

Abstract
As a sensor of cellular energy status, the AMP-activated protein kinase (AMPK) is believed to act in opposition to the metabolic phenotypes favored by proliferating tumor cells. Consequently, compounds known to activate AMPK have been proposed as cancer therapeutics. However, the extent to which the anti-neoplastic properties of these agonists are mediated by AMPK is unclear. Here we examined the AMPK-dependence of six commonly used AMPK agonists (metformin, phenformin, AICAR, 2DG, salicylate and A-769662) and their influence on cellular processes often deregulated in tumor cells. We demonstrate that the majority of these agonists display AMPK-independent effects on cell proliferation and metabolism with only the synthetic activator, A-769662, exerting AMPK-dependent effects on these processes. We find that A-769662 promotes an AMPK-dependent increase in mitochondrial spare respiratory capacity (SRC). Finally, contrary to the view of AMPK activity being tumor suppressive, we find A-769662 confers a selective proliferative advantage to tumor cells growing under nutrient deprivation. Our results indicate that many of the anti-growth properties of these agonists cannot be attributed to AMPK activity in cells, and thus any observed effects using these agonists should be confirmed using AMPK-deficient cells. Ultimately, our data urge caution, not only regarding the type of AMPK agonist proposed for cancer treatment, but also the context in which they are used.
Keywords: AMPK, agonist, activation, A-769662, metformin, phenformin, AICAR, salicylate, mTORC1, metabolism
Introduction
AMPK is a highly conserved Ser/Thr protein kinase complex that monitors the bioenergetic state of a cell. AMPK is a heterotrimeric kinase complex composed of a catalytic α subunit and two regulatory subunits, β and γ. AMP or ADP binding to the γ subunit of the AMPK complex promotes increased kinase activity of the α subunit and phosphorylation at Thr-172 by upstream kinases.1–3 AMPK activation promotes ATP conservation by inhibiting cell growth and proliferation.4 AMPK also inhibits anabolic/growth-promoting pathways including lipid biosynthesis,5 TORC1-dependent protein biosynthesis,6, 7 and cell proliferation.8, 9 Thus, under nutrient stress, AMPK promotes a metabolic and proliferative phenotype unfavorable to proliferating cancer cells and as such is thought to negatively impact tumorigenesis.10
One tangible link between AMPK and tumor suppression is the fact that the upstream AMPK kinase LKB1 is a tumor suppressor.11 LKB1 is inactivated in patients with Peutz-Jegher’s syndrome,12 a condition that predisposes these individuals to gastrointestinal polyps and malignant tumors.13, 14 Recent clinical studies have also identified LKB1 as the second most frequently mutated tumor suppressor in sporadic human lung cancer.15 AMPK activity is dramatically reduced in both human and mouse tumors lacking LKB1,16, 17 providing evidence that LKB1 loss reduces AMPK pathway activity in tumors. We have recently shown that loss of AMPK can cooperate with Myc to accelerate lymphomagenesis,18 indicating that AMPK itself can act as a tumor suppressor. Thus, stimulating the anti-tumor activity of AMPK has been proposed as a possible anti-neoplastic therapy.19
Interest in the therapeutic advantages of activating AMPK with chemical agonists has increased as more evidence has emerged supporting an anti-tumorigenic role for the kinase. The most convincing data that AMPK agonists may function as anti-cancer agents has been through experiments using biguanides, such as metformin and phenformin, in therapeutic settings. Metformin is a first-line therapy for type II diabetes that functions by inhibiting complex I of the mitochondrial electron transport chain.20, 21 Metformin-dependent inhibition of oxidative phosphorylation (OXPHOS) promotes an increase in intracellular ADP and AMP, leading to indirect AMPK activation. Retrospective analysis of tumor development in type II diabetics revealed evidence that patients on metformin were associated with a significantly lower cancer incidence than those patients on other medication.22, 23 Furthermore, treatment of animals harboring tumor xenografts with metformin or phenformin has been shown to delay progression of established tumors.24–26 Several prospective clinical trials examining anti-neoplastic effects of metformin are currently ongoing.27
Other AMPK agonists, such as the AMP mimetic AICAR and the synthetic activator A-769662, are commonly used in the laboratory to activate AMPK, and have also been explored as anti-neoplastic agents. AICAR is metabolized in cells into the monophosphorylated nucleotide ZMP, which can bind the γ subunit in place of AMP,28 leading to AMPK activation. Various studies have shown that AICAR can inhibit tumor cell proliferation.9, 29, 30 A-769662 activates AMPK directly in cell-free assays independently of adenylate levels.31 It was recently discovered that salicylate, the metabolic derivative of aspirin, can activate AMPK by binding the same site on the AMPKβ1 subunit as A-769662.32 Both A-769662 and salicylate can promote whole-body fat oxidation in mice in an AMPK-dependent manner.32 For many years salicylate has been known to possess anti-tumor properties;33 however, it has several cellular targets in addition to AMPK, and it is not known to what extent, if at all, these effects are dependent on AMPK activation.
In this study, we have systematically assessed the impact of six common AMPK agonists on a defined set of tumor cell phenotypes. We examined the effects of the chemical agonists on mTOR signalling, proliferation, apoptosis, viability, and cellular metabolism, and used non-transformed and transformed cells lacking AMPK expression to verify the specificity of these effects to AMPK. We find that while these agonists exert significant effects on proliferation and metabolism, many of these effects are AMPK-independent, with only A-769662 displaying AMPK-specific effects on proliferation and metabolism. This study provides a reference point for the use of chemical AMPK agonists as research tools, and catalogs the effects of these compounds on cellular processes and their dependence on AMPK.
Results
Agonists activate AMPK in a dose-dependent manner and stimulate downstream signalling
AMPK agonists are commonly used in the laboratory to assess the impact of AMPK signalling. Here we investigated the cellular effects of six AMPK agonists that activate AMPK either directly (A-769662 and salicylate) or indirectly (metformin, phenformin, 2DG and AICAR) (schematic in Figure 1A). We first examined the effect of these agonists on AMPK signalling in HEK293 cells and immortalized mouse embryo fibroblasts (MEFs) lacking both catalytic isoforms of AMPK (AMPKα1/α2−/−, labelled hereafter as DKO) via immunoblot. HEK293 cells were pre-treated with Ca2+/calmodulin-dependent protein kinase kinase 2 (CAMKK2) inhibitor STO-609 prior to stimulation to reduce basal AMPK phosphorylation, as has been done previously.6 In HEK293 cells, each agonist induced phosphorylation of the AMPK downstream substrate ACC, which is widely used as a biomarker for AMPK activity.34 AMPK phosphorylation at Thr-172 was minimally induced by each agonist in most cases (Figure 1B) (see discussion). In wild type (WT) MEFs, each agonist induced phosphorylation of AMPK and ACC in a dose-dependent manner, whereas AMPK and ACC phosphorylation were completely absent in DKO MEFs, as was AMPKα expression (Figure 1C).
Figure 1. Six known AMPK agonists activate AMPK in Hek293 and MEF cells.

A) Schematic summarising the mechanism of action of the 6 AMPK agonists used in this study. AICAR is metabolized in cells to ZMP, which binds the γ subunit in place of AMP, leading to AMPK activation. Metformin and phenformin inhibit complex I of the electron transport chain leading to an elevation in the AMP/ATP ratio and subsequent AMPK activation. 2DG also elevates the AMP/ATP ratio by inhibiting glycolysis. Both salicylate and A-769662 activate AMPK directly, binding to the β subunit. B) Immunoblot for ACC (Ser79 and total), AMPKα (Thr172 and total) and actin on lysates from HEK293 cells. HEK293 cells were pretreated with STO-609 (25μM) for 30min before incubation with metformin (5mM), phenformin (1.25mM), AICAR (1.25mM), 2DG (12.5mM), salicylate (2.5mM) or A-769662 (25μM) for 1h. C) Immunoblot for ACC (Ser79 and total), AMPKα (Thr172 and total) and actin on lysates from wild-type (WT) or AMPKα-deficient (DKO) MEFs. Cells were incubated with metformin, phenformin, AICAR, 2DG, salicylate or A-769662 for 1h at the indicated concentrations.
One of the main downstream effects of AMPK signalling is the inhibition of mTORC1 activity.10 Thus, we investigated the AMPK-specific effects of each agonist on mTORC1 signalling using WT and DKO MEFs. All agonists except metformin promoted increased phosphorylation of the AMPK substrate raptor in WT but not DKO MEFs (Figure 2). Phosphorylation of the downstream mTORC1 substrate p70S6K (S6K) was reduced by all agonists to varying degrees (with the exception of 2DG), with phenformin, salicylate and A-769662 promoting the greatest decreases in pS6K levels. This is consistent with the large increase in raptor phosphorylation induced by phenformin, salicylate and A-769662. Total S6K levels were also decreased by salicylate and phenformin treatment in DKO cells, despite the lack of functional AMPK activity in these cells. Treatment with all agonists, with the exception of metformin, promoted hypophosphorylation of 4E-BP1, as indicated by the presence of lower molecular weight bands on the immunoblot in WT MEFs (Figure 2). Phenformin promoted hypophosphorylation of 4E-BP1 to the greatest extent, although this effect was also partially observed in AMPK-deficient MEFs, suggesting that phenformin may also affect 4E-BP1 phosphorylation through AMPK-independent mechanisms (Figure 2).
Figure 2. AMPK agonists stimulate AMPK-dependent signal transduction in MEFs.

Immunoblot for AMPKα (Thr172 and total), ACC (Ser79 and total), raptor (Ser792 and total), p70S6K (Thr389 and total), 4E-BP1 (different phosphorylation states marked by arrows) and actin on lysates from WT or DKO MEFs. Cells were incubated with metformin (5mM), phenformin (1.25mM), AICAR (1.25mM), 2DG (12.5mM), salicylate (2.5mM) or A-769662 (25μM) for 1h.
The majority of AMPK agonists inhibit cell proliferation in an AMPK-independent manner
Activation of AMPK has previously been linked to reduced proliferation and cell cycle arrest.8, 9, 35 Thus, we assessed HEK293 cell proliferation using crystal violet staining following 48h of culture with the various AMPK agonists. Similar to our results with mTOR signaling, all agonists except metformin reduced HEK293 cell proliferation (Figure 3A). The anti-proliferative effects of these agonists were largely dose-dependent, with only AICAR failing to act in a dose-dependent manner (Figure 3A). We next assessed the AMPK-dependence of these agonists on cell proliferation by conducting growth assays using WT and DKO MEFs. Phenformin, AICAR, 2DG and salicylate reduced the proliferation of WT MEFs in a largely dose-dependent manner, but also had a dramatic suppressive effect on the proliferation of AMPKα-null cells (Figure 3B). These results were confirmed by cell counting assays for WT and DKO MEFs over a range of agonist concentrations at 24, 48 and 72h (Figure S1A).
Figure 3. The majority of AMPK agonists inhibit proliferation in an AMPK-independent manner.

A) HEK293 cells were grown for 48h in the presence of metformin, phenformin, AICAR, 2DG, salicylate or A-769662 at the concentrations indicated. Adherent cell growth was determined by crystal violet staining. Data are expressed as mean ± SEM for triplicate cultures. B) Growth of WT or DKO MEFs was determined as in (A). C) WT and DKO MEFs were incubated for 24h with phenformin (0.63mM), AICAR (0.63mM), 2DG (6.25mM) or salicylate (2.5mM) before the percentage of cells in G1, S and G2/M phase were measured by propidium iodide staining. D) Growth of WT and DKO MEFs in the presence of A-769662 was determined as in (A). E) Cell cycle profiles of WT and DKO MEFs following treatment with A-769662 (25μM) was determined as in (C).
To further examine the impact of AMPK agonists on cell proliferation we conducted cell cycle analysis of WT and DKO MEFs using propidium iodide (PI) staining (Figure 3C and S1B). To evaluate the distribution of actively dividing cells before the induction of cell death we harvested cells 24h after treatment with the various compounds. Phenformin treatment promoted an increase in the proportion of cells in G1 phase, while AICAR treatment led to a decrease in the proportion of cells G2/M, and these effects were observed in both WT and DKO MEFs (Figure 3C and S1B). Incubation with 2DG did not significantly affect the cell cycle profile in WT MEFs (slight increase of cells in S phase, with a reduction in the percentage of G2/M phase cells), whereas 2DG promoted a decrease in the proportion of DKO cells in S phase, suggesting 2DG treatment promotes cell cycle arrest in DKO cells (Figure 3C and S1B).
We next assessed the effect of A-769662 on the proliferation of WT and DKO MEFs. In contrast to other AMPK agonists, A-769662 reduced the proliferation of WT MEFs by 20–30%, but did not affect the proliferation of DKO MEFs (Figure 3D). Cell cycle analysis of WT MEFs treated with A-769662 indicated a decrease in the proportion of G2/M phase cells combined with an increase in G1 phase cells, which was not observed to the same extent in DKO cells (Figure 3E and S1C).
To assess whether AMPK agonists could induce apoptosis in HEK293 and MEF cells, we assayed for active (cleaved) caspase 3 in compound-treated cells. We first assessed the extent of caspase 3 cleavage in HEK293 cells following 48h of culture with the various AMPK agonists. All agonists except metformin led to a mild, dose-dependent increase in caspase 3 cleavage (Figure 4A), mirroring our cell proliferation results (Figure 3A). Phenformin treatment led to the greatest increase in caspase 3 cleavage in HEK293 cells (Figure 4A).
Figure 4. The majority of AMPK agonists induce apoptosis and cell death in cells lacking AMPK expression.

A) HEK293 cells were grown for 48h in the presence of metformin, phenformin, AICAR, 2DG, salicylate or A-769662 at the concentrations indicated and the extent of caspase 3 cleavage was determined. Each bar represents the mean ± SEM for triplicate cultures. B) Caspase 3 cleavage was determined for WT and DKO MEFs in response to phenformin, AICAR, 2DG and salicylate treatment as in (A). C) Viability of WT and DKO MEFs following incubation with phenformin (1.25mM), AICAR (1.25mM), 2DG (12.5mM) or salicylate (2.5mM). Cell death was measured after 48h incubation via propidium iodide uptake. Each bar represents the mean ± SEM for triplicate cultures. D) Caspase 3 cleavage was determined for WT and DKO MEFs in response to A-769662 treatment as in (A). E) Viability of WT and DKO MEFs following incubation with A-769662 (25μM) and phenformin (1.25mM) was determined as in (C).
We next assessed whether the effect of these agonists on apoptosis was AMPK-dependent by measuring levels of active caspase 3 in WT and DKO MEFs. Consistent with the reduced proliferation observed in DKO cells (Figure 3B), phenformin, AICAR, 2DG and salicylate mildly increased caspase 3 cleavage in WT MEFs, but promoted a dramatic dose-dependent increase in caspase 3 activation in DKO MEFs (Figure 4B). In addition, DKO cells displayed increased levels of dead cells in response to phenformin, AICAR, 2DG, and salicylate treatment (Figure 4C), indicating increased sensitivity to apoptosis by these agonists in AMPKα−/− cells.
We next assessed the effect of A-769662 on the viability of WT and DKO MEFs. In contrast to other AMPK agonists, A-769662 treatment led to a small increase in caspase 3 cleavage in WT MEFs and no change in DKO MEFs (Figure 4D). The reduced proliferation of WT cells induced by A-769662 was not accompanied by increased cell death (Figure 4E), suggesting an overall effect of A-769662 on cell proliferation and but not cell viability.
Finally, we assessed the impact of AMPK agonists on tumor cell proliferation using HCT116 colon carcinoma cells. Consistent with our results using HEK293 cells and MEFs, phenformin, 2DG and salicylate exhibited the greatest suppressive effect on cell proliferation under anchorage-independent conditions (Figure 5A). We next measured the level of AMPK activation and activity in HCT116 cells expressing AMPKα1/2 shRNAs18, 36, 37 to determine the AMPK-dependence of the results observed in Figure 5A. All AMPK agonists tested induced AMPKα phosphorylation at Thr-172 as well as phosphorylation of downstream substrates ACC and raptor in HCT116 cells, and the effect of these agonists on mTORC1 or AMPK signaling were reduced in HCT116 cells expressing AMPKα1/2 shRNAs (Figure 5B). AMPKα1/2 shRNA-expressing HCT116 cells displayed increased sensitivity to cell growth arrest in response to AICAR, 2DG and salicylate treatment relative to control cells (Figure 5C). Of note, A-769662 had little effect on tumor cell growth in this assay. Downregulation of AMPK signaling did not significantly change the sensitivity HCT116 cells to growth arrest induced by phenformin (Figure 5C, black bars, and Figure S2).
Figure 5. AMPK agonists inhibit anchorage-independent growth of HCT116 colon cancer cells in an AMPK-independent manner.

A) HCT116 cells were cultured under anchorage–independent conditions in the presence of metformin (5mM), phenformin (0.625mM), AICAR (1.25mM), 2DG (12.5mM), salicylate (2.5mM) or A-769662 (25μM) as indicated. After 5 days images were taken. B) Immunoblot for AMPKα (Thr172 and total), ACC (Ser79 and total), raptor (Ser792 and total) and actin on lysates from HCT116 cells expressing either control or AMPKα1/2 shRNA. Cells were incubated with metformin (5mM), phenformin (1.25mM), AICAR (1.25mM), 2DG (12.5mM), salicylate (2.5mM) or A-769662 (25μM) for 1h. C) HCT116 cells expressing either control (-) or AMPKα1/α2 shRNA (+) were cultured as in (A), here viable cells were estimated by the fluorescence of the metabolic reduction product of Alamar blue. Each bar represents the mean fluorescence of three replicate wells ± SEM.
AMPK agonists differentially affect glycolysis
We recently demonstrated that loss of AMPK promotes an increase in aerobic glycolysis,18 a metabolic profile of tumor cells also known as the Warburg effect.38 One inference from these data is that AMPK may function as a negative regulator of glycolysis. Thus, we examined the metabolic impact of AMPK agonists on proliferating cells. HEK293 cells were cultured for 48h with the various AMPK agonists, and then glucose consumption (Figure 6A) and lactate production (Figure 6B) by cells was measured. Interestingly, AMPK agonists displayed differential effects on glycolytic metabolism. Metformin and phenformin increased both glucose consumption and lactate production by HEK293 cells, while AICAR and 2DG treatment reduced glucose consumption and lactate production. In contrast, salicylate and A-769662 exerted no significant effects on glucose uptake or lactate production.
Figure 6. AMPK agonists differentially affect glycolysis.

Glucose consumption (A) and lactate production (B) were determined in HEK293 following treatment with metformin (1.25mM), phenformin (0.31mM), AICAR (0.31mM), 2DG (3.13mM), salicylate (1.25mM) or A-769662 (25μM) for 48h. Glucose consumption (C) and lactate production (D) were determined as in (A) and (B) respectively in WT and DKO MEFs following treatment with metformin (1.25mM) and phenformin (0.31mM) for 48h. E) Basal extracellular acidification rate (ECAR) was determined in WT and DKO MEFs following treatment with metformin (5mM) and phenformin (1.25mM) for 6h. Each bar represents the mean ± SEM for triplicate cultures. Glucose consumption (F), lactate production (G) and ECAR (H) were determined in WT and DKO MEFs following treatment with AICAR (0.31mM in F and G, 1.25mM in H) and 2DG (3.13mM in F and G, 12.5mM in H) as in (C), (D) and (E). In all experiments each bar represents the mean ± SEM for triplicate cultures and statistical significance is represented by: *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
We next cultured WT and DKO MEFs with AMPK agonists to assess the AMPK-dependence of the changes in glucose metabolism observed in Figures 6A and 6B. Metformin and phenformin elevated glucose consumption independent of AMPKα expression after 24h (Figure S3C) or 48h (Figure 6C) of culture, and did so in a dose-dependent manner (Figure S3A). Consistent with our previous observations,18 lactate production was elevated in DKO MEFs relative to WT MEFs (Figure 6D). However, similar to glucose consumption, lactate production was increased by biguanide treatment regardless of AMPKα expression (Figure 6D, S3B, and S3D). We also measured the extracellular acidification rate (ECAR), an indicator of glycolytic rate, of WT and DKO MEFs following 6h incubation with these compounds. Biguanide treatment promoted an increase in ECAR in both WT and DKO MEFs (Figure 6E), indicating that the pro-glycolytic effects of biguanides are AMPK-independent. In contrast, AICAR and 2DG reduced glucose consumption (Figure 6F and S3C), lactate production (Figure 6G and S3D) and ECAR (Figure 6H) in both WT and DKO MEFs in a dose-dependent manner (Figure S3A and S3B). Salicylate and A-769662 induced a small reduction in glucose consumption in HEK293 cells (Figure 6A), but this effect was not observed in MEFs (Figure S3E).
AMPK agonists differentially affect mitochondrial respiration
Given the links between AMPK and oxidative metabolism,39 we next assessed the impact of AMPK agonists on mitochondrial respiration using a Seahorse extracellular flux analyzer. We first assessed the oxygen consumption rate (OCR) and level of coupled respiration in WT and DKO MEFs treated with AMPK agonists. Coupled respiration is the proportion of mitochondrial respiration coupled to ATP production as opposed to the consequence of proton leak (uncoupled), which does not generate ATP. To differentiate between these two states, we measured the OCR of MEF cells treated acutely with oligomycin (an ATP synthase inhibitor) followed by rotenone and antimycin A (to block mitochondrial electron transport). Consistent with their roles as complex I inhibitors,20, 21 metformin and phenformin reduced the oxygen consumption rate (OCR) and proportion of coupled respiration in both WT and DKO MEFs in a dose-dependent manner (Figure 7A and B). Similar to the function of biguanides, 2DG reduced OCR in both WT and DKO MEFs (Figure S4B), while having no impact on the level of coupled respiration (Figure S4A). As reported,40, 41 salicylate promoted the uncoupling of cellular respiration expression (Figure 7A), but did not alter cellular OCR (Figure 7B). DKO MEFs displayed a slight resistance to salicylate-dependent mitochondrial uncoupling (Figure 7A).
Figure 7. AMPK agonists differentially affect mitochondrial respiration.

Coupled respiration (A) and oxygen consumption rate (OCR) (B) of WT and DKO MEFs treated with metformin, phenformin and salicylate for 24h at the concenrations indicated. OCR (C) and spare respiratory capacity (SRC) (D) of WT and DKO MEFs treated with A-769662 (25μM) for 6h and 24h respectively. E) SRC in WT MEFs upon 6h A-769662 treatment, concentration as indicated. In all experiments each bar represents the mean ± SEM for triplicate cultures and statistical significance is represented by: ****, p<0.0001.
We next examined the impact of A-769662 on mitochondrial bioenergetics. Treatment of WT MEFs with A-769662 promoted a 60% drop in OCR, while having little effect on DKO cells (Figure 7C). In addition, A-769662 promoted a 4-fold increase in mitochondrial spare respiratory capacity (SRC) in WT cells (Figure 7D). SRC is the reserve mitochondrial capacity available for energy promotion and is an index of cellular metabolic fitness.42 In contrast to WT MEFs, SRC in DKO cells was approximately 60–70% of that of control cells and was unaffected by A-769662 treatment (Figure 7D). Of the AMPK agonists tested, only A-769662 displayed AMPK-specific changes in SRC. The observed effect of A-769662 on SRC of AMPKα-expressing cells was dose-dependent and maximal between 12.5 – 25μM (Figure 7E).
A-769662 promotes tumor cell growth under low glucose conditions
To investigate the impact of AMPK activation on mitochondrial respiratory capacity in cancer cells, we examined the SRC of HCT116 colon carcinoma cells and H1299 non-small-cell lung carcinoma cells following treatment with A-769662. SRC increased in a dose-dependent manner in both HCT116 and H1299 cells, achieving a maximum level between 12.5 and 25μM (Figure 8A). To investigate whether the increase in SRC promoted by A-769662 could affect cell proliferation under nutrient-limiting conditions, we cultured HCT116 and H1299 cells under non-adherent conditions in full glucose (25mM) or glucose-free conditions and co-cultured in the presence or absence of A-769662. As seen in Figure 8B, both HCT116 and H1299 cells displayed a ~30% increase in cell proliferation under glucose-free conditions specifically when treated with A-769662. In contrast, A-769662 treatment did not affect the proliferation of HCT116 or H1299 cells expressing AMPKα1/α2 shRNAs (Figure 8B).
Figure 8. A-769662 promotes tumor cell growth under glucose-free conditions.

A) SRC of HCT116 and H1299 cells following incubation with A-769662 at the indicated concentrations for 24h. B) HCT116 and H1299 cells expressing either control or AMPKα1/2 shRNA were cultured under anchorage–independent conditions in the presence of A-769662 (25μM) and the presence or absence of glucose. After 5 days, the number of viable cells was estimated by the fluorescence of the metabolic reduction product of Alamar blue. Each bar represents the mean fluorescence of three replicate wells ± SEM and statistical significance is represented by: *, p<0.05, p<0.01; ***.
Discussion
Chemical agonists of AMPK have been frequently used to research the cellular effects of AMPK activation. Recent work linking AMPK activation to inhibitory effects on cell growth and proliferation has increased interest in exploring the use of AMPK agonists for anti-cancer therapy,19 particularly biguanides.27 In this study, we systematically examined the effect of 6 AMPK agonists (metformin, phenformin, AICAR, salicylate, 2DG and A-769662) on cellular functions associated with proliferating cells. We focused our work on processes often deregulated in tumorigenesis: mTORC1 signalling (Figure 2), proliferation, apoptosis and viability (Figures 3 – 5), glycolysis (Figure 6), and mitochondrial respiration (Figure 7). Given that many of these agonists promote AMPK activation by indirect mechanisms (with the exception of salicylate and A-769662) (Figure 1A), allowances must be made for ‘off-target’ or AMPK-independent effects of these compounds. Many of these compounds have been shown to possess anti-proliferative activity;19 thus, it is important to establish whether their cellular effects can be attributed to AMPK. We have attempted to clarify these issues using a specific AMPK agonist (A-769662) and cells lacking AMPK expression. We show here that all AMPK agonists used (with the exception of A-769662) have significant AMPK-independent effects on proliferation, cell viability and metabolism. Only A-769662 exhibited specific AMPK-dependent effects on cell growth and metabolism. Our findings are summarized in Table 1.
Table 1.
Summary of the AMPK-dependence and effects of agonists on cellular process
| mTOR signalling | Proliferation | Viability | Anchorage | Glycolysis | ATP-coupled | Oxygen consumption | Spare respiratory | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Effect | AMPK? | Effect | AMPK? | Effect | AMPK? | Effect | AMPK? | Effect | AMPK? | Effect | AMPK? | Effect | AMPK? | Effect | AMPK? | |
| Metformin | - | - |

|
No |
|
No | - | - |
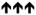
|
No |
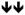
|
No |
|
No |
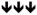
|
No |
| Phenformin |
|
Partial |
|
No |
|
No |
|
No |
|
No |
|
No |
|
No |
|
No |
| AICAR |
|
Yes |
|
No |
|
No | - | - |
|
No | - | - | - | - | - | - |
| 2DG |
|
Yes |
|
No |
|
No |
|
No |
|
No | - | - |
|
No |
|
No |
| Salicylate |
|
Partial |
|
No |
|
No |
|
No | - | - |
|
No | - | - | - | - |
| A-769662 |
|
Yes |
|
Yes | - | - | - | - | - | - | - | - |
|
Yes |
|
Yes |
The AMPK agonists used in this study all activated AMPK in a dose-dependent manner, as determined by phosphorylation of both AMPKα and its downstream target ACCα. We observed no effect of the agonists on ACCα phosphorylation in DKO MEFs, consistent with ACCα being a reliable biomarker for AMPK activity.34 We found that the dose-dependent induction of ACCα phosphorylation was often a better readout of agonist exposure than phosphorylation of AMPK itself. Indeed, a high background of AMPK phosphorylation has been observed in many cell types. For example, high AMPK Thr172 phosphorylation has been reported in HEK293T cells due to constitutive activity of CAMKK2,6 which is also an AMPK kinase.43 Moreover, the effects of A-769662 on AMPK phosphorylation are small relative to its effects on ACC phosphorylation (Figure 1C and previous reports44). Our results indicate that raptor phosphorylation at Ser792 is also a reliable readout of AMPK activation by multiple agonists. Thus, to assay affects of agonists on AMPK function, we recommend measuring phosphorylation of both AMPK and downstream targets (such as ACC and raptor), and to confirm results using cells lacking AMPK expression as specific controls for activity.
A key tumor suppressor function exerted by AMPK is the inhibition of mTORC1 activity. AMPK inhibits mTORC1 signalling through multiple mechanisms, including phosphorylation of the upstream regulator TSC27 and the mTORC1 subunit Raptor.6 A-769662, AICAR and 2DG induced AMPK-dependent inhibition of mTORC1 signalling, whereas the effects of phenformin exhibited only partial AMPK dependence. The AMPK-independent effects of phenformin on mTORC1 signalling may be due to the increased potency of the drug relative to metformin, but also argue that these compounds may function through additional mechanisms to inhibit mTOR. Indeed, biguanides (metformin and phenformin) have previously been shown to inhibit mTORC1 signaling in an AMPK-independent fashion through their effect on the Rag GTPases.45 Of note, metformin was a poor inhibitor of mTORC1 in our cell systems. Metformin has previously been shown to downregulate mTORC1 signalling, albeit at much higher doses than used here.46 Phenformin is 50-fold more potent than metformin,47 and unlike metformin, does not require the expression of the organic cation transporter 1 (OCT1) to be transported into cells,48 which may explain the limited efficacy of metformin in our assays. Thus, one must consider the dose and length of biguanide treatment when considering its effects on cellular assays.
AMPK activation has also been linked with induction of cell cycle arrest. AMPK activation has been shown to suppress cell proliferation through multiple mechanisms including stabilization of p538, 9 and regulation of the cyclin dependent kinase (CDK) inhibitors p21WAF1 and p27CIP1.35 Of the AMPK agonists tested here, we found that only the direct AMPK activator A-769662 displayed a small but AMPK-dependent effect on proliferation and cell cycle progression in MEF cells without causing apoptosis. In contrast, Zadra et al. have recently shown that MT-38, a direct chemical agonist of AMPK, induces mitotic arrest and apoptosis in prostate cancer cells,49 suggesting that the induction of apoptosis in response to AMPK activation may be cell-type dependent. Of note, A-769662 has been shown in vivo to delay tumor onset in PTEN+/− mice.50 Loss of PTEN promotes increased PI3K signalling in tumors, leading to increased Akt and mTORC1 signalling that drives tumor progression. Our data suggest that activating AMPK (via A-769662) may have a significant anti-tumor effect on PTEN-null tumors by shutting down amplified signalling networks, specifically mTORC1 signalling, in this tumor type.
In contrast to A-769662, phenformin, AICAR, salicylate and 2DG increased caspase 3 activation and reduced cellular viability in addition to effects on proliferation. Rather than this effect being dependent on AMPK, this effect was actually enhanced in cells lacking AMPK expression. Treatment of cells with phenformin, AICAR, salicylate and 2DG applies a metabolic stress to cells, either by reducing cellular ATP levels or mimicking an increase in AMP levels. We and others have shown that cells lacking AMPK are unable to adapt to energetic stress, and as a consequence are more sensitive to apoptosis induced by treatment with these compounds.17, 24, 36 Recently, Shackelford et al. have shown that KRAS-driven tumors lacking LKB1 are sensitive to the ATP-depleting effects of phenformin in vivo.51 Our results suggest that the effectiveness of these compounds on tumor growth in vivo may be due to defective responses to energetic stress rather than cell cycle arrest programs. In this light, these compounds may be specific agents for treating LKB1-deficient tumors or those with low LKB1 or AMPK activity.
One important phenotypic change in tumors is the reprogramming of cellular metabolism to support unrestrained cell growth.52 One of the primary metabolic changes associated with proliferating tumor cells is the upregulation of glycolytic metabolism.38, 53 We recently demonstrated that tumor cells lacking AMPK display enhanced rates of glycolysis at baseline, linking AMPK to control of the Warburg effect in tumors.18 We hypothesized that AMPK agonists may antagonize tumor cell growth by suppressing the Warburg effect and metabolic potential of tumor cells. However, the data here indicate that AMPK agonists have differential effects on glycolysis and oxygen consumption, and that these processes are largely AMPK-independent. Biguanides increased glycolysis, while AICAR and 2DG decreased glycolysis, despite the fact that AMPK was activated under all conditions (Table 1). Moreover, the effect of these agents on glucose metabolism was also observed in AMPKα-deficient cells. Biguanides likely trigger increased glycolysis in response to reduced OXPHOS due to complex I inhibition. Likewise, 2DG is a non-metabolizable glucose analog and functions to block glycolysis. AICAR has been shown previously to inhibit glycolysis in the liver,54, 55 which has been attributed to diminished glucose phosphorylation by glucokinase and decreased concentration of fructose-2,6-bisphosphate (reducing allosteric activation of phosphofructokinase 1), which are both induced by the accumulation of z-nucleotides.55 Notably, A-769662 treatment had no effect on glycolysis. Thus, while AMPK loss promotes increased glycolysis through increased HIF-1α activity,18 acute activation of AMPK (by A-769662) appears to have no impact on glycolysis under nutrient-replete growth conditions.
In contrast to glycolysis, our data indicate that AMPK activation can specifically impact mitochondrial bioenergetics. Treatment with A-769662 reduced mitochondrial respiration in an AMPK-dependent manner (Figure 7C). This reduction in OXPHOS may result from AMPK-dependent inhibition of mTORC1, which has recently been shown to stimulate OXPHOS by promoting the transition of nuclear-encoded components of the electron transport chain that support OXPHOS.56 However, A-769662 treatment also stimulated an AMPK-dependent increase in mitochondrial respiratory capacity. SRC is a measure of the maximal bioenergetic capacity of the cell, and indicates the cell’s ability to respond to an increase in energy demand.57 Stimulating SRC was a unique feature of A-769662, and may function to prepare cells for future energetic stress.
While A-769662 is mildly anti-proliferative under high glucose conditions, cells treated with this compound displayed a proliferative advantage when grown under glucose-free, but not in nutrient-replete, conditions. The mechanism(s) driving this phenotype remain to be determined. One possibility is that increased SRC stimulated by A-769662 may facilitate increased OXPHOS and ATP production under low nutrient conditions. Alternatively, AMPK activation may promote tumor cell viability in response to glucose deprivation by maintaining intracellular NADPH levels, which antagonize rising reactive oxygen species (ROS) in nutrient starved cells.58 In this context, AMPK activation by A-769662 may paradoxically exert pro-tumorigenic properties under nutrient limitation.52 Indeed, studies in glioblastoma59 and prostate cancer60 suggest that AMPK signalling can promote cell survival depending on environmental context. Our data argue that more extensive characterization of direct AMPK activators on tumor cell growth under conditions of nutrient limitation is warranted.
In summary, we have sought to clarify the AMPK-dependent effects of defined “AMPK agonists” on cellular function. Our results demonstrate that compounds commonly used to activate AMPK exhibit variable effects on cell proliferation, apoptosis and metabolism. Moreover, the majority of the effects of these agonists on cell physiology are AMPK-independent. Our work suggests that the anti-growth properties of many of these compounds (notably the biguanides, salicylate and 2DG) may be due to the induction of metabolic stress in cells. We believe that the use of these compounds should primarily be associated with their ability to induce cellular metabolic stress, with AMPK activation being one consequence of their global actions on cellular bioenergetics. Attributing anti-neoplastic properties of these compounds to AMPK should be done with caution, and results confirmed using cells lacking AMPK activity. Our data indicate that only A-769662 exerts AMPK-specific effects on mTORC1 suppression, proliferation, and mitochondrial function. Thus, we suggest that A-769662 be used over AICAR or biguanides to study AMPK-dependent effects on cellular function. One specific effect of acute A-769662 treatment is an AMPK-specific effect on mitochondrial SRC, suggesting a potential mechanism for AMPK-dependent bioenergetic adaptation to cells growing under metabolic stress. Together our findings suggest that AMPK can exert growth-suppressive or growth-promoting effects on tumor cells depending on context, and that AMPK activation may actually enhance tumor cell growth under certain conditions. Thus, the context of using specific AMPK activators (such as A-769662) will be important when considering its dual effects on the proliferation and metabolism of tumor cells.
Materials and methods
Materials
Metformin (1,1-Dimethylbiguanide hydrochloride), phenformin (phenethylbiguanide hydrochloride), AICAR (5-Aminoimidazole-4-carboxamide 1-β-D-ribofuranoside), sodium salicylate, STO-609 and 2DG (2-deoxyglucose) were obtained from Sigma-Aldrich (St Louis, MO, USA) and reconstituted in water. A-769662 was obtained from Abcam (Cambridge, UK) and reconstituted in DMSO.
Cell culture
AMPKα1/α2+/+ and AMPKα/α2−/− SV40-immortalized MEFs have been previously described.61. HEK293, HCT116 and H1299 cell lines were purchased from ATCC (Manassas, VA, USA). Cells were cultured ‘growth medium’ containing DMEM or RPMI (H1299) and supplemented with 10% fetal bovine serum (FBS), 20000U/ml penicillin, 7mM streptomycin and 200mM glutamine and non-essential amino acids (H1299 cells only). Cells were grown at 37°C in a humidified atmosphere supplemented with 5% (v/v) CO2. Validation of AMPKα knockdown in HCT116 and H1299 cells has previously been described.18
Western blots
Cells were lysed in CHAPS buffer (10mM Tris-HCl, 1mM MgCl2, 1mM EGTA, 0.5mM CHAPS, 10% glycerol, 5mM NaF) supplemented with protease and phosphatase tablets (Roche, Basel, Switzerland), DTT (1μg/ml), and benzamidine (1μg/ml). Cleared lysates were resolved by SDS-PAGE, transferred to nitrocellulose, and incubated with primary antibodies. Primary antibodies to AMPKα (pThr172-specific and total), ACC (Ser79-specific and total) p70 S6-kinase (pThr389-specific and total), raptor (pSer792-specific and total), 4E-BP1 (total) and Actin, as well as an HRP-conjugated anti-rabbit secondary antibody were obtained from Cell Signaling Technology (Danvers, MA, USA).
Adherent growth assays
Cells were either seeded in 96 (8000 cells/well) or 384 well plates (500 cells/well) in growth medium. After 24h medium was replaced with fresh growth medium containing AMPK agonists or vehicle. Cells were fixed with 4% paraformaldehyde at 24, 48 or 72h. Cells were either stained with crystal violet (0.05% (w/v) crystal violet and 20% (v/v) 95% ethanol) and analyzed at 595nm on a Molecular Devices (Sunnyvale, CA, USA) Spectramax plate reader or Hoechst DNA stain to determine cell number by nuclei counting. Images were taken using an Operetta High Content Imaging System and analyzed using Harmony High Content Imaging and Analysis Software, both from Perkin Elmer (Waltham, MA, USA).
Cell cycle analysis
Cell cycle analysis was carried out as previously described.62 Briefly, cells were trypsinized and fixed using 100% ethanol before staining with propidium iodide (40mg/ml). Cells were treated with RNase A (Worthington Biochemicals, Lakewood Township, NJ, USA) (0.5ug/ml) before samples were analyzed by an LSRII flow cytometry system (BD Biosciences, Franklin Lakes, NJ, USA) and FlowJo software (Tree Star, Ashland, OR).
Cleaved caspase-3 assay
Analysis of caspase 3 cleavage was carried out as described.16 Cells were seeded (8000 cells/well) in a 96-well plate. After 24h, medium was replaced with fresh growth medium containing AMPK agonists or vehicle. Cells were fixed after 48h and incubated with cleaved caspase-3 antibody (Cell Signaling Technology, #9661, Whitby, ON, Canada) followed by a secondary HRP-conjugated antibody, chemiluminescent reagent was added and detected using a Synergy HT plate reader (Biotek, Winooski, VT, USA). The resulting chemiluminescent signals were normalized to cell number, determined by crystal violet staining.
Determination of cell viability
Cells were trypsinized, harvested and stained using PI. Cell size of viable cells was measured by flow cytometry and quantified as the mean fluorescence intensity for FSC. All flow cytometry was conducted using Gallios (Beckman Coulter, Fullerton, CA) flow cytometer and analyzed with FlowJo software.
Anchorage-independent growth assay
Soft agar growth assays were carried out as described in.63 Briefly cells were trypsinized and suspended (15000cells/well) in 0.4% (w/v) noble agar in growth medium containing AMPK agonists or vehicle. Suspension cultures were incubated for 5days at which point 10% by volume of Alamar Blue (Serotec, Kidlington, UK) was added to the wells and the cultures incubated for 2–5h. Living cells convert Alamar Blue to a fluorescent indicator and this is proportionate to cell number. Fluorescence was analyzed using a Synergy HT plate reader with 544nm excitation and 590nm emission filters. Images were taken using the Operetta High Content Imaging system.
Metabolic Assays
Glucose production and lactate consumption was measured using a NOVA Bioanalysis flux analyzer or the Eton Bioscience kit (Eton Bioscience, Charlestown, MA, USA) according to the manufacturer’s instructions. OCR and the ECAR of cells were measured using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Boston, MA, USA) as described.64 In brief, cells were plated at 15000cells/well in growth medium for 24h. Cells were incubated for either a further 6h or 24h in medium containing AMPK agonists or vehicle prior to loading into the XF96 apparatus.
Statistical Analysis
Statistics were determined using paired Student’s t test using Prism software (GraphPad, San Diego, CA, USA). Data are calculated as the mean ± SEM. Statistical significance is represented in figures by: *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
Supplementary Material
Acknowledgments
Funding: Grants from McGill Integrated Cancer Research Training Program (MICRTP), CIHR (MOP-93799), Canadian Cancer Society (2010-700586), and Terry Fox Research Foundation (TEF-116128).
We acknowledge Douglas Elder and Nicole Beauchemin, and members of the Jones Lab for critical reading of this manuscript. E.E.V., P.P.C. and T.G. were funded by the McGill Integrated Cancer Research Training Program (MICRTP) and J.B. by Fonds de researche Santé Québec (FRSQ). This work was supported by grants to R.G.J. from the CIHR (MOP-93799), Canadian Cancer Society (2010-700586), and Terry Fox Research Foundation (TEF-116128).
Footnotes
Conflict of interest: The authors declare no conflict of interest
References
- 1.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 2.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 3.Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Hardie DG. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans. 2011;39:1–13. doi: 10.1042/BST0390001. [DOI] [PubMed] [Google Scholar]
- 5.Davies SP, Sim AT, Hardie DG. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur J Biochem. 1990;187:183–190. doi: 10.1111/j.1432-1033.1990.tb15293.x. [DOI] [PubMed] [Google Scholar]
- 6.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 8.Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287:562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- 9.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 10.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, et al. Complexes between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 13.Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM, Booker SV, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987;316:1511–1514. doi: 10.1056/NEJM198706113162404. [DOI] [PubMed] [Google Scholar]
- 14.Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12:3209–3215. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene. 2007;26:7825–7832. doi: 10.1038/sj.onc.1210594. [DOI] [PubMed] [Google Scholar]
- 16.Dupuy F, Griss T, Blagih J, Bridon G, Avizonis D, Ling C, et al. LKB1 is a central regulator of tumor initiation and pro-growth metabolism in ErbB2-mediated breast cancer. Cancer & metabolism. 2013;1:18. doi: 10.1186/2049-3002-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shackelford DB, Vasquez DS, Corbeil J, Wu S, Leblanc M, Wu CL, et al. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci U S A. 2009;106:11137–11142. doi: 10.1073/pnas.0900465106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK Is a Negative Regulator of the Warburg Effect and Suppresses Tumor Growth In Vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim I, He YY. Targeting the AMP-Activated Protein Kinase for Cancer Prevention and Therapy. Front Oncol. 2013;3:175. doi: 10.3389/fonc.2013.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 21.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 22.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 2010;3:1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 24.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 25.Wu N, Gu C, Gu H, Hu H, Han Y, Li Q. Metformin induces apoptosis of lung cancer cells through activating JNK/p38 MAPK pathway and GADD153. Neoplasma. 2011;58:482–490. doi: 10.4149/neo_2011_06_482. [DOI] [PubMed] [Google Scholar]
- 26.Appleyard MV, Murray KE, Coates PJ, Wullschleger S, Bray SE, Kernohan NM, et al. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br J Cancer. 2012;106:1117–1122. doi: 10.1038/bjc.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollak M. Potential applications for biguanides in oncology. J Clin Invest. 2013;123:3693–3700. doi: 10.1172/JCI67232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 29.El-Masry OS, Brown BL, Dobson PR. Effects of activation of AMPK on human breast cancer cell lines with different genetic backgrounds. Oncol Lett. 2012;3:224–228. doi: 10.3892/ol.2011.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosilio C, Lounnas N, Nebout M, Imbert V, Hagenbeek T, Spits H, et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer letters. 2013;336:114–126. doi: 10.1016/j.canlet.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 31.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2013;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elder DJ, Hague A, Hicks DJ, Paraskeva C. Differential growth inhibition by the aspirin metabolite salicylate in human colorectal tumor cell lines: enhanced apoptosis in carcinoma and in vitro-transformed adenoma relative to adenoma relative to adenoma cell lines. Cancer Res. 1996;56:2273–2276. [PubMed] [Google Scholar]
- 34.Hardie DG. AMPK: A Target for Drugs and Natural Products With Effects on Both Diabetes and Cancer. Diabetes. 2013;62:2164–2172. doi: 10.2337/db13-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 36.Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zannella VE, Cojocari D, Hilgendorf S, Vellanki RN, Chung S, Wouters BG, et al. AMPK regulates metabolism and survival in response to ionizing radiation. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2011;99:293–299. doi: 10.1016/j.radonc.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 38.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang J, Mills G. AMPK: a contextual oncogene or tumor suppressor? Cancer Res. 2013;73:2929–2935. doi: 10.1158/0008-5472.CAN-12-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pachman L, Esterly N, Peterson R. The effect of salicylate on the metabolism of normal and stimulated human lymphocytes in vitro. J Clin Invest. 1971;50:226–230. doi: 10.1172/JCI106478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brody T. Action of sodium salicylate and related compounds on tissue metabolism in vitro. The Journal of pharmacology and experimental therapeutics. 1956;117:39–51. [PubMed] [Google Scholar]
- 42.Nicholls DG. Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem Soc Trans. 2009;37:1385–1388. doi: 10.1042/BST0371385. [DOI] [PubMed] [Google Scholar]
- 43.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 44.Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007;282:32549–32560. doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 47.Dykens JA, Jamieson J, Marroquin L, Nadanaciva S, Billis PA, Will Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicology and applied pharmacology. 2008;233:203–210. doi: 10.1016/j.taap.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 48.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117:1422–1431. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO molecular medicine. 2014;6:519–538. doi: 10.1002/emmm.201302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 51.Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, et al. LKB1 Inactivation Dictates Therapeutic Response of Non-Small Cell Lung Cancer to the Metabolism Drug Phenformin. Cancer Cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer letters. 2014 doi: 10.1016/j.canlet.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 53.Garber K. Energy deregulation: licensing tumors to grow. Science. 2006;312:1158–1159. doi: 10.1126/science.312.5777.1158. [DOI] [PubMed] [Google Scholar]
- 54.Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, et al. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes. 2006;55:865–874. doi: 10.2337/diabetes.55.04.06.db05-1178. [DOI] [PubMed] [Google Scholar]
- 55.Vincent MF, Bontemps F, Van den Berghe G. Inhibition of glycolysis by 5-amino-4-imidazolecarboxamide riboside in isolated rat hepatocytes. Biochem J. 1992;281(Pt 1):267–272. doi: 10.1042/bj2810267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, et al. mTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation. Cell Metab. 2013;18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 57.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Godlewski J, Nowicki MO, Bronisz A, Nuovo G, Palatini J, De Lay M, et al. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol Cell. 2010;37:620–632. doi: 10.1016/j.molcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chhipa RR, Wu Y, Mohler JL, Ip C. Survival advantage of AMPK activation to androgen-independent prostate cancer cells during energy stress. Cell Signal. 2010;22:1554–1561. doi: 10.1016/j.cellsig.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, et al. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Crissman HA, Steinkamp JA. Rapid, simultaneous measurement of DNA, protein, and cell volume in single cells from large mammalian cell populations. The Journal of cell biology. 1973;59:766–771. doi: 10.1083/jcb.59.3.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vincent EE, Elder DJ, Curwen J, Kilgour E, Hers I, Tavare JM. Targeting non-small cell lung cancer cells by dual inhibition of the insulin receptor and the insulin-like growth factor-1 receptor. PLoS One. 2013;8:e66963. doi: 10.1371/journal.pone.0066963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci U S A. 2014;111:2554–2559. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.