

Abstract
Bombesin-receptor-subtype-3(BB3 receptor) is a G-protein-coupled-orphan-receptor classified in the mammalian Bombesin-family because of high homology to gastrin-releasing peptide(BB2 receptor)/neuromedin-B receptors(BB1 receptor). There is increased interest in BB3 receptor because studies primarily from knockout-mice suggest it plays roles in energy/glucose metabolism, insulin-secretion, as well as motility and tumor-growth. Investigations into its roles in physiological/pathophysiological processes are limited because of lack of selective ligands. Recently, a selective,peptide-antagonist,Bantag-1, was described. However, because BB3 receptor has low-affinity for all natural, Bn-related peptides, there is little known of the molecular basis of its high-affinity/selectivity. This was systematic investigated in this study for Bantag-1 using a chimeric-approach making both Bantag-1 loss-/gain-of-affinity-chimeras, by exchanging extracellular(EC) domains of BB3 /BB2 receptor, and using site-directed-mutagenesis. Receptors were transiently expressed and affinities determined by binding studies. Bantag-1 had >5000-fold selectivity for BB3 receptor over BB2/BB1 receptors and substitution of the first EC-domain(EC1) in loss-/gain-of affinity- chimeras greatly affected affinity. Mutagenesis of each amino acid difference in EC1 between BB3 receptor/BB2 receptor showed replacement of His107 in BB3 receptor by Lys107(H107K-BB3 receptor -mutant) from BB2 receptor, decreased affinity 60-fold, and three replacements [H107K,E11D,G112R] decreased affinity 500-fold. Mutagenesis in EC1’s surrounding transmembrane-regions(TMs) demonstrated TM2 differences were not important, but R127Q in TM3 alone decreased affinity 400-fold. Additional mutants in EC1/TM3 explored the molecular basis for these changes demonstrated in EC1, particularly important is the presence of aromatic-interactions by His107, rather than hydrogen-bonding or charge-charge interactions, for determining Bantag-1 high affinity/selectivity. In regard to Arg127 in TM3, both hydrogen- bonding and charge-charge interactions contribute to the high-affinity/selectivity for Bantag-1.
Keywords: Bombesin, gastrin-releasing peptide, Neuromedin B, satiety, obesity
Graphical Abstract
1. Introduction
The bombesin receptor subtype 3 (BB3 receptor) is an orphan G-protein-coupled receptor (GPCR) classified as a member of the mammalian bombesin receptor (BnR) family, because of its high homology to the known mammalian BnR members [gastrin-releasing peptide receptor (BB2 receptor) and the neuromedin B receptor (BB1 receptor)] [1–3]. However, there is little known of BB3 receptor’s roles in physiological or pathological processes [2–5]. This has occurred because its native ligand is unknown, it has low affinity for all natural Bn peptides, and unlike BB2 or BB1 receptor, for which numerous selective agonists and antagonists are described [2,6–11], until recently no agonist or antagonist with sufficient selectivity to be useful for in vivo studies, existed for BB3 receptor [2,3,12–17]. A high affinity BB3 receptor agonist has been described, [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6–14) (peptide #1), which allowed studies of BB3 receptor’s signaling cascades, demonstrating it was coupled to phospholipase C, A2 and D activation as well as tyrosine kinase cascades [4,14,18–21]. However, peptide #1 was not useful for pharmacological/pathological studies because it was nonselective, having high affinity for BB2 receptor / BB1 receptor in all species [12,22–24], as well as human BB3 receptor [25–27], but not rat/mouse BB3 receptor [27,28].
At present, some insights into the possible importance of BB3 receptor either physiologically or in pathological conditions have come from studies of mice in which BB3 receptor has been removed by targeted deletion (BB3 receptor -KO mice) [3,13,29–33]. These studies and others provide evidence that, similar to the other BnRs (i.e. BB2 receptor /BB1 receptor), BB3 receptor is important in regulation of feeding/satiety[34] in addition to regulation of various behaviors, glucose and insulin homeostasis, as well as metabolic homeostasis, and may play an important role in diabetes and obesity [3,13,15,29,30,32,33]. However, BB3 receptor selective antagonists/agonists would be invaluable to further investigate BB3 receptor role in these and other areas.
Recently, the BB3 receptor selective peptide antagonist Bantag-1 was described [14,15,35], however nothing is known of the molecular basis for its high affinity/selectivity for BB3 receptor. With other Bn receptors [36], as with other GI hormone/neurotransmitter GPCR’s [36,37], there are only limited studies of the molecular basis of high affinity, selectivity of peptide antagonists [36–39]. This has occurred principally because potent peptide antagonists have been described for only a few GI hormone/neurotransmitter GPCRs. Therefore in this study, we examined in detail the molecular basis selectivity/high affinity of the peptide antagonist Bantag-1 for the BB3 receptor.
2. Materials and methods
2.1. Materials
Polyoma large T antigen- expressing Chinese hamster ovary (CHOP) cells were a gift from James W. Dennis (Samuel Lunenfeld Research Institute, Toronto, Canada); Bombesin receptor subtype-3 antagonist (Bantag-1) was gifts from Merck, Sharp and Dohme (West Point, PA); the mammalian expression vectors, pcDNA3, custom primers were from Invitrogen (Carlsbad, CA); QuikChange Site-Directed Mutagenesis Kit was from Agilent Technologies (Santa Clara, CA); cDNA of hBB3 receptor, mBB2 receptor and mBB1 receptor were obtained as described previously[40–42]; Dulbecco’s minimum essential medium (DMEM), phosphate-buffered saline (PBS), G418 sulfate, fetal bovine serum (FBS), penicillin, streptomycin and sodium pyruvate from Gibco Life Technology (Grand Island, NY); DpnI, Phusion® HF DNA Polymerase, dNTP, 100 % DMSO and 5X Phusion HF (GC) Buffer were from New England Biolabs (Ipswich, MA); formic acid, ammonium formate, disodium tetraborate, and alumina were obtained from Sigma-Aldrich (St. Louis, MO); iodine- 125 (100 mCi/ml) was from Perkin Elmer Life Sciences (Boston, MA); Polyethylenimine lipofectamine (P.E.I) (lipofectamine) was from Polysciences, Inc. (Warrington, PA); Standard protected amino acids and other synthetic reagents were obtained from Bachem Bioscience Inc. (King of Prussia, PA); XL1-Blue Supercompetent Cells from Agilent Technologies (Santa Clara, CA).
2.2. Preparation of 125I-Labeled Peptides
125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn-(6–14), with specific activity of 2,200 Ci/mmol, was prepared by a modification of methods described elsewhere [12,14]. In brief, 0.8 μg of IODO-GEN (in 0.02 mg/ml chloroform) was transferred to a vial, dried under a stream of nitrogen, and washed with 100 μl of 0.5 M KH2PO4, pH 7.4. To the reaction vial 20 μl of 0.5 M KH2PO4, pH 7.4, 8 μg of peptide in 4 μl of water, and 2 mCi (20 μl) Na125I were added, mixed gently, and incubated at room temperature for 6 minutes. The incubation was stopped by the addition of 100 μl of distilled water. Radiolabeled peptide was separated using a Sep-Pak (Waters Associates, Milford, MA) and high-performance liquid chromatography as described previously elsewhere [12,14]. The radioligand was stored with 0.5% BSA at −20°C.
2.3. Strategy for Construction of Mutant BB3 Receptors and BB2 receptors
Human BB3 and mouse BB2 receptors were used because the two species have similar pharmacology for Bantag-1, high homology and allow comparison to results in numerous previous studies in which these two receptors have been used. cDNA from human BB3 and mouse BB2 receptors was used for all chimeras, as well as single or combination points mutations. The cDNA exon coding regions only were inserted in pCMV6-Entry (OriGene, Rockville, MD) and then subcloned into pcDNA3 at the EcoRI site. All BB3 mutant receptors were constructed by using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA), following the manufacturer’s instructions, with minor modifications as described previously [40–42]. Nucleotide sequence analysis of the entire coding region was performed using an automated DNA sequencer on the wild type and ΔH294R receptor and all mutant receptors (Applied Biosystems Inc., Foster City, CA). Three BB3 receptors loss-of-affinity and three BB2 receptor gain-of-affinity mutants for Bantag-1 were constructed as described previously [36,42,43]. The chimeric receptors were made from EC1, EC2 and EC3 regions because previous studies demonstrated the NH2 terminus was not important for high affinity peptide binding [36,44]. Three loss-of-affinity BB3 receptors chimeric receptors were made by substituting the extracellular domains of BB2 receptor for the comparable domains in BB3 receptor. [(EC1-BB2)-BB3] loss-of-affinity chimera was made substituting the first extracellular domain of BB2 receptor –from D98 to K115– for the comparable domain of BB3 receptor –from D104 to K121; [(EC2-BB2)-BB3] loss-of-affinity chimera was made substituting the second extracellular domain of mouse BB2 receptor –from F179 to A214– for the comparable domain of BB3 receptor –from F185 to L220– and [(EC3-BB2)-BB3] loss-of-affinity chimera was made substituting the third extracellular domain of BB2 receptor –from R288 to S305– for the comparable domain of BB3 receptor –from H294 to T312. Three gain-of-affinity BB3 chimeric receptors were made by substituting the extracellular domains of BB3 receptor for the comparable domains in BB2 receptor: [(EC1-BB3)-BB2] gain-of-affinity chimera was made substituting the first extracellular domain of BB3 receptor –from D104 to K121– for the comparable domain of BB2 receptor –from D98 to K115; [(EC2-BB3)-BB2] gain-of-affinity chimera was made substituting the second extracellular domain of human BB3 receptor –from F185 to L220– for the comparable domain of BB2 receptor –from F179 to A214– and [(EC3-BB3)-BB2] gain-of-affinity chimera was made substituting the third extracellular domain of BB3 receptor –from H294 to T312– for the comparable domain of BB2 receptor –From R288 to S305.
2.4. Cell culture
CHOP cells were grown in DMEM containing 10% (v/v) FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 200 μg/ml G418. Cells were maintained at 37°C in a 5% CO2 atmosphere. Cells were split every 3 to 4 days at confluence after detaching the cells with trypsin/versene solution.
2.5. Cell Transfections and Isolation of Stable Cell Lines
CHOP cells, which contain no native Bn receptors, were used for transient transfection studies performed as described previously [41,42]. Briefly, CHOP cells were seeded in a 10-cm tissue culture dish at a density of 0.2 x 106 cells/dish and grown overnight at 37°C in growth medium. On the following morning, 12 μg of plasmid DNA was transfected using the cationic lipid-mediated method with Lipofectamine for 2 h at 37°C. At the end of the incubation period, the medium was replaced with growth medium. CHOP cells were maintained at 37°C in a 5% CO2 atmosphere and were used 48 h later for binding assays.
2.6. Binding studies
Binding studies were performed using transfected CHOP cells and 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6–14) as described previously [14,41,42]. Briefly, CHOP cells (0.2 – 4.0 x 106 cells/ml) were incubated for 60 minutes at 21°C with 50 pM 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6–14) in 300 μl of binding buffer. The standard binding buffer contained 24.5 mM HEPES, pH 7.4, 98 mM NaCl, 6 mM KCl, 2.5 mM KH2PO4, 5 mM sodium pyruvate, 5 mM sodium fumarate, 5 mM sodium glutamate, 2 mM glutamine, 11.5 mM glucose, 0.5 mM CaCl2, 1.0 mM MgCl2, 0.01% (w/v) soybean trypsin inhibitor, 0.2% (v/v) amino acid mixture, 0.2% (w/v) BSA, and 0.05% (w/v) bacitracin. Although BnR expression levels varying by much as 160-fold [45] have been shown to have no effect on receptor affinity with the binding conditions used in this studies, as an added precaution to correct for any differences in ligand bound by different mutant BnR receptors, binding results with each mutant BnR receptor was compared only to results with wild type receptor-containing cells binding similar amounts of ligand. The cell concentration was adjusted between 0.2 and 4.0 x106 cells/ml for each mutant receptor so that <15% of the total added radioactive ligand was bound during the incubation and the results compared to cells transfected with wild type BB3 receptor adjusted in concentration to bind a similar amount of ligand. After the incubation, 100 μl of each sample were removed and added to 300 μl of incubation buffer in 400-μl Microfuge tubes and centrifuged for 1 minute at 10,000g (Microfuge B; Beckman Coulter, Fullerton, CA) to separate the bound radioligand from unbound radioligand. The supernatant was aspirated, and the pelleted cells were rinsed twice with a washing buffer that contained 1% (w/v) BSA in PBS. The amount of radioactivity bound to the cells was measured using a Cobra II Gamma Counter (Packard Instruments, Meriden, CT). Binding was expressed as the percentage of the total radioactivity that was associated with the cell pellet and in all cases calculations were based on the saturable binding. Nonsaturable binding was <10% of the total binding in all experiments. Each point was measured in duplicate, and each experiment was replicated at least 3 times.
To assist in interpreting the binding data, in all binding assays, in addition to determining the affinity for Bantag-1, the affinity for the agonist, peptide #1 was also determined. Previous studies [41,42] have examined the basis of high affinity interaction of peptide#1 and related peptides for the BB3 receptor and shown the molecular basis for this is due to a distinct receptor regions not demonstrated to be important for Bantag-1 selectivity in this study. Therefore, an alteration in affinity for peptide #1 with a given mutation, in the present study, was interpreted as likely a global effect on the receptor conformation and could not be interpreted as a specific effect on Bantag-1 affinity.
2.7. Statistical Analysis
The results are the mean and S.E.M. from at least three separate experiments. The Ki values were calculated from the IC50 obtained from competitive inhibition curves using peptide #1 or Bantag-1 and 50 pM 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6–14) using the Cheng-Prusoff equation [46]. IC50’s from the binding data were curve-fitted using Prism GraphPad 4.0 (nonlinear curve-fitting). An analysis of variance was used to determine the statistical significance of differences in affinity of each BB3 receptor mutant compared with its own wild type BB3 receptor control in receptors with changes ≥2-fold difference. In all experiments cell concentrations were set such that <15% of total radioactivity was bound and the amount of saturable ligand bound was similar for the mutant and wild type receptors and therefore, in the statistical analysis, only two variables (i.e. the BB3 receptor mutant and its wild type control, binding similar amount of ligand) were analyzed.
3. Results
3.1 Wild type BB3, BB3*, wild type BB2 and wild type BB1 receptors
The peptide agonist, peptide #1 had a high affinity each of the three wild type Bn receptors (Ki: 1.7- 8.1 nM, Fig. 1, Table 1), similar to results in other studies [12,23,25,47]. In contrast, the peptide antagonist, Bantag-1 had high affinity only for BB3 receptor (Ki: 5.2–5.6 nM, Fig. 1, Table 1) and did not interact with the other two Bn receptor subtypes – BB2 and BB1 receptor– even at concentrations up to 30,000 nM (Fig. 1, Table 1).
Figure 1.

Comparison of the ability of the antagonist Bantag-1 and the nonselective Bn analog agonist (peptide #1) to inhibit binding to cells containing wild type BB3, BB3*, wild type BB2 or wild type BB1 receptors. The peptides were incubated with 50 pM 125I- [D-Tyr6, β-Ala11, Phe13, Nle14Bn-(6–14) for 60 minutes at 21°C in 300 μl of binding buffer with BB3 receptor cells (3 x 106 cells/ml), BB3* receptor cells (1.1 x 106 cells/ml), BB2 receptor cells (7 x 106 cells/ml) or BB1 receptor cells (0.1 x 106 cells/ml) and the saturable binding was determined as described in Materials and Methods. The results are expressed as the percentage of saturable binding without unlabeled peptide added (percentage control). The results are the mean and S.E.M. from at least three separate experiments and in each experiment the data points were determined in duplicated. Abbreviations: Bantag-1, Boc-Phe-His-4-amino-5-cyclohexyl-2,4,5-trideoxypentonyl-Leu- (3-dimethylamino) benzylamide N-methylammonium trifluoroacetate; BB3 or BB3 receptor, Bombesin receptor subtype 3; BB3*or BB3* receptor, His294 in BB3 receptor substituted for by Arg288 in comparable position of BB2 receptor which increases expression of the receptor but does not change affinity for Bantag-1 alone; CHOP, polyoma large T antigen-expressing Chinese hamster ovary; BB2 or BB2 receptor, gastrin-related peptide receptor; BB3, BB3*, BB2 or BB1 receptors stably transfected into CHOP cells; BB1 receptor, neuromedin B receptor; peptide #1, [D-Tyr6, β-Ala11, Phe13, Nle14]Bn-(6–14).
Table 1A.
Affinities of Bantag-1 and peptide #1 for the members of the Bn receptor family.
|
Ki (nM)
|
||
|---|---|---|
| Receptor | Bantag-1 | Peptide #1 |
| BB2 | >30,000 | 1.7 ± 0.1 |
| BB1 | >30,000 | 5.1 ± 0.3 |
| BB3 | 5.6 ± 0.4 | 8.1 ± 1.0 |
| BB3* | 5.2 ± 0.5 | 4.9 ± 0.8 |
Table 1B.
Comparison of saturable binding of BB3 and BB3* receptor.
| Saturable binding (per 1 x 106 cells/ml)
|
||
|---|---|---|
| Receptor | % added ligand | Receptors/cell |
| BB3 | 1.9 ± 0.2 | 2,000 ± 211 |
| BB3* | 7.4 ± 0.1a | 8,000 ± 108a |
P <0.0001 compared to BB3 receptor.
CHOP cells type were incubated with 50 pM 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn-(6–14) for 60 minutes at 21°C and binding was determined as described in Materials and Methods. In each experiment each value was determined in duplicate, and values given are means and S.E.M from at least three separate experiments. Data are calculated from dose-inhibition curves shown in Fig. 1. Abbreviations: BB3*, His 294 in BB3 substituted for by Arg288 in comparable position of BB2 to increase expression level; Bn, bombesin; for other, see in Fig. 1 legend.
With wild type BB3 receptor, the expression level after transient transfection was low averaging only 1.9 ± 0.1% of the total radioactivity saturably bound with a cell concentration of 3.0 x106 cells/ml in the incubation. A previous study [48], reported that substitution of histidine 294 in BB3 receptor by arginine in a comparable position in BB2 receptor, increased affinity for GRP. Because of that, we wanted to know if we could include that substitution to improve the results in our study. First, we found it increased expression levels with transient transfection of BB3 receptor constructs to the extent that with a cell concentration of 1.0 x106 cells/ml in the incubation the total amount of saturable binding was increased >3-fold to 7.4 ± 0.1% of the total radioactivity saturably bound. Second, we determined the effect of its presence on affinity of peptide #1 and Bantag-1 (Fig. 1, Table 1). We found it had no effect on affinity of Bantag-1 for BB3 or BB3* receptors (Ki: 5.6 ± 0.4 and 5.2 ± 0.5 nM, respectively. Fig. 1, Table 1). However, the affinity of peptide #1 for BB3* receptor was increased 1.7-fold compared to BB3 receptor (Fig. 1, Table 1) and therefore could be used to increase the amount of binding to the BB3 receptor mutants (i.e. BB3* receptor mutants) and allow greater accuracy in determining the affinity constants.
3.2 Extracellular chimeric receptors
To explore the molecular basis for the selectivity of the BB3 receptor selective antagonist Bantag-1 both BB3 loss-of-affinity and BB2 gain-of-affinity chimeric receptors were made (Fig. 2 and 3, Table 2). Chimeric receptors were made using BB3 receptor for which Bantag-1 has a high affinity (Fig. 2 and 3, Table 2) and BB2 receptor, which has a low of affinity for Bantag-1 (Fig. 2 and 3, Table 2).
Figure 2.
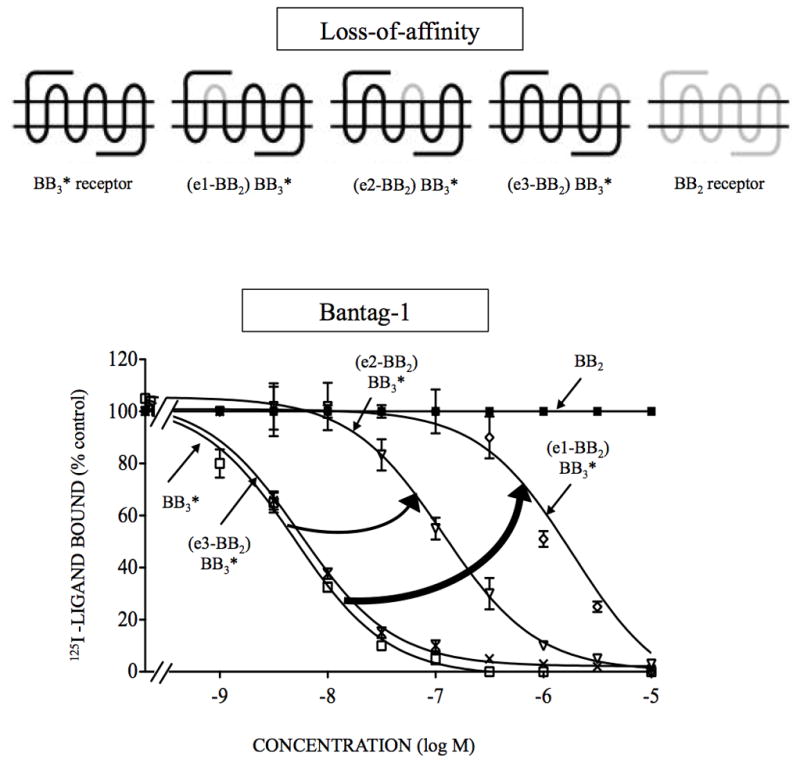
Affinities of the antagonist, Bantag-1 for loss-of-affinity BB3 chimeric receptors and BB2 expressed in CHOP cells. The diagrams of the chimeric receptors formed are shown at the top. The chimeric BB3 receptors were formed by replacing each of the extracellular domains of BB3* receptor one at a time by the comparable BB2 receptor extracellular domain as described in Material and Methods. The peptides were incubated with 50 pM 125I- [D-Tyr6, β-Ala11, Phe13, Nle14]Bn-(6–14) for 60 minutes at 21°C in 300 μl of binding buffer with BB3* receptor cells (1.1 x 106 cells/ml), (e1-BB2) BB3* cells (4.2 x 106 cells/ml), (e2-BB2) BB3* cells (4.8 x 106 cells/ml), (e3-BB2) BB3* cells (2.1 x 106 cells/ml) or BB2 receptor cells (7 x 106 cells/ml), and the saturable binding was determined as described under Materials and Methods. The results are expressed as the percentage of saturable binding without unlabeled peptide added (percentage control). The results are the mean and S.E.M. from at least three separate experiments and in each experiment the data points were determined in duplicated. The arrows indicate large changes in affinity from the BB3 receptor. Abbreviations: e or EC, extracellular; for other, Fig. 1 legend.
Figure 3.
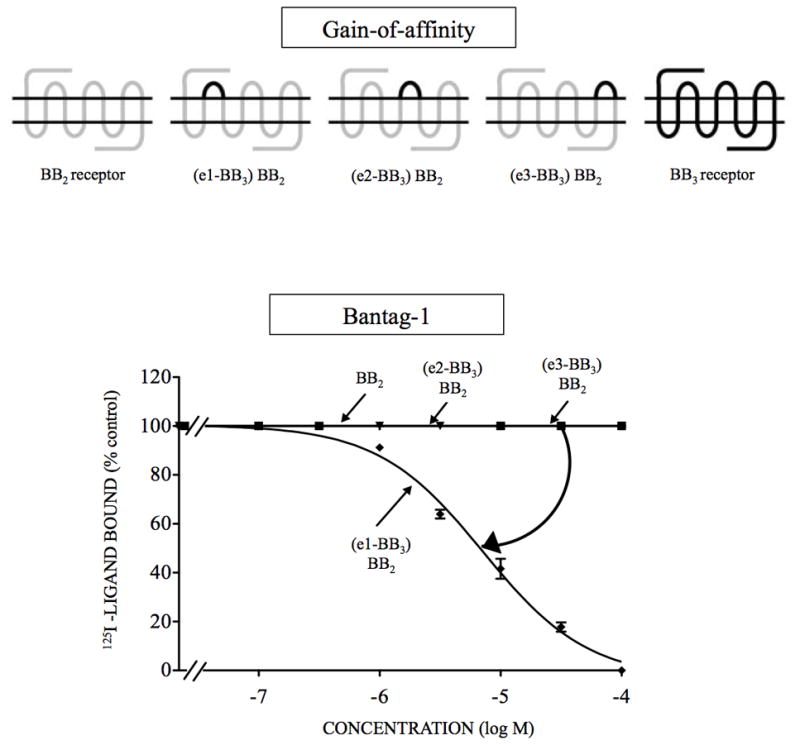
Affinities of the antagonist Bantag-1 for BB2 receptor gain-of-affinity BB2 chimeric receptors and BB3 receptor expressed in CHOP cells. The diagrams of the chimeric receptors formed are shown at the top. The chimeras BB2 receptors were formed by replacing each of the extracellular domains of BB2 receptor one at a time by the comparable BB3* receptor extracellular domain as described in Material and Methods. The different concentrations of Bantag-1 were incubated with 50 pM 125I- [D-Tyr6, β-Ala11, Phe13, Nle14]Bn- (6–14) for 60 minutes at 21°C in 300 μl of binding buffer with (e1-BB3) BB2 cells (0.6 x 106 cells/ml), (e2-BB3) BB2 cells (2 x 106 cells/ml), (eC3-BB3) BB2 cells (1.4 x 106 cells/ml) or BB2 receptor cells (7.0 x 106 cells/ml), and the saturable binding was determined as described under Materials and Methods. The results are expressed as the percentage of saturable binding without unlabeled peptide added (percentage control). The results are the mean and S.E.M. from at least three separate experiments and in each experiment the data points were determined in duplicated. The arrow indicates large change in affinity from the wild type BB2 receptor. Abbreviations: see Fig. 1 legend.
Table 2.
Affinities of Bantag-1 and peptide #1 for wild type BB3, BB3*, wild type BB2, and extracellular chimeric BB3 and BB2 receptors (loss- and gain-of-affinity).
|
Ki (nM)
|
||||
|---|---|---|---|---|
| BB3* | Wild type | |||
|
|
||||
| Receptor | Bantag-1 | Peptide #1 | Bantag-1 | Peptide #1 |
| BB3 | 5.2 ± 0.5 | 4.9 ± 0.8 | 5.6 ± 0.4 | 8.1 ± 1.0 |
| BB2 | ND | ND | >30,000 a | 1.7 ± 0.1a |
| Extracellular chimeras (loss-of-affinity) | ||||
| (e1-BB2) BB3 | 1,962 ± 432a | 9.7 ± 1.6 | N.B. | N.B. |
| (e2-BB2) BB3 | 120 ± 26a | 8.1 ± 2.9 | N.B. | N.B. |
| (e3-BB2) BB3 | 5.9 ± 0.7 b | 6.3 ± 1.2 b | 5.9 ± 0.7 | 6.3 ± 1.2 |
| Extracellular chimeras (gain-of-affinity) | ||||
| (e1-BB3) BB2 | ND | ND | >6,000 | 1.8 ± 0.2 |
| (e2-BB3) BB2 | ND | ND | >30,000 | 3.8 ± 1.0 |
| (e3-BB3) BB2 | ND | ND | >30,000 | 3.6 ± 0.2 |
P <0.0001 compared to BB3 receptor.
ND, Not done because Arg is already present in position 288 in BB2 receptor.
N.B, no detectable binding above background.
CHOP cells type were incubated with 50 pM 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn-(6–14) for 60 minutes at 21°C and binding was determined as described in Materials and Methods. In each experiment each value was determined in duplicate, and values given are means and S.E.M. from at least three separate experiments. Data are calculated from dose-inhibition curves shown in Fig. 1–3. Abbreviations: See in Fig. 1 and 2 legends.
The loss-of-affinity BB3 chimeric receptors were made by substituting the extracellular domains of BB2 receptor for the comparable domains in BB3 receptor and the gain-of-affinity chimeras constructed using the reverse strategy, substituting in BB2 receptor, the extracellular domains of BB3 receptor, to attempt to restore affinity for BB3 receptor. Chimeric BB3 receptors with (BB3* receptor) or without H294R (BB3 receptor) mutations to increase expression were made (Table 2).
The substitution of EC1 or EC2 of BB2 receptor into the comparable position in BB3 receptor resulted in chimeras with such low saturable binding that affinities could not be assessed (Table 2). In contrast, substitution of EC3 into BB3 receptor resulted in no effect on Bantag-1 affinity (Table 2). To improve expression, the same studies were performed on similar chimeras in a mutant BB3* receptor [(H294R) BB3 receptor], which alone had no effect on Bantag-1 or peptide #1 affinity (Fig. 1–3, Tables 1–2). The substitution of EC1 or EC2 in the loss-of-affinity BB3* receptor chimeras, by the comparable domain of BB2 receptor, decreased the affinity for Bantag-1 by 377- and 23-fold, respectively (Ki: 1,962 and 120 nM, Fig. 2, Table 2). As Arg288 is located in EC3 of BB2 receptor, the EC3 of BB3 is the same structure as EC3 BB2 in BB3*. Peptide #1 had similar affinities for the different chimeras (Table 2). These results suggest differences in EC1 were the most important for determining Bantag-1 selectivity for BB3 receptor.
To provide additional support for this conclusion, three gain-of-affinity BB3 chimeric receptors were made by substituting the extracellular domains of BB3 receptor for the comparable domains in BB2 receptor, to attempt to regain affinity for Bantag-1 with the gain- of-affinity wild type BB2 receptor chimeras. Only the substitution of EC1 of BB2 receptor by the comparable domain of BB3 receptor increased the affinity for Bantag-1 with an increase of 5-fold (Ki: >6,000 nM, Fig. 3, Table 2). Each of these gain-of-affinity chimeras had good expression and thus, BB3 receptor mutants were not needed to assess affinity. These results support the importance of the EC1 domain of the BB3 receptor for the high binding and selectivity of Bantag-1.
3.3 EC1 of BB3 receptor Mutants (Loss-of-affinity point mutations)
To investigate further the molecular basis for the selectivity of Bantag-1, we investigated which specific amino acids are responsible for the high affinity for BB3 receptor in EC1 by studying each of the amino acid differences in the EC1 domain of these two receptors – BB3 and BB2 receptors (Fig. 4). The two receptors in the EC1 domain differed in 4 amino acids (Fig. 4), occurring at positions 106, 107, 111 and 112 of BB3 receptor, which are comparable with positions 100, 101, 105 and 106 of BB2 receptor. To study the 4 amino acid differences, we first made 4 BB3 receptor losses-of-affinity point mutants by substituting in BB3 or BB3* receptors the comparable different amino acid from BB2 receptor [i.e. T106S, H107K, E111D, G112R-BB3*] (Fig. 4). For Bantag-1, the substitution of histidine for lysine at position 107 produced the greatest effect, decreasing the affinity by 35-fold for wild type BB3 receptor (Ki: 195 nM, Fig. 4A, Table 3) and 54-fold for BB3* receptor (Ki: 283 nM, Fig. 4A, Table 3). The point mutation [G112R] caused only a loss of 2-fold in the affinity of Bantag-1 for wild type and BB3* receptor (Ki: 11.6–16 nM, Fig. 4A, Table 3). In contrast, point substitutions [T106S] and [E111D] had no effect on the affinity of Bantag-1 for wild type (Ki: 6.2 and 6.6 nM, Fig. 4A, Table 3) or BB3* receptor (Ki: 5.5 and 7.4 nM, Fig. 4A, Table 3). For peptide #1, none of these 4 point mutations had an effect on its affinity for wild type (Ki: 5.6–10.1 nM, Table 3) or BB3* receptor (Ki: 5.3–6.7 nM, Table 3). These results support the importance of the presence of histidine at position 107 in the BB3 receptor instead of lysine in a similar position of BB2 receptor for determining affinity/selectivity of Bantag-1.
Figure 4.

Effect of single point mutations in the first extracellular domain of BB3 receptor on affinity for Bantag-1 (loss-of-affinity BB3 receptor point mutants). Top, alignment of amino acid sequences in the first extracellular domain of BB3 and BB2 receptor. The boxes indicate divergent amino acids between these two receptors in these regions. Arrows indicate the position of the point mutations made in BB3 receptor by substituting into BB3* receptor the divergent amino acid from the comparable position in BB2 receptor. (A) Results with the four BB3 receptor mutants made to explore the importance each of the four amino acid differences in EC1 of BB2 and BB3 receptor for determining the selectivity of Bantag-1. (B) Importance of the presence of a charged amino acid or with an aromatic group in position 107 of BB3 receptor for determining selectivity of Bantag-1. The experimental conditions were the same as described in Fig. 1 legend. The results are expressed as the percentage of saturable binding without unlabeled peptide added (percentage control). The results are the mean and S.E.M. from at least three separate experiments and in each experiment the data points were determined in duplicated. Abbreviations: E111D refers to the replacement of glutamic acid in EC1 position 111 in BB3 receptor by aspartic acid; TM, transmembrane; for other, see in Fig. 1 legend.
Table 3.
Affinities of Bantag-1 and peptide #1 for mutants in the first extracellular domain in wild type BB3, BB3* and wild type BB2 receptors (loss-of-affinity EC1 point mutations).
|
Ki (nM)
|
||||
|---|---|---|---|---|
| BB3* | Wild type | |||
|
|
||||
| Receptor | Bantag-1 | Peptide #1 | Bantag-1 | Peptide #1 |
| BB3 | 5.2 ± 0.5 | 4.9 ± 0.8 | 5.6 ± 0.4 | 8.1 ± 1.0 |
| BB2 | ND | ND | >30,000 a | 1.7 ± 0.1a |
| EC1 differences | ||||
| [T106S] | 7.4 ± 0.8 | 5.7 ± 0.3 | 6.6 ± 0.2 | 10.1 ± 1.0 |
| [H107K] | 283 ± 21a | 6.7 ± 0.3 | 195 ± 51a | 8.6 ± 1.9 |
| [E111D] | 5.5 ± 1.1 | 6.4 ± 0.9 | 6.2 ± 0.3 | 9.5 ± 1.0 |
| [G112R] | 11.6 ± 2.1b | 5.3 ± 0.4 | 16 ± 3b | 5.6 ± 1.5 |
| Importance of H107 | ||||
| [H107R] | 261 ± 8a | 8.7 ± 2.9 | >1,000 | <1 |
| [H107D] | 1,000± 23 a | 3.9 ± 0.9 | 11 ± 0.7b | 2.3 ± 0.3 |
| [H107F] | 83 ± 10a | 1.5 ± 0.2 | 24 ± 6b | 4.2 ± 1.0 |
| [H107N] | 425 ± 59a | 4.4 ± 0.2 | 318 ± 31a | 4.3 ± 0.5 |
| [H107Y] | 37 ± 1a | 1.9± 0.6 | 70 ± 3a | 3.1 ± 0.4 |
| [H107W] | >1,000 a | 4.9 ± 1.9 | >1,000 a | 27 ± 6a |
P <0.0001 compared with BB3 receptor;
P <0.05 compared with BB3 receptor.
ND, Not done because Arg is already present in position 288 in BB2 receptor.
CHOP cells type were incubated with 50 pM 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn-(6–14) for 60 minutes at 21°C and binding was determined as described in Materials and Methods. In each experiment each value was determined in duplicate, and values given are means and S.E.M. from at least three separate experiments. Data are calculated from dose-inhibition curves shown in Fig. 1 and 4. Abbreviations: See in Fig. 1 and 2 legends.
To investigate the molecular basis for the large effect on affinity of substituting histidine by lysine in position 107, we explored the importance of charge on the substituted amino acid, as well as the presence of an aromatic ring, or the size of the backbone substitution, for determining affinity for Bantag-1. To do this, we made 7 BB3 or BB3* receptors H107 loss-of- affinity point mutants with amino acids with different charges, backbone size and presence or absence of different aromatic rings by substituting for the positively charge histidine (Fig. 4B, Table 3). Substitution of phenylalanine or tyrosine at position 107 decreased the affinity of Bantag-1, 4- and 13-fold for wild type BB3 receptor, respectively (Ki: 24 and 70 nM, Fig. 4B, Table 3), but did not decrease the affinity for peptide #1 (Table 3). Similarly, point mutations, [H107F] and [H107Y] in BB3* receptor decreased the affinity of Bantag-1 7–16-fold (Ki: 83 and 37 nM, Fig. 4B, Table 3) with no decreased in the affinity of peptide #1 (Table 3). Replacement of histidine in either BB3 or BB3* receptor by the larger uncharged group asparagine [H107N], caused a marked decrease in Bantag-1 affinity of >58-fold (Fig. 4B, Table 3). Substitution of different amino acids with different aromatic groups demonstrated [H107Y] and [H107F] had the least effect causing a 4–16-fold decrease in affinity in either BB3 or BB3* receptor, whereas substitution of the large aromatic group in tryptophan [H107W] marked decreased affinity for Bantag-1 by >190 times (Fig. 4B, Table 3). Replacement of positively charged histidine by a larger positive group in [H107R] decreased affinity for Bantag-1 with both BB3 or BB3* receptors by 50–178-fold demonstrating the greater importance of steric factors in this position than charge, per se. The substitution of a negative charge in this position, [H107D], caused >192-fold decrease in the BB3* receptor, whereas it had only a minimal effect in BB3 receptor, demonstrating that charge of the substitution in position 107 was playing a minimal role in the wild type receptor (Fig. 4B, Table 3). With all EC1 single amino acid mutants, no decrease in affinity was seen for peptide #1, except for H107W in the wild type BB3 receptor, which demonstrate a 3-fold decrease, suggesting this substitution was having a global effect on receptor affinity, which was not seen in the [H107W] BB3* receptor mutant (Fig. 4B, Table 3).
3.4 EC1 of BB3 receptor combination mutants (EC1 Loss-of-affinity combination mutations)
None of the single EC1 amino acid substitutions alone (i.e. 1.4–54-fold decrease) that were investigated above caused a decreased in affinity equal to the >377-fold decreased in affinity seen for Bantag-1 when the entire first extracellular BB3 receptor domain was replaced by that from BB2 receptor (Table 2). Therefore, combination mutations of the single amino acid changes which decreased Bantag-1 affinity were made to identify which amino acids together were important for determining the antagonist’s selectivity (Fig. 5, Table 4).
Figure 5.

Effect of various point mutations in combination in the first extracellular domain and the third transmembrane domain of BB3 receptor on affinity for Bantag-1 (loss-of-affinity combination mutants). (A) Effect of multiple mutations in BB3 receptor in the EC1 on the affinity of Bantag-1. (B) Effect of combination mutations in TM3 of BB3 receptor on determining selectivity of Bantag-1. The experimental conditions were the same as described in Fig. 1 legend. In each case either one or multiple mutations were made in the wild type BB3 or BB3* receptor. The results are expressed as the percentage of saturable binding without unlabeled peptide added (percentage control). The results are the mean and S.E.M. from at least three separate experiments and in each experiment the data points were determined in duplicated. Abbreviations: See in Fig. 1, 2 and 4 legends.
Table 4.
Affinities of Bantag-1 and peptide #1 for mutants in first extracellular domain and third transmembrane in wild type BB3, BB3* and wild type BB2 receptors (loss-of-affinity EC1 & TM3 combination point mutants).
|
Ki (nM)
|
||||
|---|---|---|---|---|
| BB3* | Wild type | |||
|
|
||||
| Receptor | Bantag-1 | Peptide #1 | Bantag-1 | Peptide #1 |
| BB3 | 5.2 ± 0.5 | 4.9 ± 0.8 | 5.6 ± 0.4 | 8.1 ± 1.0 |
| BB2 | ND | ND | >30,000a | 1.7 ± 0.1a |
| Combination point mutants in EC1 | ||||
| [H107K] | 282 ± 21a | 6.7 ± 0.3 | 195 ± 51a | 8.6 ± 1.9 |
| [E111D] | 5.5 ± 1.1 | 6.4 ± 0.9 | 6.1 ± 0.3 | 9.5 ± 1.0 |
| [G112R] | 11.6 ± 2.1b | 5.3 ± 0.4 | 16 ± 3b | 5.6 ± 1.5 |
| [H107K, G112R] | 1,303 ± 113a | 7.1 ± 0.6 | 378 ± 43a | 9.8 ± 1.1 |
| [H107K, E111D, G112R] | 2,883 ± 98a | 16 ± 2b | 2,882 ± 98a | 7.6 ± 1.3 |
| Combination point mutants in TM3 | ||||
| [L123I] | 20 ± 1a | 10.1 ± 1.5b | >3,000 | 9.2 ± 3.6 |
| [R127Q] | 2,068 ± 258a | 7.0 ± 0.7 | 1,331 ± 182a | 5.9 ± 0.3 |
| [L123I, R127Q] | >11,000a | 7.0 ± 0.3 | >15,000a | 6.0 ± 1.4 |
P <0.0001 compared with BB3 receptor;
P <0.05 compared with BB3 receptor.
ND, Not done because Arg is already present in position 288 in BB2 receptor.
CHOP cells type were incubated with 50 pM 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn-(6–14) for 60 minutes at 21°C and binding was determined as described in Materials and Methods. In each experiment each value was determined in duplicate, and values given are means and S.E.M. from at least three separate experiments. Data are calculated from dose-inhibition curves shown in Fig. 1, 4, 5 and 6. Abbreviations: See in Fig. 1 and 2 legends.
In EC1, the combination mutant [H107K, G112R] showed a 68- and 250-fold decrease in affinity for wild type and BB3* receptor (Ki: 378 and 1,303 nM, Fig. 5A, Table 4), which was greater than the 35–54-and 2–3-fold decrease caused for each alone. In contrast, the double mutation had no effect on the affinity of peptide #1 for wild type or BB3* receptor (Ki: 9.8 and 7.1 nM, Table 4). The triple mutation [H107K, E111D, G112R] had even a greater effect on the affinity of Bantag-1, decreasing >500-fold the affinity of Bantag-1 for wild type and BB3* receptor (Ki: 2,882 and 2,883 nM, Fig. 5A, Table 4). However, this triple mutation was causing a global alteration in BB3* receptor because it caused a 3-fold decrease the affinity of the in control peptide, peptide #1 (Table 4). However, this triple mutation did not affect the affinity of peptide #1 for wild type BB3 receptor (Ki: 7.6 nM,Table 4).
3.5 TM2 and TM3 of BB3 receptor Mutants (Loss-of-affinity point mutations)
Previous studies with peptide antagonists for other GPCRs [39,49] report amino acids in upper TM regions can also be important for determining their affinity/selectivity. Even with the triple combination [H107K, E111D, G112R], the loss-of-affinity was still 10-fold less than seen between BB2 and BB3 or BB3* receptors (compared Table 1 and Table 4), suggesting other receptor areas could also be important for Bantag-1 affinity. Therefore, to investigate further the molecular basis for the selectivity of Bantag-1, we assessed whether other specific amino acid differences might be responsible for the high affinity/selectivity for BB3 receptor by analyzing the amino acid differences and similarities in the EC1 surrounding TMs, upper TM2 (UTM2) and upper TM3 (UTM3) (Fig. 6, Table 5). The BB3 and BB2 receptors in UTM2 domain differed in 2 amino acids, occurring at positions 98 and 101 of BB3* receptor, which are comparable with positions 92 and 95 of BB2 receptor. To study the 2 amino acids differences, we made 2 BB3 receptor losses-of-affinity point mutants by substituting in BB3 or BB3* receptor the comparable different amino acid from BB2 receptor (i.e. [L98V], [V101A]). The substitution of valine for alanine at position 101 [V101A] produced the greatest effect, decreasing by 179-fold the affinity of Bantag-1 for BB3 receptor (Ki: >1,000 nM, Table 5). However, it did not alter the affinity of Bantag-1 for BB3* receptor (Ki: 5.0 nM, Table 5). For peptide #1, this substitution decreased the affinity by 3-fold for wild type BB3 receptor (Ki: 21, Table 5), suggesting a global alteration in conformation in the receptor in these cells. However, the [V101A] mutation did not alter peptide #1 affinity in BB3* receptor (Table 5), supporting the conclusion it was not important for Bantag-1 affinity. In contrast, point substitution [L98V] had no effect on the affinity of Bantag-1 or peptide #1 for wild type or BB3* receptor (Table 5). These data demonstrate that amino acids in TM2 near EC1 are not important in selectivity of Bantag-1.
Figure 6.

Effect of single point mutations in the third transmembrane domain of BB3 receptor on affinity for Bantag-1 (loss-of-affinity BB3 receptor mutations). Top, alignment of amino acid sequences in the second and third transmembrane domain of BB3 and BB2 receptor. The boxes indicate divergent amino acids between these two receptors in these regions. Arrows indicate the position of the point mutations made in BB3 receptor by substituting into BB3* receptor the divergent amino acid from the comparable position in BB2 receptor. (A) Effect of four point mutants in BB3 receptor made to explore the importance of four amino acid differences in TM3 between BB3 and BB2 receptors for determining selectivity of Bantag-1. (B) Importance of the presence of a of charged amino acid in position 127 for determining selectivity of Bantag-1. The experimental conditions were the same as described in Fig. 1 legend. The results are expressed as the percentage of saturable binding without unlabeled peptide added (percentage control). The results are the mean and S.E.M. from at least three separate experiments and in each experiment the data points were determined in duplicated. Abbreviations: See in Fig. 1, 2 and 4 legends.
Table 5.
Affinities of Bantag-1 and peptide #1 for mutants in the second and third transmembrane domain in wild type BB3, BB3* and wild type BB2 receptors (loss-of-affinity TM2 & TM3 point mutations).
| Ki (nM) | |||||
|---|---|---|---|---|---|
|
| |||||
| BB3* | Wild type | ||||
|
|
|||||
| Receptor | TM Location+ | Bantag-1 | Peptide #1 | Bantag-1 | Peptide #1 |
| BB3 | 5.2 ± 0.5 | 4.9 ± 0.8 | 5.6 ± 0.4 | 8.1 ± 1.0 | |
| BB2 | ND | ND | >30,000 a | 1.7 ± 0.1a | |
| Second transmembrane domain | |||||
| [L98V] | 2.55 | 6.0 ± 1.7 | 6.0 ± 0.8 | 5.0 ± 0.6 | 6.0 ± 0.0 |
| [V101A] | 2.58 | 5.0 ± 1.7 | 8.0 ± 0.2 | >1,000 a | 21± 1.5a |
| Third transmembrane domain | |||||
| [V122L] | 3.27 | 5.0 ± 0.5 | 5.2 ± 0.2 | 5.3 ± 0.5 | 11.4 ± 3.4 |
| [L123I] | 3.28 | 20 ± 1a | 10.1 ± 1.5b | >3,000 a | 9.1 ± 3.6 |
| [S124P] | 3.29 | 288 ± 43a | 8.0 ± 0.6 | >3,000 a | 11.3 ± 1.4 |
| [R127Q] | 3.32 | 2,068 ± 258a | 7.0 ± 0.7 | 1,332 ± 182a | 5.9 ± 0.3 |
| Importance of the R127 substitution | |||||
| [R127E] | 3.32 | >1,000 | 6.2 ± 1.9 | >1,000 a | 2.0 ± 0.3 |
| [R127K] | 3.32 | 234 ± 23a | 8.5 ± 0.7 | >3,000 | 11.9 ± 1.9b |
| [R127N] | 3.32 | 415 ± 32a | 6.3 ± 0.7 | 66 ± 11a | 5.5 ± 0.3 |
| [R127H] | 3.32 | >1,000 | 3.3 ± 0.5 | >1,000 a | 2.2 ± 0.9 |
P <0.0001 greater than BB3 receptor;
P <0.05 greater than BB3 receptor.
Ballesteros-Weinstein numbering.
ND, Not done because Arg is already present in position 288 in BB2 receptor.
CHOP cells type were incubated with 50 pM 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn-(6–14) for 60 minutes at 21°C and binding was determined as described in Materials and Methods. In each experiment each value was determined in duplicate, and values given are means and S.E.M. from at least three separate experiments. Data are calculated from dose-inhibition curves shown in Fig. 1 and 6. Abbreviations: See in Fig. 1 and 4 legends.
A similar approach was used to examine the importance of amino acid difference in the EC1 adjacent upper TM3 region (Fig. 6). The two receptors in this domain differed in 4 amino acids (Fig. 6), occurring at positions 122, 123, 124 and 127 of BB3 receptor, which are comparable with positions 116, 117, 118 and 121 of BB2 receptor. To study these amino acids differences, we first made 4 BB3 receptor loss-of-affinity point mutants by substituting in BB3 or BB3* receptor the comparable different amino acid from BB2 receptor [i.e. V122L, L123I, S124P, R127Q]. Point mutations [R127Q], [L123I] and [S124P] had the greatest effect decreasing the affinity of Bantag-1 by 233–536-fold for wild type BB3 receptor (Ki: 1,332- >3,000 nM, Fig. 6A, Table 5), without changing the affinity of peptide #1 (Table 5). With BB3* receptor, [R127Q] and [S124P] showed a 54- to 390-fold decrease in affinity (Ki: 288 and 2,068 nM, Fig. 6A, Table 5), whereas [L123I] showed only a 4-fold decrease in Bantag-1 affinity (Ki: 20 nM, Fig. 6A, Table 5), however with this mutation the affinity of peptide #1 was also decreased suggesting a possible global alteration of the mutant receptor. In contrast, point substitution [V122L] had no effect on the affinity of Bantag-1 or peptide #1 for wild type or BB3* receptor. These results support the importance of the presence of arginine at position 127 in BB3 receptor instead of glutamine in a comparable position in BB2 receptor for determining the binding affinity/selectivity for Bantag-1, as well as leucine instead of isoleucine at position 123 and proline instead of serine at position 124 in the 3rd UTM.
Because the greatest effect for differences in TM3 between either BB3 and BB3* receptor compared to BB2 receptor was seen with the [R127Q] substitution, we attempted to investigate the molecular basis for this in more detail (Fig. 6B, Table 5). The effect of a possible difference in the charge or the size of the backbone substitution was explored by making 5 BB3 and BB3* receptor loss-of-affinity point mutants. To do this, the arginine –with a positive charge- was replaced with asparagine [R127N] –with a large aliphatic, uncharged group; two other positive charged groups of different size, histidine [R127H] and lysine [R127K], and a negatively charged glutamate [R127E]. Changing the charge from positive to negative in the 127 substitution by making the point mutation [R127E] had a marked effect by decreasing 188-fold the affinity of Bantag-1 for wild type and BB3* receptors (Ki: >1,000 nM, Fig. 6B, Table 5) but did not decrease the affinity of peptide #1 (Ki: 6.2 nM, Table 5). Replacing the positively charge arginine by the uncharged, polar asparagine in point mutation [R127N] decreased the affinity 12- and 78-fold of Bantag-1 for wild type and BB3* receptors (Ki: 66 and 415 nM, Fig. 6B, Table 5). Replacing arginine 127 with other positively charged amino acids ([R127K] and [R127H]), decreased the affinity of Bantag-1 >526 and 175-fold for wild type BB3 receptor (Ki: >3,000 and >1,000 nM, Fig. 6B, Table 5) and 45- and 192-fold for BB3* receptor, respectively (Ki: 234 and >1,000 nM, Fig. 6B, Table 5). Whereas, the [R127H] substitution had no effect on affinity for peptide #1 (Table 5) in BB3 or BB3* receptor or the [R127K] mutant in BB3 receptor, the latter mutation decreased affinity for peptide #1 in BB3* receptor (Table 5), demonstrating it was having a global effect on receptor conformation. These results support the conclusion that the presence of a positive charge and proper side-chain size at position 127 of the BB3 receptor are important for determining the affinity for Bantag-1.
3.6 TM3 of BB3 receptor combination mutants (TM3 Loss-of-affinity combination mutations)
To explore further the effect of multiple UTM3 mutations, we made additional combination mutants. In TM3, a double mutation in UTM3 [L123I, R127Q] had an increased effect than either of the single mutations on the affinity of Bantag-1 for wild type and BB3* receptors, decreasing the affinity by >2,000-fold (Ki: >15,000 and >11,000 nM, Fig. 5B, Table 4). In contrast, these substitutions had no effect on the affinity of peptide #1 for wild type BB3 or BB3* receptor (Ki: 6.0 and 7.0 nM, Table 4).
4. Discussion
In the present study, we found that peptide antagonist Bantag-1 had a high affinity for BB3 receptor as reported previously [14,15] and high selectivity for BB3 receptor, with a >30,000-fold higher affinity for BB3 receptor over BB2 or BB1 receptor, despite the fact that these receptors share an 51 and 47% amino acid identity, respectively [1,32]. To investigate this in detail, a chimera receptor approach was initially used substituting extracellular domains of BB3 receptor (high affinity, Bantag-1) with those from BB2 receptor (low affinity, Bantag- 1) resulting in potentialaBantag-1 loss-of-affinity chimeras. The reverse was also performed, substituting in BB2 receptor, extracellular domains of BB3 receptor to form potential Bantag-1 gain-of-affinity chimeras. Subsequently, site-directed mutagenesis was used to make potential loss-of affinity point mutants in BB3 receptor in the key affected areas identified from the chimeric studies, as well as constructing potential loss-of-affinity point BB3 receptor mutants in surrounding upper transmembrane regions (UTM) of the important extracellular domains. The chimeric approach was initially used because numerous studies investigating peptide agonist/antagonist interaction with BnRs and other GPCRs demonstrate receptor extracellular interaction plays an import role in determining high selectivity/affinity of peptide and many other non-aminergic ligands [36,50–53], which is in contrast to bioactive amines and many nonpeptide antagonists, whose high affinity is primarily determined by amino acids in the TM regions [36,37,41,51–55].
A number of our results support the conclusion that differences in the first extracellular receptor domain and the adjacent upper TM3 region between BB3 and BB2 receptors are primarily responsible for the BB3 receptor selectivity and difference in affinity for the peptide antagonist Bantag-1 for these two closely related receptors. First, the BB3 receptor loss-of affinity chimeras constructed by replacing the EC domains one at a time of BB3 receptor by those from BB2 receptor, demonstrated that differences in the EC1 domain were almost entirely responsible for Bantag-1 high affinity. Second, performing the reverse study by constructing gain-of-affinity BB2 receptor chimeras by replacing the EC domains of BB2 receptor one at a time by those of BB3 receptor, demonstrated only the exchange of EC1 of BB3 receptor into BB2 receptor resulted in a gain-of affinity for Bantag-1. Third, loss-of-affinity point mutations in BB3 receptor, made by replacing the EC1 adjacent upper TM amino acids in BB3 receptor (TM2, TM3) by those which differed in a similar position in BB2 receptor, demonstrated that three replacements in upper TM3 (L123I, S124P, R127Q) had a marked effect on Bantag-1 affinity, whereas similar replacements in TM2 had no effect. These results have both similarities and differences from studies of the molecular basis of affinity/selectivity of various ligands interacting with the other two human BnRs (BB2 and BB1 receptors), as well as other G-protein-coupled receptors. Our results are similar to findings in studies on other GPCRs which show that, even though EC1 is usually small in size and highly variable among family members, EC1 can function as a contact point for ligands, provide structure to the extracellular region of the ligand binding site and can enable movement of the transmembrane region upon ligand binding [50].Our results differ from previous studies of other BnRs (BB2 and BB1 receptors) investigating the importance of the different EC domains for ligand selectivity using a similar chimeric receptor approach. In these studies the selectivity of the native agonist ligand GRP for BB2 receptor and NMB for BB1 receptor, are primarily affected by differences in EC2 [43,56]; whereas the high BB2 receptor selectivity of two peptide antagonists (JMV591, JMV641)[42] are primarily to differences in the EC3, with a small contribution from EC1 and for the BB1 receptor peptoid antagonist, PD168368, no EC domains were involved in determining selectivity, instead it was determined primarily by differences in TM5 [53]. These results also differ from BnR chimeric studies investigating the importance of the EC domains for the selectivity of two peptide agonists for BB3 receptor over BB2 receptor /BB1 receptor, finding primarily differences in EC2 were the most important [42]. Our finding that the EC1 domain is the most important extracellular domain for Bantag-1 selectivity shows differences from the limited data from studies examining peptide antagonist selectivity for other GPCRs. This conclusion is shown by the finding that with the cyclopentapeptide antagonist, FC131 for the CXC4 chemokine receptor [57] or with the agouti-related protein antagonist of the melanocortin-4 receptor [57], high receptor affinity/selectivity was due to differences in interaction with the EC2 and EC2/EC3 respectively, and with the VPAC1 peptide receptor antagonist, Ac-His1 [DPhe2,Lys15,Arg16,Leu27] VIP(3–7)/GRF(8–27) , its selectivity was due to differences in the amino terminal VPAC receptor domain[58]. Our results are generally similar to findings in other studies on BnRs which show differences in both EC regions and in upper TM regions are important for the high affinity/selectivity of various BB3 receptor selective peptide agonists, as well as GRP and NMB for BB2 and BB1 receptors, respectively [41,44,48,56,59–61]. Our results with the peptide antagonist, Bantag-1, are also similar to findings with various peptide agonist ligand’s interaction with other gastrointestinal/neurotransmitter GPCRs, such as CCK-8 with CCK-B receptors [62], neuropeptide S for the neuropeptide S receptor [63], substance P for the neurokinin-1 receptor [64], [D-Ala2,MePhe4,Gly5-ol]encephalin (DAMGO) for μ opioid receptor [65] or CCK8 for CCKB receptor [62], all of which also require interaction with EC domains and transmembrane regions for selectivity. However, they differ from studies with other peptide antagonists for other GPCRs which demonstrate interaction with amino acids in the TMs are the important determinant of high affinity/selectivity such as interaction of the Neuropeptide Y Y1 receptor with the peptide antagonist 1229U9(TM1, 6,7) [66], melanocortin-4 receptor with SHU9119 [67](TM3) or the selectivity of the peptide antagonist centrorelix to the GHRH receptor (Tm3,5,6,7)[68].
To determine which amino acids in EC1 of BB3 receptor account for the high selectivity/ affinity of Bantag-1 for BB3 receptor over BB2 receptor, we performed a comparative alignment of the amino acids in this region and singly mutated each of the four amino acids that differed between the two receptors in this area. Our results support the conclusion that principally the presence of a histidine in position 107 of BB3 receptor in EC1, instead of a lysine in BB2 receptor, and to a lesser extent the presence of a glutamic acid and glycine at positions 111 and 112 of BB3 receptor rather than an aspartic acid and arginine in BB2 receptor, are the key amino acid differences determined in EC1, responsible for the selectivity/ high affinity of the peptide antagonist, Bantag-1 for BB3 receptor over the BB2 receptor. The importance of the E111 or G112 in BB3 receptor EC1could have been easily missed because the E111D or G112R BB3 receptor mutants demonstrated minimal changes in affinity for Bantag-1 (1 to 2.5 fold decrease) suggesting they did not contribute to the high affinity/selectivity. However, because the EC1 H107K mutant resulted in a 50-fold decrease in affinity for Bantag-1 which did not completely account for the >400 folded decrease in affinity for Bantag-1 seen with replacement of the entire EC1 domain, we made combination mutants of H107K, E111D, G112R. These had a potentiating effect on decreasing the affinity for Bantag-1 and all three together decreased affinity for Bantag-1 as much as seen with the entire EC1 domain substitution, demonstrating their importance in the Bantag-1 interaction. The histidine, aspartic acid, and glycine residues found to be an important in determining BB3 receptor selectivity/affinity for Bantag-1 in the present study have been reported in several studies in the BB2 receptor /BB1 receptor and other GPCRs, to play a critical role in determining high affinity interaction and selectivity for their ligands. Histidine in the same EC1 location in BB3 receptor is important for the selectivity/high affinity of the peptide agonist, peptide #4 [42], in EC1 of the AT1 receptor for high affinity for angiotensin [69], in EC1 of the NK1 receptor for selectivity for substance P, in EC3 for high affinity of CCKB receptors for CCK [62], and in TM3 of the CRF receptor [70] or TM5 of the NK1 receptor [71] for high affinity for their selective nonpeptide antagonists, NBI-27914 and CP-96345, respectively. The presence of glycine in in EC1 of BB2 receptor is essential for determining high affinity of human BnRs (BB2, BB1 and BB3 receptors) for the agonist, peptide #1 [D- Tyr6, βAla11,Phe13,Nle14]Bn-(6–14)] [41] and a glycine in the TM1 of the ETA receptor [72] or TM6 of the melatonin receptor [73], is important for determining high affinity for endothelin and melatonin, respectively. In the AT1 receptor the presence of a glutamic acid in TM7 is essential for high affinity interaction with angiotensin [74] as is its presence in TM1 of MCR4 receptor needed for binding and full potency of the peptide agonist, JRH887–9 [75].
In different GPCRs histidine has been shown to contribute to high affinity/selectivity of various ligands by different mechanisms. To gain insight into the molecular basis for the importance of histidine for high affinity Bantag-1 binding, a series of point mutations at position 107 in BB3 receptor were made substituting amino acids with differing characteristics. Because the imidazole side-chain in histidine is reversibly protonated at physiological pH and the un-protonated form can exist in two different tautomeric structures, histidine can simultaneously form aromatic, hydrogen bonding, and salt bridge and charge-charge interactions [76]. With respect to the affect of the possible positive charge leading to charge-charge interactions, a number of our results support the conclusion it is not important for histidine in position 107 of the EC1 of BB3 receptor for determining high affinity/selectivity for Bantag-1. Similar to replacing the histidine by a positively charged lysine in this position in BB2 receptor, replacement of histidine by a positively charge arginine, resulted in a marked decrease in affinity for Bantag-1 (i.e. 50 and 178-fold). Conversely, replacement by a negatively charged aspartic acid had only a minimal effect on Bantag-1 affinity. Similarly replacement of histidine by the polar uncharged amino acid asparagine, which similar to histidine, has hydrogen donor and acceptor groups in its side chain and can form hydrogen bonds or charge interactions [77,78] resulted in a large decrease in affinity for Bantag-1. In contrast, replacement of histidine 107 by other aromatic amino acids (phenylalanine or tyrosine) resulted in only a small decrease in binding affinity (5–17-fold) suggesting that aromatic interactions are playing a major role in the histidine107 Bantag-1 interaction.
Previous studies with peptide agonists and antagonists for both BnRs (BB2 and BB1 receptors) and other GPCRs [37,39,41,48,49,59,60,67,79] report amino acids in upper TM regions, often in proximity to an important extracellular domain, can also be important for determining their affinity/selectivity. Therefore, to investigate further their possible role in the molecular basis for the selectivity of Bantag-1, we assessed whether other specific amino acid differences than in the region of EC1 might be responsible for the high affinity/selectivity for BB3 receptor, by analyzing the amino acid differences and similarities in the EC1 surrounding TMs, upper TM2 and upper TM3. Our results support the conclusion that the differences in the amino acids in upper TM3, but not upper TM2, are critical for high affinity of the peptide antagonist Bantag-1 for BB3 receptor. Specifically, we found when each of the four amino acid differences in upper TM3 in BB3 receptor compared to BB2 receptor (V122L, L123I, S124P, R127Q) were singly mutated in either BB3 or BB3* receptor, all but the V122L mutant showed a decrease in affinity for Bantag-1, with the largest decrease seen with replacement of the arginine at position 127 of BB3 receptor by glutamine in a comparable position in in BB2 receptor (>390-fold). These results have both similarities and differences from previous studies with BnR ligands and other peptide ligands with other GPCRs. The presence of arginine 107 in TM3 of the BB3 receptor instead of a glutamine in the comparable position of the BB2 or BB1 receptor, is critical for high affinity binding/selectivity for BB3 receptor for a synthetic Bn-related peptide agonist (peptide #4, [42]). However, the reverse is true for BB1 receptor’s high affinity and selectivity for NMB [60] or BB2 receptor for GRP [48], each of which require a glutamine in this position rather than an arginine. The presence of arginine in EC3 of BB2 receptor is critical its high affinity/selectivity for GRP [48,59], in EC3 of BB1 receptor for high affinity and selectivity for NMB [60], in all BnRs (BB2 receptor < BB1 receptor < BB3 receptor) in EC3 for the high affinity interaction with the universal agonist, peptide #1[[D-Tyr6, βAla11,Phe13,Nle14]Bn-(6–14)] [41] and in EC3 of the CCKA receptor for selectivity/high affinity for the of the peptide antagonist JMV179 [37].
To provide insight into the molecular basis for importance of arginine for high affinity Bantag-1 binding, a series of point mutation at position 127 in BB3 receptor were made substituting amino acids with differing characteristics. Our data demonstrated the presence of a positive charged moiety at position 127 in TM3 of BB3 receptor had an important effect on affinity for Bantag-1 because substitution of a negatively charged glutamic acid decreased affinity >200-fold, and substitution of the uncharged glutamine, decreased affinity >400 fold. However, replacement by a positively charged, lysine decreased affinity 45-fold and a replacement by a histidine caused a >192 -fold decrease in affinity. The data with lysine and histidine suggest the size of the backbone substitution determining the placement of the positive charge in relation to the peptide backbone, is playing an important role in determining Bantag-1 interaction. Hydrogen bonding interactions are frequently with arginine [80], and our data suggest this could also contribute to Bantag-1 interaction because replacement with asparagine decreased affinity but much less than replacement with glutamine.
In conclusion, in the present study, we identified important amino acids for determining Bantag-1 binding affinity for the BB3 receptor exist in EC and UTM regions; particularly the presence of His107 in EC1 and Arg127 in TM3. Detailed substitutions at these locations demonstrate that in the EC1 area, particularly important is the presence of aromatic interactions, rather than hydrogen bonding or charge-charge interactions, in playing an important role for determining the high affinity/selectivity of this ligand. On the other hand, in regard to Arg127 in TM3, our results support the conclusion that both hydrogen bonding and charge-charge interactions contribute to the high affinity/selectivity for Bantag-1. The identification of the important amino acids for determining the high affinity of Bantag-1 for the BB3 receptor may help to provide insights into the future design of other BB3 receptor antagonists or perhaps biased agonists which could be useful for exploring both the intracellular signaling of BB3 receptor, as well as its role in physiological/pathophysiological states.
Acknowledgments
Funding: This study was supported by intramural funds of NIDDK, NIH.
Abbreviations
- Bantag-1
Boc-Phe-His-4-amino-5-cyclohexyl-2,4,5-trideoxypentonyl-Leu- (3-dimethylamino) benzylamide N-methylammonium trifluoroacetate, BB3 receptor antagonist
- BB3 receptor or BB3
Bombesin receptor subtype-3
- BB3* receptor or BB3*
His294 in BB3 receptor substituted for by Arg288 in comparable position of BB2 receptor to increase expression level
- Bn
Bombesin
- BnR
Bombesin receptor
- CHOP
Polyoma large T antigen- expressing Chinese hamster ovary cells
- DMEM
Dulbecco’s minimum essential medium
- EC or e
extracellular domain
- FBS
fetal bovine serum
- GI
gastrointestinal
- GPCR
G-protein-coupled-orphanreceptor
- GRP
gastrin-releasing peptide
- BB2 receptor or BB2
gastrin-releasing peptide receptor
- h
human
- KO
knockout
- m
mouse
- NMB
neuromedin-B
- BB1
neuromedin B receptor
- PBS
phosphate-buffered saline
- peptide #1
[D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6–14)
- r
rat
- TM
transmembrane region
- UTM
upper transmembrane region
Footnotes
The authors have no conflicts of interest with this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fathi Z, Corjay MH, Shapira H, Wada E, Benya R, Jensen R, et al. BRS-3: novel bombesin receptor subtype selectively expressed in testis and lung carcinoma cells. J Biol Chem. 1993;268(8):5979–5984. [PubMed] [Google Scholar]
- 2.Jensen RT, Battey JF, Spindel ER, Benya RV. International Union of Pharmacology. LVIII. Mammalian Bombesin Receptors: Nomenclature, distribution, pharmacology, signaling and functions in normal and disease states. Pharmacol Rev. 2008;60:1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramos-Alvarez I, Moreno P, Mantey SA, Nakamura T, Nuche-Berenguer B, Moody TW, et al. Insights into bombesin receptors and ligands: highlighting recent advances. Peptides. 2015;72:128–144. doi: 10.1016/j.peptides.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majumdar ID, Weber HC. Biology and pharmacology of bombesin receptor subtype-3. Curr Opin Endocrinol Diabetes Obes. 2012;19:3–7. doi: 10.1097/MED.0b013e32834ec77d. [DOI] [PubMed] [Google Scholar]
- 5.Majumdar ID, Weber HC. Biology of mammalian bombesin-like peptides and their receptors. Curr Opin Endocrinol Diabetes Obes. 2011;18:68–74. doi: 10.1097/MED.0b013e328340ff93. [DOI] [PubMed] [Google Scholar]
- 6.Uehara H, Gonzalez N, Sancho V, Mantey SA, Nuche-Berenguer B, Pradhan T, et al. Pharmacology and selectivity of various natural and synthetic bombesin related peptide agonists for human and rat bombesin receptors differs. Peptides. 2011;32:1685–1699. doi: 10.1016/j.peptides.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez N, Mantey SA, Pradhan TK, Sancho V, Moody TW, Coy DH, et al. Characterization of putative GRP- and NMB-receptor antagonist's interaction with human receptors. Peptides. 2009;30:1473–1486. doi: 10.1016/j.peptides.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang LH, Coy DH, Taylor JE, Jiang NY, Kim SH, Moreau JP, et al. Desmethionine alkylamide bombesin analogues: a new class of bombesin receptor antagonists with a potent antisecretory activity in pancreatic acini and antimitotic activity in Swiss 3T3 cells. Biochemistry (Mosc) 1990;29(3):616–622. doi: 10.1021/bi00455a004. [DOI] [PubMed] [Google Scholar]
- 9.Wang LH, Coy DH, Taylor JE, Jiang NY, Moreau JP, Huang SC, et al. Des-Met carboxyl-terminally modified analogues of bombesin function as potent bombesin receptor antagonists, partial agonists, or agonists. J Biol Chem. 1990;265(26):15695–15703. [PubMed] [Google Scholar]
- 10.Heinz-Erian P, Coy DH, Tamura M, Jones SW, Gardner JD, Jensen RT. [D-Phe12]bombesin analogues: a new class of bombesin receptor antagonists. Am J Physiol. 1987;252:G439–G442. doi: 10.1152/ajpgi.1987.252.3.G439. [DOI] [PubMed] [Google Scholar]
- 11.Coy DH, Taylor JE, Jiang NY, Kim SH, Wang LH, Huang SC, et al. Short-chain pseudopeptide bombesin receptor antagonists with enhanced binding affinities for pancreatic acinar and Swiss 3T3 cells display strong antimitotic activity. J Biol Chem. 1989;264:14691–14697. [PubMed] [Google Scholar]
- 12.Mantey SA, Weber HC, Sainz E, Akeson M, Ryan RR, Pradhan TK, et al. Discovery of a high affinity radioligand for the human orphan receptor, bombesin receptor subtype 3, which demonstrates it has a unique pharmacology compared to other mammalian bombesin receptors. J Biol Chem. 1997;272(41):26062–26071. doi: 10.1074/jbc.272.41.26062. [DOI] [PubMed] [Google Scholar]
- 13.Majumdar ID, Weber HC. Appetite-modifying effects of bombesin receptor subtype-3 agonists. Handb Exp Pharmacol. 2012:405–432. doi: 10.1007/978-3-642-24716-3_19. [DOI] [PubMed] [Google Scholar]
- 14.Moreno P, Mantey SA, Nuche-Berenguer B, Reitman ML, Gonzalez N, Coy DH, et al. Comparative pharmacology of bombesin receptor subtype-3, nonpeptide agonist MK-5046, a universal peptide agonist, and peptide antagonist Bantag-1 for human bombesin receptors. J Pharmacol Exp Ther. 2013;347:100–116. doi: 10.1124/jpet.113.206896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan XM, Chen H, Dobbelaar PH, Dong Y, Fong TM, Gagen K, et al. Regulation of Energy Homeostasis by Bombesin Receptor Subtype-3: Selective Receptor Agonists for the Treatment of Obesity. Cell Metab. 2010;11:101–112. doi: 10.1016/j.cmet.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Sebhat IK, Franklin C, Lo MC, Chen D, Jewell JP, Miller R, et al. Discovery of MK-5046, a potent, selective bombesin recptor subtype-3 agonist for the treatment of obesity. ACS Medicinal Chemistry Letters. 2011;2:43–47. doi: 10.1021/ml100196d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reitman ML, Dishy V, Moreau A, Denney WS, Liu C, Kraft WK, et al. Pharmacokinetics and pharmacodynamics of MK-5046, a bombesin receptor subtype-3 (BRS-3) agonist, in healthy patients. J Clin Pharmacol. 2012;52:1306–1316. doi: 10.1177/0091270011419854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryan RR, Weber HC, Mantey SA, Hou W, Hilburger ME, Pradhan TK, et al. Pharmacology and intracellular signaling mechanisms of the native human orphan receptor BRS-3 in lung cancer cells. J Pharmacol Exp Ther. 1998;287:366–380. [PubMed] [Google Scholar]
- 19.Ryan RR, Weber HC, Hou W, Sainz E, Mantey SA, Battey JF, et al. Ability of various bombesin receptor agonists and antagonists to alter intracellular signaling of the human orphan receptor BRS-3. J Biol Chem. 1998;273:13613–13624. doi: 10.1074/jbc.273.22.13613. [DOI] [PubMed] [Google Scholar]
- 20.Moody TW, Sancho V, Di Florio A, Nuche-Berenguer B, Mantey S, Jensen RT. Bombesin receptor subtype-3 agonists stimulate the growth of lung cancer cells and increase EGF receptor tyrosine phosphorylation. Peptides. 2011;32:1677–1684. doi: 10.1016/j.peptides.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moody TW, Mantey SA, Moreno P, Nakamura T, Lacivita E, Leopoldo M, et al. ML-18 is a non-peptide bombesin receptor subtype-3 antagonist which inhibits lung cancer growth. Peptides. 2015;64:55–61. doi: 10.1016/j.peptides.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mantey SA, Coy DH, Entsuah LK, Jensen RT. Development of bombesin analogs with conformationally restricted amino acid substitutions with enhanced selectivity for the orphan receptor human bombesin receptor subtype 3. J Pharmacol Exp Ther. 2004;310:1161–1170. doi: 10.1124/jpet.104.066761. [DOI] [PubMed] [Google Scholar]
- 23.Pradhan TK, Katsuno T, Taylor JE, Kim SH, Ryan RR, Mantey SA, et al. Identification of a unique ligand which has high affinity for all four bombesin receptor subtypes. Eur J Pharmacol. 1998;343:275–287. doi: 10.1016/s0014-2999(97)01527-6. [DOI] [PubMed] [Google Scholar]
- 24.Mantey SA, Coy DH, Pradhan TK, Igarashi H, Rizo IM, Shen L, et al. Rational design of a peptide agonist that interacts selectively with the orphan receptor, bombesin receptor subtype 3. J Biol Chem. 2001;276:9219–9229. doi: 10.1074/jbc.M008737200. [DOI] [PubMed] [Google Scholar]
- 25.Ryan RR, Katsuno T, Mantey SA, Pradhan TP, Weber HC, Battey JF, et al. Comparative pharmacology of a nonpeptoid neuromedin B antagonist PD 168368. J Pharmacol Exp Ther. 1999;290:1202–1211. [PubMed] [Google Scholar]
- 26.Nakamura T, Igarashi H, Ito T, Jensen RT. Important of case-reports/series, in rare diseases: Using neuroendocrine tumors as an example. World J Clin Cases. 2014;2:608–613. doi: 10.12998/wjcc.v2.i11.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramos-Alvarez I, Nakamura T, Mantey SA, Moreno P, Nuche-Berenguer B, Jensen RT. Novel chiral-diazepines function as specific, selective receptor agonists with variable coupling and species variability in human, mouse and rat BRS-3 receptor cells. Peptides. 2015;75:8–17. doi: 10.1016/j.peptides.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J, Lao ZJ, Zhang J, Schaeffer MT, Jiang MM, Guan XM, et al. Molecular basis of the pharmacological difference between rat and human bombesin receptor subtype-3 (BRS-3) Biochemistry (Mosc) 2002;41:8954–8960. doi: 10.1021/bi0202777. [DOI] [PubMed] [Google Scholar]
- 29.Ohki-Hamazaki H, Watase K, Yamamoto K, Ogura H, Yamano M, Yamada K, et al. Mice lacking bombesin receptor subtype-3 develop metabolic defects and obesity. Nature. 1997;390(6656):165–169. doi: 10.1038/36568. [DOI] [PubMed] [Google Scholar]
- 30.Yamada K, Wada E, Imaki J, Ohki-Hamazaki H, Wada K. Hyperresponsiveness to palatable and aversive taste stimuli in genetically obese (bombesin receptor subtype-3-deficient) mice. Physiol Behav. 1999;66:863–867. doi: 10.1016/s0031-9384(99)00032-3. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez N, Moody TW, Igarashi H, Ito T, Jensen RT. Bombesin-related peptides and their receptors: recent advances in their role in physiology and disease states. Curr Opin Endocrinol Diabetes Obes. 2008;15:58–64. doi: 10.1097/MED.0b013e3282f3709b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez N, Moreno P, Jensen RT. Bombesin receptor -subtype 3 as a potential target for obesity and diabetes. Exp Opin Ther Targets. 2015;19:1153–1170. doi: 10.1517/14728222.2015.1056154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada K, Wada E, Wada K. Bombesin-like peptides: studies on food intake and social behaviour with receptor knock-out mice. Ann Med. 2000;32:519–529. doi: 10.3109/07853890008998831. [DOI] [PubMed] [Google Scholar]
- 34.Sayegh AI. The role of bombesin and bombesin-related peptides in the short-term control of food intake. Prog Mol Biol Transl Sci. 2013;114:343–370. doi: 10.1016/B978-0-12-386933-3.00010-8. [DOI] [PubMed] [Google Scholar]
- 35.Feng Y, Guan XM, Li J, Metzger JM, Zhu Y, Juhl K, et al. Bombesin Receptor Subtype-3 (BRS-3) Regulates Glucose-Stimulated Insulin Secretion in Pancreatic Islets across Multiple Species. Endocrinology. 2011;152:4106–4115. doi: 10.1210/en.2011-1440. [DOI] [PubMed] [Google Scholar]
- 36.Tokita K, Katsuno T, Hocart SJ, Coy DH, Llinares M, Martinez J, et al. Molecular basis for selectivity of high affinity peptide antagonists for the gastrin-releasing peptide receptor. J Biol Chem. 2001;276:36652–36663. doi: 10.1074/jbc.M104566200. [DOI] [PubMed] [Google Scholar]
- 37.Gigoux V, Escrieut C, Fehrentz JA, Poirot S, Maigret B, Moroder L, et al. Arginine 336 and asparagine 333 of the human cholecystokinin-A receptor binding site interact with the penultimate aspartic acid and the C-terminal amide of cholecystokinin. J Biol Chem. 1999;274:20457–20464. doi: 10.1074/jbc.274.29.20457. [DOI] [PubMed] [Google Scholar]
- 38.Krystek SR, Jr, Patel PS, Rose PM, Fisher SM, Kienzle BK, Lach DA, et al. Mutation of peptide binding site in transmembrane region of a G protein-coupled receptor accounts for endothelin receptor subtype selectivity. J Biol Chem. 1994;269:12383–12386. [PubMed] [Google Scholar]
- 39.Nardone J, Hogan PG. Delineation of a region in the B2 bradykinin receptor that is essential for high-affinity agonist binding. Proc Natl Acad Sci U S A. 1994;91:4417–4421. doi: 10.1073/pnas.91.10.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benya RV, Fathi Z, Battey JF, Jensen RT. Serines and threonines in the gastrin-releasing peptide receptor carboxyl terminus mediate internalization. J Biol Chem. 1993;268:20285–20290. [PubMed] [Google Scholar]
- 41.Uehara H, Hocart SJ, Gonzalez N, Mantey SA, Nakagawa T, Katsuno T, et al. The molecular basis for high affinity of a universal ligand for human bombesin receptor (BnR) family members. Biochem Pharmacol. 2012;84:936–948. doi: 10.1016/j.bcp.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez N, Hocart SJ, Portal-Nunez S, Mantey SA, Nakagawa T, Zudaire E, et al. Molecular basis for agonist selectivity and activation of the orphan bombesin receptor subtype 3 receptor. J Pharmacol Exp Ther. 2008;324:463–474. doi: 10.1124/jpet.107.132332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tokita K, Hocart SJ, Coy DH, Jensen RT. Molecular basis of the selectivity of the gastrin-releasing peptide receptor for gastrin-releasing peptide. Mol Pharmacol. 2002;61:1435–1443. doi: 10.1124/mol.61.6.1435. [DOI] [PubMed] [Google Scholar]
- 44.Fathi Z, Benya RV, Shapira H, Jensen RT, Battey JF. The fifth transmembrane segment of the neuromedin B receptor is critical for high affinity neuromedin B binding. J Biol Chem. 1993;268(#20):14622–14626. [PubMed] [Google Scholar]
- 45.Tsuda T, Kusui T, Hou W, Benya RV, Akeson MA, Kroog GS, et al. Effect of gastrin-releasing peptide receptor number on receptor affinity, coupling, degradation and receptor modulation. Mol Pharmacol. 1997;51(5):721–732. doi: 10.1124/mol.51.5.721. [DOI] [PubMed] [Google Scholar]
- 46.Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 47.Sancho V, Moody TW, Mantey SA, Di Florio A, Uehara H, Coy DH, et al. Pharmacology of putative selective hBRS-3 receptor agonists for human bombesin receptors (BnR): Affinities, potencies and selectivity in multiple native and BnR transfected cells. Peptides. 2010;31:1569–1578. doi: 10.1016/j.peptides.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akeson M, Sainz E, Mantey SA, Jensen RT, Battey JF. Identification of four amino acids in the gastrin-releasing peptide C receptor that are required for high affinity agonist binding. J Biol Chem. 1997;272:17405–17409. doi: 10.1074/jbc.272.28.17405. [DOI] [PubMed] [Google Scholar]
- 49.Bastian S, Pruneau D, Loillier B, Robert C, Bonnafous JC, Paquet JL. Identification of a key region of kinin B(1) receptor for high affinity binding of peptide antagonists. J Biol Chem. 2000;275:6107–6113. doi: 10.1074/jbc.275.9.6107. [DOI] [PubMed] [Google Scholar]
- 50.Peeters MC, van Westen GJ, Li Q, Ijzerman AP. Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol Sci. 2011;32:35–42. doi: 10.1016/j.tips.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 51.Lee SM, Booe JM, Pioszak AA. Structural insights into ligand recognition and selectivity for classes A, B, and C GPCRs. Eur J Pharmacol. 2015;763:196–205. doi: 10.1016/j.ejphar.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Audet M, Bouvier M. Restructuring G-protein- coupled receptor activation. Cell. 2012;151:14–23. doi: 10.1016/j.cell.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 53.Tokita K, Hocart SJ, Katsuno T, Mantey SA, Coy DH, Jensen RT. Tyrosine 220 in the fifth transmembrane domain of the neuromedin B receptor is critical for the high selectivity of the peptoid antagonist PD168368. J Biol Chem. 2001;276:495–504. doi: 10.1074/jbc.M006059200. [DOI] [PubMed] [Google Scholar]
- 54.Strader CD, Fong TM, Graziano MP, Tota MR. The family of G-protein-coupled receptors. FASEB J. 1995;9:745–754. [PubMed] [Google Scholar]
- 55.Shang Y, Filizola M. Opioid receptors: Structural and mechanistic insights into pharmacology and signaling. Eur J Pharmacol. 2015;763:206–213. doi: 10.1016/j.ejphar.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gonzalez N, Nakagawa T, Mantey SA, Sancho V, Uehara H, Katsuno T, et al. Molecular basis for the selectivity of the mammalian bombesin peptide, neuromedin B, for its receptor. J Pharmacol Exp Ther. 2009;331:265–276. doi: 10.1124/jpet.109.154245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang YK, Dickinson CJ, Zeng Q, Li JY, Thompson DA, Gantz I. Contribution of melanocortin receptor exoloops to Agouti-related protein binding. J Biol Chem. 1999;274:14100–14106. doi: 10.1074/jbc.274.20.14100. [DOI] [PubMed] [Google Scholar]
- 58.Juarranz MG, Van Rampelbergh J, Gourlet P, De Neef P, Cnudde J, Robberecht P, et al. Vasoactive intestinal polypeptide VPAC1 and VPAC2 receptor chimeras identify domains responsible for the specificity of ligand binding and activation. Eur J Biochem. 1999;265:449–456. doi: 10.1046/j.1432-1327.1999.00769.x. [DOI] [PubMed] [Google Scholar]
- 59.Nakagawa T, Hocart SJ, Schumann M, Tapia JA, Mantey SA, Coy DH, et al. Identification of key amino acids in the gastrin-releasing peptide receptor (GRPR) responsible for high affinity binding of gastrin-releasing peptide (GRP) Biochem Pharmacol. 2005;69:579–593. doi: 10.1016/j.bcp.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 60.Sainz E, Akeson M, Mantey SA, Jensen RT, Battey JF. Four amino acid residues are critical for high affinity binding of neuromedin B to the neuromedin B receptor. J Biol Chem. 1998;273:15927–15932. doi: 10.1074/jbc.273.26.15927. [DOI] [PubMed] [Google Scholar]
- 61.Maughfling EJ, Boden P, Hall MD. Construction of chimeric human bombesin receptors to identify neuromedin B and gastrin-releasing peptide receptor binding sites. Biochem Soc Trans. 1997;25:455S. doi: 10.1042/bst025455s. [DOI] [PubMed] [Google Scholar]
- 62.Silvente-Poirot S, Escrieut C, Wank SA. Role of the extracellular domains of the cholecystokinin receptor in agonist binding. Mol Pharmacol. 1998;54:364–371. doi: 10.1124/mol.54.2.364. [DOI] [PubMed] [Google Scholar]
- 63.Clark SD, Tran HT, Zeng J, Reinscheid RK. Importance of extracellular loop one of the neuropeptide S receptor for biogenesis and function. Peptides. 2010;31:130–138. doi: 10.1016/j.peptides.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fong TM, Huang RR, Strader CD. Localization of agonist and antagonist binding domains of the human neurokinin-1 receptor. J Biol Chem. 1992;267:25664–25667. [PubMed] [Google Scholar]
- 65.Wang WW, Shahrestanifar M, Jin J, Howells RD. Studies on mu and delta opioid receptor selectivity utilizing chimeric and site-mutagenized receptors. Proc Natl Acad Sci U S A. 1995;92:12436–12440. doi: 10.1073/pnas.92.26.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kannoa T, Kanatani A, Keen SL, rai-Otsuki S, Haga Y, Iwama T, et al. Different binding sites for the neuropeptide Y Y1 antagonists 1229U91 and J-104870 on human Y1 receptors. Peptides. 2001;22:405–413. doi: 10.1016/s0196-9781(01)00350-3. [DOI] [PubMed] [Google Scholar]
- 67.Yang Y, Chen M, Lai Y, Gantz I, Georgeson KE, Harmon CM. Molecular determinants of human melanocortin-4 receptor responsible for antagonist SHU9119 selective activity. J Biol Chem. 2002;277:20328–20335. doi: 10.1074/jbc.M201343200. [DOI] [PubMed] [Google Scholar]
- 68.Soderhall JA, Polymeropoulos EE, Paulini K, Gunther E, Kuhne R. Antagonist and agonist binding models of the human gonadotropin-releasing hormone receptor. Biochem Biophys Res Commun. 2005;333:568–582. doi: 10.1016/j.bbrc.2005.05.142. [DOI] [PubMed] [Google Scholar]
- 69.Hjorth SA, Schambye HT, Greenlee WJ, Schwartz TW. Identification of peptide binding residues in the extracellular domains of the AT1 receptor. J Biol Chem. 1994;269:30953–30959. [PubMed] [Google Scholar]
- 70.Liaw CW, Grigoriadis DE, Lorang MT, De Souza EB, Maki RA. Localization of agonist- and antagonist-binding domains of human corticotropin-releasing factor receptors. Mol Endocrinol. 1997;11:2048–2053. doi: 10.1210/mend.11.13.0034. [DOI] [PubMed] [Google Scholar]
- 71.Fong TM, Cascieri MA, Yu H, Bansal A, Swain C, Strader CD. Amino-aromatic interaction between histidine 197 of the neurokinin-1 receptor and CP 96345. Nature. 1993;362:350–353. doi: 10.1038/362350a0. [DOI] [PubMed] [Google Scholar]
- 72.Breu V, Hashido K, Broger C, Miyamoto C, Furuichi Y, Hayes A, et al. Separable binding sites for the natural agonist endothelin-1 and the non-peptide antagonist bosentan on human endothelin-A receptors. Eur J Biochem. 1995;231:266–270. [PubMed] [Google Scholar]
- 73.Gubitz AK, Reppert SM. Chimeric and point-mutated receptors reveal that a single glycine residue in transmembrane domain 6 is critical for high affinity melatonin binding. Endocrinology. 2000;141:1236–1244. doi: 10.1210/endo.141.3.7356. [DOI] [PubMed] [Google Scholar]
- 74.Pignatari GC, Rozenfeld R, Ferro ES, Oliveira L, Paiva AC, Devi LA. A role for transmembrane domains V and VI in ligand binding and maturation of the angiotensin II AT1 receptor. Biol Chem. 2006;387:269–276. doi: 10.1515/BC.2006.036. [DOI] [PubMed] [Google Scholar]
- 75.Maiga A, Merlin J, Marcon E, Rouget C, Larregola M, Gilquin B, et al. Orthosteric binding of rho-Da1a, a natural peptide of snake venom interacting selectively with the alpha1A-adrenoceptor. PLoS ONE. 2013;8:e68841. doi: 10.1371/journal.pone.0068841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mayevu NM, Choe H, Abagyan R, Seong JY, Millar RP, Katz AA, et al. Histidine(7. 36(305)) in the conserved peptide receptor activation domain of the gonadotropin releasing hormone receptor couples peptide binding and receptor activation. Mol Cell Endocrinol. 2015;402:95–106. doi: 10.1016/j.mce.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 77.Liao SM, Du QS, Meng JZ, Pang ZW, Huang RB. The multiple roles of histidine in protein interactions. Chem Cent J. 2013;7:44. doi: 10.1186/1752-153X-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yokota A, Tsumoto K, Shiroishi M, Nakanishi T, Kondo H, Kumagai I. Contribution of asparagine residues to the stabilization of a proteinaceous antigen-antibody complex, HyHEL-10-hen egg white lysozyme. J Biol Chem. 2010;285:7686–7696. doi: 10.1074/jbc.M109.089623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chai BX, Pogozheva ID, Lai YM, Li JY, Neubig RR, Mosberg HI, et al. Receptor-antagonist interactions in the complexes of agouti and agouti-related protein with human melanocortin 1 and 4 receptors. Biochemistry (Mosc) 2005;44:3418–3431. doi: 10.1021/bi0478704. [DOI] [PubMed] [Google Scholar]
- 80.Borders CL, Jr, Broadwater JA, Bekeny PA, Salmon JE, Lee AS, Eldridge AM, et al. A structural role for arginine in proteins: multiple hydrogen bonds to backbone carbonyl oxygens. Protein Sci. 1994;3:541–548. doi: 10.1002/pro.5560030402. [DOI] [PMC free article] [PubMed] [Google Scholar]