

Abstract
It is unknown if a lower size limit exists for human blood coagulation under flow over physiologically vessel wall triggers as small as a single collagen fiber. Prior determinations of the smallest sized surface stimuli necessary for clotting of human blood, defined as the patch size threshold, have not deployed whole blood, hemodynamic flow, and platelet adhesive stimuli. For whole blood perfused in microfluidic devices, we report that steady venous flow (wall shear rate, 100 s−1) was sufficient to drive platelet deposition on 20-micron long zones of collagen fibers or on a single fiber. With tissue factor (TF)-coated collagen, flowing blood generated robust platelet deposits, platelet-localized thrombin, and fibrin on a single collagen fiber, thus demonstrating the absence of a physiological patch size threshold under venous flow. In contrast, at arterial wall shear rate (1000 s−1) with TF present, essentially no platelet or fibrin deposition occurred on 20-micron collagen zones or on a single collagen fiber, demonstrating a patch threshold, which was overcome by pre-coating the collagen with von Willebrand factor (VWF). For venous flows, human blood can clot on one of the smallest biological units of a single collagen fiber presenting TF. For arterial flows, VWF together with TF allows human blood to generate thrombin and fibrin on a patch stimulus as limited as a single collagen fiber. VWF-dependent platelet adhesion represents a particle-based sensing mechanism of micron-scale stimuli that then allows amplification of the molecular components of TF-driven thrombin and fibrin production under arterial flow.
Keywords: platelet, hemostasis, thrombosis, diffusion, convection, thrombin
INTRODUCTION
Blood coagulation in healthy humans is tightly regulated such that hemostatic clot formation is rapid but self-limiting at sites of vascular injury. Vascular damage involves exposure of flowing blood to collagen and tissue factor (TF), components that platelets and plasma do not normally encounter in healthy vessels. TF binds Factor VIIa to generate Factor Xa (FXa) and Factor IXa (FIXa), leading to amplified thrombin generation and consequent fibrin polymerization. Platelet deposition is driven by collagen and thrombin and autocrinic release of ADP and thromboxane. The extracellular triggers, platelet receptors, and intracellular signaling events of platelet activation as well as the TF-triggered extrinsic pathway of coagulation are well defined in terms of molecular components.
However, it is unknown if a lower size limit of a surface defect exists for human blood coagulation under flow. Prior experimental determinations of the smallest sized surface stimuli necessary for clotting of human blood, defined as the patch size threshold, have not deployed the combination of whole blood, hemodynamic flow, and a platelet adhesive stimulus. Observations with plasma indicate that a surface trigger may be small enough to escape hemostatic responses because diffusible and rapidly inhibited species might not reach a local critical concentration. Thus, transport effects may sufficiently damp the amplification reactions of the coagulation cascade. Plasma incubated under static conditions has great difficulty in generating fibrin when placed on lipid/TF features smaller than 50 μm in diameter, but rapidly generates fibrin on features larger than 100 μm in diameter.1 In such measurements, the rate of generation of active FXa and Factor Va, thrombin, and fibrin monomer in proximity to the discrete lipid surface must overcome (i) diffusion that dilutes local concentrations and (ii) inhibitors such as tissue factor pathway inhibitor and antithrombin that quench active species. A similarly sized patch threshold was detected with static incubation of platelet rich plasma over lipid-TF features smaller than 100 μm in diameter,2 but a strong patch size threshold was less apparent when whole blood was incubated on such features. Convective flow adds an additional transport mechanism to remove reactive species from a discrete triggering zone. Perfusion of plasma through a capillary with a 200-μm long zone of lipid/TF displayed difficulty in clotting at even sub-physiological wall shear rates above 30 s−1.3 Similarly, capillary perfusion of platelet rich plasma over 200-μm long zones of lipid/TF displayed difficulty in clotting at a venous wall shear rates of 80 to 120 s−1. However, a platelet adhesive and stimulatory surface such as collagen was not part of that measurement. In contrast, human whole blood readily clots on 250-μm diameter spots of microprinted collagen/TF 4 or 250-μm long zones of collagen/TF in microfluidic channels 5, 6 at venous and arterial flow conditions.
In the present microfluidic study of human blood, surface feature size was reduced to one of the smallest possible physiological procoagulant trigger, a single collagen fiber. Lipidated TF and von Willebrand factor (VWF) were used to decorate collagen fibers to mimic the surface stimuli expected in vivo.7, 8 Human whole blood was minimally perturbed with 4 μg/mL corn trysin inhibitor (CTI, a βFXIIa inhibitor) and was immediately perfused over patterned small patches after phlebotomy within 5 min in order to minimize contact-pathway triggered clotting while maintaining normal blood responsiveness of the extrinsic pathway of TF initiated coagulation.5, 9 By taking this microfluidic approach, we determined if a patch size threshold exists for human blood. We determined under what biochemical and hemodynamic conditions a single collagen fiber can support a clotting response of platelet deposition, platelet-localized thrombin generation,10 and fibrin polymerization.
RESULTS
Micropatterned zones of collagen fibers
Using a micropatterning microfluidic device, a 20-μm long zone of multiple collagen fibers was deposited on glass, followed by placement perpendicular to the fiber orientation of the 8-channel microfluidic blood perfusion device with 8 independent 250-μm wide flow channels for blood perfusion (Fig. 1A). Typically, 4 to 8 individual collagen fibers were present in this 20-μm long zone (Fig. 1B, C) which were then coated with lipidated TF (Fig. 1D) and/or VWF in some experiments. No fibers were detected outside the 250-μm wide × 20-μm long patterning region. Similarly, a single collagen fiber was deposited across the confined 250-μm wide region (Fig. 1 E-G). Annexin V staining confirmed that collagen fibers were able to bind by physisorption the TF liposomes containing phosphatidylserine (Fig. 1G). TF liposomes covered 30.4 ± 4.1 % (n = 11) of total collagen fiber surface, which corresponded to a relatively high TF surface concentration of ~1-10 TF molecules/μm2 5. This TF surface concentration is comparable to that expected in adventitial regions of vessels or in human atherosclerotic carotid artery plaques.7, 11
Fig. 1. Microfluidic perfusion and micropatterned collagen fiber surfaces.
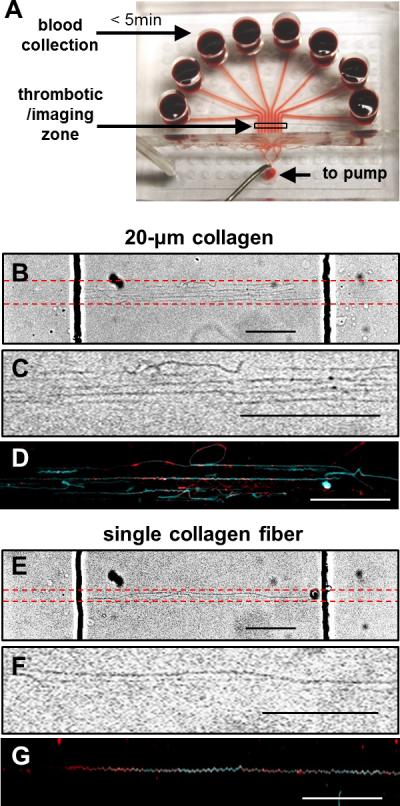
(A) An 8-channel microfluidic device (each channel: 250-μm wide × 60-μm high) for perfusion of whole blood over a patterned collagen feature. CTI (4 μg/ml)-treated whole blood was transferred to each reservoir within 5 min of phlebotomy and was immediately perfused over the collagen zone at venous or arterial wall shear rates (100 or 1000 s−1) controlled by a syringe pump connected at the outlet. Red dashed lines indicate location of patterned collagen (B, E). Collagen fibers were oriented perpendicularly to the flow channels (dark vertical lines are channel side walls). A 20-μm collagen zone was typically composed of 4 to 8 aligned collagen fibers (B, C). Single collagen fibers were confined within the 250-μm wide zone and no upstream or downstream collagen deposition was detected (E, F). Annexin V staining demonstrated TF liposomes (red) absorbed on fibers in the 20-μm collagen zone (D, cyan) or on a single collagen fiber (G, cyan). All scale bars: 50 μm in B-G.
A single collagen fiber with tissue factor triggers coagulation at venous wall shear rate
The combination of TF-coated collagen fibers, CTI-treated whole blood, and a venous shear rate (100 s−1) resulted in a favorable condition for thrombin generation. Under this condition, platelets were able to adhere and form a substantial clot buildup of platelets and fibrin on either a 20-μm long collagen/TF zone (20-μm collagen/TF) or on a single collagen/TF fiber (Fig. 2A). For the 20-μm collagen/TF, platelet accumulation began in the first 100 sec of flow and continued over the entire 600 sec experiment, resulting in a clot mass that essentially filled the channel (60 μm high) over the collagen surface (Fig. 2C). Within the clot formed on the 20-μm collagen/TF, thrombin was robustly detected using the thrombin activity biosensor bound to the deposited platelets (Fig. 2A), while fibrin polymerization proceeded over the entire course of the experiment (Fig. 2C) within the platelet deposit. For zones mimicking the defect size of a single endothelial cell (~20 μm), human blood robustly clots under venous flow conditions when presented the essential triggers of collagen and tissue factor. On a single collagen/TF fiber, platelets were able to adhere and accumulate, generate thrombin and fibrin under a venous flow condition (Fig. 2A, C). For a single collagen fiber, the platelet deposits did not grow across the entire channel height, but reached a height of about 30 microns by 600 sec. This reduction in platelet accumulation (per unit area) on the single collagen/TF fiber was apparent throughout the entire time course of the experiment (Fig. 2C). However, fibrin generation per unit area was not significantly affected by the reduction of stimulus size to the limit of a single collagen fiber that captured platelets (Fig. 2C). Consistent with prior studies with lipid coated surfaces presenting TF, the use of lipidated TF alone without collagen was not able to trigger or support a clotting response in the wall shear rate range of 100 to 1000 s−1 1, 2.
Fig. 2. Thrombus formation on collagen/TF at venous and arterial flow conditions.

CTI-treated (4 μg/ml) whole blood was perfused over collagen/TF at venous wall shear rate (100 s−1, A) or arterial wall shear rate (1000 s−1, B). Blood coagulation occurred on the 20-μm long collagen/TF zone and the single collagen fiber at 100 s−1, but not at 1000 s−1 (excluding side-wall accumulation where shear rates are reduced). For the center two-thirds of the channel (excluding the side-wall), platelet and fibrin fluorescence were imaged dynamically for 8 replicate clots at each condition (B, D). Vertical dashed lines represent flow channel side walls. Flow direction: top to bottom. Scale bar: 50 μm. (***p < 0.005; ns, not significant).
The production of thrombin and fibrin in this experiment was highly dependent on the lipidated TF bound to the collagen fibers. Fibrin generation was not detected on a 20-μm collagen zone or the single collagen fiber lacking TF (Figs. S1B,E and S2). In the absence of TF, thrombin generation in minimally perturbed blood (4 μg/mL CTI) requires a Factor XIIa/Factor XIa-dependent mechanism via the contact pathway, which is substantially inhibited by CTI during the short duration of the 600-sec experiment. Also, contact activation is generally slower and less efficient than the extrinsic pathway under flow condition.9 Even with a low level of CTI (4 μg/mL), a small amount of thrombin likely was generated to enhance platelet activation and deposition on the 20-μm collagen zone lacking TF, but was insufficient to generate any fibrin (Fig. S1E). Relative to CTI-treated blood, complete inhibition of thrombin generation using PPACK/apixaban caused a significant reduction of platelet deposition on the 20-μm collagen lacking TF (Fig. S1D). For perfusion of CTI-treated whole blood at venous shear rate, there was substantial platelet deposition on 20-μm collagen lacking TF, but much smaller deposits were seen on a single collagen fiber lacking TF (Fig. S2). Platelet deposition on a single collagen fiber (no TF) was essentially negligible upon thrombin inhibition with PPACK/apixaban (Fig. S2C). Thus, the combination of a platelet adhesive and stimulatory surface along with local thrombin generation triggered by TF (or much less potently by contact activation in CTI-treated blood) was required for platelet deposition on a trigger as small as a single collagen fiber.
Patch threshold on collagen/TF at arterial flows when von Willebrand Factor is absent
Under arterial flow with wall shear rate of 1000 s−1, reactive species washout in the boundary layer is greater (Péclet number > 103), platelet collision times are shorter (<5 msec) and shear forces per platelet are substantially greater (>1000 pN).12, 13 Severely impaired thrombus formation was consistently observed on both 20-μm long zones of collagen and single collagen fibers, even in the presence of TF coating the collagen (Fig. 2B, D). On the 20-μm collagen/TF zone, some platelets deposited near the channel side wall where the no-slip condition applies and the flow velocities were lower. However, in the center two-thirds of the rectangular channel (neglecting the side walls) where the wall shear rate was 1000 s−1, platelet and fibrin deposition was negligible (Fig. 2B,D and Fig. S2B). Even with immobilized TF, a single collagen fiber was neither able to capture platelets nor able to support thrombin generation at 1000 s−1 (Fig. 2B,D and Fig. S2). This result was fully consistent with the essential role of VWF required for platelet GPIbα-dependent capture to collagen at arterial shear rates14 that proceeds firm arrest mediated by GPVI-engagement and subsequent α2β1 integrin activation. In the absence of VWF pre-adsorbed to collagen, a patch size threshold existed on 20-μm collagen/TF and on a single collagen fiber/TF due to a fundamental defect in platelet attachment at arterial wall shear rates, even with TF present. The level of wall shear stress (10 dyne/cm2) at this arterial condition was insufficient to cause plasma vWF to unfold and form fiber aggregates on the collagen, as previously observed at pathological wall shear stresses of >300 dyne/cm2 (at wall shear rate of >30,000 s−1) typical of stenosis.15
Single collagen fiber with TF and VWF supports coagulation at arterial shear rate
At an arterial wall shear rate of 1000 s−1, pre-adsorption of VWF to collagen eliminated the patch size threshold and promoted platelet and fibrin deposition on both the 20-μm collagen/TF/VWF and the single collagen fiber/TF/VWF feature (Fig. 3). On collagen/TF/VWF, platelet deposition began immediately after flow initiation. The rate of platelet deposition on the 20-μm collagen/TF/VWF was relatively constant over the 600 sec experiment while a gradual slowing in platelet deposition was observed on a single collagen/TF/VWF fiber after the first 100 sec (Fig. 3B,E). Under arterial flow, fibrin generation was less abundant than that observed for venous flow conditions (Fig. 3C), consistent with previously observations for 250-μm collagen/TF features lacking pre-adsorbed VWF.6 Fibrin generation was however no longer confined near the side walls of the device with reduced flow, as was seen for collagen/TF (no VWF). Fluorescent staining revealed that the captured platelets also released platelet VWF and the clot localized VWF signal increased with time (Fig. 4). In contrast, the single collagen/TF fiber lacking pre-adsorbed VWF did not exhibit the ability to capture either flowing platelets or sufficient plasma VWF from blood (Fig. 4, Fig. 5C). Clearly, pre-adsorbed VWF mediated more efficient platelet deposition than collagen alone, as expected for high shear conditions 16. Furthermore, the presence of TF significantly promoted the probability of platelet deposition on a single VWF coated collagen fiber. Unlike the complete coverage of the single collagen/TF/VWF fiber by platelets and fibrin (Fig. 5A), scattered platelet microaggregation but not fibrin deposition was observed on the single collagen/VWF fiber without immobilized TF (Fig. 5B). The maximum height of the formed clot on a single collagen/TF/VWF fiber was limited to ~10 μm over the collagen fiber on the surface (Fig. S3), considerably smaller than the nearly occlusive clot heights seen over the 20-μm collagen/TF/VWF zone under venous flow conditions. Scanning electron microscopy (SEM) images show only three to four layers of platelet deposition on the single collagen/TF/VWF fiber (Fig. 5 D,E). The bottom layer platelets were fully spread and adherent to the surface, whereas the top layers platelets displayed an activated and rounded morphology with pseudopods.
Fig. 3. For arterial flow, coagulation on collagen/TF fibers requires pre-adsorbed VWF.

CTI-treated whole blood was perfused at arterial wall shear rate 1000 s−1 over 20-μm collagen/TF zone plus VWF or over a single collagen/TF fiber plus VWF. With VWF, the collagen/TF accumulated platelets (red) and fibrin (green) for the 20-μm zone (A) or the single fiber (D) by 600 sec. Onset of platelet deposition on collagen/TF/VWF fibers was instantaneous after flow initiation (B, E). There was ~ 300 sec delay in fibrin production on both 20-μm zone and a single collagen/TF/VWF (C, F). In comparison, both platelet deposition and fibrin formation were essentially absent on collagen/TF lacking VWF (B–E). Vertical dashed lines represent flow channel side walls. Flow direction: top to bottom. Scale bar: 50 μm. (***p < 0.005).
Fig. 4. Incorporation of plasma vWF into formed thrombus on single collagen/TF/VWF fiber.

Detected VWF (cyan) on a single collagen/TF/VWF fiber is co-localized with platelet (red) (A, B). VWF signal on single collagen/TF/VWF increased significantly over time (C). Negative control images confirm that a single collagen/TF fiber lacking pre-adsorped VWF is not able to capture VWF from plasma. All images were taken at the end of flow experiments (t = 600 sec). Vertical dashed lines outline flow channels. Flow direction: top to bottom. Scale bar: 50 μm. (*** p < 0.005).
Fig. 5. VWF and TF synergistically promote coagulation on single collagen fiber at arterial flow.
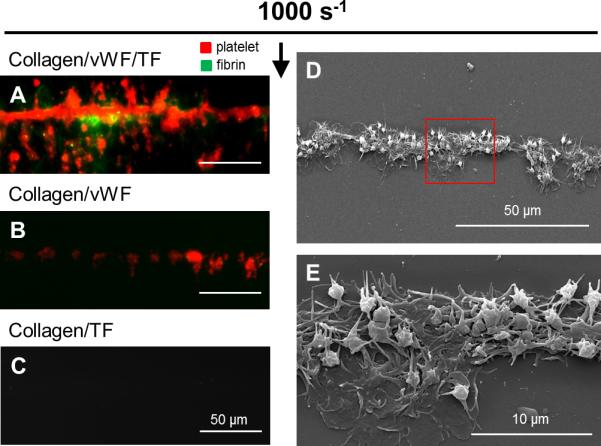
CTI-treated whole blood was perfused at arterial wall shear rate 1000 s−1 over single collagen fibers. Platelets (red) and fibrin (green) coated the single collagen/vWF/TF fiber by the end of 400 sec (A). Scattered small platelet aggregates but not fibrin deposited on the single collagen/vWF fiber (B). No platelet or fibrin was captured on the single collagen/TF fiber lacking VWF (C). Scale bars represent 50 μm in A-C. SEM image showing the structure of a thrombus formed on a single collage/vWF/TF fiber (D). Closer view of the area enclosed in the red box showing three to four layers of platelet deposition on the collagen/vWF/TF fiber (E). Bottom layer platelets were flat and fully adherent to the surface, whereas top layer platelets were activated but unspread. Flow direction: top to bottom.
DISCUSSION
In this microfluidic study with human blood, we define the conditions for flowing blood to clot on features as small as a single collagen fiber. At venous whole blood flow, a single TF-coated collagen fiber promotes robust coagulation in the absence of any apparent patch size threshold. For arterial blood flow over collagen/TF lacking VWF (and platelets), the convective removal of FXa and FIXa from the TF liposomes quenched any observable thrombin production and subsequent fibrin polymerization. To overcome patch size thresholding at arterial flows, human blood exploits VWF to initiate a full coagulation response on a biological unit as small as a single collagen fiber.
Numerous mouse studies,17-19 ex vivo studies with human blood in microfluidic assays,10, 20, 21 and multiscale numerical modeling 22-25 have quantified the complex hemodynamic and transport interactions during coagulation.26 Beyond the highly regulated biochemical networks associated with platelet signal transduction and the coagulation protease cascade, relevant physical processes during hemostasis include: (i) platelet margination to the wall due to red blood cell motions, (ii) the diffusivity of reactive species and their enhanced removal by convection or intraclot permeation, (iii) modulation of intraclot diffusion and permeation by platelet retraction, and (iv) the effect of flow on the rate and adhesive success of platelet encounters with the surface.18, 19, 21, 27-29 Importantly, the structural and kinetic properties of VWF facilitate platelet capture at high shear conditions 14 and VWF deficiency is strongly linked to bleeding phenotypes.30
A plasma or platelet rich plasma (PRP)-based static experimental system does not fully reflect the complexity of whole blood clotting under hemodynamic conditions. With flow, red blood cells drive elevated platelet levels near the wall and this phenomenon does not occur in flowing platelet rich plasma. With flowing blood, the platelet deposition density greatly exceeds that expected under static conditions. Platelet surface area in the dense retracted core of a clot can reach levels as high as 100 cm2/μL of clot.26 With platelet deposition, the membrane surface area is greatly increased for accumulation of coagulation factors (such as FXa), a process absent in plasma studies of patch size thresholding. Deposited platelets can also create restricted transport zones to facilitate assembly of coagulation components in the core of the hemostatic thrombus 10, 17-19 as well as offering α2bβ3 binding sites to anchor fibrin polymerization under flow.
These studies address the minimum length scale and biochemical criteria necessary for a hemostatic response and are fully consistent with the known bleeding risks linked to deficiencies in platelets, thrombin generation, or VWF. Consistent with our findings with human whole blood, in vivo laser injury of the mouse cremaster arteriole causes a micron-scale wall defect 31 to drive platelet deposition, thrombin generation,10, 19 and fibrin polymerization, especially in the core of the clot.17, 18 Clot production under flow is also highly relevant to dangerous thrombotic events of coronary artery occlusion. An important issue related to the patch size threshold is whether a surface defect is of sufficient size and potency to cause vessel occlusion at a given prevailing flow condition. In coronary stenosis, pathological shear rates are many fold greater than the physiological levels of the current study. Collagen triggers plasma VWF fiber formation upon acute exposure of platelet free plasma to a pathological shear of over 30,000 s−1.15 Also, these large insoluble fibers can capture and support shear induced platelet activation.32 The observation of a single collagen fiber coated with TF and VWF supporting clotting is also consistent with an earlier study of whole blood perfused over VWF pre-coated microspheres (2-μm diameter) at a pathological shear rate of 10,000 s−1 with platelet aggregation extending over 5 bead diameters downstream of the initial platelet-bead adhesion.33
The maximum height of microthrombi on single collagen/TF/VWF fibers was ~10 μm with three to four layers of platelets, indicating a self-limiting response at arterial flow condition. The rapid flow-enhanced elution of platelet agonists may be a cause of self-limited clot growth on a single collagen fiber. Even though endothelium was not included in this study, evidence for microthrombi formation at extremely small sites of endothelial erosion on developing coronary plaque is also consistent with our findings.34, 35 Spatial heterogeneity of platelet activation is well documented in laser injury of mouse cremaster arterioles 17 where thrombin 10 and fibrin are localized in the platelet P-selectin positive core. SEM images (Fig. 5) show platelets in individual layers exhibit different morphologies, even for a single fiber triggering event, suggesting heterogeneity in the level of platelet activation along the height of the deposit. Furthermore, we found TF and VWF synergistically promoted thrombus formation on single collagen fibers at arterial flow condition. By capturing platelets with VWF at flow arterial conditions to a single fiber presenting TF, the generated FXa and FIXa were kinetically significant and sufficiently localized to promote continued platelet capture, thrombin generation, and fibrin polymerization. For defects as small as a single collagen fiber, the molecular components of the extrinsic pathway function at arterial flow conditions because of local VWF-dependent platelet deposition. For exceedingly small arterial defects, platelets are particle-based sensors that allow engagement of the coagulation protease cascade under high flow.
METHODS
PDMS patterning and flow devices
Poly(dimethylsiloxane) (PDMS, Ellsworth Adhesives, Germantown, WI) microfluidic devices were fabricated as previous described.36 Single channel (10 or 20 μm in width, 60 μm in height) devices were used for protein patterning on glass slides. The device used for microfluidic thrombosis assay has 8 evenly spaced flow channels (250 μm in width, 60 μm in height) that connect individual cylindrical reservoirs to a single outlet. Both devices can be reversibly mounted on glass slides by vacuum bonding.
Preparation of small collagen patches
Glass slides were treated with Sigmacote® (Sigma-Aldrich, St. Louis, MO) to impede blood clotting outside the patterned prothrombotic surfaces before they were rinsed with DI water and were dried with compressed filtered air. Perfusion of 5 μL of collagen type I (1 mg/mL, Chronolog Corp, Havertown, PA) followed by 20 μL of bovine serum albumin (0.5% BSA in HBS) through the main channel on the patterning device that was mounted on a glass slide resulted in immobilized patches of aligned collagen fibers on the glass slide.36, 37 In order to capture single collagen fibers, collagen type I was diluted (10x dilution, 100 μg/mL) in isotonic glucose buffer before being perfused through 10 μm wide channels. Lipidated TF was added by incubating Dade® Innovin® recombinant human TF (VWR Corp, Radnor, PA) over patterned collagen for at least 30 min before a BSA rinse. VWF was added by incubating collagen fibers with human plasma VWF (30 μg/mL, FVIII free, Haematologic Technologies, Essex Junction, VT) prior to perfusion and an immediate rinse with BSA. Both bright field imaging and fluorescent post-staining were used to ensure the precision of the patterning technique (Fig. 1). For collagen staining, micropatterned zones were stained with biotinylated anti-collagen I antibody (Abcam, Cambridge, MA), which was then detected with Alexa Fluor 488 streptavidin (Life Technologies, Grand Island, NY) before a buffer wash. Annexin V-PE (1% in 5mM CaCl2 buffer, BD Bioscience, San Jose, California) and anti-VWF (0.5% in HBS buffer, Abcam, Cambridge, MA) were subsequently added for fluorescent staining when collagen fibers were precoated with TF liposomes and VWF, respectively.
Blood collection and preparation
Blood was collected via venipuncture from health donors (who were self-claimed to be free of alcohol and medication for at least 72 hr prior to donation) into D-Phe-Pro-Arg-CMA (PPACK, 100 μM, Haemtech, Essex Junction, VT) and apixaban (1 μM, Selleckchem, Houston, TX) or into CTI (4 or 40 μg/mL WB, Haematologic Technologies, Essex Junction, VT). Informed consent was obtained for each donor and performed in accordance with the University of Pennsylvania's IRB approval and the Declaration of Helsinki. Minimal amount of CTI (4 μg/mL) was used to block surface induced clotting during blood collection while still allowing thrombin generation through both the contact and/or the extrinsic pathways depending on TF surface concentration in patterned prothrombotic patches.5, 9 The combination of 100 μM PPACK (irreversible thrombin inhibitor) and 1 μM apixaban (direct Xa inhibitor) was used to achieve complete thrombin inhibition.
Blood was treated with anti-human CD61 antibody (BD Biosciences, San Jose, California) and Alexa Fluor 647 fluorescent fibrinogen (Life Technologies, Grand Island, NY) for platelet and fibrin(ogen) detection, respectively. All experiments were initiated within 5 min after venous phlebotomy. Platelet thrombin biosensor was added into blood in a 1:9 ratio for the observation of thrombin generation in some experiments 10. In these experiments, anti-human CD41a antibody was used for platelet detection.
Microfluidic thrombosis model
The 8-channel flow device was mounted perpendicularly to patterned collagen patches. Blood was perfused over collagen patches at either venous (100 s−1) or arterial initial wall shear rates (1000 s−1). Initial wall shear rate in flow channels was controlled with a syringe pump withdrawal at constant volumetric flow rate at the outlet on the device. Platelet accumulation, fibrin formation, and thrombin generation were monitored with a fluorescence microscope (IX81, Olympus America Inc., Center Valley, PA). Images were captured with a CCD camera (Hamamatsu, Bridgewater, NJ) and were analyzed with ImageJ (NIH, Bethesda, DC). To avoid edge-wall effects, average fluorescence after background subtraction in the center 65% region of the collagen patches were collected and recorded for data analysis.
Scanning electronic microscopy
Thrombi formed on single collagen/TF/VWF fibers were fixed under flow in situ with 2% glutaraldehyde in 0.1 M sodium cacodylate buffer for at least 3 hr at room temperature before they were removed from the PDMS microfluidic devices. The fixed thrombi were then incubated in the same buffer overnight at 4 °C, dehydrated in graded ethanol (with balance of sodium cacodylate buffer), finalized with hexamethyldisilane, air dried and stored under vacuum before sputter coating with gold/palladium and imaged by scanning electron microscopy.
Statistical analysis
Data were compared to controls using two-tail Student's t-test. P-value < 0.05 was considered statistical significant.
CONCLUSIONS
In conclusion, we demonstrated in a microfluidic system that initiation of clotting of flowing human whole blood overcomes a patch threshold response to stimuli patch size when TF and VWF are present. At venous shear rate, the presence of tissue factor can trigger a full clotting response with platelet deposition, thrombin generation, and fibrin polymerization on a single collagen fiber. Pre-adsorbed VWF and TF enabled clotting on a single collagen fiber at arterial shear rate. Blood coagulates on surface triggers as small as a single collagen fiber to obviate any physiological patch size threshold.
Supplementary Material
Insight.
Blood must operate under flow conditions in order to seal vessel wall defects and maintain vasculature integrity. Observations with plasma and platelet rich plasma suggest that a defect may be small enough to escape hemostatic responses because diffusible species cannot reach a local critical concentration, termed a patch size threshold. For blood perfused at venous shear rate across a single collagen fiber coated with tissue factor to trigger coagulation, a full clotting response occurs with platelet deposition, thrombin generation, and fibrin polymerization. For arterial flows, von Willebrand Factor pre-adsorbed on a single collagen fiber with tissue factor enabled clotting. Blood coagulates on surface triggers as small as a single collagen fiber to obviate any physiological patch size threshold.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health (NIH) R01 HL103419 to S.L.D.
Footnotes
† Electronic supplementary information (ESI) available: Supplementary data.
Conflict of interest: The authors have no conflicts of interest to declare.
REFERENCES
- 1.Kastrup CJ, Runyon MK, Shen F, Ismaqilov RF. Proc Natl Acad Sci U S A. 2006;103:15747–15752. doi: 10.1073/pnas.0605560103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kastrup CJ, Shen F, Runyon MK, Ismaqilov RF. Biophys J. 2007;93:2969–2977. doi: 10.1529/biophysj.107.109009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen F, Kastrup CJ, Liu Y, Ismaqilov RF. Arterioscl Thromb Vasc Biol. 2008;28:2035–2041. doi: 10.1161/ATVBAHA.108.173930. [DOI] [PubMed] [Google Scholar]
- 4.Okorie UM, Denney WS, Chatterjee MS, Neeves KB, Diamond SL. Blood. 2008;111:3507–3513. doi: 10.1182/blood-2007-08-106229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu S, Travers RJ, Morrissey JH, Diamond SL. Blood. 2015;126:1494–1502. doi: 10.1182/blood-2015-04-641472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colace TV, Muthard RW, Diamond SL. Arterioscl Thromb Vasc Biol. 2012;32:1466–1476. doi: 10.1161/ATVBAHA.112.249789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonderman D, Teml A, Jakowitsch J, Adlbrecht C, Gyongyosi M, Sperker W, Lass H, Mosgoeller W, Glogar DH, Probst P, Maurer G, Nemerson Y, Lang IM. Blood. 2002;99:2794–2800. doi: 10.1182/blood.v99.8.2794. [DOI] [PubMed] [Google Scholar]
- 8.Walski M, Chlopicki S, Celary-Walska R, Frontczak-Baniewicz M. J Physiol Pharmacol. 2002;53:713–723. [PubMed] [Google Scholar]
- 9.Zhu S, Diamond SL. Thromb Res. 2014;134:1335–1343. doi: 10.1016/j.thromres.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welsh JD, Colace TV, Muthard RW, Stalker TJ, Brass LF, Diamond SL. J Thromb Haemost. 2012;10:2344–2353. doi: 10.1111/j.1538-7836.2012.04928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilcox JN, Smith KM, Schwartz SM, Gordon D. Proc Natl Acad Sci U S A. 1989;86:2839–2843. doi: 10.1073/pnas.86.8.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doggett TA, Girdhar G, Lawshe A, Schmidtke DW, Laurenzi IJ, Diamond SL, Diacovo TG. Biophys J. 2002;83:194–205. doi: 10.1016/S0006-3495(02)75161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mody NA, King MR. Biophys J. 2008;95:2539–2555. doi: 10.1529/biophysj.107.127670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruggeri ZM. Thromb Res. 2007;120(Suppl 1):S5–9. doi: 10.1016/j.thromres.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colace TV, Diamond SL. Arterioscler Thromb Vasc Biol. 2013;33:105–113. doi: 10.1161/ATVBAHA.112.300522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franchini M, Lippi G. Ann Hematol. 2006;85:415–423. doi: 10.1007/s00277-006-0085-5. [DOI] [PubMed] [Google Scholar]
- 17.Stalker TJ, Traxler EA, Wu J, Wannemacher KM, Cermignano SL, Voronov R, Diamond SL, Brass LF. Blood. 2013;121:1875–1885. doi: 10.1182/blood-2012-09-457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stalker TJ, Welsh JD, Tomaiuolo M, Wu J, Colace TV, Diamond SL, Brass LF. Blood. 2014;124:1821–1831. doi: 10.1182/blood-2014-01-550319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welsh JD, Stalker TJ, Voronov R, Muthard RW, Tomaiuolo M, Diamond SL, Brass LF. Blood. 2014;124:1808–1815. doi: 10.1182/blood-2014-01-550335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muthard RW, Diamond SL. Lab Chip. 2013;13:1883–1891. doi: 10.1039/c3lc41332b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muthard RW, Diamond SL. Arterioscl Thromb Vasc Biol. 2012;32:2938–2945. doi: 10.1161/ATVBAHA.112.300312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flamm MH, Colace TV, Chatterjee MS, Jing H, Zhou S, Jaeger D, Brass LF, Sinno T, Diamond SL. Blood. 2012;120:190–198. doi: 10.1182/blood-2011-10-388140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leiderman K, Fogelson AL. Bull Math Biol. 2013;75:1255–1283. doi: 10.1007/s11538-012-9784-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leiderman K, Fogelson AL. Math Med Biol. 2011;28:47–84. doi: 10.1093/imammb/dqq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beltrami E, Jesty J. Math Biosci. 2001;172:1–13. doi: 10.1016/s0025-5564(01)00064-5. [DOI] [PubMed] [Google Scholar]
- 26.Brass LF, Diamond SL. J Thromb Haemost. 2016 doi: 10.1111/jth.13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fogelson AL, Neeves KB. Annu Rev Fluid Mech. 2015;47:377–403. doi: 10.1146/annurev-fluid-010814-014513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeh C, Eckstein EC. Biophys J. 1994;66:1706–1716. doi: 10.1016/S0006-3495(94)80962-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vahidkhah K, Diamond SL, Bagchi P. Biophys J. 2014;106:2529–2540. doi: 10.1016/j.bpj.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mannucci PM. N Engl J Med. 2004;351:683–694. doi: 10.1056/NEJMra040403. [DOI] [PubMed] [Google Scholar]
- 31.Hechler B, Nonne C, Eckly A, Magnenat S, Rinckel JY, Denis CV, Freund M, Cazenave JP, Lanza F, Gachet C. J Thromb Haemost. 2010;8:173–184. doi: 10.1111/j.1538-7836.2009.03666.x. [DOI] [PubMed] [Google Scholar]
- 32.Herbig BA, Diamond SL. J Thromb Haemost. 2015;19:1699–1708. doi: 10.1111/jth.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nesbitt WS, Westein E, Tovar-Lopez FJ, Tolouei E, Mitchell A, Fu J, Carberry J, Fouras A, Jackson SP. Nat Med. 2009;15:665–673. doi: 10.1038/nm.1955. [DOI] [PubMed] [Google Scholar]
- 34.Ross R. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 35.Davies MJ. N Engl J Med. 1997;336:1312–1314. doi: 10.1056/NEJM199705013361809. [DOI] [PubMed] [Google Scholar]
- 36.Neeves KB, Maloney SF, Fong KP, Schmaier AA, Kahn ML, Brass LF, Diamond SL. J Thromb Haemost. 2008;6:2193–2201. doi: 10.1111/j.1538-7836.2008.03188.x. [DOI] [PubMed] [Google Scholar]
- 37.Maloney SF, Brass LF, Diamond SL. Integr Biol. 2009;2:183–192. doi: 10.1039/b919728a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.