

Abstract
Buprenorphine has long been classified as a mu analgesic, although its high affinity for other opioid receptor classes and the orphanin FQ/nociceptin ORL1 receptor may contribute to its other actions. The current studies confirmed a mu mechanism for buprenorphine analgesia, implicating several subsets of mu receptor splice variants. Buprenorphine analgesia depended upon the expression of both exon 1-associated traditional full length 7 transmembrane (7TM) and exon 11-associated truncated 6 transmembrane (6TM) MOR-1 variants. In genetic models, disruption of delta, kappa1 or ORL1 receptors had no impact on buprenorphine analgesia, while loss of the traditional 7TM MOR-1 variants in an exon 1 knockout (KO) mouse markedly lowered buprenorphine analgesia. Loss of the truncated 6TM variants in an exon 11 KO mouse totally eliminated buprenorphine analgesia. In distinction to analgesia, the inhibition of gastrointestinal transit and stimulation of locomotor activity were independent of truncated 6TM variants. Restoring expression of a 6TM variant with a lentivirus rescued buprenorphine analgesia in an exon 11 KO mouse that still expressed the 7TM variants. Despite a potent and robust stimulation of 35S-GTPγS binding in MOR-1 expressing CHO cells, buprenorphine failed to recruit β-arrestin-2 binding at doses as high as 10 μM. Buprenorphine was an antagonist in DOR-1 expressing cells and an inverse agonist in KOR-1 cells. Buprenorphine analgesia is complex and requires multiple mu receptor splice variant classes, but other actions may involve alternative receptors.
Keywords: MOR-1, opioid, 6TM
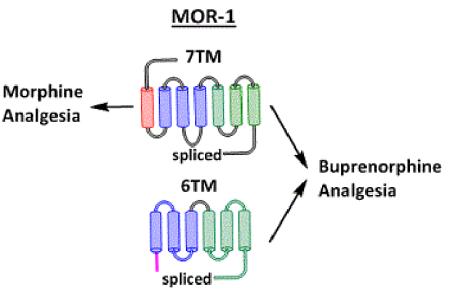
The mu opioid receptor gene Oprm1 undergoes alternatively splicing to generate several sets of splice variants. Traditional 7TM MOR-1 variants mediate morphine actions. The current study shows that buprenorphine produces analgesia through a combination of 7TM and 6TM splice variants.
Introduction
Buprenorphine, first synthesized and evaluated over 40 years ago, has a long clinical history (Bentley and Hardy, 1967; Campbell and Lovell, 2012; Lewis, 1985; Pergolizzi et al., 2010). Early studies reported an intriguing pharmacological profile (Cowan et al., 1977; Lewis, 1985), with its actions extending beyond analgesia, possibly influenced by its high binding affinity for all the traditional opioid receptors, including the ORL1 receptor (Khroyan et al., 2015). It has been studied extensively (Cowan et al., 1977; Ding and Raffa, 2009; Dum and Herz, 1981; Huang et al., 2001; Kamei et al., 1995a; Kamei et al., 1995b; Kamei et al., 1997; Leander, 1987; Lewis, 1985; Lutfy and Cowan, 2004; Lutfy et al., 2003; Pick et al., 1997; Romero et al., 1999; Virk et al., 2009; Walker et al., 1995). Its analgesic activity has long been classified as mu. In some paradigms, buprenorphine is a partial mu agonist, antagonizing some morphine actions and precipitating withdrawal signs in morphine dependent mice and monkeys, but not rats. There also is some controversy regarding its sensitivity to reversal by naloxone, with some observations suggesting the need for higher doses for reversal than prevention. A recent report suggests two mechanisms of buprenorphine’s actions are based upon the greater sensitivity of its spinal, but not supraspinal, actions to naloxone (Ding and Raffa, 2009). Loss of the ORL1 receptor reportedly enhances buprenorphine analgesia, implying an inhibitory mu/ORL1 interaction (Lutfy et al., 2003) and raising the possibility that activation of ORL1 receptors by higher buprenorphine doses may explain its biphasic dose-response.
Our understanding of the molecular pharmacology of mu receptors has evolved since the early studies of buprenorphine pharmacology. The mu opioid receptor gene contains two distinct promoters and undergoes extensive splicing to generate three classes of splice variants (Table 1) (Pasternak and Pan, 2013). The promoter associated with exon 1 generates full length, 7 transmembrane (7TM) variants and a single transmembrane (1TM) protein due to exon skipping with both also undergoing 3’ splicing to yield differing C-terminal sequences. A second promoter is associated with exon 11, which is located approximately 30 kb upstream of exon 1. The exon 11 promoter generates a set of truncated 6 transmembrane (6TM) domain variants (Pan et al., 2001). Similar splicing profiles exist in mice, rats and humans (Pasternak and Pan, 2013). Each of these sets of splice variants is pharmacologically distinct and important. The full length 7TM variants mediate the analgesic actions of traditional mu opioids such as morphine and methadone. Although the 1TM variants do not bind opioids, they potentiate opioid action through a chaperone-like action that enhances the stability and thereby the levels of 7TM receptors (Xu et al., 2013). The role of the truncated 6TM variants was initially uncovered using 3-iodobenzoyl-6β-naltrexamide (IBNtxA) that revealed a unique pharmacological profile (Majumdar et al., 2011; Majumdar et al., 2012; Pasternak and Pan, 2013). Although IBNtxA is a potent analgesic, it lacks respiratory depression, does not produce physical dependence with chronic administration and lacks reward behavior in the conditioned place preference assay. Furthermore, IBNtxA analgesia can be readily antagonized by levallorphan, but not as easily by naloxone. On the surface, some of these pharmacological characteristics resemble the original descriptions of buprenorphine, such as the diminished sensitivity to naloxone. Furthermore, buprenorphine has high affinity for a 125I-IBNtxA binding site in brain corresponding to a 6TM target that is distinct from traditional opioid receptors or from ORL1 and that does not bind morphine or orphinan FQ/nociceptin (Majumdar et al., 2011). The current study focuses upon the receptor mechanisms of buprenorphine analgesia.
Table 1.
Classification of mouse knockout models
| Knockout model | ||||||
|---|---|---|---|---|---|---|
| E1 | E11 | E1/E11 | KOR-1 KO | Triple KO | ORL1 KO | |
| Mu (MOR-1) | ||||||
| 7TM | Lost | Retained | Lost | Retained | Lost | Retained |
| 6TM | Retained | Lost | Lost | Retained | Retained | Retained |
| 1TM | Retained | Retained | Lost | Retained | Lost | Retained |
| Kappa (KOR-1) | Retained | Retained | Retained | Lost | Lost | Retained |
| Delta (DOR-1) | Retained | Retained | Retained | Retained | Lost | Retained |
| ORL1 | Retained | Retained | Retained | Retained | Retained | Lost |
Mice have been generated that lack exon 1 (E1 KO) (Schuller et al., 1999), exon 11 (E11 KO) (Pan et al., 2009) or both exon 1 and exon 11(E1/E11 KO) (Lu et al., 2015) of the Oprm1 gene. The E1 KO mice lack all the 7TM and 1TM variants due to the absence of exon 1, but still express the 6TM exon 11 variants. Conversely, the E11 KO mice lack the 6TM variants and still express the 7TM and 1TM variants. The E1/E11 KO mice lack all mu opioid receptor splice variants due to the loss of both exons 1 and 11 and their promoters. The delta receptor knockout targeted exon 2 of the Oprd1 gene ((Zhu et al., 1999) while another mouse targeted the kappa receptor (KOR-1 KO) (Zhang et al., 1998). Triple KO mice were generated by backcrossing E1 KO, KOR-1 KO and DOR-1 KO mice. The ORL1 KO has a disruption in exons 2 and 3 of Oprl1 gene. The summary represents KO mice specifically referenced. Other Oprm1, kappa, delta and ORL1 KO mice may differ in their receptor variant expression profiles and pharmacology.
Materials and Methods
Drugs
Buprenorphine HCl, morphine sulfate, naloxone, DAMGO ([D-Ala2,D-MePhe4,Gly(ol)5]enkephalin), DPDPE ([D-Pen2,D-Pen5]enkephalin), and U50,488H were provided by the Research Technology Branch of the National Institute on Drug Abuse (Rockville, MD). Drugs were dissolved in 0.9% sterile saline at a concentration of 0.1 - 1 mg/mL for behavioral studies. [35S]-GTPγS was purchased from Perkin Elmer (Waltham, MA).
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee of the Memorial Sloan-Kettering Cancer Center or UMDNJ-RWJMS, and were conducted in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals in facilities accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). Animals were maintained on a 12-h light/dark cycle with Purina rodent chow and water available ad libitum, and were housed in groups of 5 until testing.
CD-1 and C57BL/6 mice were obtained from Charles River Laboratories. Exon 11 MOR-1 KO animals were generated by our laboratory as previously described (Pan et al 2009), and were backcrossed to a C57BL/6 background through speed congenic breeding (The Jackson Laboratory). Exon 1 MOR-1, KOR-1, DOR-1, ORL1, and triple KO (TKO) were generated in the laboratory of John Pintar (Zhang et al 1998, Schuller et al 1999, Zhu et al 1999, Clarke 2002) and were maintained on an inbred 129/Sv background. Double exon 1/exon 11 knockout mice (E1/E11 KO) on a mixed 129-C57BL/6 background were generated as previously reported (Lu et al., 2015). All in vivo testing of KO models used the corresponding background strain for comparison. Since the E1/E11 KO were not congenic, we used mixed background controls for these animals.
Lentivirus production and injection
Lentiviral constructs and lentivirus production were produced as described previously (Lu et al., 2015). Briefly, the 6TM mMOR-1G cDNA was subcloned into a modified pWPI vector that independently expresses an enhanced green fluorescence protein (EGFP) (a gift from Dr. Didier Trono, Ecole Polytechnique Federale de Lausanne, Switzerland) to construct the lenti-mMOR-1G. Two lentiviruses, one expressing mMOR-1G and EGFP and the other, which expresses only EGFP (as a vector control), were generated in human embryonic kidney (HEK) 293T cells by co-transfecting the lenti-mMOR-1G construct or pWPI vector with PAX2 (a packaging vector) and pMD2 (an envelope vector) using FuGENE HD transfection reagent (Promega). The viral titer of the concentrated lentiviral particles was determined by quantifying EGFP-expressing cells in infected HEK293T cells with different dilutions using fluorescent microscope. Two μl of the lentiviral particles expressing mMOR-1G or vector alone without insertion (1.5 × 109 transducing units/ml) were administrated intrathecally or intracerebroventricularly in E11 KO or E1/E11 KO mice on days 1, 3, and 5 under general halothane anesthesia. Under these conditions, protein expression from the virus progressively increases over a month and then remains stable for at least 14 weeks (Lu et al., 2015). All drug testing was carried out between 5 and 14 weeks post viral injection.
Tail Flick Analgesia
ED50 values were determined using a cumulative dose-response approach to measure tail flick latency following a radiant heat stimulus (Majumdar et al., 2011; Majumdar et al., 2012; Rossi et al., 1996). Baseline latencies were typically around 2.5 s. Following baseline latency determinations, each animal was injected subcutaneously with escalating doses of buprenorphine and tested 30 minutes after the previous injection at peak effect. A maximal latency of 10 s was employed to minimize tissue damage.
Tailflick latencies were converted to %MPE by the formula %MPE = (Observed latency – Baseline latency) / (10 – Baseline Latency) * 100%. Analgesia also was assessed quantally as a doubling or greater of the baseline latency for the individual animal. Dose-response curves were fit by nonlinear regression by GraphPad Prism (Carlsberg, CA). Dose-response curves were compared using an extra sum-of-squares F test.
Gastrointestinal motility assay
Gastrointestinal transit was measured as previously described (Pan et al., 2009). Briefly, animals (n=6-7 per group) were injected with either saline or buprenorphine (0.3 mg/kg in saline, s.c.) and 10 minutes later received a charcoal meal (10% charcoal and 2.5% gum tragacanth in distilled water) by gavage. Thirty minutes after administration of the charcoal meal, animals were sacrificed by cervical dislocation and the charcoal meal transit distance was measured and expressed as a fraction of the total distance from the pyloric sphincter to the cecum.
Respiratory rate assay
Respiratory rates were determined using the MouseOx system (Starr Life Sciences Corp, Oackmont, PA). In brief, the neck region of the mice were shaved approximately 24hrs prior to respiratory testing. On the day of testing, a sensor that was connected to the MouseOx system (Rev. 6.3) was placed around the neck and a baseline respiratory depression determined. For the baseline, measurements were taken for 5 sec every 5 min until a stable baseline was obtained for at least 25 min. Mice then received the indicated drug or saline subcutaneously and measurements taken for 5 sec every 5 min over at least 50 min.
Locomotor activity assay
Locomotor activity was assessed using open field activity chambers (MED-OFA-510, MedAssociates, St. Albans, VT). Animals (n=7 per genotype) were habituated to the testing room for at least 1 hour prior to testing. On Day 1, animals were injected with saline and immediately placed in the activity chamber; at the same time on Day 2, the same animals received buprenorphine (3 mg/kg in saline, s.c.). Their movements were recorded for 1 hour following injection using MedAssociates’ Activity Monitor software.
Stimulation of 35S-GTPγS binding
[35S]-GTPγS Assays were performed based upon published methods (Bolan et al., 2004; Pan et al., 2005; Selley et al., 1998). Membrane homogenates from C57 mouse brain (25 μg protein) or CHO cells stably transfected with mMOR, mDOR, or mKOR (50 μg protein) were incubated for 1 hr at 30oC with the indicated drug, 35S-GTPγS (0.05 nM) and GDP (60 μM in cell lines and 40 μM in brain) in a final volume of 1mL assay buffer containing Tris HCl (50 mM; pH 7.4 at 37oC), MgCl2 (3 mM), EGTA (0.2 mM), NaCl (100 mM), and a protease inhibitor cocktail (leupeptin, bestatin, aprotinin, and peptstatin). GDP concentrations were optimized for each receptor assay: DOR-1 and KOR-1, 10μM; MOR-1, 30μM; brain, 60μM). Nonspecific binding was assessed by the addition of 10 μM cold GTPγS. Binding was terminated by vacuum filtration through Whatman GF/C glass fiber filters which were rinsed 3x2mL with cold Tris HCl. Filters were cut out and 3mL of scintillation fluor (Liquiscint, National Diagonistics, Atlanta, GA) was added to each tube and incubated at room temperature for at least 2 hours before being counted on a Packard Tri-Carb TR-2900 liquid scintillation counter.
β-Arrestin-2 recruitment assay
β-arrestin-2 recruitment was determined using the PathHunter© enzyme complementation assay (DiscoveRx, Fremont, CA) using an engineered MOR-1 in CHO cells (DiscoveRx, Fremont, CA). Cells were plated at a density of 2500 cells/well in a 384-well plate as described in the manufacturer’s protocol. The following day, cells were treated with the indicated compound for 90 minutes at 37°C followed by incubation with PathHunter© detection reagents for 60 minutes. Chemiluminescence was measured with an Infinite M1000 Pro plate reader (Tecan, Männedorf, Switzerland).
Results
Buprenorphine analgesia
Buprenorphine was a potent analgesic in CD-1 mice regardless of whether the effects were assessed quantally or as %MPE (Fig. 1). Quantal analysis of the dose-response yielded an ED50 of 0.033 mg/kg, s.c. while %MPE gave an ED50 value of 0.051 mg/kg, s.c. It has been reported that buprenorphine analgesia is not easily reversed by naloxone (Cowan et al., 1977; Ding and Raffa, 2009; Lewis, 1985), but this may be dependent both upon the site of administration, the species/strain of animal and whether or not it was administered prior to or after buprenorphine. In our studies, prior naloxone readily blocked systemic buprenorphine analgesia in CD-1 mice (Fig. 2).
Figure 1. Buprenorphine analgesia in CD-1 mice.
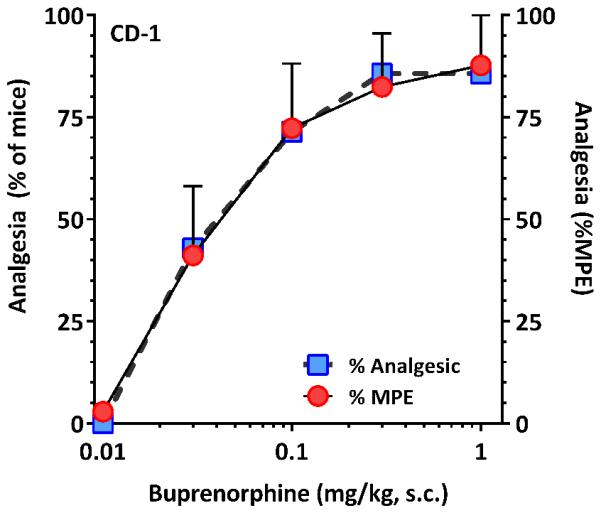
A group of male mice (n = 7) were injected with escalating doses of buprenorphine (0.01, 0.3, 0.1, 0.3, 1 mg, kg, s.c.) and their tail flick latencies tested 30 minutes after the injection to generate dose-response curves. Analysis of the data using a quantal approach in which analgesia was defined as a doubling or greater of the individual subject’s baseline value gave ED50 (95% CL) of 0.033 mg/kg (0.015, 0.073). Analysis of the same curves using graded %MPE approach yielded an ED50 value of 0.051 mg/kg (0.026, 0.103).
Figure 2. Reversal of buprenorphine analgesia by opioid antagonists.
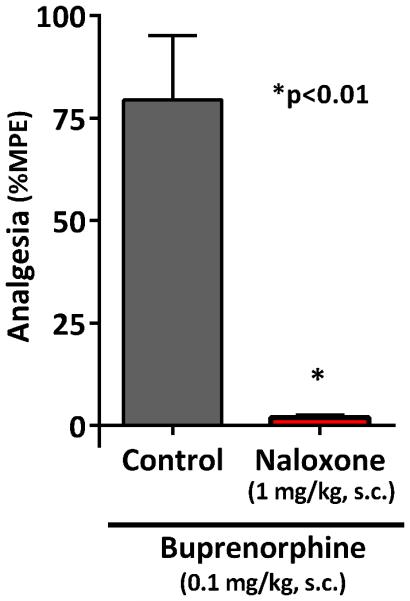
Groups of male CD-1 mice (n = 10 for each group) received buprenorphine alone (0.1mg/kg, s.c.) or in combination with naloxone (1 mg/kg, s.c). Naloxone readily prevented buprenorphine analgesia (p < 0.01, two-tailed t-test).
Knockout mice offer an approach to genetically assess the contributions of various receptors in behavioral actions. We recently generated a full MOR-1 knockout mouse by disrupting both exon 11 and exon 1 (E1/E11 KO) (Lu et al., 2015). Like all the mu opioids tested, buprenorphine was inactive in these mice, confirming that buprenorphine analgesia is completely dependent upon mu receptors, similar to a previous report in an exon 2 MOR-1 KO mouse (Lutfy et al., 2003). We then independently examined the role of the two major sets of MOR-1 splice variants, those associated with exon 1 and those associated with exon 11. Disrupting exon 11 eliminates the truncated 6TM variants, leaving the traditional full length 7TM variants, which explains why morphine and methadone retain full analgesic potency in the E11 KO mice (Pan et al., 2009). Conversely, disruption of exon 1 in the Pintar E1 MOR-1 KO (Schuller et al., 1999) mice removes all the 7TM and 1TM variants while 6TM variants continue to be expressed. Buprenorphine was analgesic in wildtype C57 mice with a potency (ED50 0.028 mg/kg, s.c.) similar to that in the CD-1 mice (Fig. 3a). However, the buprenorphine response in the E11 KO mice was not significantly different from baseline values at all doses except 1 mg/kg. Buprenorphine analgesic efficacy was diminished by approximately 65% in the E1 KO mice, with no appreciable change in potency (ED50 0.064 mg/kg, s.c.) (Fig. 3b).
Figure 3. Buprenorphine analgesia in MOR-1 exon 11 and exon 1 knockout mice.

Mice were injected with escalating doses of buprenorphine and their tail flick latencies tested 30 minutes later to generate dose-response curves. Results are pooled from 2 independent experiments giving similar results and are expressed as mean ± SEM. A) Buprenorphine analgesia (0.012, 0.05, 0.1, 0.3, 1 3, 10 mg/kg, s.c.) is lost in MOR-1 exon 11 knockout mice. Buprenorphine was a potent analgesic in wildtype C57BL/6 mice (n = 9), with an ED50 value (95%CL) 0.028 mg/kg (0.015 – 0.051), while in MOR exon 11-knockout animals (n = 20) it showed only a slight elevation over baseline latency that was not statistically significant. There was a significant difference between curves (p<0.0001, Extra sum-of-squares F test). B) Buprenorphine analgesia (0.03, 0.1, 0.3, 1 mg/kg, s.c.) was significantly reduced in MOR-1 exon 1 knockout mice. Buprenorphine was also a potent analgesic in wildtype 129S6 mice (n = 20-24) with an ED50 value (95%CL) of 0.079 mg/kg (0.048 – 0.13). In MOR exon 1 KO (n = 12), buprenorphine significantly increased latencies over baseline at 0.1 (p<0.003), 0.3 (p<0.001) and 1.0 (p<0.0001) as determined by ANOVA with a ceiling effect of approximately 35% of MPE with an ED50 (95%CL) value of 0.064 mg/kg (0.011 – 0.37).
To further define buprenorphine effects on traditional 7TM mu opioid receptors, we took advantage of the fact that morphine retains full analgesic activity in E11 KO mice while buprenorphine alone is inactive (Majumdar et al., 2011; Pan et al., 2009). In these E11 KO mice, the initial dose of morphine (10 mg/kg, s.c.) elicited nearly a full response that was not significantly influenced by the subsequent dose of saline, but was lowered, but not eliminated, by buprenorphine (Fig. 4). This would be consistent with partial agonist actions at the 7TM target.
Figure 4. Effect of buprenorphine on morphine analgesia in MOR exon 11 knockout mice.
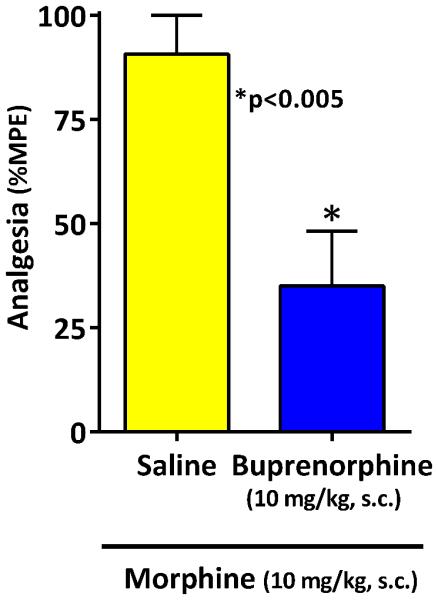
E11 KO mice (21) were given morphine (10 mg/kg) and their tail flick latencies tested 30 minutes later at peak effect. Animals were assessed and were randomly assigned to receive either saline (n=10) or buprenorphine (n=11; 10 mg/kg). 15 minutes after receiving the second injection, animals were retested in the tail flick test. Buprenorphine significantly decreased morphine analgesia relative to saline control (**, p<0.003, 2-tailed t-test). Results are pooled from 2 independent experiments with similar results and are expressed as mean ± SEM.
Delta, kappa1 and ORL1 receptors did not play an observable role in buprenorphine analgesia. Buprenorphine analgesia was not significantly altered in knockout mice with disruptions of kappa (KOR-1) or the orphanin FQ/nociceptin (ORL1) receptors (Fig. 5). A prior report suggested that loss of ORL1 receptors enhanced the buprenorphine response due to the loss of the pronociceptive effect of ORL1 stimulation by buprenorphine (Lutfy et al., 2003). This would be consistent with earlier reports showing that ORL1 receptors mediate both pronociceptive and antinociceptive responses depending upon the dose and assay (Meunier et al., 1995; Reinscheid et al., 1995; Rossi et al., 1997). We did not observe an increased response in the ORL1 knockout mice, but this likely reflected differences in the experimental paradigms. The increased response in the Lufty et al report reflected in increase in efficacy (i.e. an increased maximal reponse) rather than a shift in the dose reponse curve. We would not have been able to detect these differences since buprenorphine was a full agonist in our paradigm. All traditional 7TM opioid receptors can be eliminated by crossing exon 1 MOR-1 KO mice with KOR-1 and delta (DOR-1) knockout mice to generate a triple KO mouse (Cox et al., 2005), although the animals still express truncated exon 11-associated MOR-1 variants (Majumdar et al., 2011). Buprenorphine analgesia in the triple KO mice was similar to that seen in the E1 MOR-1 KO animals, indicating that the additional loss of delta and kappa1 receptors did not influence the response. Again, buprenorphine was a partial agonist with a maximal response of approximately 30% with no change in ED50. Thus, the residual analgesia seen in these E1 knockout animals could not result from activation of delta, kappa1 or traditional mu receptors.
Figure 5. Buprenorphine analgesia in DOR, KOR, ORL-1, and triple opioid receptor knockout mice.
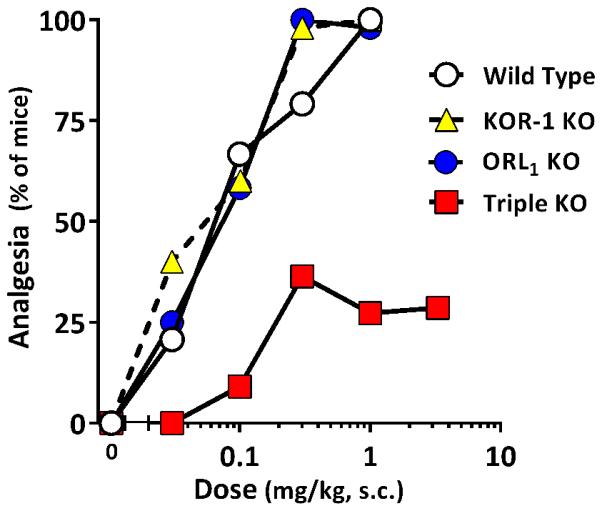
Mice were injected with escalating doses of buprenorphine and their tail flick latencies tested 30 minutes later to generate dose-response curves. KOR-1 (n=5; 0.03, 0.1, .3, 1 mg/kg, s.c.), and ORL1 (n=12; 0.03, 0.1, .3, 1 mg/kg, s.c.) knockout animals displayed no difference from wildtype 129S6 controls, while a triple opioid receptor knockout animal (n = 7-11; 0.03, 0.1, .3, 1, 3 mg/kg, s.c.) lacking MOR exon 1, as well as DOR-1 and KOR-1, reached a plateau with an ED50 (95%CL) 0.067 mg/kg (0.017 – 0.26) and a maximal response of 30% of animals, results similar to the MOR exon 1 alone knockout mice. There was no significant difference between dose response curves for MOR exon 1 and triple KO animals, and no significant difference between the ED50 value of the triple KO and the other genotypes, but the maximal effect for the triple knockout was significantly lower (p<0.0001; Extra sum-of-squares F test).
Rescue of buprenorphine analgesia in an exon 11 knockout mouse
Buprenorphine was inactive in the double E1/E11 KO mice, as were the other opioids tested (Lu et al., 2015). IBNtxA (3’-iodobenzoyl-6β-naltrexamide) analgesia is dependent upon exon 11-associated variants and independent of all the classical 7TM mu, delta and kappa1 opioid receptors (Majumdar et al., 2011). In the E1/E11 KO mice, IBNtxA analgesia could be rescued by a lentivirus expressing the 6TM variant mMOR-1G given intrathecally (Lu et al., 2015). The failure of this paradigm to rescue buprenorphine analgesia in that study, along with the knockout studies described above, suggested that buprenorphine analgesia required both exon 1 and exon 11-associated variants. To assess this possibility, we administered the lentivirus expressing the 6TM mMOR-1G into E11 KO mice, which still natively expressed exon 1-associated variants, enabling the reconstitution of the necessary repertoire of both 7TM and 6TM MOR-1 variants in the E11 KO mice (Fig 6). We initially examined intrathecal administration of the lentivirus followed by systemic buprenorphine since the 6TM variants are most highly expressed at the spinal level (Xu et al., 2015; Xu et al., 2014). As expected, buprenorphine analgesia was markedly reduced in the E11 KO mice alone. Administration of the lentivirus control vector lacking the 6TM variant failed to rescue the response. However, the lentivirus expressing the 6TM variant mMOR-1G at the spinal level fully restored buprenorphine analgesia (Fig. 6a). Similarly, the lentivirus expressing mMOR-1G injected intracerebroventricularly fully rescued buprenorphine analgesia (Fig. 6b).
Figure 6. Rescue of buprenorphine analgesia in an exon 11 knockout mouse by a lentivirus expressing mMOR-1G.

A lentivirus expressing EGFP and mMOR-1G or EGFP alone (vector) was injected and tested for analgesia by buprenorphine given systemically (1 mg/kg, s.c.) at least 5 weeks later. a) Intrathecal lentivirus: Groups of mice (WT: n=9; E11 KO: n=7; E11 KO/Vector: n=8; E11 KO/MOR-1G: n=10) received the indicated treatment. Data was analyzed with one way ANOVA followed by Tukey’s multiple comparison test. Buprenorphine analgesia was significantly lowered in E11 KO animals relative to wildtype (WT) controls (p <0.05). E11 animals treated with vector control lentivirus were no different from untreated E11 KO animals. However, analgesia was restored in E11 KO animals treated with viral mMOR-1G (p <0.001 vs E11 KO/Vector, p<0.001 vs E11 KO). There was no significant difference between analgesia in the WT or in mice give the lentivirus with the mMOR-1G. b) Intracerebroventricular lentivirus: Groups of mice (WT: n= 9; E11 KO: n= 9; E11 KO/MOR-1G: n= 8) received the indicated treatment and were tested at least 5 weeks later. Data was analyzed by one way ANOVA followed by Tukey’s multiple comparison test. Buprenorphine analgesia was significantly lowered in the E11 KO compared to both WT (p<0.001) and lentivirus with mMOR-1G (p<0.01). There was no significant difference between analgesia in the WT and mice given the lentivirus with the mMOR-1G.
Buprenorphine’s effects on respiratory depression
Respiratory depression is a major problem with opioid use. Buprenorphine showed a slight decrease in respiratory rates of up to about 25% at a dose 5-fold greater than its analgesic ED50 (Fig. 7). However, this effect was less pronounced than an equipotent dose of morphine that lowered rates by almost 50% (Fig. 7).
Figure 7. Effect of buprenorphine on respiratory rate.
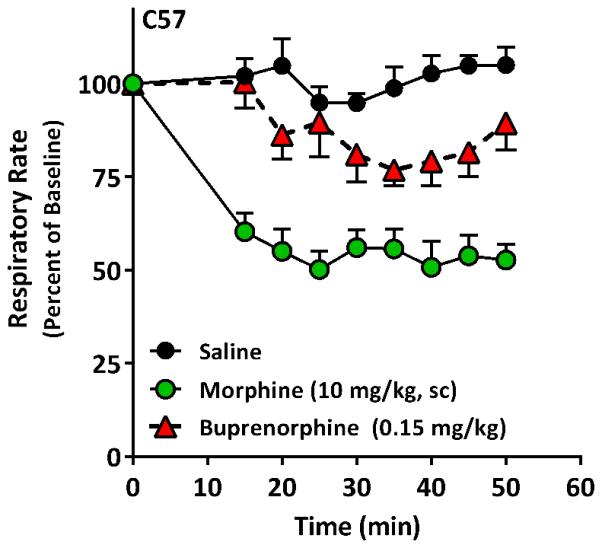
A group of male C57 mice received buprenorphine (0.15 mg/kg, n=5), morphine (10 mg/kg, n=5) or saline (n=6). Respiratory rate was determined using a MouseOx system. After ensuring a stable baseline for 25 min, the animals received the indicated drug s.c.. Both buprenorphine and morphine doses were equivalent to 5-fold their respective analgesic ED50 in previous experiments. Significance was determined by repeated measures ANOVA followed by Bonferroni multiple comparison test. All morphine points were significantly different from saline (p<0.001). Buprenorphine was significantly different from saline at 20 min (p<0.05) nad at time points 35, 40 and 45 (p<0.01).
Buprenorphine’s effects on gastrointestinal transit
Like other opioids, buprenorphine inhibits gastrointestinal transit, a major contributor to constipation. Despite the full loss of analgesia in exon 11 KO mice, buprenorphine continued to potently inhibit gastrointestinal transit (Fig. 8). Thus, buprenorphine inhibition of gastrointestinal transit does not involve exon 11-associated variants. The transit distance in the saline groups was increased in the E11 KO mice relative to wildtype, raising the possibility of a tonic inhibition of transit through this target.
Figure 8. Effect of buprenorphine on GI transit in MOR exon 11 knockout mice.
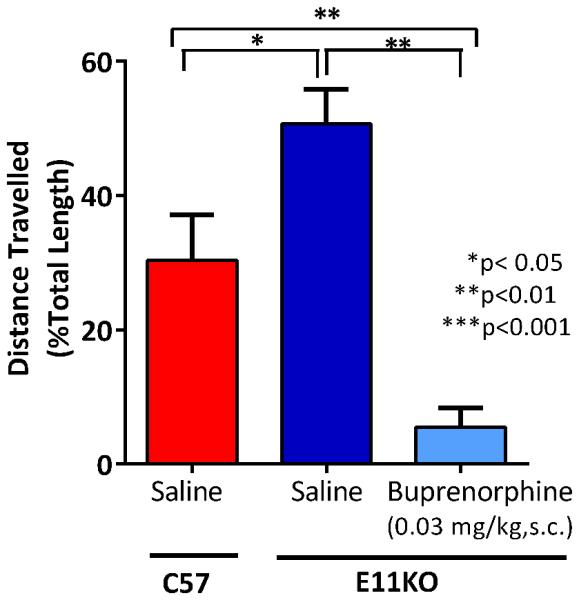
The control level of GI transit was assessed in wildtype C57 mice (n = 6). In exon 11 knockout mice groups (n = 7) were injected with either saline or buprenorphine (0.3 mg/kg s.c.) and 10 minutes before a charcoal meal by gavage. Transit was measured 30 minutes after administration of the charcoal meal, animals were sacrificed by cervical dislocation and the distance traveled by the charcoal meal was measured as a fraction of the total distance from the pyloric sphincter to the cecum. Buprenorphine significantly decreased gastrointestinal motility relative to both saline-treated Exon 11 knockout controls (p < 0.0001, post-hoc Bonferroni multiple comparisons test following 1-way ANOVA) and saline-treated wildtype C57 controls (p < 0.01). The saline groups in control and E11 KO mice also were different (p < 0.05). Results are mean ± SEM.
Buprenorphine’s effects on locomotion
Mu opioids classically increase locomotion in mice, presumably through the disinhibition of dopamine release in the mesolimbic dopamine pathway (Johnson and North, 1992; Joyce and Iversen, 1979; Urs et al., 2011). Buprenorphine stimulated locomotion is lost in an exon 2 KO mouse which presumably lacks both 7TM and 6TM variants (Marquez et al., 2007). In our studies, buprenorphine increased locomotion in wildtype C57 mice (Fig. 9), with a peak effect at 10 min that gradually declined over an hour. A similar response was observed in the exon 11 knockout mouse. The cumulative distance traveled also did not differ between the C57 and the knockout mice in either treatment group. Coupled with the earlier observation in the exon 2 KO mouse, these findings indicate locomotion was mediated through 7TM variants and not 6TM ones.
Figure 9. Effect of buprenorphine on locomotor activity in MOR exon 11 knockout mice.

Animals (n = 7 for each group) were injected with saline or buprenorphine (3 mg/kg s.c.), and their locomotor behavior recorded in an open field activity chamber for 60 minutes post-injection. Results are mean ± SEM. A) Total distance traveled during the 60 min session was significantly increased by buprenorphine injection in both genotypes relative to saline vehicle control (p < 0.0001 for a main effect of drug, 2-way repeated measures ANOVA; p<0.001 for C57 and p<0.0001 for E11KO, post-hoc Tukey’s multiple comparisons test); however, there was no difference between genotypes for either saline or buprenorphine injection (p = 0.39 for a main effect of genotype, p = 0.12 for drug x genotype interaction; p = 0.95 for saline comparison, p = 0.31 for buprenorphine comparison) B) Time course of locomotor activity, 5 minute intervals. Within 10 minutes post-injection, buprenorphine significantly increased locomotor behavior in both MOR exon 11 knockout animals and wildtype C57 controls (p < 0.001 for each point minutes 10-60; post-hoc Tukey’s multiple comparisons test). There was no significant difference between genotypes at any time point for either saline or buprenorphine condition.
Buprenorphine’s effects on 35S-GTPγS binding
Buprenorphine analgesia involves a combination of mu receptors, with little contribution from kappa1 or delta receptors despite its high binding affinity for these sites. This is consistent with our findings in cellular functional assays. In CHO (Chinese Hamster Ovary) cells stably expressing MOR-1, buprenorphine potently stimulated 35S-GTPγS binding (EC50 1.7 nM) with a maximal response (102%) indistinguishable from DAMGO alone (Fig. 10). In contrast, buprenorphine alone had no effect on cells expressing the delta receptor DOR-1 and reversed the stimulation induced by the delta agonist DPDPE (IC50 1.5 nM). Buprenorphine was an inverse agonist at KOR-1, lowering basal levels by more than 50% (IC50 0.36 nM). Buprenorphine also reversed the stimulation of 35S-GTPγS binding by U50,488H, eventually lowering binding below basal levels at higher concentrations (IC50 0.52 nM).
Figure 10. Effect of buprenorphine on 35S-GTPγS binding in cell lines.

35S-GTPγS binding assays were performed with membrane homogenates from CHO cells stably expressing the indicated opioid receptor. Results are pooled from 3 independent replications and are expressed as mean ± SEM. A) At MOR-1, buprenorphine acted as a full agonist relative to 1 μM DAMGO control with an EC50 (95%CL) of 1.8 nM (1.3, 2.3). B) At DOR-1, buprenorphine produced little or no stimulation of 35S-GTPγS binding above basal levels, and C) potently antagonized 30 nM DPDPE stimulation (IC50 (95%CL) 1.5 nM [0.67, 3.5]). D) At KOR-1, buprenorphine behaved as an inverse agonist, potently reducing 35S-GTPγS binding more than 50% below basal levels (IC50 (95%CL) 0.36 nM [0.11, 1.4]) and E) reversing stimulation produced by 100 nM U50,488 (IC50 (95%CL) 0.52 nM [0.23, 1.2]) with higher concentrations further reducing binding below basal levels.
Buprenorphine’s effects in brain membranes were less clear, failing to show activity at concentrations up to 0.1 μM, which induced near maximal effects in the cell lines (Fig 11). Since buprenorphine is an inverse kappa agonist, we questioned whether the lack of effect might reflect the summation of stimulation of mu receptors and lowering of kappa activity. To assess this possibility, we examined the effects of the mu antagonist CTAP. If the lack of an observable effect were due to the summation of opposing actions, antagonizing the mu receptors should ‘uncover’ the inverse agonist activity of buprenorphine. In controls, CTAP reversed the activity of the mu agonist DAMGO, but it did not appreciably lower 35S-GTPγS binding levels seen with buprenorphine, indicating that in this system buprenorphine failed to stimulate mu receptors in brain tissue.
Figure 11. Effect of buprenorphine on 35S-GTPγS binding in C57 mouse brain.
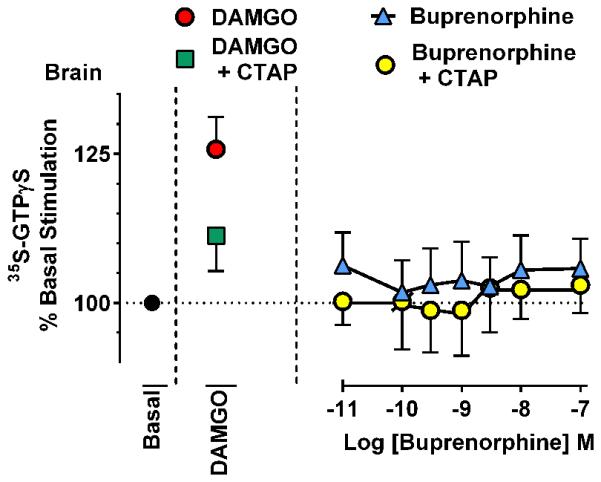
35S-GTPγS binding assays were performed with membrane homogenate from C57 mouse brain. Although DAMGO stimulated 35S-GTPγS binding, buprenorphine did not appear active concentrations up to 100 nM, a concentration which produced near-maximal effects in cell lines. The addition of CTAP failed to unmask any inverse agonist effect produced by kappa opioid receptors, although it substantially lowered DAMGO stimulation. Results are the mean ± s.e.m. of 4 independent replicates, each showing similar results.
Buprenorphine’s effects on β-arrestin-2 recruitment
Using the PathHunter© assay in CHO cells stably expressing MOR-1, buprenorphine failed to stimulate β-arrestin-2 recruitment at any dose up to 10 μM, despite the robust stimulation of the cells to DAMGO (Fig. 12), confirming a previous report (McPherson et al., 2010). Furthermore, buprenorphine antagonized in a dose-dependent manner the stimulation of β-arrestin-2 recruitment by a fixed DAMGO concentration (1 μM), lowering the response to basal levels (IC50 1.1 nM).
Figure 12. Buprenorphine recruitment of β-arrestin-2.

A) Buprenorphine failed to stimulate recruitment of β-arrestin-2 at concentrations up to 10 μM in a PathHunter© assay using MOR-1 cells. B) Buprenorphine potently antagonized the β-arrestin-2 recruitment produced by a fixed dose of 1 μM DAMGO with an IC50 (95%CL) of 1.1 nM [0.12, 11.0]. Results are pooled from at least 2 independent experiments with similar results, and are expressed as means ± SEM.
Discussion
Buprenorphine is a potent analgesic and its clinical use is increasing, both as an analgesic and in maintenance programs. However, it has a complex pharmacology. Buprenorphine has high affinity for all the traditional opioid receptors in receptor binding assays and labels ORL1 receptors with moderate affinity (Khroyan et al., 2015; Pergolizzi et al., 2010). It also labels a target involving truncated 6TM splice variants of the mu opioid receptor MOR-1 (Majumdar et al., 2011). However, its analgesic actions are dependent only upon mu receptors, as revealed by the use of a series of knockout mice. The absence of any buprenorphine analgesia in the double E1/E11 KO mouse established that mu receptors are essential for its activity. Conversely, the full analgesic response in the kappa1 or ORL1 KO mice indicates that neither receptor contributes to its analgesic activity. Delta receptors also do not significantly contribute to buprenorphine analgesia based upon the results in the triple KO mice, where the additional loss of DOR-1 and KOR-1 receptors did not alter the analgesic response beyond that seen with the E1 KO alone.
The maximal buprenorphine analgesic response requires both full length and truncated 6TM MOR-1 splice variants. Removal of either of these two sets of variants impacted the analgesic response. The ability of buprenorphine to lower morphine responses in the E11 KO mice supported the concept that it was a partial agonist at the 7TM receptors mediating morphine actions in those mice. The need for both sets of MOR-1 variants was further supported by rescue experiments. Although IBNtxA analgesia was rescued in the E1/E11 KO by the lentivirus with mMOR-1G, buprenorphine-induced analgesia was not (Lu et al., 2015), suggesting that a 6TM variant alone was insufficient to elicit analgesia. The E11 KO mice still express the exon 1-associated variants, so repletion of the 6TM mMOR-1G with the lentivirus should restore both sets of MOR-1 variants. The rescue of buprenorphine analgesia in E11 KO mice by the lentivirus with 6TM mMOR-1G confirms the need for both E1 and E11 variants.
The buprenorphine receptor sensitivity profile is distinct and contrasts with morphine and methadone, which are solely dependent upon the full length 7TM variants (Pan et al., 2009) and with IBNtxA (3-iodobenzoylnaltrexamide), which depends only upon 6TM variants (Majumdar et al., 2011; Majumdar et al., 2012). Thus, buprenorphine falls into a third category dependent upon both. Several other opioids fall into this category based upon their partial dependence upon both sets of variants, including heroin, M6G, and fentanyl (Pan et al., 2009). Differences in their receptor mechanisms among these mu opioids may help explain why many of them display incomplete cross tolerance, likely an important mechanism involved in Opioid Rotation in pain management (Chou et al., 2009).
Unlike analgesia, other buprenorphine actions were independent of E11-associated variants. Its inhibition of gastrointestinal transit persisted in the E11 knockout mouse as did its ability to increase locomotor activity, a common observation with traditional mu opioids.
The effects of buprenorphine on signal transduction pathways were particularly interesting. Selly and co-workers reported that buprenorphine was a partial agonist in MOR-1 expressing CHO cells, achieving a maximal response of slightly less than 50% with an ED50 of 4 nM (Selley et al., 1998). However, they found different results with the natively expressed receptor. In SK-N-SH cells, buprenorphine was over 10-fold more potent, but only stimulated 35S-GTPγS binding 15%. Similarly, in rat thalamus, buprenorphine potently stimulated binding only 10%, results similar to those from Rothman’s group (Romero et al., 1999). We observed similar results in brain tissue, but a far greater stimulation of 35S-GTPγS binding in CHO cells expressing MOR-1. Natively expressing systems may provide a better model for in vivo actions since they contain the other proteins/factors normally involved with receptor activation. Our failure to observe stimulation in brain and our knockout studies suggest that buprenorphine is a partial agonist at traditional mu receptors in vivo. However, both brain and SK-N-SH express a number of E1 and E11 splice variants, which may complicate the interpretation of the results.
While buprenorphine analgesia is limited to mu opioid receptors, other opioid systems may play a role in its overall pharmacological profile. Our results confirmed earlier reports that buprenorphine is an antagonist at delta and kappa1 receptors (Leander, 1987; Negus et al., 1989; Pergolizzi et al., 2010; Romero et al., 1999). Blockade of delta receptor action prevents the development of morphine tolerance (Abdelhamid et al., 1991; Kest et al., 1996; King et al., 1998; Schiller et al., 1999) while kappa antagonists have been implicated in a range of functions, including treatment of addiction (Chavkin, 2011; Rothman et al., 2000).
Mu opioid signaling bias has been implicated in both tolerance and respiratory depression (Bohn et al., 2004; Bohn et al., 1999; Dewire et al., 2013). Disrupting the β-arrestin-2 gene significantly enhanced morphine analgesia and DAMGO stimulation of 35S-GTPγS binding, shifting both dose-response curves to the left, despite no significant change in mu opioid receptor binding (Bohn et al., 1999). Furthermore, mice lacking β-arrestin-2 failed to show respiratory depression (Dewire et al., 2013; Kelly, 2013; Pradhan et al., 2012; Raehal et al., 2011; Reiter et al., 2012). Thus, it is interesting to consider whether buprenorphine’s failure to recruit β-arrestin-2 recruitment also may impact its pharmacology along with its antagonist actions on delta and kappa opioid receptors.
Mice have been generated that lack exon 1 (E1 KO) (Schuller et al., 1999), exon 11 (E11 KO) (Pan et al., 2009) or both exon 1 and exon 11(E1/E11 KO) (Lu et al., 2015) of the Oprm1 gene. The E1 KO mice lack all the 7TM and 1TM variants due to the absence of exon 1, but still express the 6TM exon 11 variants. Conversely, the E11 KO mice lack the 6TM variants and still express the 7TM and 1TM variants. The E1/E11 KO mice lack all mu opioid receptor splice variants due to the loss of both exons 1 and 11 and their promoters. The delta receptor knockout targeted exon 2 of the Oprd1 gene ((Zhu et al., 1999) while another mouse targeted the kappa receptor (KOR-1 KO) (Zhang et al., 1998). Triple KO mice were generated by backcrossing E1 KO, KOR-1 KO and DOR-1 KO mice. The ORL1 KO has a disruption in exons 2 and 3 of Oprl1 gene. The summary represents KO mice specifically referenced. Other Oprm1, kappa, delta and ORL1 KO mice may differ in their receptor variant expression profiles and pharmacology.
Acknowledgements
We thank Drs. Tom Wehrman and Elizabeth Quinn of DiscoveRx for their assistance. No authors have a conflict of interest. This work was supported in part by grants from Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, The Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center, The Harrington Discovery Institute, the Tri-Institutional Therapeutics Discovery Institute, the Peter F. McMannus Charitable Trust and grants from the National Institutes on Drug Abuse of the National Institutes of Health (DA06241 and DA07242) to GWP and (DA15237) to JP, a core grant from the National Cancer Institute of the National Institutes of Health (CA08748) to MSKCC, a fellowship from the PhRMA Foundation to SGG and a National Science Foundation Graduate Research Fellowship Grant (DGE-1257284) to GFM.
Abbreviations
- IBNtxA
3’-iodobenzoyl-6β-naltrexamide
- DAMGO
[D-Ala2,MePhe4,Gly(ol)5]enkephalin
- U50,488H
(±)-(5α,7α,8β)-3,4-Dichloro-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]benzeneacetamide
- CTAP
D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2
- MOR-1
the clone for the mu opioid receptor (Oprm1)
- KOR-1
the clone for the kappa1 opioid receptor (Oprk1)
- DOR-1
the clone for the delta opioid receptor (Oprd1)
- ORL1
the clone for the orphanin FQ/nociceptin receptor (Oprn1)
Footnotes
The authors have no conflict of interest.
References
- Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. JPharmacolExpTher. 1991;258(1):299–303. [PubMed] [Google Scholar]
- Bentley KW, Hardy DG. Novel analgesics and molecular rearrangements in the morphine-thebaine group. III. Alcohols of the 6,14-endo-ethenotetrahydrooripavine series and derived analogs of N-allylnormorphine and -norcodeine. JAmChemSoc. 1967;89(13):3281–3292. doi: 10.1021/ja00989a032. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. MolPharmacol. 2004;66(1):106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science. 1999;286(5449):2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bolan EA, Pasternak GW, Pan Y-X. Functional analysis of MOR-1 splice variants of the mu opioid receptor gene, Oprm. Synapse. 2004;51(1):11–18. doi: 10.1002/syn.10277. [DOI] [PubMed] [Google Scholar]
- Campbell ND, Lovell AM. The history of the development of buprenorphine as an addiction therapeutic. Annals of the New York Academy of Sciences. 2012;1248(1):124–139. doi: 10.1111/j.1749-6632.2011.06352.x. [DOI] [PubMed] [Google Scholar]
- Chavkin C. The therapeutic potential of kappa-opioids for treatment of pain and addiction. Neuropsychopharmacology. 2011;36(1):369–370. doi: 10.1038/npp.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou R, Fanciullo GJ, Fine PG, Adler JA, Ballantyne JC, Davies P, Donovan MI, Fishbain DA, Foley KM, Fudin J, Gilson AM, Kelter A, Mauskop A, O'Connor PG, Passik SD, Pasternak GW, Portenoy RK, Rich BA, Roberts RG, Todd KH, Miaskowski C. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. JPain. 2009;10(2):113–130. doi: 10.1016/j.jpain.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan A, Lewis JW, Macfarlane IR. Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. BrJPharmacol. 1977;60(4):537–545. doi: 10.1111/j.1476-5381.1977.tb07532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox V, Clarke S, Czyzyk T, Ansonoff M, Nitsche J, Hsu MS, Borsodi A, Tomboly C, Toth G, Hill R, Pintar J, Kitchen I. Autoradiography in opioid triple knockout mice reveals opioid and opioid receptor like binding of naloxone benzoylhydrazone. Neuropharmacology. 2005;48(2):228–235. doi: 10.1016/j.neuropharm.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Dewire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G Protein-Biased Ligand at the mu-Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphine. JPharmacolExpTher. 2013;344(3):708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- Ding Z, Raffa RB. Identification of an additional supraspinal component to the analgesic mechanism of action of buprenorphine. BrJPharmacol. 2009;157(5):831–843. doi: 10.1111/j.1476-5381.2009.00209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dum JE, Herz A. In vivo receptor binding of the opiate partial agonist, buprenorphine, correlated with its agonistic and antagonistic actions. BrJPharmacol. 1981;74:627–633. doi: 10.1111/j.1476-5381.1981.tb10473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Kehner GB, Cowan A, Liu-Chen LY. Comparison of pharmacological activities of buprenorphine and norbuprenorphine: norbuprenorphine is a potent opioid agonist. J Pharmacol Exp Ther. 2001;297(2):688–695. [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12(2):483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce EM, Iversen SD. The effect of morphine applied locally to mesencephalic dopamine cell bodies on spontaneous motor activity in the rat. Neurosci Lett. 1979;14(2-3):207–212. doi: 10.1016/0304-3940(79)96149-4. [DOI] [PubMed] [Google Scholar]
- Kamei J, Saitoh A, Morita K, Nagase H. Antagonistic effect of buprenorphine on the antitussive effect of morphine is mediated via the activation of μ1-opioid receptors. Life Sci. 1995a;57(16):PL231–PL235. doi: 10.1016/0024-3205(95)02115-y. [DOI] [PubMed] [Google Scholar]
- Kamei J, Saitoh A, Suzuki T, Misawa M, Nagase H, Kasuya Y. Buprenorphine exerts its antinociceptive activity via μ1- opioid receptors. Life Sci. 1995b;56:PL285–PL290. doi: 10.1016/0024-3205(95)00078-x. [DOI] [PubMed] [Google Scholar]
- Kamei J, Sodeyama M, Tsuda M, Suzuki T, Nagase H. Antinociceptive effect of buprenorphine in μ1-opioid receptor deficient CXBK mice. Life Sci. 1997;60(22):PL333–PL337. doi: 10.1016/s0024-3205(97)00170-7. [DOI] [PubMed] [Google Scholar]
- Kelly E. Efficacy and ligand bias at the mu-opioid receptor. BrJPharmacol. 2013;169(7):1430–1446. doi: 10.1111/bph.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kest B, Lee CE, McLemore GL, Inturrisi CE. An antisense oligodeoxynucleotide to the delta opioid receptor (DOR-1) inhibits morphine tolerance and acute dependence in mice. Brain ResBull. 1996;39(3):185–188. doi: 10.1016/0361-9230(95)02092-6. [DOI] [PubMed] [Google Scholar]
- Khroyan TV, Wu J, Polgar WE, Cami-Kobeci G, Fotaki N, Husbands SM, Toll L. BU08073 a buprenorphine analogue with partial agonist activity at mu-receptors in vitro but long-lasting opioid antagonist activity in vivo in mice. Br J Pharmacol. 2015;172(2):668–680. doi: 10.1111/bph.12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M, Chang A, Pasternak GW. Functional blockade of opioid analgesia by orphanin FQ/nociceptin. Biochemical Pharmacology. 1998;55(9):1537–1540. doi: 10.1016/s0006-2952(98)00023-9. [DOI] [PubMed] [Google Scholar]
- Leander JD. Buprenorphine has potent kappa opioid receptor antagonist activity. Neuropharmacology. 1987;26:1445–1447. doi: 10.1016/0028-3908(87)90112-2. [DOI] [PubMed] [Google Scholar]
- Lewis JW. Buprenorphine. Drug Alcohol Depend. 1985;14(3-4):363–372. doi: 10.1016/0376-8716(85)90067-5. [DOI] [PubMed] [Google Scholar]
- Lu Z, Xu J, Rossi GC, Majumdar S, Pasternak GW, Pan YX. Mediation of opioid analgesia by a truncated 6-transmembrane GPCR. J Clin Invest. 2015;125(7):2626–2630. doi: 10.1172/JCI81070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K, Cowan A. Buprenorphine: a unique drug with complex pharmacology. CurrNeuropharmacol. 2004;2(4):395–402. doi: 10.2174/1570159043359477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K, Eitan S, Bryant CD, Yang YC, Saliminejad N, Walwyn W, Kieffer BL, Takeshima H, Carroll FI, Maidment NT, Evans CJ. Buprenorphine-induced antinociception is mediated by mu-opioid receptors and compromised by concomitant activation of opioid receptor-like receptors. J Neurosci. 2003;23(32):10331–10337. doi: 10.1523/JNEUROSCI.23-32-10331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, Grinnell S, Le R, Burgman M, Polikar L, Ansonoff M, Pintar J, Pan YX, Pasternak GW. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. ProcNatlAcadSciUSA. 2011;108(49):19776–19783. doi: 10.1073/pnas.1115231108. V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, Subrath J, Le Rouzic V, Polikar L, Burgman M, Nagakura K, Ocampo J, Haselton N, Pasternak AR, Grinnell S, Pan Y-X, Pasternak GW. Synthesis and evaluation of aryl-naloxamide opiate analgesics targeting truncated exon 11-associated mu opioid receptor (MOR-1) splice variants. JMedChem. 2012;55(14):6352–6362. doi: 10.1021/jm300305c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez P, Baliram R, Kieffer BL, Lutfy K. The mu opioid receptor is involved in buprenorphine-induced locomotor stimulation and conditioned place preference. Neuropharmacology. 2007;52(6):1336–1341. doi: 10.1016/j.neuropharm.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey CP, Rosethorne EM, Charlton SJ, Henderson G, Kelly E. mu-opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. MolPharmacol. 2010;78(4):756–766. doi: 10.1124/mol.110.066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B, Mazargull H, Vassart G, Parmentier M, Costentin J. Isolation and structure of the endogenous agonist of opioid receptor- like ORL1 receptor. Nature. 1995;377(6549):532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- Negus SS, Picker MJ, Dykstra LA. Kappa antagonist properties of buprenorphine in non-tolerant and morphine-tolerant rats. Psychopharmacology (Berl) 1989;98(1):141–143. doi: 10.1007/BF00442021. [DOI] [PubMed] [Google Scholar]
- Pan Y-X, Xu J, Mahurter L, Bolan EA, Xu MM, Pasternak GW. Generation of the mu opioid receptor (MOR-1) protein by three new splice variants of the Oprm gene. ProcNatlAcadSciUSA. 2001;98(24):14084–14089. doi: 10.1073/pnas.241296098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YX, Xu J, Bolan E, Moskowitz HS, Xu M, Pasternak GW. Identification of four novel exon 5 splice variants of the mouse mu-opioid receptor gene: functional consequences of C-terminal splicing. MolPharmacol. 2005;68(3):866–875. doi: 10.1124/mol.105.011858. [DOI] [PubMed] [Google Scholar]
- Pan YX, Xu J, Xu M, Rossi GC, Matulonis JE, Pasternak GW. Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin, but not morphine, actions. ProcNatlAcadSciUSA. 2009;106(12):4917–4922. doi: 10.1073/pnas.0811586106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW, Pan Y-X. Mu opioids and their receptors: Evolution of a concept. PharmacolRev. 2013;65(4):1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pergolizzi J, Aloisi AM, Dahan A, Filitz J, Langford R, Likar R, Mercadante S, Morlion B, Raffa RB, Sabatowski R, Sacerdote P, Torres LM, Weinbroum AA. Current knowledge of buprenorphine and its unique pharmacological profile. Pain Pract. 2010;10(5):428–450. doi: 10.1111/j.1533-2500.2010.00378.x. [DOI] [PubMed] [Google Scholar]
- Pick CG, Peter Y, Schreiber S, Weizman R. Pharmacological characterization of buprenorphine, a mixed agonist-antagonist with kappa3 analgesia. Brain Res. 1997;744(1):41–46. doi: 10.1016/s0006-8993(96)01069-4. [DOI] [PubMed] [Google Scholar]
- Pradhan AA, Smith ML, Kieffer BL, Evans CJ. Ligand-directed signalling within the opioid receptor family. BrJPharmacol. 2012;167(5):960–969. doi: 10.1111/j.1476-5381.2012.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Schmid CL, Groer CE, Bohn LM. Functional selectivity at the mu-opioid receptor: implications for understanding opioid analgesia and tolerance. PharmacolRev. 2011;63(4):1001–1019. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Civelli O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. AnnuRevPharmacolToxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero DV, Partilla JS, Zheng QX, Heyliger SO, Ni Q, Rice KC, Lai J, Rothman RB. Opioid peptide receptor studies. 12. Buprenorphine is a potent and selective μ/kappa antagonist in the [35S]-GTP-gamma-S functional binding assay. Synapse. 1999;34(2):83–94. doi: 10.1002/(SICI)1098-2396(199911)34:2<83::AID-SYN1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Rossi GC, Brown GP, Leventhal L, Yang K, Pasternak GW. Novel receptor mechanisms for heroin and morphine-6β -glucuronide analgesia. NeurosciLett. 1996;216:1–4. doi: 10.1016/0304-3940(96)12976-1. [DOI] [PubMed] [Google Scholar]
- Rossi GC, Leventhal L, Bolan EA, Pasternak GW. Pharmacological characterization of orphanin FQ/nociceptin and its fragments. JPharmacolExpTher. 1997;282:858–865. [PubMed] [Google Scholar]
- Rothman RB, Gorelick DA, Heishman SJ, Eichmiller PR, Hill BH, Norbeck J, Liberto JG. An open-label study of a functional opioid kappa antagonist in the treatment of opioid dependence. Journal of substance abuse treatment. 2000;18(3):277–281. doi: 10.1016/s0740-5472(99)00074-4. [DOI] [PubMed] [Google Scholar]
- Schiller PW, Fundytus ME, Merovitz L, Weltrowska G, Nguyen TM, Lemieux C, Chung NN, Coderre TJ. The opioid mu agonist/delta antagonist DIPP-NH(2)[Psi] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. JMedChem. 1999;42(18):3520–3526. doi: 10.1021/jm980724+. [DOI] [PubMed] [Google Scholar]
- Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ, Chang A, Czick ME, Unterwald EM, Pasternak GW, Pintar JE. Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. NatNeurosci. 1999;2(2):151–156. doi: 10.1038/5706. [DOI] [PubMed] [Google Scholar]
- Selley DE, Liu QX, Childers SR. Signal transduction correlates of Mu opioid agonist intrinsic efficacy: Receptor-stimulated [35S]GTPgammaS binding in mMOR-CHO cells and rat thalamus. Journal of Pharmacology and Experimental Therapeutics. 1998;285(2):496–505. [PubMed] [Google Scholar]
- Urs NM, Daigle TL, Caron MG. A dopamine D1 receptor-dependent beta-arrestin signaling complex potentially regulates morphine-induced psychomotor activation but not reward in mice. Neuropsychopharmacology. 2011;36(3):551–558. doi: 10.1038/npp.2010.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virk MS, Arttamangkul S, Birdsong WT, Williams JT. Buprenorphine is a weak partial agonist that inhibits opioid receptor desensitization. JNeurosci. 2009;29(22):7341–7348. doi: 10.1523/JNEUROSCI.3723-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EA, Zernig G, Woods JH. Buprenorphine antagonism of mu opioids in the rhesus monkey tail-withdrawal procedure. JPharmacolExpTher. 1995;273:1345–1352. [PubMed] [Google Scholar]
- Xu J, Faskowitz AJ, Rossi GC, Xu M, Lu Z, Pan YX, Pasternak GW. Stabilization of morphine tolerance with long-term dosing: association with selective upregulation of mu-opioid receptor splice variant mRNAs. Proc Natl Acad Sci U S A. 2015;112(1):279–284. doi: 10.1073/pnas.1419183112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lu Z, Xu M, Rossi GC, Kest B, Waxman AR, Pasternak GW, Pan YX. Differential Expressions of the Alternatively Spliced Variant mRNAs of the mu Opioid Receptor Gene, OPRM1, in Brain Regions of Four Inbred Mouse Strains. PLoS ONE. 2014;9(10):e111267. doi: 10.1371/journal.pone.0111267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Xu M, Brown T, Rossi GC, Hurd YL, Inturrisi CE, Pasternak GW, Pan YX. Stabilization of the mu opioid receptor by truncated single transmembrane splice variants through a chaperone-like action. JBioChem. 2013;288:21211–21227. doi: 10.1074/jbc.M113.458687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, King M, Pasternak GW, Pintar JE. Production and analgesic characterization fo KORO-1 deficient mice. Society for Neuroscience. 1998;24:525. [Google Scholar]
- Zhu YX, King MA, Schuller AGP, Nitsche JF, Reidl M, Elde RP, Unterwald E, Pasternak GW, Pintar JE. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in δ opioid receptor knockout mice. Neuron. 1999;24(1):243–252. doi: 10.1016/s0896-6273(00)80836-3. [DOI] [PubMed] [Google Scholar]