

Abstract
Proteases are essential for normal physiology as well as multiple diseases, e.g., playing a causative role in cancer progression, including in tumor angiogenesis, invasion, and metastasis. Identification of dynamic alterations in protease activity may allow us to detect early stage cancers and to assess the efficacy of anti-cancer therapies. Despite the clinical importance of proteases in cancer progression, their functional roles individually and within the context of complex protease networks have not yet been well defined. These gaps in our understanding might be addressed with: 1) accurate and sensitive tools and methods to directly identify changes in protease activities in live cells, and 2) pathomimetic avatars for cancer that recapitulate in vitro the tumor in the context of its cellular and non-cellular microenvironment. Such avatars should be designed to facilitate mechanistic studies that can be translated to animal models and ultimately the clinic. Here, we will describe basic principles and recent applications of live-cell imaging for identification of active proteases. The avatars optimized by our laboratory are three-dimensional (3D) human breast cancer models in a matrix of reconstituted basement membrane (rBM). They are designated mammary architecture and microenvironment engineering (MAME) models as they have been designed to mimic the structural and functional interactions among cell types in the normal and cancerous human breast. We have demonstrated the usefulness of these pathomimetic avatars for following dynamic and temporal changes in cell:cell interactions and quantifying changes in protease activity associated with these interactions in real-time (4D). We also briefly describe adaptation of the avatars to custom-designed and fabricated tissue architecture and microenvironment engineering (TAME) chambers that enhance our ability to analyze concomitant changes in the malignant phenotype and the associated tumor microenvironment.
Keywords: Protease, Live-cell imaging, Breast cancer, Tumor microenvironment, 3D models
1. Proteases, cancer and the tumor microenvironment
Protease (or proteolytic) activity mediates both physiological and pathological processes. Alterations in protease activity, involving members of all five major classes of endopeptidases [i.e., aspartic, cysteine, matrix metalloproteinases (MMPs), serine, and threonine proteases], are associated with malignant progression including tumor angiogenesis, invasion and metastasis [for review see Refs. [1,2]]. Proteases within the tumor microenvironment originate from both tumor and associated stromal cells (e.g., fibroblasts, macrophages, and endothelial cells). Typically, dysregulation of proteolysis at the tumor:stroma interface is not due to changes in any one particular protease; rather, it is the result of changes in the collective activity of a variety of proteases from multiple classes. Complex pathophysiological alterations within the local tumor microenvironment can lead to increased basal levels of proteolysis thereby augmenting the invasive capacity of the tumor. There are several different mechanisms by which proteolysis can be increased during malignant progression including aberrant localization of proteases, gene amplification, increased mRNA stability, altered expression or stability of endogenous inhibitors, and increased acidity at tumor margins leading to pH dependent protease activation.
Changes within the non-cellular tumor microenvironment, such as a decrease in extracellular pH or oxygen, can also promote the proteolytic breakdown of stromal tissues surrounding tumors. The tumor microenvironment is often acidic due to shifts in tumor metabolism (i.e., the Warburg Effect) [3,4]. Moreover, alterations in ion pumps/exchangers such as the V-ATPase proton pumps, which can be found in the plasma membrane, and NHE-1, the sodium-hydrogen ion exchanger, can contribute to the acidification of the tumor microenvironment [5,6]. Another contributing factor is poor tumor perfusion that generates a hypoxic tumor microenvironment. Oxidative stress within this hypoxic environment shifts the metabolic state of tumor cells from normal oxidative phosphorylation to glycolytic metabolism, resulting in the secretion of acidifying byproducts, activation of hypoxia-inducible factor (HIF)-1α, and transcription of target genes involved in angiogenesis and metastasis [7]. Acidification has been linked to an aggressive tumor phenotype and drug resistance [8–11]. Gillies and colleagues [5] have hypothesized that acidification of the tumor microenvironment promotes tumor invasion and metastasis by stimulating stromal remodeling. The general understanding is that localized tumor acidity creates an environment in which acid-resistant cells have a selective advantage. Tumor acidification has been shown to increase peritumoral protease activity [12] as illustrated in Fig. 1. For example, under normal conditions, procathepsin B (immature zymogen) is localized primarily to late endosomes/lysosomes, in which the acidic environment favors autoactivation or enzymatic activation by the aspartic protease cathepsin D of the zymogen into mature, active cathepsin B. Asparaginyl endopeptidase (AEP)/legumain, an endolysosomal cysteine protease, further processes cathepsin B from its active single chain form to its active double chain form [13]. During tumor progression, however, both procathepsin B and cathepsin B can be secreted or redirected to the plasma membrane. In conjunction with the Gillies laboratory [5], we have shown that an acidified tumor microenvironment also allows for extracellular activation of procathepsin B, and subsequent proteolysis of its substrates (e.g., collagen IV), which may provide space for tumor cell proliferation and invasion [12]. Using a murine window chamber model, we have shown that neutralizing the acidic pH of the tumor microenvironment with sodium bicarbonate can reduce the invasive potential of tumors [5]. Elucidating the dynamic nature of proteolysis within the tumor microenvironment will be key to understanding stromal remodeling, local invasion, and potential drug response.
Fig. 1.

Live cell imaging of proteolysis in 3D MAME cultures. MDA-MB-231 cell were grown in 3D rBM overlay cultures, at pH 6.8, in the presence of either DQ-collagen IV (A–C) or GB123 activity-based probe for cysteine cathepsins (D–F). Insets illustrate cultures grown at pH 7.4. Top panels depict images of entire 3D volume of cells (red), nuclei (blue) and DQ-collagen IV degradation products (green) (A); entire 3D volume of DQ-collagen IV degradation products only (B), and intensity maps for DQ-collagen IV degradation products (white, most intense; violet, least intense) (C). Bottom panels depict images of the entire volume of cells (red) and total (pericellular plus intracellular) active cysteine cathepsins bound to GB123 (cyan) (D); pericellular bound GB123 only (E) and intracellular bound GB123 only (F). Bars, 22.6 μm. Adapted from Rothberg et al., 2013. Neoplasia [12].
Some roles of individual or classes of proteases in malignant progression are clear; however, the complexity of proteolytic networks in cancer is overwhelming, calling into question whether targeting proteases will be efficacious. The lackluster results of clinical trials testing MMP inhibitors, nearly fifteen years ago, underscore this point. Although many MMPs have been implicated in malignant progression (e.g., MMP-1, 2, 3, 7, 9, 10, 11, 13, 14, and 26) [14], and thus considered potential therapeutic targets, their roles within the proteolytic network of the tumor microenvironment are more complex than originally thought. For example, during tumorigenesis some MMPs promote tumor progression while others are tumor suppressing [2,15,16]. In addition, tumor cells are capable of circumventing the effects of MMP inhibitors by activating alternative proteolytic pathways [17–19]. Thus, targeting only one class of proteases involved in proteolysis at the tumor:- stroma interface, may have only limited effects. Compensatory mechanisms further contribute to the complexity of proteolytic networks in cancer. Moreover, these networks are dynamic in regard to changes in protease expression, localization and activity depending on cancer type and stage of disease. Finally, a variety of proteases are also secreted from a multitude of tumor-associated cells, thereby contributing to the complex cancer degradome.
Over the past decade there has been an explosion in research to describe the dynamic and complex nature of protease interactions, regulation, and signaling. Overall and colleagues [20] have constructed a human protease web based upon computational analyses of data available from 23 tissue types in which they identified 1230 human proteases, inhibitors, and substrates as well as ~141,000 connections between them. They identify both interaction pairs and general hierarchies/nodes that interconnect all classes of proteases. Their work has important implications for the study of proteases in cancer since few current methods analyze the complex and dynamic nature of tumor proteolysis. Indeed, the sheer number of proteases and cell types involved in promoting malignant progression limits the scientific value of using traditional 2D culture systems to study protease activity since gene and protein expression profiles of cells under 2D culture conditions are greatly different from ones under 3D culture conditions [21,22]. We argue that this is the case because proteases from multiple cell types (e.g., tumor cells, fibroblasts, macrophages) all contribute to the collective proteolytic network of tumors. Thus, using cancer models that do not recapitulate the native tumor microenvironment may yield only a partial narrative. We propose that live-cell imaging of protease activity in complex, 3D pathomimetic culture models that include more than one cell type will provide a more complete understanding of: 1) the role(s) of protease networks in malignant progression; 2) how protease interactions contribute to stromal remodeling; and 3) how interactions between tumor and stromal cells within the tumor microenvironment modulate proteolysis.
2. Protease-targeted probes for live-cell imaging
Conventional methods to measure total protease and/or proteolytic levels provide limited information on dynamic changes in proteolytic activity that occur in live cells. To address these limitations, fluorescently labeled substrates and probes have been developed to identify and quantify active proteases in live cells as well as in whole animals. These imaging probes are advantageous because they can detect both total levels and/or spatio-temporal changes in active proteases. Moreover, these probes have been useful for biomarker discovery and the detection of cancer progression [23]. There are a number of reviews on using imaging to study proteases [24–26]. Here we review applications of substrate-based and activity-based probes for the detection of active proteases associated with cancers.
2.1. Substrate-based probes
Substrate-based probes (SBPs), one of the most common tools to monitor protease activity, contain peptide sequences that can be cleaved by the targeted proteases. SBPs generally consist of two parts: peptides cleaved by the protease(s) of interest, and a fluorophore that generates visible signals when the probe is cleaved by proteases. Various fluorescent dyes such as aminomethylcoumarin (AMC), boron-dipyrromethene (BODIPY), rhodamine, cyanine, dansyl, and nitrobenz-2-oxa-1,3-diazole (NBD) have been commonly used as fluorophores [27]. Near-infrared fluorochromes are used preferably for in vivo imaging applications because they exhibit greater depths of penetration into tissues and have a higher signal to background ratio [28]. Synthetic substrates are often used for imaging as well as other biochemical assays due to their enhanced selectivity for target proteases as compared to natural substrates [29,30]. The protease recognition site within the probes is crucial for conferring selectivity. For example, SBPs with high selectivity toward cathepsin B have been used to detect cathepsin B activity in live cancer cells [31] and in dysplastic intestinal adenomas in mice [32].
Recently, many SBPs have been constructed to contain a quencher along with the fluorophore. The quencher and the fluorophore are positioned at opposite ends of a recognition site to prevent high levels of background fluorescence. Upon cleavage of the quenching functional group, the fluorescent signal is released, enabling their detection. Some SBPs contain two or more fluorophores that are self-quenched when in close proximity. These fluorophores are linked by peptides containing protease-selective recognition sites. When the target protease cleaves the recognition site between fluorophores, free monomers that are released emit fluorescent signals. This class of SBPs has been widely used in animal models for the detection of cysteine cathepsins [32,33]. These probes function on the basis of fluorescence resonance energy transfer (FRET) technology. FRET monitors changes in proteineprotein interactions and protein conformation in a distance-dependent manner by using two proximal fluorophores (a donor and an acceptor). Typically, the emission wavelength of a donor is similar to the excitation wavelength of an acceptor, so that in close proximity the acceptor quenches the fluorescence emitted by the donor. Once a peptide sequence between these pairs is hydrolyzed and/or conformationally changed by proteases, the distance between the donor and acceptor fluorophores increases resulting in the emission of fluorescence. Changes in the fluorescence intensity are correlated with proteolytic activity and can be detected and quantified by live imaging or other biochemical assays of protease activity [34]. These probes can also be used to determine the proximity of the donor and acceptor within a cell. Using this method, many proteases including MMPs [35–39] and cathepsin B [38] have been visualized in live cancer cells. For more detailed descriptions of FRET-based protease probes including dye quenched protein substrates, please see Dive et al. [25] and Hu et al. [40].
Dye quenched (DQ) protein substrates have been employed to detect and distinguish pericellular proteolytic activity from intra-cellular proteolytic activity (Fig. 1A–C) [12,41]. These substrates are analogs of proteins commonly found in the extracellular matrix (ECM) (e.g., collagens). Labeling of these substrates with excessive numbers of fluorescent dyes quenches the signal, and allows them to remain undetected prior to proteolytic degradation. Upon hydrolysis of the substrate, the dyes are separated from each other and detected as a fluorescent signal. Our laboratory has established methods to quantify degradation of DQ-collagens on a per cell basis, including quantification of total, pericellular and intracellular degradation products [41]. Moreover, through the use of protease inhibitors we further revealed that cysteine cathepsins, MMPs and serine proteases are involved in DQ-collagen degradation by breast and colon cancer cells [42]. These probes have also been used to monitor ECM degradation associated with cancer cell migration and invasion [12,42–45].
A major advantage of SBPs is the signal amplification and therefore their ability to detect less abundant proteases. On the contrary, one major drawback of SBPs is that they do not allow direct identification of the target proteases since they are selective rather than specific for any single protease. Furthermore they do not remain bound to the target proteases after proteolysis and thus fluorescent signals can diffuse away from the cleavage site, making accurate localization of the target protease(s) difficult.
2.2. Activity-based probes
Activity-based probes (ABPs), unlike SBPs, directly label active proteases through activity-dependent covalent modification. Typical ABPs are composed of three critical components: 1) a reactive functional group, referred to as a warhead, for binding at the active site of a target enzyme, 2) a tag for visualization, and 3) a linker that separates the reactive functional group from the tag, allowing for increased accessibility of probes to target proteases [46]. New generation ABPs contain a quencher in close proximity to the tag to reduce non-specific, fluorescent signals from unbound probes. Once the quenched ABPs (qABPs) covalently bind to the active site of the target enzyme, the quencher is released by the active enzyme and a fluorescent signal is generated. Hence, qABPs have a higher signal to background ratio as compared to non-quenched ABPs, allowing for imaging of more defined proteolytic activity in live cells [reviewed in Refs. [25,26]].
ABPs have been developed for the detection of cysteine, serine, threonine, and metallo-proteases, both in live cells and animals [28,37,47–51]. Methods to monitor active caspases [52] and cysteine cathepsins [12,28,51,53] in cancers and inflammation using ABPs are well-developed. In Fig. 1D–F, we illustrate the use of an ABP for cysteine cathepsins to image active forms of cathepsins B and L [12]. Blum et al. [28] generated and developed a series of nonquenched (GB123) and quenched ABPs (GB137) that target active cysteine cathepsins. In breast cancer animal models, GB123 tagged with Cy5 and GB137 with near-infrared fluorescent tags reached a maximal signal to background ratio in 6 h and the signal then lasted for an extended period. Edgington and coworkers [52] demonstrated optical imaging of apoptosis by caspases-3 and -7 with ABPs, AB50-Cy5 and tAB50-Cy5, in mouse xenograft models bearing human colorectal tumor cells. The caspase ABPs are stable in vivo and show highly selective labeling of target enzymes. Such applications of ABPs show their potential for the detection of protease as biomarkers and protease-targeted drugs in diseases including cancers [54].
Compared to SBPs, a major advantage of ABPs is that they bind covalently to the active site of a target protease. This facilitates evaluation of their spatial distribution. Since the target proteases are covalently tagged with a fluorescent label, ABPs can also be used to identify the labeled proteases by posthoc testing (e.g., by Western blotting and flow cytometry) and thereby profiling of the biological activity of target proteases in live cells [55]. Since an ABP will covalently bind to the active site of only one target protease, a potential disadvantage of using ABPs is the low signal output. Furthermore, ABPs are based on inhibitors and thus there may be possible structural and functional alterations in the target protease that have unexpected biological consequences (e.g., pharmacological knock-down of the target protease). Nonetheless, ABPs have been shown to have a minimal effect on cysteine cathepsin activity in vivo, likely due to the low doses used for imaging [56]. To our knowledge, this has not been verified for live-cell imaging of protease activity in vitro. This needs to be considered if live-cell proteolysis assays are to be used in MAME cultures as a surrogate readout for efficacy of drug treatments over extended periods of time (see below). Over time such assays and other pathobiological endpoints may be affected even if an ABP only minimally reduces protease activity.
Evaluation of collective protease activity using SBPs and of specific protease activity using ABPs has provided valuable information on the role of proteases in cancer both in vitro and in vivo [25,26,46]. Nonetheless, current imaging probes for the detection of proteolytic activities still lack high spatial and temporal resolution, sensitivity and selectivity for target proteases. Hence, next-generation probes should be developed with consideration for greater selectivity, photo-stability, and increased signal to background ratios.
3. MAME cultures: avatars for live-cell imaging of protease activity
In vitro and in vivo studies using imaging probes to detect proteolytic activity have greatly advanced the areas of cancer detection, patient prognosis, and prediction of responses to various therapies, but both have their limitations. It is difficult to obtain a comprehensive understanding of the vast complexity of proteolysis solely by using animal models. The myriad aspects of cross-talk between cancer cells and associated stromal cells cannot be easily assessed in animal models, nor do animal models support tightly regulated and reproducible parametric studies. Furthermore, the findings in animal models may not translatable to human cancers.
On the other hand, in vitro two-dimensional (2D) cell cultures, although advantageous due to their highly controllable culture environments, lack the 3D architectural context including the cell microenvironment. Consequently, results from 2D culture experiments are often untranslatable and irreproducible in vivo. The difficulty of studying tumor-associated protease activity upon the backdrop of a 2D culture system underscores this point. Moreover, it has been well established that the proteases contributing to collective tumor proteolysis originate from a variety of cell types including tumor cells, fibroblasts, macrophages, and endothelial cells. Thus, the use of models that do not mimic the native tumor microenvironment may produce intrinsically limited insights in relation to the identification of mechanisms and pathways involved in malignant progression.
The use of 2D cell cultures also restricts our understanding of the mechanisms regulating the remodeling of the tumor micro-environment that accompanies local invasion. Conventional in vitro 2D cultures cannot reproduce morphogenesis and cellecell interactions in physiological conditions that occur by either physical contact or cytokine autocrine/paracrine mechanisms in a complex tumor microenvironment comprised of multiple cell types. The absence of the microenvironment in 2D cultures also has an effect on gene expression, protein synthesis and trafficking/secretion of proteases. We have previously shown an increase in secretion of procathepsin B through integrin signaling (α1, α2, and β1) by human breast fibroblasts grown in 3D collagen I gels as compared to 2D cultures grown on plastic or glass culture dishes [57]. Studies of pre-invasive breast carcinoma cells (MCF10.DCIS) and fibroblasts revealed that fibroblasts grown in 3D cultures secreted more MMP14, hepatocyte growth factor (HGF), cyclooxygenase (COX) 2, and CXCL12, as compared to those grown in 2D. The secretions by fibroblasts increased the invasive phenotype of co-cultured MCF10.DCIS cells [22].
Many proteases are redirected to the plasma membrane and/or secreted into the tumor microenvironment of cancers, yet changes in trafficking of proteases may not necessarily increase proteolytic activity unless other factors, including proper localization of protease receptors and activators (such as uPAR and MMP14), the absence of endogenous protease inhibitors [such as tissue inhibitor of metalloproteinases (TIMPs) and serpins], and pericellular acidification also occur. These factors can be altered during tumor progression by hypoxia and acidification of the tumor microenvironment, and cellular interactions with various ECM components, such as laminin, collagens I and IV, and fibronectin. Thus, to better understand the ways in which proteolysis contributes to stromal remodeling and local invasion, models should be: 1) composed of ECM components that best mimic in vivo conditions, 2) developed for co-culturing with tumor-associated cells, and 3) controlled to allow monitoring of changes in the tumor microenvironment such as hypoxia and acidification. Ideal models for the multistep process of tumor development would allow for phenotypic heterogeneity of and genetic changes in tumor cells, biological contexts, heterotypic cell crosstalk and non-cellular factors (e.g., hypoxia and acidosis) of the microenvironment.
Three-dimensional in vitro models have been developed as an important alternative that takes advantage of both the in vivo tumor microenvironment, by the use of rBM, and the refined and reproducible conditions of traditional 2D cultures [58]. Many studies have shown that tumor architecture and complexity can be reproduced using 3D cultures of human-derived cells or organoids, which enable the investigations of specific communications between neighboring cells and the effects of non-cellular factors (e.g., hypoxia and acidosis) within defined tumor microenvironments. Here, we are introducing a 3D in vitro breast cancer avatar and discussing the integration of these avatars into environmentally controlled chambers to perform live-cell imaging and secretome (i.e., the total organic and inorganic molecules secreted by cells) analysis that accompany cell:cell interactions over extended periods of time.
We developed a tractable 3D/4D (3D + time) rBM overlay culture model designated mammary architecture and microenvironment engineering (MAME) as a compromise between 2D in vitro culture and in vivo models. Our MAME models are extended and refined from those of Bissell and Brugge [59,60]. The MAME models are pathomimetic avatars of breast cancers and are being used for functional imaging of proteolysis and morphometric analyses in real time during progression (Fig. 2). Detailed protocols for establishing MAME cultures have been described previously [61]. In MAME cultures [62], normal breast epithelial cells and tumor cells show strikingly similar characteristics to the same cells grown as orthotopic xenografts in vivo [63]. We have also observed degradation of DQ-collagens in MAME models and identified cysteine cathepsins associated with tumor progression by using ABPs (for example, see Fig. 1) [12,64]. Our MAME models also enable the user to study cells grown in 3D in mixed or parallel co-cultures and analyze dynamic and temporal changes in proteolytic activity in real-time, i.e., 4D.
Fig. 2.

Schematic illustrations of normal versus cancerous human breast cells within the in vivo breast microenvironment (A) and as modeled in our 3D MAME avatars (B). In vivo, normal human breast ducts, comprised of myoepithelial cells and luminal epithelial cells (i.e., epithelial cells lining the lumen) (A, gray dashed box), grow in a controlled normoxic and neutral pH microenvironment. In contrast, breast cancer cells interact with adjacent and infiltrating stromal cells (e.g., fibroblasts, inflammatory cells, endothelial cells, and adipocytes) (A, orange dashed box) and grow irregularly in a hypoxic and acidic tumor microenvironment. Our 3D MAME avatars are designed to mimic in vivo normal and cancerous breast tissues (B). Normal epithelial cells grown in 3D MAME cultures form acinar structures with lumens resembling normal breast tissue (B, top image) and exhibit a basal level of DQ-collagen IV degradation (green). In comparison, breast ductal carcinoma in situ cells grown in 3D MAME cultures form larger, irregular and dysplastic structures (B, middle image). Co-cultures of breast carcinoma cells and fibroblasts result in an invasive morphology and increased degradation of DQ-collagen IV (B, bottom image). A was modified from Nelson and Bissell, 2005. Seminars in Cancer Biology [80].
We investigated whether the growth of breast epithelial cells in the MAME model could replicate in vivo-like structures. Thus, we analyzed morphogenesis and oncogenic transformation of human breast epithelial cells using MCF-10A cells. The MCF-10A cells, non-tumorigenic breast epithelial cells, grown in MAME culture conditions proliferated and formed unique 3D structures not seen in 2D culture conditions. Immunostaining for the basal receptor, integrin α6, shows the formation of spheroids that develop into acini with central lumens, resembling acinar structures of mammary lobules [62]. When the isogenic MCF-10A progression series of cell lines, c-H-ras transformed MCF10AneoT [63,65] and non-invasive MCF10AT1 [63], were analyzed under the same conditions, they formed simple duct-like structures with no central lumens and multi-acinar structures, which are typical of in vivo-like phenotypes of benign hyperplastic and atypical hyperplastic lesions, respectively. In MAME models, during the transition of ductal carcinoma in situ (DCIS) cells, MCF10.DCIS, to invasive ductal carcinomas cells, MCF10.CA1d, there was a change a change in morphology from large dysplastic structures to invasive structures, along with loss of the expression of integrin α6 [62]. Indeed, the morphologies of these cells in MAME cultures accurately imitate their in vivo behavior. Additionally, experiments with GB111, an ABP for the detection of cysteine cathepsins [62], revealed that cathepsin B promotes proliferation and malignant progression of breast epithelial cells, which is consistent with animal studies showing reduced tumorigenic capacity in cathepsin B-deficient mouse models [66] and increased tumor growth in cathepsin B-overexpressing mouse models [67].
One important non-cellular factor that affects tumor progression in vivo is acidosis. Adjusting acidic conditions to mimic levels found in the in vivo tumor microenvironment (i.e., pH 6.8) in conjunction with the use of DQ-collagen IV showed an increase in degradation of this substrate in the peritumoral regions of MDA-MB-231 breast carcinoma cells as compared to those grown at a neutral pH of 7.4 (Fig. 1A–C) [12]. Moreover, using GB123, an ABP for cysteine cathepsins, we demonstrated increases in the secretion of active cathepsin B at pH 6.8, which contributed to increased degradation of collagen IV (Fig. 1). These results are consistent with the fact that acidity generated within the tumor microenvironment enhances tumor invasion in vivo by inducing secretion of active cathepsin B [68].
We have also addressed specific cell crosstalk between breast carcinoma cells and surrounding stromal cells including fibroblasts, adipocytes, and endothelial cells within an in vivo-like human tumor microenvironment using MAME models. Among these cells, fibroblasts are the most abundant cell types and their infiltration into tumors with extensive loss of basement membrane is observed during malignant progression from pre-invasive DCIS to invasive breast carcinoma. Co-culturing of MCF10.DCIS cells in MAME models with mammary fibroblasts overexpressing stromal-derived hepatocyte growth factor (HGF) (MF:HGF) or in the presence of conditioned media from the fibroblasts results in increased invasiveness and development of invasive outgrowths by the MCF10.DCIS cells [69]. Enhanced degradation of extracellular DQ-collagen IV was also observed in MCF10.DCIS cells co-cultured with MF:HGF. The mechanism by which HGF promotes the invasive nature of these cells is through activation of c-Met and stimulation of secretion of urokinase plasminogen activator (uPA) and its receptorm uPAR. Xenografts formed by co-injection of MCF10.DCIS cells and MF:HGF exhibit significant increases in progression to invasive ductal carcinomas, consistent with the results in vitro in MAME cultures [69]. In other studies, we have shown that degradation of DQ-collagen IV by tumor cells co-cultured with fibroblasts was further augmented by addition of macrophages [70], consistent with studies by others showing the importance of stromal cells, and in particular inflammatory cells, for the study of tumor proteolysis [47,66,71].
Other applications in which we are using MAME models are for the analysis of immunotherapeutic approaches with armed activated T cells targeting breast carcinoma cells, photodynamic therapy against inflammatory breast cancer cells, and therapy with small molecule kinase inhibitors or cytokine blocking antibodies against breast carcinoma cells (unpublished results). We contend that MAME models provide a tractable and controllable environment into which one can incorporate specific cells, growth factors, and probes for functional imaging including imaging in real-time of proteolysis by live cells. In addition to delineating biological mechanisms during tumor development, MAME models also serve as reliable platforms for generating predictive preclinical results before in vivo evaluation of drugs.
4. Applications of tissue architecture and microenvironment engineering (TAME) chambers beyond live-cell imaging of proteolytic activity
We saw the need to develop an imaging modality, which not only maintains the well-defined microenvironment present in our MAME models, but also allows the ability to grow cultures undisturbed for extended periods of time. Our MAME models have some limitations as an avatar for cancer research, including the: 1) inability to examine the effects of secreted factors on cell:cell interactions over extended periods; and 2) difficulty in evaluating cell migration in complex co-cultures. Therefore, the MAME models restrict study of the dynamic nature of tumor proteolysis over extended periods of time. In addition, we also had an interest in developing a model to monitor changes in secretome proteins, cell:cell interactions, migration, morphology, and other factors implicated in tumor growth (e.g., hypoxia and acidosis) in real-time.
In line with the MAME models that our lab has developed, we designed a new imaging platform, the tissue architecture and microenvironment engineering (TAME) chamber. The TAME chamber supports long-term growth of cell cultures and is designed for live-cell imaging. We have developed platforms with single well chambers and dual-well chambers connected by a small channel that can support monoculture, direct co-culture, or parallel co-culture studies. To date, we have cultured human breast carcinoma cells in monoculture and parallel co-cultures with human breast carcinoma-associated fibroblasts (CAFs) and normal breast fibroblasts (NFs) on our platform for >60 days. These cultures were allowed to grow undisturbed for the entire experiment only requiring media changes every seven days at which time conditioned media was collected for secretome analysis. Cells were labeled with vital dyes thus enabling live-cell imaging by confocal microscopy over the entire length of the experiment. We were able to detect differences in fibroblast migration toward tumor cells as well as changes in tumor cell morphology over time depending on whether they were in monoculture or parallel co-culture with CAFs or NFs. CAFs, but not NFs, could be detected infiltrating into tumor-like structures when grown in parallel co-culture with tumor cells, as shown in the cartoons of Fig. 3. Planned studies with TAME chambers include the study of dynamic changes in the secretome of breast cancer cells in monoculture as compared to parallel co-culture with various fibroblast cell lines, as well as, determining whether CAFs are capable of preferentially migrating toward other breast cancer cell lines in our TAME chamber model. Since one focus of our lab is the study of proteolysis by live-cell imaging, we are also interested in imaging proteolysis in these same cultures over 60 days. In an effort to make our models better able to mimic the conditions of the tumor microenvironment, we are in the process of developing novel pH and oxygen sensors that will allow us to monitor the effects of acidic and hypoxic microenvironment conditions over extended periods of time in TAME chambers.
Fig. 3.
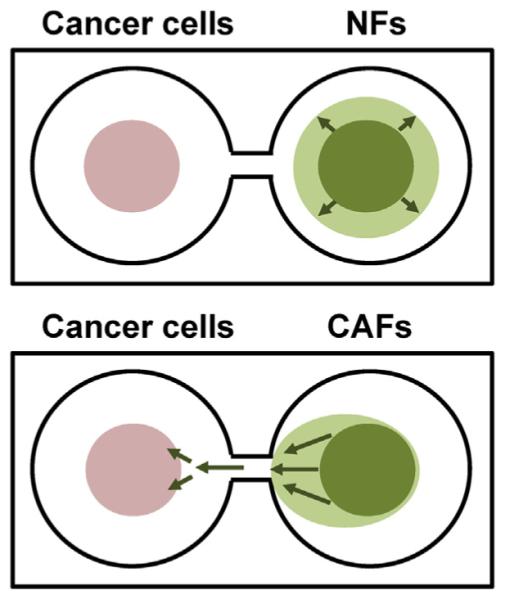
Cartoons illustrating distribution of breast cancer cells and normal fibroblasts (NFs; upper) or carcinoma-associated fibroblasts (CAFs; lower) grown in parallel cocultures in TAME chambers. Breast cancer cells (pink circles) and fibroblasts (green) were imaged live to follow cell:cell interactions and movement over a 63 day period. Green arrows indicate the observed migration of fibroblasts over this period. CAFs, but not NFs, migrated preferentially toward breast cancer 3D structures.
We believe that the TAME chamber represents a powerful new tool for studying the complex and dynamic nature of tumor kinetics and could potentially be developed as a drug screening platform. Indeed, this model can support drug screening studies in which cultures are seeded onto rBM and allowed to form 3D structures prior to adding therapeutic agents. Furthermore, the analyses can be carried out in real-time and over extended periods of time. This can include quantification of dynamic responses of cell:cell and cell:matrix interactions, growth, cytotoxicity, and cytokine/chemokine secretion. Since the TAME chamber is designed to support imaging studies, its use as a drug screening platform allows for the study of therapies that directly target tumor cells, tumor proteolysis, other cells associated with tumors such as fibroblasts and inflammatory cells or non-cellular features of the tumor microenvironment such as acidosis. It also could be useful as a model for studying tumor re-growth after drug treatment and thus a key to better understanding cancer drug-resistance and recurrence. One could potentially use the platform as a modality for developing new immunotherapies: cancer vaccine formulation could be supported by the arrangement of complex co-cultures between antigen-presenting cells and T-cells to test the ability of a particular antigen to activate T-cell response.
Some have proposed the development of personalized patient-derived xenograft models for the purposes of testing drug therapies concurrently with patient treatment as a method of predicting patient drug responses [72,73]. The limits of these models include the cost and number of mice required for such analyses. It may also be legitimate to question the ability of personalized mouse models to produce life-saving results and insights since xenograft models are notorious for being biased toward false positive results [74]. Also important to note is that tumors are intrinsically heterogeneous and genetically unstable, and so the feasibility and reliability of such models may be of concern. Many recent studies and reviews have been published using patient derived organoids to support genetic, microenvironment and drug screening studies [75–77]. Many of these platforms require organoid splitting every week therefore disturbing the organoid cultures. While this may have minimal effects on studying certain aspects of organoid culture or drug efficacy, others analyses such as proteolysis, secretion and cell:cell interactions require that the cultures not be disturbed. Our TAME chambers should be useful for establishing patient derived organoids for long term, undisturbed live-cell imaging studies on drug efficacy, phenotypic changes, and tumor proteolysis.
5. Perspectives
The association between protease activity and cancer progression was first reported in 1946 [78]. Protease-targeted imaging probes including SBPs and ABPs to monitor and visually identify active proteases, or proteolytic activities, have been utilized and applied in the study of many pathologies including cancer. Several challenges for these probes remain to be addressed: 1) SBPs can be used to measure accumulated proteloytic activities with signal amplification over time, but do not allow direct identification of a target protease(s), and 2) ABPs bind covalently to a single target protease, but may modify proteases and do not amplify signals. Thus, comparative studies using these two types of probes could provide valuable information with each compensating for the limitations of the other. In addition, improved cell permeability, selectivity, stability, and imaging resolution need to be considered for the development of future imaging probes.
The limits of traditional 2D cell culture models and even in vivo models for cancer research have become evident over the last few decades. Thus, the development of 3D pathomimetic avatars for studying various aspects of tumor biology, including proteolytic activity, has enabled the robust recapitulation in vitro of a wide array of factors of the in vivo tumor microenvironment. Hence, avatars such as the MAME and TAME models developed by our group, represent a powerful modality enabling study of complex co-cultures, long-term analysis of proteolysis and secreted factors (e.g., cytokines), and have the potential to be useful as a drug screening platform. Our convergence of MAME culture models and TAME chambers has many advantages: 1) dynamic analysis of spatio-temporal cell:cell interactions, 2) co-culturing of multiple cell types, 3) introduction of engineered cells, scaffold matrices, and conditioned medium, 4) real-time imaging of proteolysis using SBP and/or ABPs in live cells, 5) quantification of real-time changes in cell volume and proteolysis, 6) replenishment and/or collection of media without disturbing cultures, and 7) manipulation of the extracellular pH and/or oxygen of the cultures in a controlled manner. Moreover, while our current use of this model has been primarily in studying breast cancer, this integrated system can be extended to other cell culture models representing both normal and pathological conditions.
We have used MAME avatars to determine the roles of cellular and non-cellular (i.e., pH) components of the tumor microenvironment in progression of premalignant DCIS cells to an invasive phenotype and reversion therefrom [(12, 69); M. Sameni and B.F. Sloane, unpublished results]. In addition, we have used MAME avatars to demonstrate the ability of neutralizing antibodies to interleukin 6 to reduce the invasive phenotype of tumor cells [79]; this is a class of antibodies that have been FDA-approved for treatment of Castleman's disease. We have also used MAME avatars to determine the efficacy of drugs targeting MET, vascular endothelial growth factor receptor (VEGFR) 2 and mitogen-activated protein kinase (MEK) [(69); M. Sameni and B.F. Sloane, unpublished results], photocaged inhibitors of cathepsin B (S. Ramalho and B.F. Sloane, unpublished results), and photodynamic therapy sequentially targeting lysosomes and mitochondria (N, Aggarwal and B.F. Sloane, unpublished results). These studies serve as proof-of-principle that MAME avatars will be useful for screening of drugs and other therapeutic approaches. Moving the MAME avatars into TAME chambers will increase the throughput and reproducibility of our analyses. Moreover, it will allow us to manipulate the non-cellular aspects of microenvironment (e.g., pH and oxygen) in real-time, alter conditions of drug delivery at selected time points and collect media for analysis of the secretome without disturbing the cultures. Ongoing efforts to adapt the TAME chambers for high content analysis should further increase the value of the converged MAME:TAME system as a valuable and versatile screening platform.
Acknowledgments
We would like to thank members of the Sloane laboratory for their discussions and contributions to the development of MAME avatars, and Dr. Yong Xu's lab for technical support for fabrication of TAME chambers. This work was supported in part by R01 CA131990 and R21 CA175931 from the National Institutes of Health to Dr. Sloane and an award from the President's Research Enhancement Program of Wayne State University to Drs. Sloane and Xu. Imaging was performed in the Microscopy, Imaging and Cytometry Resources Core, which is supported, in part, by NIH Center grant P30 CA022453 to the Karmanos Cancer Institute at Wayne State University, and the Perinatology Research Branch of the National Institutes of Child Health and Development at Wayne State University.
Abbreviations
- ABPs
activity-based probes
- CAFs
carcinoma-associated fibroblasts
- DCIS
ductal carcinoma in situ
- DQ
dye quenched
- ECM
extracellular matrix
- FRET
fluorescence resonance energy transfer
- HGF
hepatocyte growth factor
- MAME
mammary architecture and microenvironment engineering
- MMP
matrix metalloproteinase
- NFs
normal human breast fibroblasts
- rBM
reconstituted basement membrane
- SBPs
substrate-based probes
- TAME
tissue architecture and microenvironment engineering
- uPA
urokinase plasminogen activator
References
- [1].Edwards D, Hoyer-Hansen G, Blasi F, Sloane BF. The Cancer Degradome: Proteases and Cancer Biology. Springer; USA: 2008. [Google Scholar]
- [2].Sloane BF, List K, Fingleton B, Matrisian L. Proteases in cancer: significance for invasion and metastasis. In: Brix K, Stocker W, editors. Proteases: Structure and Function. Springer-Verlag Wien; USA: 2013. pp. 491–550. [Google Scholar]
- [3].Gillies RJ, Schornack PA, Secomb TW, Raghunand N. Causes and effects of heterogeneous perfusion in tumors. Neoplasia. 1999;1:197–207. doi: 10.1038/sj.neo.7900037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kato Y, Ozawa S, Miyamoto C, Maehata Y, Suzuki A, Maeda T, Baba Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013;13:89. doi: 10.1186/1475-2867-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, Johnson J, Gatenby RA, Gillies RJ. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524–1535. doi: 10.1158/0008-5472.CAN-12-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cotter K, Capecci J, Sennoune S, Huss M, Maier M, Martinez-Zaguilan R, Forgac M. Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J. Biol. Chem. 2015;290:3680–3692. doi: 10.1074/jbc.M114.611210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Semenza GL. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene. 2013;32:4057–4063. doi: 10.1038/onc.2012.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Raghunand N, Gillies RJ. pH and drug resistance in tumors. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2000;3:39–47. doi: 10.1054/drup.2000.0119. [DOI] [PubMed] [Google Scholar]
- [9].Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006;66:6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- [10].Moellering RE, Black KC, Krishnamurty C, Baggett BK, Stafford P, Rain M, Gatenby RA, Gillies RJ. Acid treatment of melanoma cells selects for invasive phenotypes. Clin. Exp. Metastasis. 2008;25:411–425. doi: 10.1007/s10585-008-9145-7. [DOI] [PubMed] [Google Scholar]
- [11].Wojtkowiak JW, Verduzco D, Schramm KJ, Gillies RJ. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol. Pharm. 2011;8:2032–2038. doi: 10.1021/mp200292c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rothberg JM, Bailey KM, Wojtkowiak JW, Ben-Nun Y, Bogyo M, Weber E, Moin K, Blum G, Mattingly RR, Gillies RJ, Sloane BF. Acid-mediated tumor proteolysis: contribution of cysteine cathepsins. Neoplasia. 2013;15:1125–1137. doi: 10.1593/neo.13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shirahama-Noda K, Yamamoto A, Sugihara K, Hashimoto N, Asano M, Nishimura M, Hara-Nishimura I. Biosynthetic processing of cathepsins and lysosomal degradation are abolished in asparaginyl endopeptidase-deficient mice. J. Biol. Chem. 2003;278:33194–33199. doi: 10.1074/jbc.M302742200. [DOI] [PubMed] [Google Scholar]
- [14].Hadler-Olsen E, Winberg JO, Uhlin-Hansen L. Matrix metalloproteinases in cancer: their value as diagnostic and prognostic markers and therapeutic targets. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2013;34:2041–2051. doi: 10.1007/s13277-013-0842-8. [DOI] [PubMed] [Google Scholar]
- [15].Gutierrez-Fernandez A, Fueyo A, Folgueras AR, Garabaya C, Pennington CJ, Pilgrim S, Edwards DR, Holliday DL, Jones JL, Span PN, Sweep FC, Puente XS, Lopez-Otin C. Matrix metalloproteinase-8 functions as a metastasis suppressor through modulation of tumor cell adhesion and invasion. Cancer Res. 2008;68:2755–2763. doi: 10.1158/0008-5472.CAN-07-5154. [DOI] [PubMed] [Google Scholar]
- [16].Shay G, Lynch CC, Fingleton B. Moving targets: emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. J. Int. Soc. Matrix Biol. 2015;44–46C:200–206. doi: 10.1016/j.matbio.2015.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- [18].Tholen S, Biniossek ML, Gansz M, Ahrens TD, Schlimpert M, Kizhakkedathu JN, Reinheckel T, Schilling O. Double deficiency of cathepsins B and L results in massive secretome alterations and suggests a degradative cathepsin-MMP axis. Cell. Mol. Life Sci. CMLS. 2014;71:899–916. doi: 10.1007/s00018-013-1406-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cathcart J, Pulkoski-Gross A, Cao J. Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis. 2015;2:26–34. doi: 10.1016/j.gendis.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fortelny N, Cox JH, Kappelhoff R, Starr AE, Lange PF, Pavlidis P, Overall CM. Network analyses reveal pervasive functional regulation between proteases in the human protease web. PLoS Biol. 2014;12:e1001869. doi: 10.1371/journal.pbio.1001869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007;8:839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- [22].Sung KE, Su X, Berthier E, Pehlke C, Friedl A, Beebe DJ. Understanding the impact of 2D and 3D fibroblast cultures on in vitro breast cancer models. PLoS One. 2013;8:e76373. doi: 10.1371/journal.pone.0076373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fonovic M, Bogyo M. Activity based probes for proteases: applications to biomarker discovery, molecular imaging and drug screening. Curr. Pharm. Des. 2007;13:253–261. doi: 10.2174/138161207779313623. [DOI] [PubMed] [Google Scholar]
- [24].Baruch A, Jeffery DA, Bogyo M. Enzyme activity e it's all about image. Trends Cell Biol. 2004;14:29–35. doi: 10.1016/j.tcb.2003.11.002. [DOI] [PubMed] [Google Scholar]
- [25].Dive V, Paulick MG, McIntyre JO, Matrisian LM, Bogyo M. Activity-based imaging and biochemical profiling tools for analysis of the cancer degradome. In: Edwards D, Hoyer-Hansen G, Blasi F, Sloane BF, editors. The Cancer Degradome: Proteases and Cancer Biology. Springer; USA: 2008. pp. 101–135. [Google Scholar]
- [26].Edgington LE, Verdoes M, Bogyo M. Functional imaging of proteases: recent advances in the design and application of substrate-based and activity-based probes. Curr. Opin. Chem. Biol. 2011;15:798–805. doi: 10.1016/j.cbpa.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sadaghiani AM, Verhelst SH, Bogyo M. Tagging and detection strategies for activity-based proteomics. Curr. Opin. Chem. Biol. 2007;11:20–28. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- [28].Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat. Chem. Biol. 2007;3:668–677. doi: 10.1038/nchembio.2007.26. [DOI] [PubMed] [Google Scholar]
- [29].McIntyre JO, Fingleton B, Wells KS, Piston DW, Lynch CC, Gautam S, Matrisian LM. Development of a novel fluorogenic proteolytic beacon for in vivo detection and imaging of tumour-associated matrix metalloproteinase-7 activity. Biochem. J. 2004;377:617–628. doi: 10.1042/BJ20030582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bullok KE, Maxwell D, Kesarwala AH, Gammon S, Prior JL, Snow M, Stanley S, Piwnica-Worms D. Biochemical and in vivo characterization of a small, membrane-permeant, caspase-activatable far-red fluorescent peptide for imaging apoptosis. Biochemistry. 2007;46:4055–4065. doi: 10.1021/bi061959n. [DOI] [PubMed] [Google Scholar]
- [31].Chowdhury MA, Moya IA, Bhilocha S, McMillan CC, Vigliarolo BG, Zehbe I, Phenix CP. Prodrug-inspired probes selective to cathepsin B over other cysteine cathepsins. J. Med. Chem. 2014;57:6092–6104. doi: 10.1021/jm500544p. [DOI] [PubMed] [Google Scholar]
- [32].Marten K, Bremer C, Khazaie K, Sameni M, Sloane B, Tung CH, Weissleder R. Detection of dysplastic intestinal adenomas using enzyme-sensing molecular beacons in mice. Gastroenterology. 2002;122:406–414. doi: 10.1053/gast.2002.30990. [DOI] [PubMed] [Google Scholar]
- [33].Tung CH, Mahmood U, Bredow S, Weissleder R. In vivo imaging of proteolytic enzyme activity using a novel molecular reporter. Cancer Res. 2000;60:4953–4958. [PubMed] [Google Scholar]
- [34].Sekar RB, Periasamy A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003;160:629–633. doi: 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ouyang M, Lu S, Li XY, Xu J, Seong J, Giepmans BN, Shyy JY, Weiss SJ, Wang Y. Visualization of polarized membrane type 1 matrix metalloproteinase activity in live cells by fluorescence resonance energy transfer imaging. J. Biol. Chem. 2008;283:17740–17748. doi: 10.1074/jbc.M709872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Packard BZ, Artym VV, Komoriya A, Yamada KM. Direct visualization of protease activity on cells migrating in three-dimensions. Matrix Biol. J. Int. Soc. Matrix Biol. 2009;28:3–10. doi: 10.1016/j.matbio.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Meyer BS, Rademann J. Extra- and intracellular imaging of human matrix metalloprotease 11 (hMMP-11) with a cell-penetrating FRET substrate. J. Biol. Chem. 2012;287:37857–37867. doi: 10.1074/jbc.M112.371500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yhee JY, Kim SA, Koo H, Son S, Ryu JH, Youn IC, Choi K, Kwon IC, Kim K. Optical imaging of cancer-related proteases using near-infrared fluorescence matrix metalloproteinase-sensitive and cathepsin B-sensitive probes. Theranostics. 2012;2:179–189. doi: 10.7150/thno.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lu S, Wang Y, Huang H, Pan Y, Chaney EJ, Boppart SA, Ozer H, Strongin AY. Quantitative FRET imaging to visualize the invasiveness of live breast cancer cells. PLoS One. 2013;8:e58569. doi: 10.1371/journal.pone.0058569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hu HY, Gehrig S, Reither G, Subramanian D, Mall MA, Plettenburg O, Schultz C. FRET-based and other fluorescent proteinase probes. Biotechnol. J. 2014;9:266–281. doi: 10.1002/biot.201300201. [DOI] [PubMed] [Google Scholar]
- [41].Jedeszko C, Sameni M, Olive MB, Moin K, Sloane BF. Bonifacino Juan S., et al., editors. Visualizing protease activity in living cells: from two dimensions to four dimensions. Current Protocols in Cell Biology. 2008 doi: 10.1002/0471143030.cb0420s39. (Chapter 4: Unit 4 20) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sameni M, Dosescu J, Yamada KM, Sloane BF, Cavallo-Medved D. Functional live-cell imaging demonstrates that beta1-integrin promotes type IV collagen degradation by breast and prostate cancer cells. Mol. Imaging. 2008;7:199–213. [PMC free article] [PubMed] [Google Scholar]
- [43].Bengsch F, Buck A, Gunther SC, Seiz JR, Tacke M, Pfeifer D, von Elverfeldt D, Sevenich L, Hillebrand LE, Kern U, Sameni M, Peters C, Sloane BF, Reinheckel T. Cell type-dependent pathogenic functions of overexpressed human cathepsin B in murine breast cancer progression. Oncogene. 2014;33:4474–4484. doi: 10.1038/onc.2013.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sameni M, Cavallo-Medved D, Dosescu J, Jedeszko C, Moin K, Mullins SR, Olive MB, Rudy D, Sloane BF. Imaging and quantifying the dynamics of tumor-associated proteolysis. Clin. Exp. Metastasis. 2009;26:299–309. doi: 10.1007/s10585-008-9218-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shieh AC, Rozansky HA, Hinz B, Swartz MA. Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res. 2011;71:790–800. doi: 10.1158/0008-5472.CAN-10-1513. [DOI] [PubMed] [Google Scholar]
- [46].Serim S, Haedke U, Verhelst SH. Activity-based probes for the study of proteases: recent advances and developments. ChemMedChem. 2012;7:1146–1159. doi: 10.1002/cmdc.201200057. [DOI] [PubMed] [Google Scholar]
- [47].Joyce JA, Baruch A, Chehade K, Meyer-Morse N, Giraudo E, Tsai FY, Greenbaum DC, Hager JH, Bogyo M, Hanahan D. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443–453. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- [48].Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF. Activity-based probes for the proteomic profiling of metalloproteases. Proc. Natl. Acad. Sci. U. S. A. 2004;101:10000–10005. doi: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Berkers CR, van Leeuwen FW, Groothuis TA, Peperzak V, van Tilburg EW, Borst J, Neefjes JJ, Ovaa H. Profiling proteasome activity in tissue with fluorescent probes. Mol. Pharm. 2007;4:739–748. doi: 10.1021/mp0700256. [DOI] [PubMed] [Google Scholar]
- [50].Fonovic M, Bogyo M. Activity-based probes as a tool for functional proteomic analysis of proteases. Expert Rev. Proteomics. 2008;5:721–730. doi: 10.1586/14789450.5.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Withana NP, Blum G, Sameni M, Slaney C, Anbalagan A, Olive MB, Bidwell BN, Edgington L, Wang L, Moin K, Sloane BF, Anderson RL, Bogyo MS, Parker BS. Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Res. 2012;72:1199–1209. doi: 10.1158/0008-5472.CAN-11-2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Edgington LE, Berger AB, Blum G, Albrow VE, Paulick MG, Lineberry N, Bogyo M. Noninvasive optical imaging of apoptosis by caspase-targeted activity-based probes. Nat. Med. 2009;15:967–973. doi: 10.1038/nm.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Segal E, Prestwood TR, van der Linden WA, Carmi Y, Bhattacharya N, Withana N, Verdoes M, Habtezion A, Engleman EG, Bogyo M. Detection of intestinal cancer by local, topical application of a quenched fluorescence probe for cysteine cathepsins. Chem. Biol. 2015;22:148–158. doi: 10.1016/j.chembiol.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sanman LE, Bogyo M. Activity-based profiling of proteases. Annu. Rev. Biochem. 2014;83:249–273. doi: 10.1146/annurev-biochem-060713-035352. [DOI] [PubMed] [Google Scholar]
- [55].Edgington LE, Bogyo M. In vivo imaging and biochemical characterization of protease function using fluorescent activity-based probes. Curr. Protoc. Chem. Biol. 2013;5:25–44. doi: 10.1002/9780470559277.ch120235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Blum G, Weimer RM, Edgington LE, Adams W, Bogyo M. Comparative assessment of substrates and activity based probes as tools for non-invasive optical imaging of cysteine protease activity. PLoS One. 2009;4:e6374. doi: 10.1371/journal.pone.0006374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Koblinski JE, Dosescu J, Sameni M, Moin K, Clark K, Sloane BF. Interaction of human breast fibroblasts with collagen I increases secretion of procathepsin B. J. Biol. Chem. 2002;277:32220–32227. doi: 10.1074/jbc.M204708200. [DOI] [PubMed] [Google Scholar]
- [58].Hutmacher DW, Loessner D, Rizzi S, Kaplan DL, Mooney DJ, Clements JA. Can tissue engineering concepts advance tumor biology research? Trends Biotechnol. 2010;28:125–133. doi: 10.1016/j.tibtech.2009.12.001. [DOI] [PubMed] [Google Scholar]
- [59].Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- [60].Schmeichel KL, Bissell MJ. Modeling tissue-specific signaling and organ function in three dimensions. J. Cell Sci. 2003;116:2377–2388. doi: 10.1242/jcs.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sameni M, Anbalagan A, Olive MB, Moin K, Mattingly RR, Sloane BF. MAME models for 4D live-cell imaging of tumor: microenvironment interactions that impact malignant progression. J. Vis. Exp. 2012 Feb 17;60:3661. doi: 10.3791/3661. pii. doi: 10.3791/3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mullins SR, Sameni M, Blum G, Bogyo M, Sloane BF, Moin K. Three-dimensional cultures modeling premalignant progression of human breast epithelial cells: role of cysteine cathepsins. Biol. Chem. 2012;393:1405–1416. doi: 10.1515/hsz-2012-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Miller FR, Soule HD, Tait L, Pauley RJ, Wolman SR, Dawson PJ, Heppner GH. Xenograft model of progressive human proliferative breast disease. J. Natl. Cancer Inst. 1993;85:1725–1732. doi: 10.1093/jnci/85.21.1725. [DOI] [PubMed] [Google Scholar]
- [64].Moin K, Sameni M, Victor BC, Rothberg JM, Mattingly RR, Sloane BF. 3D/4D functional imaging of tumor-associated proteolysis: impact of microenvironment. Methods Enzymol. 2012;506:175–194. doi: 10.1016/B978-0-12-391856-7.00034-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Basolo F, Elliott J, Tait L, Chen XQ, Maloney T, Russo IH, Pauley R, Momiki S, Caamano J, Klein-Szanto AJ, et al. Transformation of human breast epithelial cells by c-Ha-ras oncogene. Mol. Carcinog. 1991;4:25–35. doi: 10.1002/mc.2940040106. [DOI] [PubMed] [Google Scholar]
- [66].Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, Deussing J, Augustin N, Nielsen BS, Almholt K, Bogyo M, Peters C, Reinheckel T. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006;66:5242–5250. doi: 10.1158/0008-5472.CAN-05-4463. [DOI] [PubMed] [Google Scholar]
- [67].Sevenich L, Werner F, Gajda M, Schurigt U, Sieber C, Muller S, Follo M, Peters C, Reinheckel T. Transgenic expression of human cathepsin B promotes progression and metastasis of polyoma-middle-T-induced breast cancer in mice. Oncogene. 2011;30:54–64. doi: 10.1038/onc.2010.387. [DOI] [PubMed] [Google Scholar]
- [68].Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69:2260–2268. doi: 10.1158/0008-5472.CAN-07-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Jedeszko C, Victor BC, Podgorski I, Sloane BF. Fibroblast hepatocyte growth factor promotes invasion of human mammary ductal carcinoma in situ. Cancer Res. 2009;69:9148–9155. doi: 10.1158/0008-5472.CAN-09-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sameni M, Dosescu J, Moin K, Sloane BF. Functional imaging of proteolysis: stromal and inflammatory cells increase tumor proteolysis. Mol. Imaging. 2003;2:159–175. doi: 10.1162/15353500200303136. [DOI] [PubMed] [Google Scholar]
- [71].Akkari L, Gocheva V, Kester JC, Hunter KE, Quick ML, Sevenich L, Wang HW, Peters C, Tang LH, Klimstra DS, Reinheckel T, Joyce JA. Distinct functions of macrophage-derived and cancer cell-derived cathepsin Z combine to promote tumor malignancy via interactions with the extracellular matrix. Genes Dev. 2014;28:2134–2150. doi: 10.1101/gad.249599.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Malaney P, Nicosia SV, Dave V. One mouse, one patient paradigm: new avatars of personalized cancer therapy. Cancer Lett. 2014;344:1–12. doi: 10.1016/j.canlet.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Whittle JR, Lewis MT, Lindeman GJ, Visvader JE. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. BCR. 2015;17:17. doi: 10.1186/s13058-015-0523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am. J. Pathol. 2007;170:793–804. doi: 10.2353/ajpath.2007.060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, Wongvipat J, Kossai M, Ramazanoglu S, Barboza LP, Di W, Cao Z, Zhang QF, Sirota I, Ran L, MacDonald TY, Beltran H, Mosquera JM, Touijer KA, Scardino PT, Laudone VP, Curtis KR, Rathkopf DE, Morris MJ, Danila DC, Slovin SF, Solomon SB, Eastham JA, Chi P, Carver B, Rubin MA, Scher HI, Clevers H, Sawyers CL, Chen Y. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sachs N, Clevers H. Organoid cultures for the analysis of cancer phenotypes. Curr. Opin. Genet. Dev. 2014;24:68–73. doi: 10.1016/j.gde.2013.11.012. [DOI] [PubMed] [Google Scholar]
- [77].Shamir ER, Ewald AJ. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat. Rev. Mol. Cell Biol. 2014;15:647–664. doi: 10.1038/nrm3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Fischer A. Mechanism of the proteolytic activity of malignant tissue cells. Nature. 1946;157:442. doi: 10.1038/157442c0. [DOI] [PubMed] [Google Scholar]
- [79].Osuala KO, Sameni M, Shah S, Aggarwal N, Simonait ML, Franco OE, Hong Y, Hayward SW, Behbod F, Mattingly RR, Sloane BF. Il-6 signaling between ductal carcinoma in situ cells and carcinoma-associated fibroblasts mediates tumor cell growth and migration. BMC Cancer. 2015;15:584. doi: 10.1186/s12885-015-1576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Nelson CM, Bissell MJ. Modeling dynamic reciprocity: engineering three-dimensional culture models of breast architecture, function, and neoplastic transformation. Seminars Cancer Biol. 2005;15:342–352. doi: 10.1016/j.semcancer.2005.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]