infection in human myeloma model. NCr nude mice (4 weeks old; n = 5) bearing subcutaneous KAS6/1 myeloma tumors were treated with a single i.v. dose (1 × 107 pfu) of (a) MuV-UC-wt; (b) rMuV-UC-LUC; or (c) rMuV-UC-L13328-GFP; (d) control saline. Tumor size was measured by serial caliper measurements. Saline versus Luc, P = 0.0191; Saline versus GFP-L13328, P = 0.0412; Saline versus WT, P = 0.0143 (e) Kaplan–Meier survival curves for the mice are shown in the survival curve. GFP, green fluorescent protein.
Discussion
Advancement of technology and approval of oncolytic viruses for cancer treatment has prompted the researchers around the globe to explore many different virus species for their usefulness in human cancer treatment. After the clinical trials in Japan, no further attempt was made to utilize the mumps virus for human cancer treatment. Currently, the FDA requires stringent preclinical efficacy and safety studies before oncolytic clinical trials are approved. Even though animal studies do not necessarily correlate well with clinical outcomes in human patients, it is unavoidable and essential to establish proof of principle in preclinical models. In order to fulfill these requirements and to bring the mumps virus back to the clinic, we established a reverse genetics system for the Urabe stain used by Asada and coworkers in cancer clinical trials.
Recombinant virus rescued from infectious clone, did not show any reduction in growth potential and even grow better than parental virus. In this report, we characterized oncolytic efficacy of recombinant Urabe mumps virus both in in vitro and in vivo studies. Oncolytic activity of recombinant mumps virus (rMuV-UC-GFP) was tested in various human cancer cell lines. The rate of infectivity differs between the cell lines which is likely due to influence of one or more factors; interferon pathway-related genes, various cellular factors (cell cycle modulators, apoptotic machinery, NF-κB, and APOBEC3) or receptors and coreceptors expression levels.14–18 More detailed study is warranted to understand the interaction between virus and host cell. Another interesting phenomenon of mumps virus infection is cytopathic effect (CPE). Although cell monolayer is fully infected with mumps virus, significant cell death or disruption of monolayer is noticed only around day 7. This is probably due to interaction of viral proteins with cellular factors that involve in apoptosis pathway. It was shown that mumps virus V protein and cellular STAT protein both play a major role in apoptosis.19–22 It has also been demonstrated that mumps virus small hydrophobic (SH) protein blocks TNF-α-mediated apoptosis pathway.23 Cells normally initiate apoptosis as soon as it detects viral infection. On the other hand viral proteins suppress apoptosis to maximize its own production. In case of mumps virus, V and SH proteins probably block the apoptosis and prevent early cell death.20
Even though humans are primary host for mumps virus, other animals can also be infected at least in experimental conditions. So we tested the infectivity and oncolytic efficacy of mumps virus in mouse and rat cancer cell lines. Researchers previously demonstrated that many mouse cell lines support the replication of mumps virus.24 They also showed that variation existed in infectivity between different strains of mumps virus. In another study, researchers also demonstrated that mumps virus replication is restricted in mouse cell lines due to interferon machinery or inefficient penetration into the cell.12 In this study we tested the infectivity of MuV-UC mumps virus in a panel of mouse cell lines. While most cells were not permissive to MuV-UC mumps virus infection, significant viral replication was observed in colon carcinoma and neuroblastoma cells. But the viral titer is significantly lower compared with human cell lines. It suggests the restricted replication of mumps virus in mouse cell lines and is due to one or combination of factors suggested above. At the same time mumps virus replicates well in one of the two rat cell lines tested, which warrants further study on mechanism of mumps virus infectivity in various rat cancer cell lines.
To preliminarily assess the oncolytic activity of MuV-UC viruses for both tumor cell killing and a subsequent immune activation in vivo, immune compromised and immunocompetent mouse models were tested. Although MuV-UC viruses did not replicate well in most mouse tumor cell lines, there was a reasonable infection and spread of the MuV-UC viruses in CT-26-LacZ mouse colon carcinoma cell and N2A mouse neuroblastoma cells in-vitro. In the CT-26-LacZ immunocompetent model, wild-type and recombinant MuV-UC viruses showed significant tumor suppression, delay in tumor growth, and also statistically significant increase in survival rate. But we could not detect any significant viral replication in the tumor tissue by immunostaining (data not shown) probably due to the cellular restriction of mumps virus infection in the tumor tissue. The tumor suppression we observed might be due to an antitumor immunity induced by the initial viral replication in the tumor tissue and it requires more detailed investigation. We noticed that wild-type MuV-UC performed better than the GFP expressing recombinant rMuV-UC-GFP virus. We suspected that GFP might have slowed down the replication of virus in vivo even though its replication rate is better in in-vitro. We have noticed this kind of phenomenon in other oncolytic studies. To improve the performance of GFP expressing mumps virus, we made a mutation in the polymerase gene (N13328H). The mutant virus, rMuV-UC-L13328-GFP replicated to a higher titer compared with rMuV-UC-GFP in Vero cells and we subsequently used this recombinant virus.
We then tested the oncolytic efficacy of mumps virus in mouse neuroblastoma immunocompetent model. Since tumor growth is extremely aggressive, most of the mice reached sacrifice criteria within a week. Due to the aggressive nature of this tumor, immunotherapeutic potential of mumps virus could not be observed. No difference was noticed in tumor growth or survival rate compared with the control group, although one mouse survived in the rMuV-UC-L13328-GFP treated group. This study demonstrated that this particular neuroblastoma tumor model may not be suitable for mumps virus oncolytic studies due to its inherent, biologically specific properties like growth rate, host species, antiviral status, etc.
Finally, in an immune compromised human xenograft model, subcutaneously implanted KAS6/1 myeloma cells were treated with single i.v. injection of mumps virus. Although wild-type MuV-UC virus caused significant delay in tumor growth, the survival rate is similar to that of recombinant viruses. In this study, KAS6/1 cells took more than a month to establish recognizable subcutaneous tumor, we suspected that there might be an issue with the particular clone of KAS6/1 cells. In our future studies we plan to use different clone of KAS6/1 cells and also other human cancer lines, especially in systemic tumor models.
In this study, we present initial data demonstrating the recombinant mumps viruses based on the Urabe strain which was used to treat cancer patients in Japan, have oncolytic activity in various tumor cell lines in vitro and some efficacy in preliminary pilot animal tumor models. More detailed in-vitro and in-vivo studies should be carried out to understand the true nature of this virus. We have isolated various clones of Urabe mumps virus from the initial stock and we will compare those clones for the growth potential and oncolytic efficacy. Initial clinical study conducted by Dr. Asada showed highly significant oncolytic activity in various human cancers. Although later trials demonstrated oncolytic and immunotherapeutic potential of mumps virus, nothing was as dramatic as the initial trial. We assume that this was probably due to loss of virulence while passaging the virus in cell culture. Initial studies utilized wild-type virus with minimal passage, which might have maintained virulent species that had replicated better in cancer patients. Later trials might have used highly passaged virus which probably might have lost its virulence that lead to lower efficacy compared with the initial trial. In this study, we have used plaque purified single clone which is most probably less virulent than the original stock. This may be one of the reasons why we could not see better efficacy in our animal studies. We have to consider making clone from different plaques and combining them for in vivo studies to increase the oncolytic efficacy.
Urabe mumps virus caused aseptic meningitis among vaccinated children from age 1–11 during 1990’s in Japan, UK, Brazil and other countries.25–28 Lately Jeryl Lynn strain replaced Urabe in MMR vaccine. It leads to the question: How safe Urabe mumps virus will be in cancer patients? The question has already been answered by the clinical trials conducted in Japan with virulent wild-type virus. No adverse events were observed in those trials except transient fever. Young children who are neither immunized nor exposed to the mumps virus probably will be in risk of developing meningitis. And it seems adults were well tolerated the high dose of virulent Urabe virus. Given the situation, it will be prudent to conduct oncolytic clinical trials in adult cancers rather than children cancers.
Various animal models have also to be investigated to study safety and efficacy of mumps virus. Because of poor replication of mumps virus in mouse tumor cells, immunocompetent syngeneic mouse models may not be efficient and reliable. Since most of the human oncolytic viruses have been tested in immunodeficient mice models effectively, it will be better to use those models for human cancers which are naturally permissive to mumps virus. At the same time it may not be possible to test the immunotherapeutic potential of the mumps virus, which is one of the major components of oncolytic virotherapy. In this case rat model could be highly feasible because the rat-based neurovirulence safety test that was developed at the Center for Biologics Evaluation and Research at the Food and Drug Administration (FDA) is efficient and reproducible and correctly assesses the neurovirulence potential of mumps viruses in humans.29,30 In this study, it is shown that mumps virus replicates better in one of the rat cell lines tested. This suggests that rat model could be more useful in mumps virus oncolytic studies.
In summary, we developed a reverse genetics system for one of the clones of Urabe mumps virus which has undergone three clinical trials for human cancer treatment. We also tested the oncolytic potential of recombinant mumps viruses both in in-vitro and in-vivo studies. This study paves the way for further research on Urabe mumps virus and translation of this virus into the clinic.
Materials and Methods
Viruses and cells
MuV-U was originally collected from saliva of a child with mumps symptoms in Japan, isolated after replication in cultured primary human embryonic kidney cells, and then the virus was amplified in human embryonic kidney to produce a seed stock.7 This seed stock then went through an unknown number of amplifications on human embryonic kidney and/or human amnion cells (AV3) to produce virus lots for several clinical trials in Japan in the 1970’s and 1980’s. An aliquot of the MuV-U stock used in these clinical trials was obtained from Dr. Koichi Yamanishi (Osaka University). The Viral Vector Production Laboratory at the Mayo Clinic isolated individual virus plaques from this stock using limited dilution in Vero cells, and determined the nucleotide sequences from reverse transcriptase-polymerase chain reaction (PCR) products from the viral RNA genomes. In this study, we used a representative clone, MuV-U 1-C-3 (MuV-UC-WT), isolated from this stock.
The cell lines used in this study were obtained from American Type Culture Collection (ATCC, Manassas, VA) and were maintained in medium recommended by ATCC in 5% CO2. The following cell lines were used in this study; baby hamster kidney cell line, Vero-African green monkey kidney cell line, KAS6/1, JJN3, MM1, RPMI6225-human myeloma, ARH-77- plasma cell leukemia, Skov3-ovarian cancer, A549-lung adenocarcinoma, Hela-cervical cancer; mouse cancer cell lines: N2A-neuroblastoma, CT-26-colon carcinoma, LLC- lung carcinoma, NB41A3-neuroblastoma, MPC11-plasmacytoma, 4T1-breast cancer, AB12-mesothelioma, EL-4-lymphoma, RENCA-renal carcinoma, XS-63-myeloma; C6, and RG2-rat glioma. The oncolytic activities of the mumps virus infections were quantitated using MTS Cell Proliferation Colorimetric Assay Kit (BioVision, Milpitas, CA).
Infectious clone construction and virus recovery
An infectious molecular cDNA clone of MuV-UC was produced by first reverse transcribing RNA isolated from MuV-UC virus, and then amplifying overlapping regions of the genome using PCR, and the PCR products were sequentially cloned into the pSMART vector (Lucigen, Middleton, WI). The rMuV-UC full-length genome was assembled between artificially introduced SnaBI and NotI restriction sites. Additional restriction sites were generated in the genome by overlapping PCR. The enhanced green fluorescent protein (eGFP) ORF was amplified from plasmid pIRES2-EGFP (Promega, Madison, WI). A translation unit that comprises the transcription start and end signals of mumps P and M intergenic region were introduced flanking the GFP coding region was constructed using overlapping PCR, and cloned between the G and L genes using introduced unique NheI and SmaI restriction sites. A T7 promoter and terminator were introduced at the beginning and end of the genome respectively. A hepatitis delta virus ribozyme was added to the 5′terminus to get the precise virus genome upon transcription.31 This completes the full-length infectious molecular clone of MuV-UC in plasmid pMuV-UC-GFP. GFP was replaced with firefly luciferase or NIS ORFs by PCR to create pMuV-UC-LUC and pMuV-UC-NIS plasmids respectively. pMuV-UC-mIFNβ-GFP was created by introduction of mIFNβ between M and F genes using SbfI and MluI restriction sites. pMuV-UC-L13328-GFP is created by overlapping PCR using primers with mutated nucleotides. To complete the MuV-UC reverse genetics system, three helper plasmids were constructed to express the MuV-UC N, P, and L proteins: pMuV-UC-N, pMuV-UC-P, and pMuV-UC-L. These sequences were PCR amplified and cloned into pCI mammalian expression vector (Promega) between NheI and NotI restriction sites.
Recombinant MuV-UCs (rMuV-UCs) were rescued as follows. Baby hamster kidney cells were plated at a density of 1 × 106 cells/well in 6-well plates. The cells were infected with the VTF-7 of vaccinia virus encoding the T7 polymerase gene at a multiplicity of infection of 10. After an hour, the supernatant containing the vaccinia virus was removed, and the cells were transfected with 5 μg pMuV, 0.5 μg pMuV-UC-N, 0.05 μg pMuV-UC-P, and 0.2 μg pMuV-UC-L using 12 μl of Fugene 6 transfection reagent (Promega) in Opti-MEM according to the manufacturer’s instructions. The cells were incubated overnight at 37°C, and then the medium replaced with growth medium. After 7 days, the culture medium was harvested, filtered twice through a 0.2-μm filter, and the filtrate overlaid onto Vero cells for virus amplification. After 5 days, the culture medium was harvested and clarified by low-speed centrifugation, and the infectious mumps virus stock titrated on fresh Vero cells. When necessary the recombinant viruses were further passaged on Vero cells to amplify the viral titer. Rescued viruses were titered using a standard plaque assay described earlier.31 The identities of the recombinant viruses were verified by determining the nucleotide sequence of cDNA products synthesized using reverse-transcriptase PCR and viral genomic RNA.
Growth curve analysis
Growth curve analysis was carried out as described earlier.31 For multistep growth curves, Vero cells were incubated with rMuV at an multiplicity of infection = 0.01 for 1 hour at 37°C. Following this incubation, supernatant was removed, the monolayer was washed, and fresh growth medium was added. Supernatant was collected at predetermined time points (24, 48, 72, 96, and 120 hours), and the virus titer was determined in a standard plaque assay.
Immunofluorescence
Fluorescence microscope was used to analyze and image GFP-expressing cells.
In vitro assays
To measure in vitro radio-iodide uptake, cells were incubated in Hanks-buffered salt solution with 10 mmol/l HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.3) in the presence of radio-labeled NaI (I125 at 1 × 105 cpm) ± 100 µmol/l potassium perchlorate (KCl04). After 1-hour incubation, the medium was removed and cells were washed twice. The remaining cells were resuspended in sodium chloride. Radioactivity was measured in a gamma-counter. The assays were performed in triplicate and the means plotted. Interferon-β secretion in supernatant of infected cells was determined using enzyme-linked immunosorbent assay against murine IFNβ (VeriKine mIFNβ ELISA Kit, PBL, Piscataway, NJ). Luciferase production was measured using Luciferase Assay Systems kit (Promega) according to the manufactures protocol.
In vivo experiments
All animal protocols were reviewed and approved by the Mayo Clinic Institutional Care and Use Committee. BALB/c mice, females, 4–6 weeks old, were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were implanted with 5 × 106 mouse colon carcinoma (CT-26-LacZ) cells in the right flank. When tumors reached an average size of 0.2–0.5 cm3, mice were treated with a single i.v. injection of mumps virus via tail vein. Tumor volume was measured using a hand-held caliper. The mice were monitored daily until the end of the study (60 days) or when they reached the euthanasia criteria. The euthanasia criteria were as follows; clinical signs of neurotoxicity, tumor ulceration, tumor volume > 2,000 mm3, weight loss >10%, or mice unable to gain access to food or water.
NCr nude mice, 4–6 weeks old, were purchased from Taconic (Hudson, NY). One day before implantation of xenografts, mice were whole body irradiated (2 Gy). The next day, 5 × 106 KAS6/1 cells were implanted subcutaneously in the right flank. When tumors reached a volume of 100 mm3, 1 × 107 pfu of mumps virus, or an equal volume of saline, was injected i.v. via tail vein. The rest of the study was conducted as the Balb/C mice experiment.
A/J mice females, 4–6 weeks old, were purchased from Jackson Laboratories. Mice were implanted with 5 × 106 mouse neuroblastoma (N2A) cells in the right flank. When tumors reached an average size of 0.5 cm3, mice were treated with a single i.v. injection of mumps viruses (1 × 107 pfu) via tail vein. The rest of the study was conducted as above.
Immunohistochemistry
Harvested tumors were frozen in optimal cutting medium for cryo-sectioning. Tumor sections were analyzed by immunofluorescence for mumps virus antigens using a polyclonal rabbit anti-MuV-U serum, followed by Alexa-labeled anti-rabbit IgG secondary antibody (Life Technologies, Grand Island, NY), and cellular nuclei were stained with Hoechst 33342 (Life Technologies).
Statistical analyses
Two-way analysis of variance model using GraphPad Prism was utilized to compare the tumor growth between groups. An unpaired two-tailed Student’s t-test was carried out to compare the values. Survival curve analysis was done using the Prism 4.0 program (GraphPad Software, San Diego, CA). Survival curves were plotted according to the Kaplan–Meier method, and survival function across treatment groups was compared using log rank test analyses.
Acknowledgments
We acknowledge the work of the Mayo Clinic Viral Vector Production Laboratory in generating and characterizing the initial mumps virus isolates, in particular Kirsten Langfield, Sharon Stephan, Deborah Melder, Henry Walker and Gennett Pike Funding was provided by the Mayo Foundation.
The authors declare no conflict of interest.
References
- Kelly, E and Russell, SJ (2007). History of oncolytic viruses: genesis to genetic engineering. Mol Ther 15: 651–659. [DOI] [PubMed] [Google Scholar]
- Singh, PK, Doley, J, Kumar, GR, Sahoo, AP and Tiwari, AK (2012). Oncolytic viruses & their specific targeting to tumour cells. Indian J Med Res 136: 571–584. [PMC free article] [PubMed] [Google Scholar]
- Ledford, H (2015). Cancer-fighting viruses win approval. Nature 526: 622–623. [DOI] [PubMed] [Google Scholar]
- Russell, SJ, Federspiel, MJ, Peng, KW, Tong, C, Dingli, D, Morice, WG et al. (2014). Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc 89: 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango, N, Varsanyi, TM, Kövamees, J and Norrby, E (1988). Molecular cloning and characterization of six genes, determination of gene order and intergenic sequences and leader sequence of mumps virus. J Gen Virol 69 (Pt 11): 2893–2900. [DOI] [PubMed] [Google Scholar]
- (2012). Mumps virus nomenclature update: 2012. Wkly Epidemiol Rec 87: 217–224. [PubMed] [Google Scholar]
- Asada, T (1974). Treatment of human cancer with mumps virus. Cancer 34: 1907–1928. [DOI] [PubMed] [Google Scholar]
- Okuno, Y, Asada, T, Yamanishi, K, Otsuka, T, Takahashi, M, Tanioka, T et al. (1977). [Mumps virus therapy of neoplasms (2)]. Nihon Rinsho 35: 3820–3825. [PubMed] [Google Scholar]
- Minton, JP (1973). Mumps virus and BCG vaccine in metastatic melanoma. Arch Surg 106: 503–506. [DOI] [PubMed] [Google Scholar]
- Okuno, Y, Asada, T, Yamanishi, K, Otsuka, T, Takahashi, M, Tanioka, T et al. (1978). Studies on the use of mumps virus for treatment of human cancer. Biken J 21: 37–49. [PubMed] [Google Scholar]
- Shimizu, Y, Hasumi, K, Okudaira, Y, Yamanishi, K and Takahashi, M (1988). Immunotherapy of advanced gynecologic cancer patients utilizing mumps virus. Cancer Detect Prev 12: 487–495. [PubMed] [Google Scholar]
- Yamada, A, Tsurudome, M, Hishiyama, M and Ito, Y (1984). Abortive infection of mumps virus in murine cell lines. J Gen Virol 65 (Pt 5): 973–980. [DOI] [PubMed] [Google Scholar]
- Escobar-Zarate, D, Liu, YP, Suksanpaisan, L, Russell, SJ and Peng, KW (2013). Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther 20: 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, DF, Galiano, MC, Lemon, K, Chen, YH, Andrejeva, J, Duprex, WP et al. (2009). Mumps virus Enders strain is sensitive to interferon (IFN) despite encoding a functional IFN antagonist. J Gen Virol 90(Pt 11): 2731–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tortorec, A, Denis, H, Satie, AP, Patard, JJ, Ruffault, A, Jégou, B et al. (2008). Antiviral responses of human Leydig cells to mumps virus infection or poly I:C stimulation. Hum Reprod 23: 2095–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brgles, M, Bonta, M, Šantak, M, Jagušić, M, Forčić, D, Halassy, B et al. (2016). Identification of mumps virus protein and lipid composition by mass spectrometry. Virol J 13: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrholz, M, Kendl, S, Prifert, C, Weissbrich, B, Lemon, K, Rennick, L et al. (2012). The innate antiviral factor APOBEC3G targets replication of measles, mumps and respiratory syncytial viruses. J Gen Virol 93(Pt 3): 565–576. [DOI] [PubMed] [Google Scholar]
- Okeoma, CM, Petersen, J and Ross, SR (2009). Expression of murine APOBEC3 alleles in different mouse strains and their effect on mouse mammary tumor virus infection. J Virol 83: 3029–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Murrieta, NH, Santos-López, G, Reyes-Leyva, J, Jurado, FS and Herrera-Camacho, I (2011). Modulation of apoptosis by V protein mumps virus. Virol J 8: 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri, M, Lemon, K, Duprex, WP, Rima, BK and Horvath, CM (2009). A point mutation, E95D, in the mumps virus V protein disengages STAT3 targeting from STAT1 targeting. J Virol 83: 6347–6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulane, CM, Rodriguez, JJ, Parisien, JP and Horvath, CM (2003). STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J Virol 77: 6385–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, P, Luthra, P, Li, Z, Fuentes, S, D’Andrea, JA, Wu, J et al. (2012). The V protein of mumps virus plays a critical role in pathogenesis. J Virol 86: 1768–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, RL, Fuentes, SM, Wang, P, Taddeo, EC, Klatt, A, Henderson, AJ et al. (2006). Function of small hydrophobic proteins of paramyxovirus. J Virol 80: 1700–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurudome, M, Yamada, A, Hishiyama, M and Ito, Y (1984). Replication of mumps virus in murine cells. Arch Virol 81: 13–24. [DOI] [PubMed] [Google Scholar]
- Dourado, I, Cunha, S, Teixeira, MG, Farrington, CP, Melo, A, Lucena, R et al. (2000). Outbreak of aseptic meningitis associated with mass vaccination with a urabe-containing measles-mumps-rubella vaccine: implications for immunization programs. Am J Epidemiol 151: 524–530. [DOI] [PubMed] [Google Scholar]
- Kimura, M, Kuno-Sakai, H, Yamazaki, S, Yamada, A, Hishiyama, M, Kamiya, H et al. (1996). Adverse events associated with MMR vaccines in Japan. Acta Paediatr Jpn 38: 205–211. [DOI] [PubMed] [Google Scholar]
- Sugiura, A and Yamada, A (1991). Aseptic meningitis as a complication of mumps vaccination. Pediatr Infect Dis J 10: 209–213. [DOI] [PubMed] [Google Scholar]
- Miller, E, Goldacre, M, Pugh, S, Colville, A, Farrington, P, Flower, A et al. (1993). Risk of aseptic meningitis after measles, mumps, and rubella vaccine in UK children. Lancet 341: 979–982. [DOI] [PubMed] [Google Scholar]
- Rubin, SA, Pletnikov, M, Taffs, R, Snoy, PJ, Kobasa, D, Brown, EG et al. (2000). Evaluation of a neonatal rat model for prediction of mumps virus neurovirulence in humans. J Virol 74: 5382–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin, SA, Afzal, MA, Powell, CL, Bentley, ML, Auda, GR, Taffs, RE et al. (2005). The rat-based neurovirulence safety test for the assessment of mumps virus neurovirulence in humans: an international collaborative study. J Infect Dis 191: 1123–1128. [DOI] [PubMed] [Google Scholar]
- Ammayappan, A, Nace, R, Peng, KW and Russell, SJ (2013). Neuroattenuation of vesicular stomatitis virus through picornaviral internal ribosome entry sites. J Virol 87: 3217–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.