

Abstract
The development of numerous types of cardiovascular disease is associated with alteration of the vascular smooth muscle cell (SMC) phenotype. We have previously shown that abdominal aortic aneurysm progression in a mouse model of the disease is associated with reduced differentiation of SMCs within the lesion and that cyclooxygenase-2 (COX-2) is critical to initiation and progression of the aneurysms. The current studies utilized human aortic SMC (hASMC) cultures to better characterize mechanisms responsible for COX-2-dependent modulation of the SMC phenotype. Depending on the culture conditions, hASMCs expressed multiple characteristics of a differentiated and contractile phenotype, or a de-differentiated and secretory phenotype. The pharmacological inhibition of COX-2 promoted the differentiated phenotype whereas treatment with the COX-2-derived metabolite prostaglandin E2 (PGE2) increased characteristics of the de-differentiated phenotype. Furthermore, pharmacological inhibition or siRNA-mediated knockdown of microsomal prostaglandin E synthase-1 (mPGES-1), the enzyme that functions down-stream of COX-2 during the synthesis of PGE2, significantly increased expression of characteristics of the differentiated SMC phenotype. Therefore, our findings suggest that COX-2 and mPGES-1 -dependent synthesis of PGE2 contributes to a de-differentiated hASMC phenotype and that mPGES-1 may provide a novel pharmacological target for treatment of cardiovascular diseases where altered SMC differentiation has a causative role.
INTRODUCTION
Alteration of the vascular smooth muscle cell (SMC) phenotype plays a key role in pathologies that occur during the development of numerous cardiovascular diseases. The transition of vascular SMCs from a differentiated, contractile phenotype to a de-differentiated, synthetic phenotype has been shown to occur following development of atherosclerosis, restenosis, and hypertension.(1–4) In addition, defects in specific contractile proteins or reduced contractile protein expression have been identified in SMCs of ascending thoracic aortic aneurysms in humans.(5–8) In a mouse model of abdominal aortic aneurysms, we have previously determined that the effectiveness of cyclooxygenase-2 (COX-2) inhibition for limiting the progression of these aneurysms is associated with maintenance of a differentiated SMC phenotype.(9, 10) Therefore, targeting the specific COX-2-dependent pathway responsible for reducing differentiation may provide a strategy for reversing alterations to the differentiated SMC phenotype that contribute to the development of vascular pathologies.
COX-2 is the inducible COX isoform and is primarily responsible for the increased synthesis of prostaglandins (PGs) that occurs during disease development. The COX isoforms are required for the synthesis of PGs, which are lipid mediators involved in numerous physiological and pathological processes. In the regulation of vascular SMC differentiation, a previously well characterized PG effect is the promotion a differentiated, contractile phenotype by PGI2, also known as prostacyclin. Activation of the prostacyclin receptor by exogenous treatment with a stable prostacyclin analog increases differentiation of cultured human aortic SMCs to a contractile phenotype.(11) The increased synthesis of prostacyclin by endothelial cells co-cultured with SMCs also promotes SMC differentiation.(12) In contrast to the promotion of differentiation by prostacyclin, the effects of other PGs on the SMC phenotype are less clear. The induction of COX-2 expression has been identified as a key marker associated with SMC de-differentiation following vascular injury in mice.(13, 14) Furthermore, in a model of SMC de-differentiation induced by increased blood flow into the abdominal aorta of rats, COX-2 inhibitor treatment increases flow, smooth muscle disorganization and intimal hyperplasia, and decreases contractile responses and medial thickness.(4) Therefore, although the previously described role for PGs in regulating the SMC phenotype has been primarily the function of prostacyclin to enhance differentiation, there has also been evidence that COX-2 functions in the de-differentiation of SMCs.
The PG that is often associated with the over-expression of COX-2 that occurs during disease development is prostaglandin E2 (PGE2). The production of PGE2 involves a PGE2 synthase which utilizes the substrate PGH2 formed by the COXs.(15) Microsomal prostaglandin E synthase-1 (mPGES-1) is the PGE2 synthase that is often co-expressed and functionally coupled with COX-2, and is thought to contribute to the increased production of PGE2 during the development of a variety of pathological conditions including inflammation, cardiovascular disease, and cancer.(16–22) In the current studies, we utilized human aortic SMCs to identify a role for PGE2 produced by the activity of COX-2 and mPGES-1 in promoting the de-differentiated SMC phenotype.
METHODS
Cell culture
Human aortic smooth muscle cells (hASMCs) were purchased from Cascade Biologics (Invitrogen) and cultured according to manufacturer instructions using recommended media (Medium 231) with supplements, penicillin (100 U/ml) and streptomycin (100 µg/ml) to induce either differentiation or de-differentiation. De-differentiation-promoting media contained 4.9% fetal bovine serum (FBS), heparin (5 ng/ml), human basic fibroblast growth factor (2 ng/ml), human epidermal growth factor (0.5 ng/ml), recombinant human insulin-like growth factor-1 (2 µg/ml), and bovine serum albumin (0.2 µg/ml). Differentiation-promoting media contained Medium 231 supplemented with 1% FBS, and heparin (30 µg/ml). Cells used in all experiments were from passage 2 through 8 and were cultured in a humidified 5% CO2 atmosphere at 37°C.
Cell lysis and protein collection
After cell culture and treatment, cell culture media were either collected for further analyses or discarded at pre-determined time points and adherent cells were rinsed briefly two times with 1XPBS. Cells were lysed with equal volumes of RIPA lysis buffer made up of 200 mM Tris with pH 7.6, glycerol, 10% SDS, 10% deoxycholate, Triton X-100, 200 mM EDTA containing protease and phosphatase inhibitors (PMSF and leupeptin). Cells lysates were subsequently scraped and pipetted into 1.5 ml Eppendorf tubes for storage at −20°C until further use. Prior to protein separation via SDS PAGE, protein content in each sample was analyzed using the Bicinchoninic Acid (BCA) method and specific volumes corresponding to equal amounts of protein was added to fixed volumes of loading buffer and heated at 100°C for 10 minutes. The loading buffer was made up of 0.5 M Tris (pH 6.8), glycerol, 10% SDS, 0.5% bromophenol and 5% β-mercaptoethanol. After heating, the tubes were vortexed briefly, spun-down using a microcentrifuge and then loaded unto polyacrylamide gels for protein separation via western blotting.
Western blot analysis
Adherent cells were briefly rinsed with PBS and lysed in RIPA buffer (200 mM Tris pH 7.6, glycerol, 10% SDS, 10% deoxycholate, 1% Triton X-100, 200 mM EDTA, 1 mM PMSF, and 20 µM leupeptin) then heated at 100°C for 10 minutes. Protein content was determined using the BCA method (Pierce) and equal amounts of protein were loaded onto 12% polyacrylamide gels, separated via SDS-PAGE and transferred onto nitrocellulose membranes (GE Healthcare). Membranes were blocked for 1 hour with 5% bovine serum albumin in Tris-buffered saline containing 1% Tween 20 and incubated overnight at 4°C with primary antibodies. Antibodies against SM α-actin (Sigma Aldrich), and SM22α (Abcam Inc.), were used as markers of differentiation and either α-tubulin (Cell signaling) or calnexin (Cell signaling) were used as loading controls. Monoclonal α-actin primary antibody was conjugated with Cy3, while biotinylated anti-rabbit secondary antibodies together with Alexa Fluor 488-conjugated streptavidin complexes were used for detection of SM22α, α-tubulin and calnexin. For COX-2 (Vector laboratories) and mPGES-1 (Oxford biomedical research) detection, horse-radish peroxidase (HRP)-conjugated anti-rabbit IgG was used as the secondary antibody. Protein detection of fluorophore-conjugated antibodies was done using the Typhoon laser scanner (GE Healthcare), while HRP-conjugated antibodies were detected by enhanced chemiluminescence. Images were analyzed and quantified via densitometry using the ImageQuant analysis software (GE Healthcare).
mRNA isolation and quantitation
mRNA was isolated from the hASMCs using the RNeasy kit (Qiagen Inc., CA, USA) according to manufacturer’s instructions. Briefly, cultured cells were lysed and homogenized with QIAshredder spin columns in the presence of guanidine-thiocyanate-containing buffer. One volume of 70% ethanol was added to the homogenized lysate, transferred to RNeasy spin columns, and RNA was eluted for further analysis. Total RNA was reverse transcribed in a two-step process according to manufacturer’s instructions (Invitrogen). Real-Time Polymerase Chain Reaction (RT-PCR) was performed using Taqman primer/probe assays for smooth muscle α-actin, SM22α, COX-2, microsomal PGE2 synthase (mPGES-1), matrix metalloproteinase −2 (MMP-2), smoothelin, myosin heavy chain 2 (MyhII), and hyaluronan synthase 2 (Has2) (Applied Biosystems). mRNA expression of the constitutively expressed housekeeping gene hypoxanthine phospho-ribosyl transferase (HPRT) was quantitated as an internal normalizing control gene. Relative gene expression levels were calculated by comparing cycle times for each target PCR using the ΔΔct method. The target PCR Ct values were normalized by subtracting the HPRT Ct value, to give the ΔCt value. The relative expression levels between treatments were then calculated using the equation: relative gene expression = 2−(ΔCtsample-ΔCtcontrol).
Immunocytochemistry
hASMCs plated in 12 or 24 well plates, were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, blocked with 1% bovine serum albumin (BSA) for 1 hour, and incubated overnight with Cy3-conjgated SM α-actin antibody in 1% BSA at 4°C. Cellular DNA content was quantitated using SYBR green (AB systems) after incubation for 1 hour at room temperature. Cells were washed with PBS and plates were imaged using a Typhoon fluorescence imager (GE Healthcare) after each incubation. Fluorescence was quantitated using ImageJ analytical software, and the ratio of α-actin to SYBR green detection was determined for each well of the plate.
mPGES-1 RNA interference using siRNA transfection
hASMCs were transfected with mPGES-1 siRNA (15540, Life Technologies) using the Lipofectamine RNAiMAX transfection reagent according to the manufacturer’s instructions (Invitrogen, NY USA). After two days, the transfection media was replaced with serum-free media for the designated times, followed by collection for PGE2 analysis. PGE2 concentrations in cell culture media were determined using an enzyme immunoassay (EIA) monoclonal kit according to the manufacturer’s protocol (Cayman chemical company). Protein and mRNA samples were isolated from replicate treatments and analyzed by Western blotting or RT-PCR, respectively.
Statistical analyses
Each experiment was repeated at least three times and statistical analyses were carried out using the GraphPad Prism software (GraphPad Software Inc). The mean and SEM were calculated for each parameter with a single treatment considered as n of 1. hASMC phenotype comparisons between differentiated and de-differentiated cells were analyzed by two-way repeated measures ANOVA with Bonferroni post-tests. To compare the significance of the difference between 3 or more treatment groups, one-way ANOVA, followed by the Dunnett’s multiple comparison post-hoc tests was utilized. Unpaired Student’s t test was used to compare the effects observed between two groups. Statistical significance was defined as p < 0.05.
RESULTS
Time-course of contractile and synthetic marker expression during differentiation or de-differentiation culture conditions
The hASMC differentiation characteristics that resulted from two different culture conditions were compared by measuring expression of the differentiation markers α-actin and SM22α, and the marker of de-differentiation, HAS-2. In culture conditions with reduced serum (1% v/v) and increased heparin (30 µg/ml) (differentiation conditions, DC), there was significantly greater expression of α-actin mRNA throughout the 4-day time-course, as compared to culture conditions containing increased serum (5% v/v) and growth factors (de-differentiation conditions, d-DC) (Figure 1A). Beginning at the 2-day time-point, there was also a significant increase in α-actin protein expression under differentiating conditions, which continued throughout the 5-day time-course (Figure 1B). The comparison of an additional differentiation maker SM22α showed an increase in expression with differentiation which became significantly different at the 3-day time-point (Figure 1C).
Figure 1. Time-course of hASMC phenotypic modulation in response to differentiation or de-differentiation culture conditions.
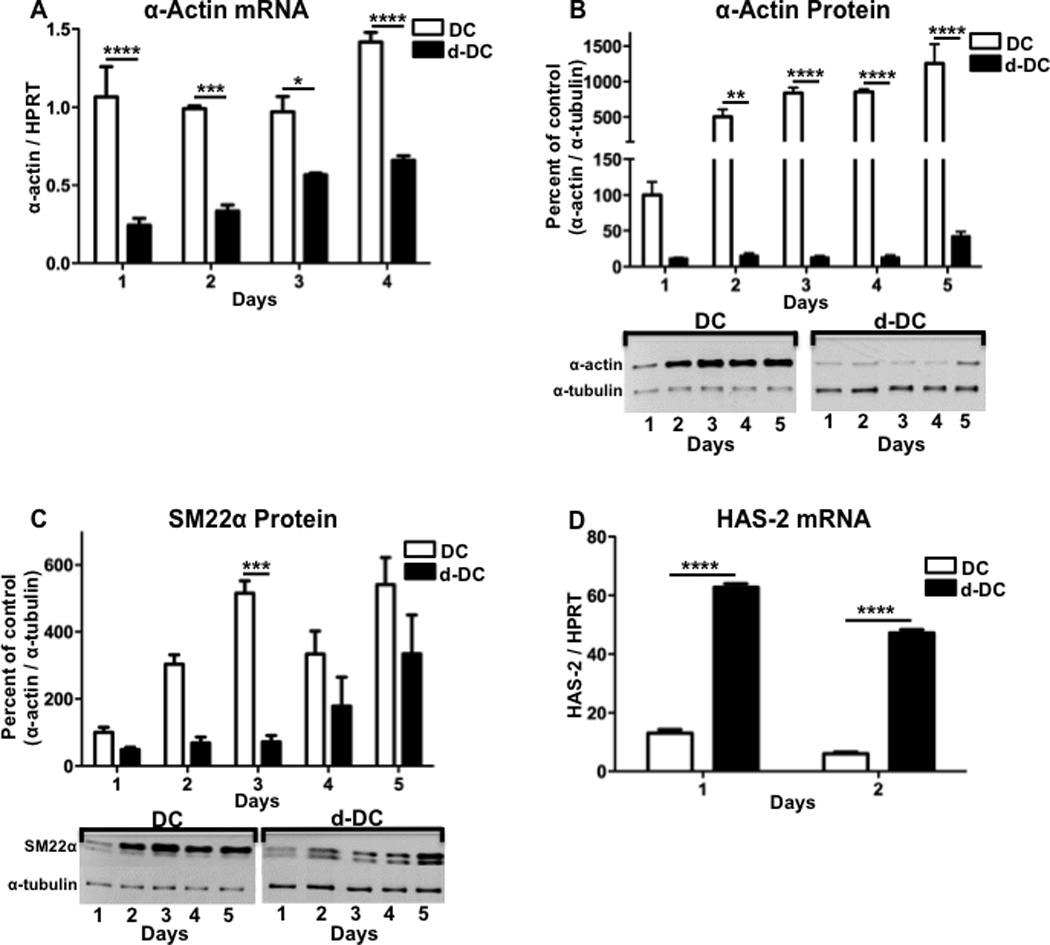
hASMCs were cultured for up to 5 days under conditions promoting either differentiation (DC) or de-differentiation (d-DC) and analyzed for (A) α-actin mRNA, (B) α-actin protein, (C) SM22α protein or (D) HAS-2 mRNA. For quantitation of protein expression, densitometry was performed on bands from Western blots and a representative Western blot is shown for each graph. The data represent mean + SEM (n=4–5), * = p < 0.05, ** = p < 0.01, *** = p < 0.001, and **** = p < 0.0001; two-way ANOVA used to determine differences due to culture condition and time.
The increased synthesis of the extracellular matrix component hyaluronic acid is a well-described characteristic of the de-differentiated SMC phenotype.(23–25) In cultured human aortic SMCs, hyaluronic acid synthase 2 (HAS-2) is the predominant isoform responsible for increased production of hyaluronic acid.(26) Our current findings show that in contrast to the differentiation markers, HAS-2 mRNA expression was significantly greater under de-differentiation conditions as compared to differentiation conditions, following either 1 day or 2 days of culture (Figure 1D). Therefore, depending on the culture conditions, hASMCs show evidence of phenotypic modulation with varying characteristics of the contractile or synthetic state.
COX-2 contributes to reduced differentiation of hASMCs
With our finding that the level of differentiation was dependent on culture conditions, we compared the expression of COX-2 in hASMCs cultured with either differentiation-promoting (DC) or de-differentiation-promoting (d-DC) conditions. Throughout a 4-day time-course, COX-2 mRNA expression was significantly greater in de-differentiated cells, as compared to differentiated cells (Figure 2A). Similar to the mRNA expression, COX-2 protein expression showed a continual decline during a 5-day time-course for cells cultured in differentiating conditions where α-actin and SM22α were increased (Figure 2B). In contrast, hASMCs cultured in the de-differentiating conditions showed greater COX-2 protein expression, which increased over the 5-day time-course (Figure 2B). The finding that increased COX-2 expression was inversely correlated with the expression of α-actin and SM22α suggests that COX-2 expressing hASMCs show a reduced level of differentiation.
Figure 2. COX-2 contributes to reduced differentiation of hASMCs.
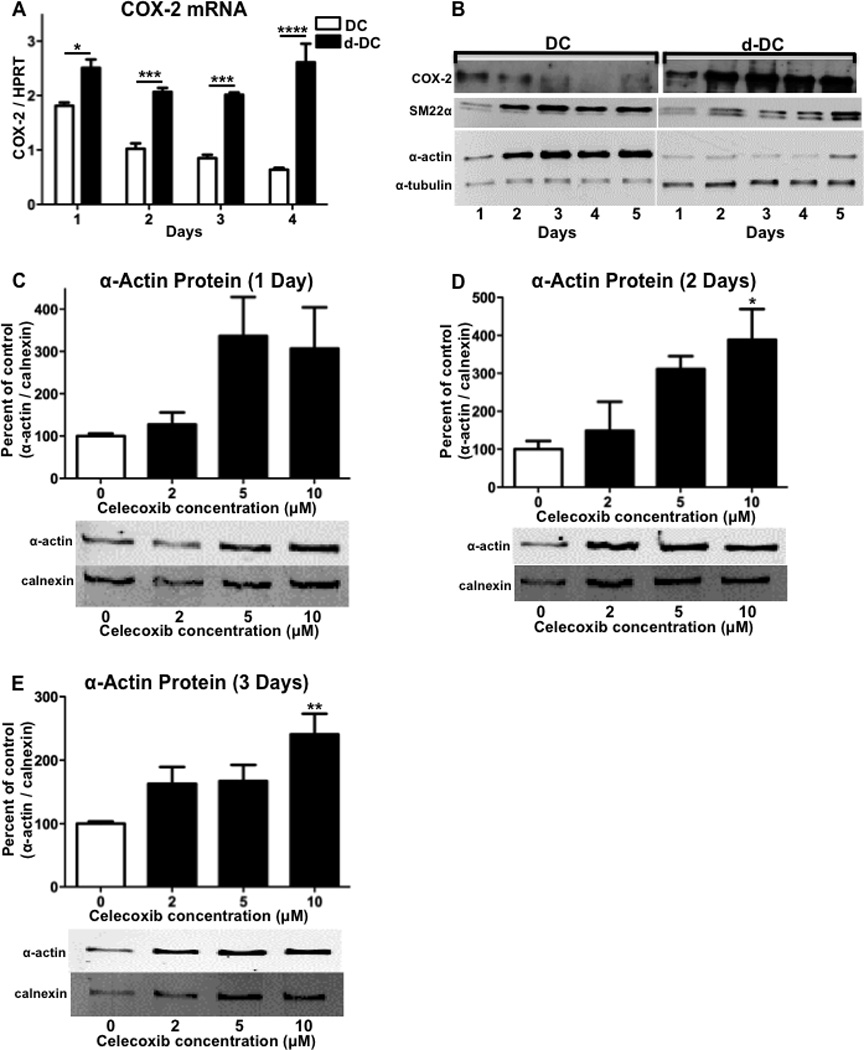
hASMCs were cultured for up to 5 days under conditions promoting either differentiation (DC) or de-differentiation (d-DC) and analyzed for (A) COX-2 mRNA, (B) COX-2 protein, SM22α protein, α-actin protein, and α-tubulin loading control. hASMCs were treated with varying concentrations of the COX-2 inhibitor celecoxib for (C) 1 day, (D) 2 days, or (E) 3 days and analyzed for expression of the differentiation marker α-actin. For quantitation of α-actin and the loading control calnexin, densitometry was performed on bands from Western blots and a representative Western blot is shown for each graph. The data represent mean + SEM (n=4–5), * = p < 0.05, ** = p < 0.01, *** = p < 0.001, and **** = p < 0.0001; (A) two-way ANOVA used to determine differences due to culture condition and time; (C–D) one-way ANOVA.
To examine the role of COX-2 in contributing to reduced differentiation of hASMCs, we examined the effect of pharmacological inhibition of COX-2. hASMCs were treated with various concentrations of celecoxib over a 3-day time-course and analyzed for α-actin protein expression by Western blot. Following a 1-day treatment, α-actin protein expression showed a trend for increased expression with higher celecoxib concentrations that was not statistically significant (Figure 2C). Treatment with celecoxib for either 2 or 3 days resulted in a dose-dependent increase in α-actin expression that was statistically significant at the higher dose of 10 (Figure 2, D and E). These findings indicate that inhibiting the activity of COX-2 promotes the differentiation of hASMCs.
Prostaglandin E2 promotes a de-differentiated hASMC phenotype
With the activity of COX-2 in human aortic SMCs known to contribute to the synthesis of PGE2,(27) we examined the effect of exogenous treatment with PGE2 on the expression of multiple differentiation markers. The treatment of hASMCs with PGE2 for 24 hours at concentrations of either 0.2 or 1 µM significantly reduced mRNA expression of α-actin, SM22α, myosin heavy chain, and smoothelin, as compared to vehicle-treated controls (Figure 3, A–D). In addition, the protein expression of both α-actin and SM22α showed significant decreases following treatments with 0.2 or 1 µM PGE2 (Figure 3, E and F). These findings indicate that PGE2 acts on hASMCs to decrease the characteristics of a differentiated phenotype.
Figure 3. PGE2 treatment decreases hASMC differentiation and increases de-differentiation.
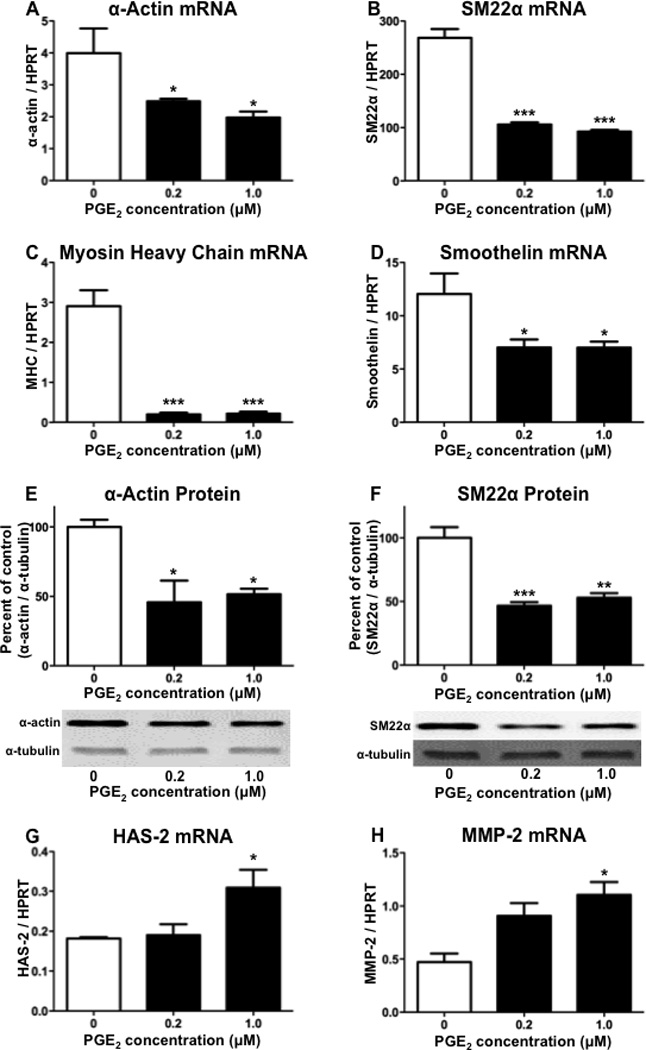
Following treatment with PGE2 for 1 day, hASMCs were analyzed for expression of (A) α-actin mRNA, (B) SM22α mRNA, (C) myosin heavy chain mRNA, (D) smoothelin mRNA, (E) α-actin protein, (F) SM22α protein, (G) HAS-2 mRNA, or (H) MMP-2 mRNA. For quantitation of protein expression, densitometry was performed on bands from Western blots and a representative Western blot is shown for each graph. The data represent mean + SEM (n=4–5), * = p < 0.05, ** = p < 0.01, *** = p < 0.001; one-way ANOVA.
The increased expression of HAS-2 and matrix metalloproteinase-2 (MMP-2) are widely used markers of SMC de-differentiation.(26, 28–30) We examined the effect of treating hASMCs with PGE2 on expression of these de-differentiation markers. Following culture in differentiation media, the SMCs were treated with either 0.2 or 1 µM PGE2 for two days. As compared to vehicle-treated controls, the higher PGE2 concentration of 1 µM significantly increased mRNA expression of both HAS-2 and MMP-2 (Figure 3, G and H). Therefore, the effect of PGE2 on modulating the SMC phenotype not only includes a reduction in differentiation, but also an increase in de-differentiation.
Pharmacological inhibition of mPGES-1 increases hASMC differentiation
mPGES-1 functions downstream of COX-2 in the synthesis of PGE2, and in human vascular SMCs, mPGES-1 is important for the synthesis of PGE2.(27, 31) In order to examine the effect of pharmacological inhibition of mPGES-1, hASMCs were treated with varying concentrations of the mPGES-1 inhibitor 15-deoxy-delta 12,14-prostaglandin J2 (15d-PGJ2) under conditions promoting de-differentiation.(32, 33) Treatment with 15d-PGJ2 at concentrations of 0.1, 1 or 10 µM significantly reduced the levels of PGE2 detected in the media from hASMC cultures (Figure 4A). There was also a significant reduction in PGE2 following treatment of hASMCs with the COX-2 inhibitor celecoxib (Figure 4A), thereby suggesting that the synthesis of PGE2 in these cells is dependent on both COX-2 and mPGES-1. With the observed reduction of PGE2, we examined whether mPGES-1 inhibition by 15d-PGJ2 would affect SMC differentiation. hASMCs were treated with 15d-PGJ2 under conditions promoting de-differentiation and analyzed for α-actin expression via quantitative immunocytochemistry. Although the lower treatment concentrations of 0.1 or 1 µM did not significantly affect α-actin expression, the higher concentration of 10 µM significantly increased α-actin expression, as compared to vehicle-treated controls (Figure 4B). We also examined the effect of treatment with 15d-PGJ2 in serum-free conditions and determined that the lower concentration of 2 µM was also effective at significantly increasing the expression α-actin (Figure 4C).
Figure 4. mPGES-1 inhibition increases hASMC differentiation.
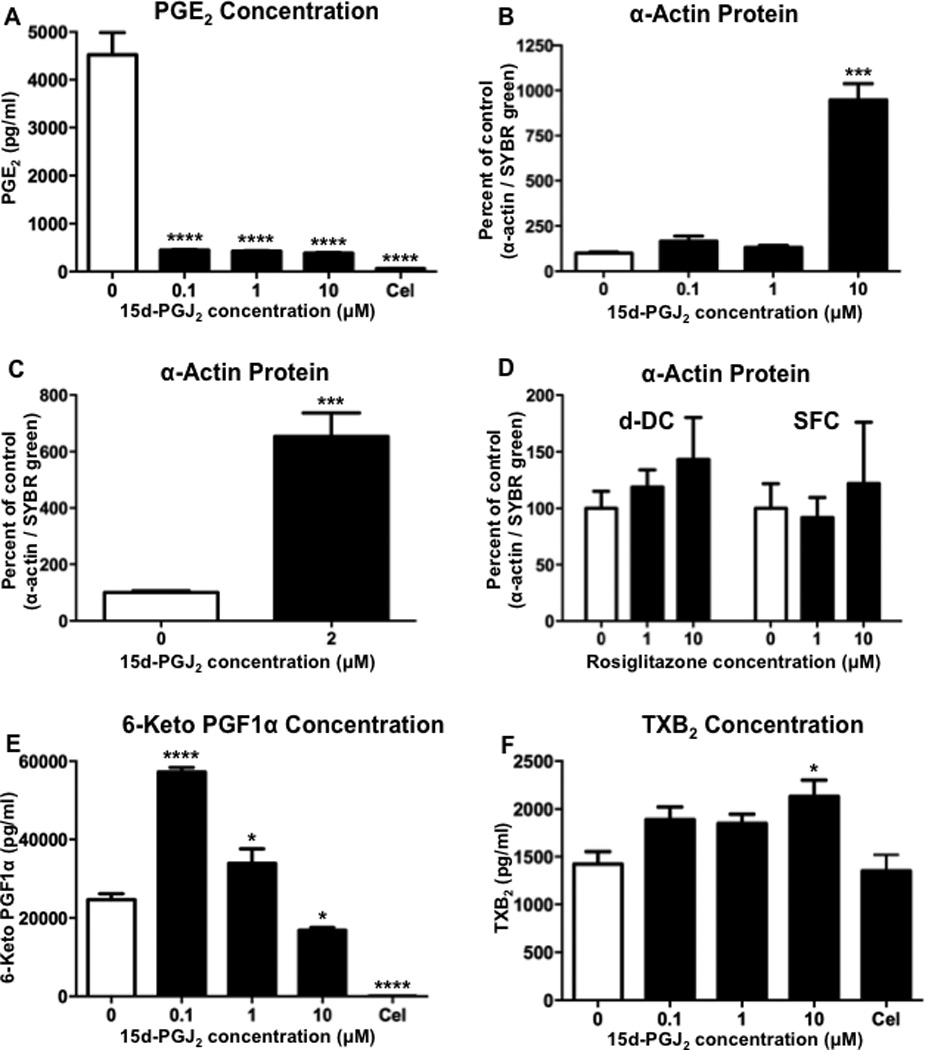
Following treatment with the mPGES-1 inhibitor 15d-PGJ2 or the COX-2 inhibitor celecoxib (Cel, 2 µM) for 1 day in de-differentiation conditions, hASMCs were analyzed for (A) PGE2 in the culture media, or (B) α-actin protein expression by immunohistochemistry. (C) hASMCs were also treated with 15d-PGJ2 in serum-free conditions and analyzed for α-actin protein expression by immunohistochemistry. (D) hASMCs were treated with rosiglitazone under de-differentiation (d-DC) and serum-free conditions (SFC) and analyzed for α-actin protein expression by immunohistochemistry. (E) hASMCs were treated for 1 day with 15d-PGJ2 or celecoxib (Cel, 2 µM) under de-differentiation conditions and analyzed for the stable PGI2 product 6-keto PG F1α in the culture media. (F) hASMCs were treated with 15d-PGJ2 or celecoxib (Cel, 2 µM) under de-differentiation conditions and analyzed for the stable TXA2 product TXB2. The data represent mean + SEM (n=4), * = p < 0.05, *** = p < 0.001, and **** = p < 0.0001; one-way ANOVA.
Previous studies using different cell types have shown that concentrations of 15d-PGJ2 which range from 3 to 10 µM are effective for activating the transcription factor peroxisome proliferator-activated receptor gamma (PPARγ).(34–37) Because the activation of PPARγ has the potential to alter the expression of differentiation markers, we examined the effect of the potent PPARγ ligand rosiglitazone. The treatment of hASMCs with rosiglitazone under conditions similar to the previous 15d-PGJ2 treatment conditions in either de-differentiating or serum-free conditions did not significantly alter α-actin expression (Figure 4D). Therefore, the increase in hASMC differentiation that we observed following treatment with 15d-PGJ2 is consistent with the inhibition of mPGES-1, rather than the activation of PPARγ.
In addition to reducing the levels of PGE2, the inactivation of mPGES-1 also has the potential to alter the levels of other prostanoids as a result of diversion of the PGH2 substrate. The genetic inactivation of mPGES-1 in mice has been shown to increase production of PGI2 and thromboxane A2 (TXA2) by VSMCs following LPS stimulation, but only PGI2 production under basal conditions.(38, 39) Pharmacological inhibition of mPGES-1 in humans has also been shown to increase production of PGI2 and TXA2.(40) Our current findings show that inhibition of mPGES-1 in hASMCs with 0.1 or 1 µM 15d-PGJ2 resulted in a significant increase in the detection of the stable PGI2 product 6-keto PG F1α, whereas the higher concentration of 10 µM significantly reduced 6-keto PG F1α levels (Figure 4E). In contrast, only the highest concentration of 10 µM 15d-PGJ2 produced a significant increase in the level of the stable TXA2 product TXB2 (Figure 4F). Furthermore, PGI2 metabolite production, but not TXB2 production, was significantly reduced by COX-2 inhibition, thereby suggesting that the PGI2 synthesis was dependent on COX-2 and the TXA2 synthesis was dependent on COX-1.
siRNA-mediated knockdown of mPGES-1 expression increases hASMC differentiation
To further examine the role of mPGES-1 in altering the SMC phenotype, we used siRNA to specifically reduce mPGES-1 expression, and compared the effects to mock-transfected cells. Following the treatment of hASMCs with mPGES-1 siRNA, there was a significant reduction in the expression of mPGES-1 mRNA (Figure 5A) and protein (Figure 5B). The reduced expression of mPGES-1 was associated with a significant reduction of PGE2 levels that were detected in the culture media (Figure 5C). mPGES-1 siRNA transfection also decreased the levels of the stable PGI2 product 6-keto PG F1α that were detected in the culture media (Figure 5D), whereas there was no significant effect of reduced mPGES-1 expression on the stable TXA2 product TXB2 (Figure 5E).
Figure 5. mPGES-1 knock-down decreases PGE2 and PGI2 synthesis.
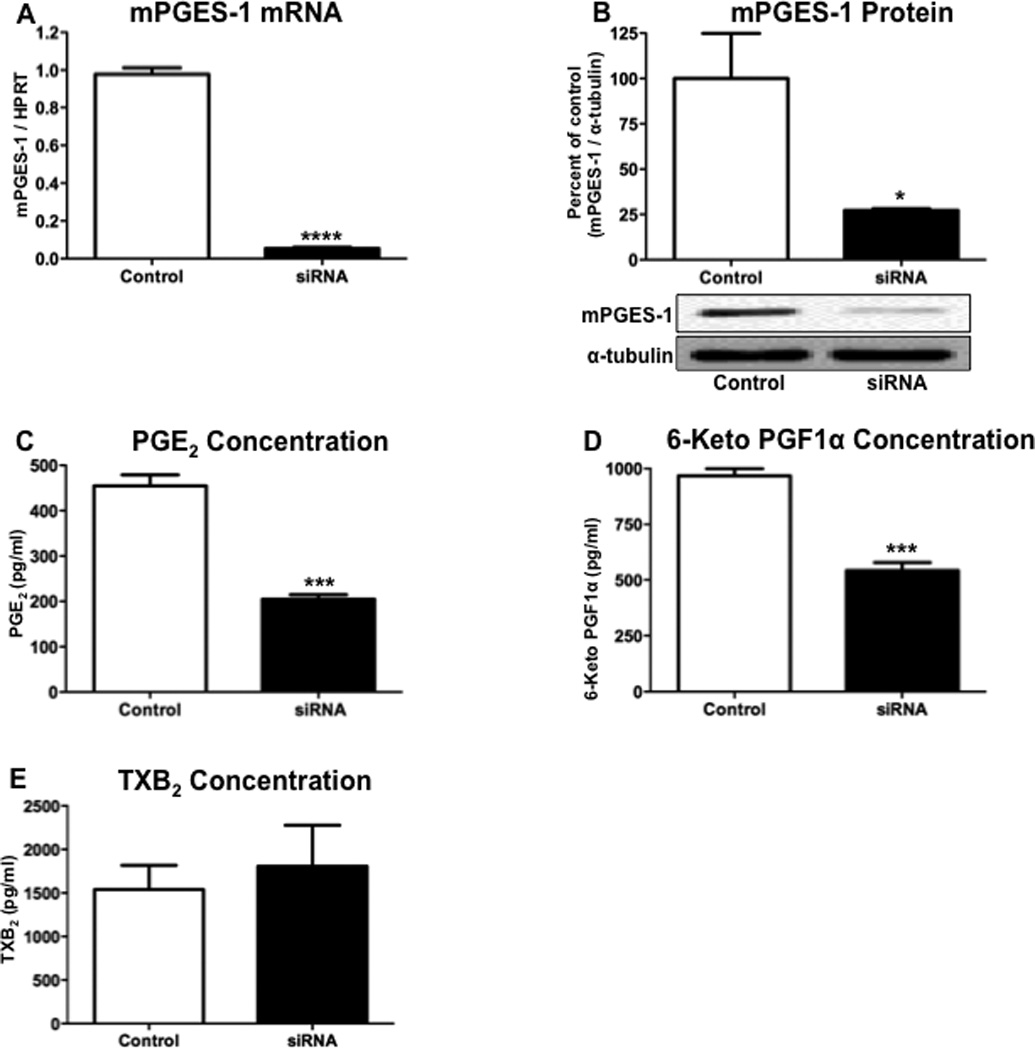
Following incubation with mPGES-1 siRNA for 3 days, hASMCs were analyzed for expression of (A) mPGES-1 mRNA, (B) mPGES-1 protein, (C) PGE2 in the culture media, (D) 6-keto PG F1α, or (E) TXB2. The data represent mean + SEM (n=3–5), * = p < 0.05, *** = p < 0.001, **** = p < 0.0001; unpaired two-tailed t-test.
With our finding that siRNA-mediated attenuation of mPGES-1 expression significantly reduced the synthesis of PGE2, we examined the effects of mPGES-1 siRNA transfection on hASMC differentiation and de-differentiation. Reducing the expression of mPGES-1 significantly increased mRNA expression of the differentiation markers α-actin and smoothelin (Figure 6, A and B). Although there was no detectable effect on the mRNA expression of SM22α (Figure 6C), siRNA-mediated mPGES-1 knockdown did significantly increase expression of the contractile proteins α-actin and SM22α (Figure 6, D and E). Therefore, the reduction of mPGES-1 expression and activity in hASMCs increases characteristics of the contractile SMC phenotype.
Figure 6. mPGES-1 knock-down increases differentiation markers and decreases de-differentiation markers.

Following incubation with mPGES-1 siRNA for 3 days, hASMCs were analyzed for expression of (A) α-actin mRNA, (B) smoothelin mRNA, (C) SM22α mRNA, (D) α-actin protein, (E) SM22α protein, (F) HAS-2 mRNA, or (G) MMP-2 mRNA. The data represent mean + SEM (n=3–5), ** = p < 0.01, *** = p < 0.001; unpaired two-tailed t-test.
Our previous findings show that PGE2 acts on hASMCs to promote the expression of the de-differentiation markers HAS-2 and MMP-2 (Figures 3G and 3H). Thus, we examined the effect on the de-differentiated phenotype that resulted from reduced levels of PGE2 following siRNA-mediated knockdown of mPGES-1. The treatment of hASMCs with mPGES-1 siRNA significantly decreased mRNA expression of HAS-2 and MMP-2, as compared to vehicle-transfected cells (Figures 6F and 6G). Therefore, reducing the expression mPGES-1 attenuates characteristics of the de-differentiated SMC phenotype.
Differential regulation of COX-2 and mPGES-1 expression by AngII in hASMCs
In different cell types, PGE2 has previously been shown to either increase or decrease the expression of COX-2.(41–43) A negative feedback mechanism would suggest that reducing the synthesis of PGE2 by mPGES-1 would result in the increased expression of COX-2, and the potential for synthesis of an altered PG profile. Thus, we examined the effects on COX-2 mRNA after treating with PGE2 or reducing mPGES-1 expression. The treatment of hASMCs with either 0.2 or 1 µM PGE2 resulted in the significant increase in COX-2 mRNA expression, as compared to the vehicle-treated control (Figure 7A). In addition, siRNA-mediated knockdown of mPGES-1 significantly decreased the levels of COX-2 mRNA, as compared to vehicle-transfected cells (Figure 7B). These findings suggest that in hASMCs decreasing the synthesis of PGE2 by mPGES-1 results in the reduced expression of COX-2.
Figure 7. Regulation of COX-2 and mPGES-1 expression in hASMCs.
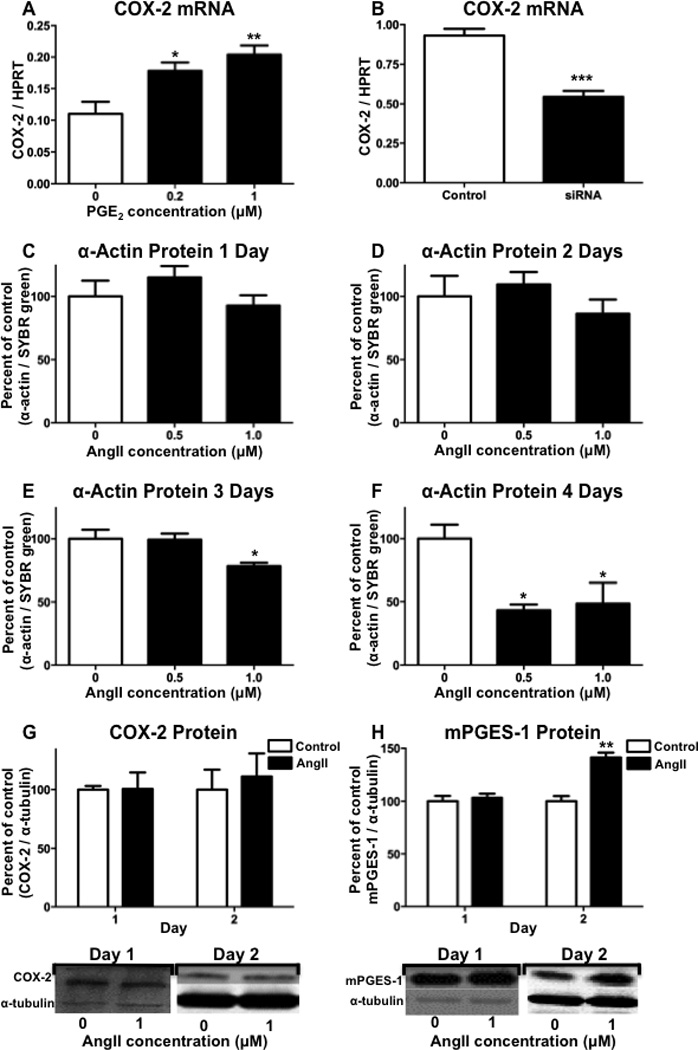
(A) Following treatment with PGE2 for 1 day, hASMCs were analyzed for expression of COX-2 mRNA. (B) Following incubation with mPGES-1 siRNA for 3 days, hASMCs were analyzed for expression of COX-2 mRNA. hASMCs were incubated with angiotensin II (AngII), for (C) 1 day, (D) 2 days, or (E) 3 days, or (F) 4 days, hASMCs were analyzed for expression of α-actin protein by immunohistochemistry. Following incubation with AngII, hASMCs were analyzed for (G) COX-2 protein or (H) mPGES-1 protein. For quantitation of protein expression, densitometry was performed on bands from Western blots and a representative Western blot is shown for each graph. The data represent mean + SEM (n=3–5), * = p < 0.05, ** = p < 0.01, *** = p < 0.001; (A, C–F) one-way ANOVA, (B, G–H) unpaired two-tailed t-test, (G–H).
Angiotensin II treatment reduces α-actin expression and increases expression of mPGES-1
We have previously shown that chronic infusion of Angiotensin II (AngII) in mice decreases expression of differentiation markers in aortic smooth muscle.(9, 10) In the current study, hASMCs were treated with AngII over a 4-day time-course and analyzed for α-actin expression by quantitative immunocytochemistry. The treatment of hASMCs with AngII for either 1 or 2 days did not significantly alter α-actin expression (Figure 7C and 7D). Following 3 days of treatment, however, 1 µM AngII significantly decreased α-actin expression, and following 4 days of treatment, there was a significant reduction in α-actin expression with AngII concentrations of 0.5 and 1 µM (Figure 7E and 7F). We also examined the effect of AngII treatment on the protein expression of COX-2 and mPGES-1 in hASMCs. Although there was no significant effect of AngII treatment on the expression of COX-2, following 2 days of AngII treatment there was a significant increase in the expression of mPGES-1 (Figure 7G and 7H). At the time-point of increased mPGES-1 expression following 2 days of AngII incubation, we also measured PGE2 levels in the media, but did not detect a significant increase in PGE2 production following AngII treatment (Control: 1328 pg/ml ± 112; AngII treated: 1647 pg/ml ± 111). Therefore, at the 2-day time-point, the absence of induced COX-2 expression may limit increased synthesis of PGE2 following AngII induction of mPGES-1 expression.
DISCUSSION
Normal mature blood vessels under physiological conditions possess vascular SMCs that exist in the contractile, differentiated, and quiescent state. During the progression of numerous types of cardiovascular diseases, including atherosclerosis, intimal hyperplasia following vascular injury, and aortic aneurysms, pathological remodeling of the vessel wall involves a change to the differentiated SMC phenotype.(1) During these disease processes, a phenotypic switch from the contractile to the synthetic phenotype with decreased expression of SMC markers of differentiation has been reported.(24, 44, 45) We have previously shown in a mouse model of abdominal aortic aneurysms (AAAs), that progression of the disease is associated with a significant reduction in the expression of markers of SMC differentiation.(10) To model the phenotypic modulation that we have previously observed, as well as characterize the mechanisms responsible for contributing to attenuation of SMC differentiation, the current report has utilized hASMCs cultured under different conditions that promote either differentiation or de-differentiation.
Smooth muscle α-actin is the most abundant and the most studied protein in differentiated SMCs, and its expression has been shown to decrease as SMCs acquire a de-differentiated phenotype during the development of cardiovascular disease.(44) The current studies utilized the quantitation of α-actin expression by hASMCs to characterize the level of differentiation in response to various culture conditions. Our findings show that differentiation-promoting culture conditions (DC) significantly increased α-actin mRNA expression at each point over a 4-day time-course and α-actin protein expression beginning on the second day of a 5-day time-course, as compared to de-differentiation-promoting conditions (d-DC) (Fig. 1A and B). We also examined the effect of the differentiation or de-differentiation culture conditions on the protein expression of SM22α. Although, the differences in SM22α expression were similar to those observed for α-actin expression, the statistical significance over the 5-day time-course was greater for the expression of α-actin (Fig. 1C). In addition to the markers of differentiation, we examined the effect of the different culture conditions on expression of HAS-2, a marker of de-differentiation that we have previously shown to be significantly increased during the development of in vivo vascular pathology.(9, 10, 23, 24) The culture of hASMCs with de-differentiation conditions for either 1 day or 2 days resulted in a significant increase in HAS-2 mRNA expression, as compared to the differentiation conditions (Fig. 1D). Our findings show that depending on the specific culture conditions hASMCs undergo phenotypic modulation between differentiated and de-differentiated states that is maintained up to 5 days of culture.
COX-2 has been shown to provide opposing functions that may increase or decrease pathological processes during the development of different types of cardiovascular disease. In models of atherosclerosis, inactivation of COX-2 has been shown to increase, decrease, or have no effect on severity of the disease, which may occur because of differences in the animal models and the cell types expressing COX-2.(46–50) Similarly, in models of vascular injury, there have also been varying conclusions as to the role of COX-2, as the result of COX-2 inhibition either increasing or decreasing intimal hyperplasia and the identification of multiple COX-2-expressing cell types at the site of injury.(51, 52) In contrast to these other cardiovascular disease models, we have previously shown by both genetic and pharmacological inactivation that COX-2 is critical to the development of AAAs in a mouse model of the disease.(53, 54) Furthermore, COX-2 inhibition that is initiated after AAAs have formed significantly delays further AAA progression.(9, 10) In this AAA model, COX-2 expression is specifically induced in the SMCs of the abdominal aorta at the site of the developing lesion.(9, 54) We also determined that the induced expression of COX-2 co-localizes in SMCs with reduced expression of markers of differentiation.(9) The analysis of numerous SMC differentiation markers shows that COX-2 inhibition attenuates the de-differentiation of SMCs in the abdominal aorta that occurs during AAA progression.(10) Therefore, in a disease model such as AAAs where the induction of COX-2 expression is localized specifically to SMCs, the decisively pathological role for COX-2 may be dependent on enhancing SMC de-differentiation.
Genome-wide mRNA expression analysis has previously shown a correlation between the induced expression of COX-2 and modulation of the vascular SMC phenotype both in vitro and in vivo.(14) In addition to these correlative findings, COX-2 inhibition has been shown to increase expression of the SMC differentiation marker MHC following 1 week of celecoxib treatment, which was then attenuated following 3 weeks of treatment, in contrast to the expression of desmin which was only increased after 3 weeks of treatment.(4) Furthermore, in this previous study, celecoxib treatment did not alter the expression of α-actin, but COX-2 inhibitor treatment did decrease contractile responses and increase intimal hyperplasia,(4) effects not necessarily expected from enhanced differentiation. In the current report, we used the manipulation of hASMC differentiation in culture to characterize mechanisms by which COX-2 regulates the SMC phenotype. We observed significantly greater mRNA expression of COX-2 in de-differentiation, as compared to differentiation conditions, thereby producing an inverse correlation between COX-2 expression and differentiation marker expression (Figure 2, A). For protein expression analysis, hASMCs were cultured to near confluence in de-differentiation conditions prior to being passaged and changed to either differentiation conditions or de-differentiation conditions. During a 5-day time-course in differentiation conditions (which lack growth factor supplements and contain reduced serum and increased heparin concentrations), there was a decline in COX-2 protein expression and an increase in contractile protein expression (Figure 2, B). At the beginning of the time-course after 1 day in differentiation conditions, COX-2 protein was detected which suggests an initial de-differentiated state which then declined over time (Figure 2, B). To determine whether there was a causative effect of COX-2 activity on regulating the hASMC phenotype, we examined the effects of pharmacological inhibition of COX-2 by celecoxib. Treatment with varying concentrations of celecoxib for multiple days identified a significant increase in α-actin expression following 2 or 3 days of treatment with the 10 µM concentration (Figure 2, C–E). Although there appeared to be an earlier trend for an increase, it is not known why 2 days of treatment with celecoxib were required to significantly increase α-actin expression. Nevertheless, prolonged treatment with a COX-2 inhibitor over multiple days of culture significantly increases hASMCs differentiation.
With our finding that the activity of COX-2 contributed to reduced differentiation of hASMCs, we utilized exogenous treatment with the two PGs primarily produced by vascular SMCs,(39) to identify PGs capable of producing de-differentiation. In contrast to the effects of celecoxib, we observed a significant increase in α-actin expression following treatment of hASMCs with the PGI2 mimetic iloprost (data not shown), similar to findings previously reported.(11) However, unlike the effects of PGI2, exogenous treatment with PGE2 significantly reduced the mRNA and protein expression of multiple differentiation markers important for contractile properties of smooth muscle (Figure 3, A–F).
In addition to the reduction in contractile protein expression, SMC de-differentiation is characterized by increased secretion of extracellular components important for pathological vascular wall remodeling. The increased expression of MMP-2 is a well-defined marker of SMC de-differentiation and has been shown to be induced by factors which promote the secretory phenotype and down-regulated by factors which promote the contractile phenotype. (29, 30, 55) In human aortic aneurysms, the increased expression of MMP-2 that occurs during SMC de-differentiation is an early characteristic of the disease.(8, 56) In both human aortic aneurysms and mouse aortic aneurysm models, the increased expression of MMP-2 is primarily derived from resident cells of the vascular wall, and not from the infiltration of inflammatory cells.(57–60) Our previous findings in mice have shown the increased aortic aneurysm progression that is dependent on the induction of COX-2 in aortic SMCs, directly correlates with increased expression of MMP-2.(9) Our current findings show that PGE2 treatment significantly increased mRNA expression of MMP-2 (Figure 3, H), thereby suggesting that COX-2-dependent SMC de-differentiation that we have observed results from increased production of PGE2.
Pathological vascular remodeling that is dependent on SMC de-differentiation is in part characterized by changes to the extracellular matrix. Remodeling of the extracellular matrix augments the migratory and proliferative characteristics of de-differentiated SMCs thereby promoting vascular lesion development. An early remodeling event that occurs during the development of atherosclerosis and vascular restenosis in humans is SMC deposition of the extracellular matrix component hyaluronic acid, which is therefore used as a marker of SMC de-differentiation. (23, 25, 61, 62) The overproduction of hyaluronic acid in vascular SMCs that results from SMC-specific transgenic overexpression of HAS-2 significantly increases atherosclerosis in a mouse model of the disease.(28) We have previously shown that COX-2-dependent de-differentiation of aortic SMCs that occurs during the development of aortic aneurysms in mice is associated with increased production of hyaluronic acid and HAS-2 mRNA expression.(10) The increased expression of HAS-2 mRNA and resulting increased production of hyaluronic acid requires the activity of COX-2 and is induced by exogenous treatment with vasodilatory PGs, including PGE2.(63) In SMCs induced to de-differentiate, PGE2 treatment enhances actin filament disorganization, although changes in the expression of contractile proteins used as established markers of differentiation were not described in this report.(64) With our current finding that PGE2 treatment of hASMCs significantly increased HAS-2 mRNA expression (Figure 3, G), the COX-2-dependent promotion of SMC de-differentiation that we have observed both in vitro and in vivo may result from increased production of PGE2.
To better characterize the contribution of PGE2 synthesized by hASMCs in promoting de-differentiation, we examined the effect of inhibiting PGE2 synthesis independent of inhibiting COX activity. mPGES-1 is the PGE2 synthase that functionally couples with COX-2 and contributes to the increased synthesis of PGE2 in a variety of pathological processes.(65) Although there is significant interest in developing inhibitors of mPGES-1, there are a limited number of reports describing mPGES-1 inhibitors that are effective in whole cells.(66) Recently, the metabolite of PGD2, 15d-PGJ2, has been shown to be active for inhibiting mPGES-1.(32, 33) To confirm the activity of 15d-PGJ2 for inhibiting the PGE2 synthesis by hASMCs, we analyzed PGE2 levels produced by hASMCs following treatment with varying concentrations of 15d-PGJ2. A concentration of 10 µM 15d-PGJ2 resulted in significant reduction of PGE2 synthesis and a significant increase in α-actin protein expression (Figure 4, A and B). 15d-PGJ2 was originally identified as a ligand for PPARγ, an effect which has been shown to occur at a concentration of approximately 10 µM,(34, 35, 37, 67) whereas the inhibition of mPGES-1 in serum-free conditions occurs in the low micro-molar range.(32, 33) We observed that in serum-free conditions a lower 15d-PGJ2 concentration of 2 µM was also effective at significantly increasing α-actin expression, whereas treatment with the specific PPARγ ligand rosiglitazone in either serum-containing or serum-free conditions did not affect α-actin expression (Figure 4, C and D). Therefore, the inhibition of mPGES-1 rather than activation of PPARγ may provide a mechanism for increased differentiation marker expression following 15d-PGJ2 treatment of hASMCs.
Although PGE2 may be synthesized by 3 different synthases depending on the cell type, in human aortic SMCs, mPGES-1 expression is required for the formation of PGE2.(27) The expression of mPGES-1 has been shown to be induced by a variety of agents, including growth factors and cytokines.(16, 17) In addition, mPGES-1 expression is potently induced by Egr-1, a transcription factor that increases target gene expression in response to serum stimulation.(68) Because the culture media used in the current studies for de-differentiation conditions contained serum and growth factors, these conditions allow for the detection of mPGES-1 mRNA and protein. In addition to the pharmacological inhibition of mPGES-1, we also utilized RNA interference to specifically reduce the expression of mPGES-1 and the synthesis of PGE2. Following mPGES-1 siRNA-mediated transfection, there were significant reductions in mPGES-1 mRNA and protein levels, together with reduced PGE2 levels (Figure 5, A–C). This attenuation of mPGES-1-specific PGE2 production resulted in a significant increase in mRNA and protein expression of markers of differentiation (Figure 6, A–E), as well as a significant reduction in de-differentiation marker expression (Figure 6, F and G). These findings are in agreement with our results showing that pharmacological inhibition of mPGES-1 with 15d-PGJ2 increased differentiation, whereas treatment with PGE2 decreased differentiation. Therefore, our current findings have demonstrated that the synthesis of PGE2 that results from the combined activities of COX-2 and mPGES-1 may be a prominent mechanism contributing to hASMC de-differentiation.
In vascular smooth muscle cells, the prostanoids that have been shown to increase as a result of the genetic deficiency of mPGES-1 are the stable PGI2 product 6-keto PG F1alpha and the stable TXA2 product TXB2.(38) Pharmacological inhibition of mPGES-1 in humans has also been shown to increase production of PGI2 and TXA2.(40) Our current finding show that at the lower concentrations of mPGES-1 inhibitor treatment, PGI2 significantly increased, whereas the highest concentration significantly reduced PGI2 levels (Figure 4E). Furthermore, the mPGES-1 siRNA transfection produced effects similar to the highest concentration of the mPGES-1 inhibitor. With our finding that the production of PGI2 is dependent on the activity of COX-2 (Figure 4E), the reduction in PGI2 that occurs following treatment with higher concentrations of the mPGES-1 inhibitor or following mPGES-1 siRNA transfection, may result from reduced expression of COX-2, which also occurs following mPGES-1 inactivation (Figure 7B).
The regulation of COX-2 expression in response to PGE2 has been shown to vary in vascular SMCs isolated from different tissue sources. In human coronary artery SMCs, exogenous treatment with PGE2 reduces the expression of COX-2, an effect that does not occur in venous SMCs.(69) A similar down-regulation of COX-2 expression occurs in porcine aortic SMCs in response to PGE2.(43) The effectiveness of inhibiting the activity of mPGES-1 as a mechanism to limit SMC de-differentiation could potentially be diminished by a feedback mechanism that resulted in the increased expression of COX-2 in response to decreased levels of PGE2. However, we determined that treatment of hASMCs with PGE2 significantly increased COX-2 mRNA expression, whereas RNA interference-mediated attenuation of mPGES-1 expression significantly decreased COX-2 mRNA expression (Figure 7, A–B). Therefore, in the hASMCs used in the current studies, the effectiveness of mPGES-1 inhibition for increasing SMC differentiation is not expected to be limited by a feedback mechanism that results in the increased expression of COX-2.
AngII contributes to the development of multiple cardiovascular diseases including hypertension, atherosclerosis, restenosis and aortic aneurysms. The hypertension promoting pathology of AngII results from contractile and hypertrophic effects of AngII on SMCs to increase expression of contractile proteins.(70–72) In contrast, AngII promotes the contribution of SMCs to the pathology of atherosclerosis and restenosis following vascular injury by increasing migration and proliferation, characteristics associated with decreased contractile protein expression and a de-differentiated phenotype.(1, 2, 73, 74) Similar to atherosclerosis, the characteristics of SMCs contributing to aortic aneurysm development in humans involves conversion to a de-differentiated phenotype.(8) In mice, the chronic infusion of AngII for 28 days results in a significant incidence of abdominal aortic aneurysms, and we have previously shown that under these conditions AngII-dependent aneurysm formation is dependent on the expression of COX-2.(54) We have also shown that AngII infusion in mice significantly down-regulates expression of markers of differentiation and increases de-differentiation marker expression.(10) Our current studies with hASMCs show that in order for AngII to induce a reduction in differentiation marker expression, chronic AngII treatment for either 3 or 4 days was required (Figure 7, C–F). Prior to the attenuation of differentiation, we did not observe an effect on COX-2 expression, but there was a significant increase in the expression of mPGES-1 protein (Figure 7, G–H). Therefore, increased expression of mPGES-1 and resulting PGE2 formation may contribute to the de-differentiation that occurs following chronic exposure of hASMCs to AngII.
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute, NIH (HL083122 to C.D.L).
Footnotes
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004 Jul;84(3):767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 2.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest. 2000 Nov;106(9):1139–1147. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007 Jun;(45 Suppl A):A25–A32. doi: 10.1016/j.jvs.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Roan JN, Tsai YC, Chen IW, Chang SW, Huang CC, Lam CF. Inhibition of cyclooxygenase-2 modulates phenotypic switching of vascular smooth muscle cells during increased aortic blood flow. Heart and vessels. 2012 May;27(3):307–315. doi: 10.1007/s00380-011-0148-y. [DOI] [PubMed] [Google Scholar]
- 5.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006 Mar;38(3):343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 6.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007 Dec;39(12):1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 7.Pannu H, Tran-Fadulu V, Papke CL, Scherer S, Liu Y, Presley C, Guo D, Estrera AL, Safi HJ, Brasier AR, Vick GW, Marian AJ, Raman CS, Buja LM, Milewicz DM. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet. 2007 Oct 15;16(20):2453–2462. doi: 10.1093/hmg/ddm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, Lau CL, Owens GK. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. 2009 Dec;138(6):1392–1399. doi: 10.1016/j.jtcvs.2009.07.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghoshal S, Loftin CD. Cyclooxygenase-2 inhibition attenuates abdominal aortic aneurysm progression in hyperlipidemic mice. PLoS One. 2012;7(11):e44369. doi: 10.1371/journal.pone.0044369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukherjee K, Gitlin JM, Loftin CD. Effectiveness of cyclooxygenase-2 inhibition in limiting abdominal aortic aneurysm progression in mice correlates with a differentiated smooth muscle cell phenotype. J Cardiovasc Pharmacol. 2012 Dec;60(6):520–529. doi: 10.1097/FJC.0b013e318270b968. [DOI] [PubMed] [Google Scholar]
- 11.Fetalvero KM, Shyu M, Nomikos AP, Chiu YF, Wagner RJ, Powell RJ, Hwa J, Martin KA. The prostacyclin receptor induces human vascular smooth muscle cell differentiation via the protein kinase A pathway. Am J Physiol Heart Circ Physiol. 2006 Apr;290(4):H1337–H1346. doi: 10.1152/ajpheart.00936.2005. [DOI] [PubMed] [Google Scholar]
- 12.Tsai MC, Chen L, Zhou J, Tang Z, Hsu TF, Wang Y, Shih YT, Peng HH, Wang N, Guan Y, Chien S, Chiu JJ. Shear stress induces synthetic-to-contractile phenotypic modulation in smooth muscle cells via peroxisome proliferator-activated receptor alpha/delta activations by prostacyclin released by sheared endothelial cells. Circ Res. 2009 Aug 28;105(5):471–480. doi: 10.1161/CIRCRESAHA.109.193656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pritchard KA, Jr, O’Banion MK, Miano JM, Vlasic N, Bhatia UG, Young DA, Stemerman MB. Induction of cyclooxygenase-2 in rat vascular smooth muscle cells in vitro and in vivo. J Biol Chem. 1994 Mar 18;269(11):8504–8509. [PubMed] [Google Scholar]
- 14.Lee MY, Garvey SM, Baras AS, Lemmon JA, Gomez MF, Schoppee Bortz PD, Daum G, LeBoeuf RC, Wamhoff BR. Integrative genomics identifies DSCR1 (RCAN1) as a novel NFAT-dependent mediator of phenotypic modulation in vascular smooth muscle cells. Hum Mol Genet. 2010 Feb 1;19(3):468–479. doi: 10.1093/hmg/ddp511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007 Sep;59(3):207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 16.Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999 Jun 22;96(13):7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000 Oct 20;275(42):32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 18.Wu YC, Su LJ, Wang HW, Jeff Lin CF, Hsu WH, Chou TY, Huang CY, Lu CL, Hsueh CT. Co-overexpression of cyclooxygenase-2 and microsomal prostaglandin E synthase-1 adversely affects the postoperative survival in non-small cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2010 Aug;5(8):1167–1174. doi: 10.1097/JTO.0b013e3181e2f4f5. [DOI] [PubMed] [Google Scholar]
- 19.Gomez-Hernandez A, Martin-Ventura JL, Sanchez-Galan E, Vidal C, Ortego M, Blanco-Colio LM, Ortega L, Tunon J, Egido J. Overexpression of COX-2, Prostaglandin E synthase-1 and prostaglandin E receptors in blood mononuclear cells and plaque of patients with carotid atherosclerosis: regulation by nuclear factor-kappaB. Atherosclerosis. 2006 Jul;187(1):139–149. doi: 10.1016/j.atherosclerosis.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 20.Catley MC, Chivers JE, Cambridge LM, Holden N, Slater DM, Staples KJ, Bergmann MW, Loser P, Barnes PJ, Newton R. IL-1beta-dependent activation of NF-kappaB mediates PGE2 release via the expression of cyclooxygenase-2 and microsomal prostaglandin E synthase. FEBS Lett. 2003 Jul 17;547(1–3):75–79. doi: 10.1016/s0014-5793(03)00672-0. [DOI] [PubMed] [Google Scholar]
- 21.Claveau D, Sirinyan M, Guay J, Gordon R, Chan CC, Bureau Y, Riendeau D, Mancini JA. Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J Immunol. 2003 May 1;170(9):4738–4744. doi: 10.4049/jimmunol.170.9.4738. [DOI] [PubMed] [Google Scholar]
- 22.Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, Sugiura H, Cao C, Watanabe Y, Kobayashi S. Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J Neurosci. 2001 Apr 15;21(8):2669–2677. doi: 10.1523/JNEUROSCI.21-08-02669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain M, He Q, Lee WS, Kashiki S, Foster LC, Tsai JC, Lee ME, Haber E. Role of CD44 in the reaction of vascular smooth muscle cells to arterial wall injury. J Clin Invest. 1996 Feb 1;97(3):596–603. doi: 10.1172/JCI118455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papakonstantinou E, Roth M, Block LH, Mirtsou-Fidani V, Argiriadis P, Karakiulakis G. The differential distribution of hyaluronic acid in the layers of human atheromatic aortas is associated with vascular smooth muscle cell proliferation and migration. Atherosclerosis. 1998 May;138(1):79–89. doi: 10.1016/s0021-9150(98)00006-9. [DOI] [PubMed] [Google Scholar]
- 25.Riessen R, Wight TN, Pastore C, Henley C, Isner JM. Distribution of hyaluronan during extracellular matrix remodeling in human restenotic arteries and balloon-injured rat carotid arteries. Circulation. 1996 Mar 15;93(6):1141–1147. doi: 10.1161/01.cir.93.6.1141. [DOI] [PubMed] [Google Scholar]
- 26.Evanko SP, Johnson PY, Braun KR, Underhill CB, Dudhia J, Wight TN. Platelet-derived growth factor stimulates the formation of versican-hyaluronan aggregates and pericellular matrix expansion in arterial smooth muscle cells. Arch Biochem Biophys. 2001 Oct 1;394(1):29–38. doi: 10.1006/abbi.2001.2507. [DOI] [PubMed] [Google Scholar]
- 27.Camacho M, Gerboles E, Escudero JR, Anton R, Garcia-Moll X, Vila L. Microsomal prostaglandin E synthase-1, which is not coupled to a particular cyclooxygenase isoenzyme, is essential for prostaglandin E(2) biosynthesis in vascular smooth muscle cells. Journal of thrombosis and haemostasis : JTH. 2007 Jul;5(7):1411–1419. doi: 10.1111/j.1538-7836.2007.02555.x. [DOI] [PubMed] [Google Scholar]
- 28.Chai S, Chai Q, Danielsen CC, Hjorth P, Nyengaard JR, Ledet T, Yamaguchi Y, Rasmussen LM, Wogensen L. Overexpression of hyaluronan in the tunica media promotes the development of atherosclerosis. Circ Res. 2005 Mar 18;96(5):583–591. doi: 10.1161/01.RES.0000158963.37132.8b. [DOI] [PubMed] [Google Scholar]
- 29.Hultgardh-Nilsson A, Lovdahl C, Blomgren K, Kallin B, Thyberg J. Expression of phenotype- and proliferation-related genes in rat aortic smooth muscle cells in primary culture. Cardiovasc Res. 1997 May;34(2):418–430. doi: 10.1016/s0008-6363(97)00030-8. [DOI] [PubMed] [Google Scholar]
- 30.Risinger GM, Updike DL, Bullen EC, Tomasek JJ, Howard EW. TGF-beta suppresses the upregulation of MMP-2 by vascular smooth muscle cells in response to PDGF-BB. Am J Physiol Cell Physiol. 2010 Jan;298(1):C191–C201. doi: 10.1152/ajpcell.00417.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamei D, Yamakawa K, Takegoshi Y, Mikami-Nakanishi M, Nakatani Y, Oh-Ishi S, Yasui H, Azuma Y, Hirasawa N, Ohuchi K, Kawaguchi H, Ishikawa Y, Ishii T, Uematsu S, Akira S, Murakami M, Kudo I. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J Biol Chem. 2004 Aug 6;279(32):33684–33695. doi: 10.1074/jbc.M400199200. [DOI] [PubMed] [Google Scholar]
- 32.Quraishi O, Mancini JA, Riendeau D. Inhibition of inducible prostaglandin E(2) synthase by 15-deoxy-Delta(12,14)-prostaglandin J(2) and polyunsaturated fatty acids. Biochem Pharmacol. 2002 Mar 15;63(6):1183–1189. doi: 10.1016/s0006-2952(02)00844-4. [DOI] [PubMed] [Google Scholar]
- 33.Prage EB, Morgenstern R, Jakobsson PJ, Stec DF, Voehler MW, Armstrong RN. Observation of two modes of inhibition of human microsomal prostaglandin E synthase 1 by the cyclopentenone 15-deoxy-Delta(12,14)-prostaglandin J(2) Biochemistry. 2012 Mar 20;51(11):2348–2356. doi: 10.1021/bi2019332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J(2) metabolite binds peroxisome proliferator- activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 35.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-Delta(12,14)-prostaglandin J(2) is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 36.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998 Jan 1;391(6662):79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 37.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998 Jan 1;391(6662):82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 38.Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, FitzGerald GA. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci U S A. 2006 Sep 26;103(39):14507–14512. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M, Ihida-Stansbury K, Kothapalli D, Tamby MC, Yu Z, Chen L, Grant G, Cheng Y, Lawson JA, Assoian RK, Jones PL, Fitzgerald GA. Microsomal prostaglandin e2 synthase-1 modulates the response to vascular injury. Circulation. 2011 Feb 15;123(6):631–639. doi: 10.1161/CIRCULATIONAHA.110.973685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin Y, Smith CL, Hu L, Campanale KM, Stoltz R, Huffman LG, Jr, McNearney TA, Yang XY, Ackermann BL, Dean R, Regev A, Landschulz W. Pharmacodynamic Comparison of LY3023703, a Novel Microsomal Prostaglandin E Synthase 1 Inhibitor, with Celecoxib. Clinical pharmacology and therapeutics. 2015 Sep 9; doi: 10.1002/cpt.260. [DOI] [PubMed] [Google Scholar]
- 41.Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011 Nov 17;118(20):5498–5505. doi: 10.1182/blood-2011-07-365825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vio CP, Quiroz-Munoz M, Cuevas CA, Cespedes C, Ferreri NR. Prostaglandin E2 EP3 receptor regulates cyclooxygenase-2 expression in the kidney. Am J Physiol Renal Physiol. 2012 Aug 1;303(3):F449–F457. doi: 10.1152/ajprenal.00634.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karim S, Berrou E, Levy-Toledano S, Bryckaert M, MacLouf J. Regulatory role of prostaglandin E2 in induction of cyclo-oxygenase-2 by a thromboxane A2 analogue (U46619) and basic fibroblast growth factor in porcine aortic smooth-muscle cells. Biochem J. 1997 Sep 1;326(Pt 2):593–599. doi: 10.1042/bj3260593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995 Jul;75(3):487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 45.Fatigati V, Murphy RA. Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. J Biol Chem. 1984 Dec 10;259(23):14383–14388. [PubMed] [Google Scholar]
- 46.Burleigh ME, Babaev VR, Oates JA, Harris RC, Gautam S, Riendeau D, Marnett LJ, Morrow JD, Fazio S, Linton MF. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation. 2002;105:1816–1823. doi: 10.1161/01.cir.0000014927.74465.7f. [DOI] [PubMed] [Google Scholar]
- 47.Narasimha A, Watanabe J, Lin JA, Hama S, Langenbach R, Navab M, Fogelman AM, Reddy ST. A novel anti-atherogenic role for COX-2--potential mechanism for the cardiovascular side effects of COX-2 inhibitors. Prostaglandins Other Lipid Mediat. 2007 Aug;84(1–2):24–33. doi: 10.1016/j.prostaglandins.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rott D, Zhu J, Burnett MS, Zhou YF, Zalles-Ganley A, Ogunmakinwa J, Epstein SE. Effects of MF-tricyclic, a selective cyclooxygenase-2 inhibitor, on atherosclerosis progression and susceptibility to cytomegalovirus replication in apolipoprotein-E knockout mice. J Am Coll Cardiol. 2003 May 21;41(10):1812–1819. doi: 10.1016/s0735-1097(03)00304-8. [DOI] [PubMed] [Google Scholar]
- 49.Pratico D, Tillmann C, Zhang ZB, Li H, Fitzgerald GA. Acceleration of atherogenesis by COX-1-dependent prostanoid formation in low density lipoprotein receptor knockout mice. ProcNatlAcadSciUSA. 2001;98:3358–3363. doi: 10.1073/pnas.061607398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hui Y, Ricciotti E, Crichton I, Yu Z, Wang D, Stubbe J, Wang M, Pure E, FitzGerald GA. Targeted deletions of cyclooxygenase-2 and atherogenesis in mice. Circulation. 2010 Jun 22;121(24):2654–2660. doi: 10.1161/CIRCULATIONAHA.109.910687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rudic RD, Brinster D, Cheng Y, Fries S, Song WL, Austin S, Coffman TM, FitzGerald GA. COX-2-derived prostacyclin modulates vascular remodeling. Circ Res. 2005 Jun 24;96(12):1240–1247. doi: 10.1161/01.RES.0000170888.11669.28. [DOI] [PubMed] [Google Scholar]
- 52.Wang K, Tarakji K, Zhou Z, Zhang M, Forudi F, Zhou X, Koki AT, Smith ME, Keller BT, Topol EJ, Lincoff AM, Penn MS. Celecoxib, a selective cyclooxygenase-2 inhibitor, decreases monocyte chemoattractant protein-1 expression and neointimal hyperplasia in the rabbit atherosclerotic balloon injury model. J Cardiovasc Pharmacol. 2005 Jan;45(1):61–67. doi: 10.1097/00005344-200501000-00011. [DOI] [PubMed] [Google Scholar]
- 53.King VL, Trivedi DB, Gitlin JM, Loftin CD. Selective cyclooxygenase-2 inhibition with celecoxib decreases angiotensin II-induced abdominal aortic aneurysm formation in mice. Arterioscler Thromb Vasc Biol. 2006 May;26(5):1137–1143. doi: 10.1161/01.ATV.0000216119.79008.ac. [DOI] [PubMed] [Google Scholar]
- 54.Gitlin JM, Trivedi DB, Langenbach R, Loftin CD. Genetic deficiency of cyclooxygenase-2 attenuates abdominal aortic aneurysm formation in mice. Cardiovasc Res. 2007 Jan 1;73(1):227–236. doi: 10.1016/j.cardiores.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 55.Jung HO, Uhm JS, Seo SM, Kim JH, Youn HJ, Baek SH, Chung WS, Seung KB. Angiotensin II-induced smooth muscle cell migration is mediated by LDL receptor-related protein 1 via regulation of matrix metalloproteinase 2 expression. Biochem Biophys Res Commun. 2010 Nov 26;402(4):577–582. doi: 10.1016/j.bbrc.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 56.Goodall S, Porter KE, Bell PR, Thompson MM. Enhanced invasive properties exhibited by smooth muscle cells are associated with elevated production of MMP-2 in patients with aortic aneurysms. Eur J Vasc Endovasc Surg. 2002 Jul;24(1):72–80. doi: 10.1053/ejvs.2002.1675. [DOI] [PubMed] [Google Scholar]
- 57.Freestone T, Turner RJ, Coady A, Higman DJ, Greenhalgh RM, Powell JT. Inflammation and matrix metalloproteinases in the enlarging abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 1995 Aug;15(8):1145–1151. doi: 10.1161/01.atv.15.8.1145. [DOI] [PubMed] [Google Scholar]
- 58.Davis V, Persidskaia R, Baca-Regen L, Itoh Y, Nagase H, Persidsky Y, Ghorpade A, Baxter BT. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 1998 Oct;18(10):1625–1633. doi: 10.1161/01.atv.18.10.1625. [DOI] [PubMed] [Google Scholar]
- 59.Goodall S, Crowther M, Hemingway DM, Bell PR, Thompson MM. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation. 2001 Jul 17;104(3):304–309. doi: 10.1161/01.cir.104.3.304. [DOI] [PubMed] [Google Scholar]
- 60.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002 Sep;110(5):625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kolodgie FD, Burke AP, Farb A, Weber DK, Kutys R, Wight TN, Virmani R. Differential accumulation of proteoglycans and hyaluronan in culprit lesions: insights into plaque erosion. Arterioscler Thromb Vasc Biol. 2002 Oct 1;22(10):1642–1648. doi: 10.1161/01.atv.0000034021.92658.4c. [DOI] [PubMed] [Google Scholar]
- 62.Fischer JW, Schror K. Regulation of hyaluronan synthesis by vasodilatory prostaglandins. Implications for atherosclerosis. Thromb Haemost. 2007 Aug;98(2):287–295. [PubMed] [Google Scholar]
- 63.Sussmann M, Sarbia M, Meyer-Kirchrath J, Nusing RM, Schror K, Fischer JW. Induction of hyaluronic acid synthase 2 (HAS2) in human vascular smooth muscle cells by vasodilatory prostaglandins. Circ Res. 2004 Mar 19;94(5):592–600. doi: 10.1161/01.RES.0000119169.87429.A0. [DOI] [PubMed] [Google Scholar]
- 64.Clement N, Glorian M, Raymondjean M, Andreani M, Limon I. PGE2 amplifies the effects of IL-1beta on vascular smooth muscle cell de-differentiation: a consequence of the versatility of PGE2 receptors 3 due to the emerging expression of adenylyl cyclase 8. J Cell Physiol. 2006 Sep;208(3):495–505. doi: 10.1002/jcp.20673. [DOI] [PubMed] [Google Scholar]
- 65.Hara S, Kamei D, Sasaki Y, Tanemoto A, Nakatani Y, Murakami M. Prostaglandin E synthases: Understanding their pathophysiological roles through mouse genetic models. Biochimie. 2010 Jun;92(6):651–659. doi: 10.1016/j.biochi.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 66.Koeberle A, Werz O. Inhibitors of the microsomal prostaglandin E(2) synthase-1 as alternative to non steroidal anti-inflammatory drugs (NSAIDs)--a critical review. Current medicinal chemistry. 2009;16(32):4274–4296. doi: 10.2174/092986709789578178. [DOI] [PubMed] [Google Scholar]
- 67.Yamakawa K, Hosoi M, Koyama H, Tanaka S, Fukumoto S, Morii H, Nishizawa Y. Peroxisome proliferator-activated receptor-gamma agonists increase vascular endothelial growth factor expression in human vascular smooth muscle cells. Biochem Biophys Res Commun. 2000 May 19;271(3):571–574. doi: 10.1006/bbrc.2000.2665. [DOI] [PubMed] [Google Scholar]
- 68.Sukhatme VP, Cao XM, Chang LC, Tsai-Morris CH, Stamenkovich D, Ferreira PC, Cohen DR, Edwards SA, Shows TB, Curran T, et al. A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell. 1988 Apr 8;53(1):37–43. doi: 10.1016/0092-8674(88)90485-0. [DOI] [PubMed] [Google Scholar]
- 69.Bishop-Bailey D, Pepper JR, Larkin SW, Mitchell JA. Differential induction of cyclooxygenase-2 in human arterial and venous smooth muscle: role of endogenous prostanoids. Arterioscler Thromb Vasc Biol. 1998 Oct;18(10):1655–1661. doi: 10.1161/01.atv.18.10.1655. [DOI] [PubMed] [Google Scholar]
- 70.Owens GK. Differential effects of antihypertensive drug therapy on vascular smooth muscle cell hypertrophy, hyperploidy, and hyperplasia in the spontaneously hypertensive rat. Circ Res. 1985 Apr;56(4):525–536. doi: 10.1161/01.res.56.4.525. [DOI] [PubMed] [Google Scholar]
- 71.Gariepy J, Massonneau M, Levenson J, Heudes D, Simon A. Evidence for in vivo carotid and femoral wall thickening in human hypertension. Groupe de Prevention Cardio-vasculaire en Medecine du Travail. Hypertension. 1993 Jul;22(1):111–118. doi: 10.1161/01.hyp.22.1.111. [DOI] [PubMed] [Google Scholar]
- 72.Geisterfer AA, Peach MJ, Owens GK. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ Res. 1988 Apr;62(4):749–756. doi: 10.1161/01.res.62.4.749. [DOI] [PubMed] [Google Scholar]
- 73.Sugiyama F, Haraoka S, Watanabe T, Shiota N, Taniguchi K, Ueno Y, Tanimoto K, Murakami K, Fukamizu A, Yagami K. Acceleration of atherosclerotic lesions in transgenic mice with hypertension by the activated renin-angiotensin system. Laboratory investigation; a journal of technical methods and pathology. 1997 Jun;76(6):835–842. [PubMed] [Google Scholar]
- 74.Prescott MF, Webb RL, Reidy MA. Angiotensin-converting enzyme inhibitor versus angiotensin II, AT1 receptor antagonist. Effects on smooth muscle cell migration and proliferation after balloon catheter injury. Am J Pathol. 1991 Dec;139(6):1291–1296. [PMC free article] [PubMed] [Google Scholar]