

ABSTRACT
Osteoarthritis (OA) is a common complex disease of high public health burden. OA is characterized by the degeneration of affected joints leading to pain and reduced mobility. Over the last few years, several studies have focused on the genomic changes underpinning OA. Here, we provide a comprehensive overview of genome‐wide, non‐hypothesis‐driven functional genomics (methylation, gene, and protein expression) studies of knee and hip OA in humans. Individual studies have generally been limited in sample size and hence power, and have differed in their approaches; nonetheless, some common themes have started to emerge, notably the role played by biological processes related to the extracellular matrix, immune response, the WNT pathway, angiogenesis, and skeletal development. Larger‐scale studies and streamlined, robust methodologies will be needed to further elucidate the biological etiology of OA going forward. © 2016 Orthopaedic Research Society. Published by Wiley Periodicals, Inc. J Orthop Res 34:1105–1110, 2016.
Keywords: osteoarthritis, functional genomics, gene expression, methylation, proteomics
Osteoarthritis (OA) is a degenerative joint disease characterized by joint pain and stiffness. OA mostly affects the knee, hip, and hand joints, and leads to changes in multiple joint tissues. Radiographic evidence of osteoarthritis is present in over 40% of individuals over the age of 70, and the incidence of OA increases with age.1 Treatment of OA is often unsuccessful in alleviating joint pain, leading to total joint replacement (TJR). Known epidemiological risk factors for OA include obesity (especially for knee OA), joint injuries, and bone morphology.2 Twin and family studies have documented that genetic factors also play an important role in the etiology of OA. The heritability of knee OA is estimated to be about 40%, while the heritability of hip and hand OA is estimated to be about 60%.3
Genetic association studies have to date identified 17 OA risk loci,4, 5, 6 many of which are specific to the joint, sex, or ethnic group (European or Asian). All of these variants are common in frequency, and have only a small to moderate effect on risk of OA. The genetic studies have shown that OA is a complex disease, so that many different genetic risk variants will contribute to the susceptibility. This is in line with many other human traits, such as height7 and BMI,8 and many diseases, such as type 2 diabetes and schizophrenia.
For OA, the known associations have not yet proven sufficient to pinpoint biological pathways involved in disease etiology. However, the disease tissue is accessible through TJR surgery. This offers an opportunity to carry out functional genomics studies, examining the methylation, RNA, and protein level signatures of OA. Notably, genetics studies of OA aim to identify heritable risk factors. By contrast, functional genomics studies can establish how molecular processes change in disease; such changes can reflect causes or consequences of the disease and are of a dynamic nature.
The techniques to interrogate methylation, RNA, or protein levels genome‐wide have recently become more affordable and scalable, leading to an increase in the number of corresponding studies (Fig. 1).9, 10, 11, 12, 13, 14 To pave the way for future progress, we review here the existing results, as well as the challenges that need to be addressed.
Figure 1.
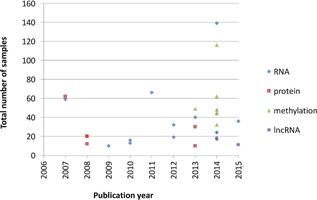
OA —omics studies included in this review.
We consider systematic, non‐hypothesis‐driven functional genomics studies of OA, focusing on knee and hip OA as the two major joints. Many studies of OA have been carried out in animal models (e.g., mice, reviewed by Fang and Beie,15 or zebrafish16). The studies reviewed were identified from PubMed searches (carried out in February 2015) for hip or knee OA, together with the terms “gene expression array” or “RNA sequencing” or “methylation” or “proteomics,” and focus exclusively on human data. We only consider studies with at least five OA and five control samples (the latter can be unaffected tissue from individuals with OA) (Supplementary Tables S1–3). Given epidemiological and genetic differences between affected joint sites, we first review knee OA and hip OA studies separately, and then discuss cross‐joint studies. Notably, studies use different methodological approaches and different thresholds for declaring novel findings (e.g., different significance threshold, multiple‐testing correction, minimum fold‐change between healthy and disease tissue), making direct comparison of results difficult. We therefore, discuss general insights gleaned from within and across studies, and conclude with opportunities and challenges for the future.
KNEE OA
Methylation
All genome‐wide methylation studies in knee OA have focused on cartilage (Supplementary Table S1) and have used arrays ranging from ∼27,00010 to ∼450,000 probes.12, 13 The gene sets found to be enriched among differentially methylated loci (DMLs) or genes include development and differentiation pathways12 and genes relating to the inflammatory response.10 Moreover, two studies observed subclustering of OA samples10, 13: in one,10 the between‐cluster DMLs were enriched in immune system or extracellular matrix (ECM) genes; and in the other, the between‐cluster DMLs were enriched in immune pathways.13 In a different non‐array‐based approach, levels of a particular methylation mark (5‐Hydroxymethylcytosine) were found to be increased in OA chondrocytes.14
Gene Transcription
Numerous studies have used microarrays to investigate gene expression changes in knee OA. The majority considered cartilage17, 18, 19, 20 (Supplementary Table S1) and two studies each highlighted ECM‐related genes19, 20 and angiogenesis.17, 19 A further study identified two subclusters of samples10 pointing to immune response pathways. Compared to methylation‐based clustering in an independent set of patients, the overlap and direction of change of cluster‐distinguishing genes was only moderate. Two further studies21, 22 specifically considered the expression of long non‐coding RNAs (lncRNAs) in cartilage (Supplementary Table S1).
Three microarray studies considered synovial tissue, subchondral bone, or peripheral blood (Supplementary Table S1). Gene sets associated with OA based on the synovial membrane include inflammation, cartilage metabolism, WNT signalling, and angiogenesis.23 By contrast, from a study of subchondral bone, the enriched gene sets included lipid metabolism, mineral metabolism, connective tissue disorders, cellular growth and proliferation, connective tissue development and function.24 Finally, in an analysis of blood leukocytes, OA samples were subdivided into two clusters based on cytokine expression.25 The “increased inflammation” cluster characterized by elevated expression of IL‐1β, TNF‐α, IL‐6, IL‐8, and COX‐2 was associated with both increased pain and increased risk of progression.
Protein Expression
Thus far, studies have mostly used gels to identify protein spots that differ between OA and control samples, or between intact and damaged OA samples. The number of differentially expressed proteins has been consistently low (Supplementary Table S1). The gene sets implicated from three cartilage‐based studies show little overlap: cellular metabolism, structure, or protein targeting gene sets in one study,26 oxidative stress response, metabolic, and apoptotic pathways in a second study,27 inflammatory response and response to wounding in the third study.28 Two further studies were based on synovial fluid. One of these found an association with the acute‐phase response signalling pathway, the complement pathway, and the coagulation pathway.29 The second study did not find differences between what they classified as “early” and “late” OA samples, but found two different subgroups of OA samples based on protein expression profiles.30
Common Pathways
Despite the differences in the tissues, molecular phenotypes, and analytical methods across studies, some biological gene sets or pathways have been implicated multiple times. Gene sets involved in immune response were implicated across all molecular phenotypes and in multiple tissues (cartilage and synovium).10, 13, 23, 28, 29 Several studies identified genes related to the extracellular matrix19, 20 or angiogenesis.17, 19 Moreover, several studies have found that knee OA samples form two different clusters10, 13, 30; two of these studies related differences between the clusters to the immune response.10, 13
HIP OA
Methylation
Three studies have investigated methylation patterns in hip OA to date; two considered cartilage (Supplementary Table S2) and found that DMLs included 24 genes previously associated with OA,11 and that they were enriched in genes annotated to the extracellular matrix, collagen, angiogenesis, and the TGF‐β pathway.13 Hip OA samples were also found to form two clusters, with genes in the between‐cluster DMLs enriched among inflammation and immunity pathways.13 A study of trabecular bone31 identified enrichment of genes associated with bone phenotypes in the GWAS catalogue,32 genes annotated to the skeleton, glycoprotein, neuronal differentiation, adherence, homeobox, and cell proliferation pathways.
Gene Transcription
Each of the three gene expression studies of hip OA have focused on different joint tissues (Supplementary Table S2). In a study of cartilage,33 the enriched pathways included stress response, cell death, regulation of development, skeletal development, WNT/β‐catenin signalling, HIF1α signalling, and p38 MAPK signalling. A study of trabecular bone34 suggested a role for genes involved in angiogenesis, as well as osteoblast, osteocyte, and osteoclast function. Some evidence was also found for association of the WNT pathway and TGF‐β/BMP signalling. Finally, a study of bone marrow mesenchymal stem cells35 found differential expression of genes annotated to signal transduction, cell development and differentiation, the WNT/catenin pathway, and collagen.
Protein Expression
Protein expression studies in hip OA have so far used gels followed up by mass spectrometry (MS), as described for knee OA above. A study of adult mesenchymal stem cells36 identified a small number of differentially expressed proteins, which included metabolic enzymes and proteins involved in the cytoskeleton or motility.
Common Pathways
The common themes emerging in functional genomics studies of hip OA have primarily included gene sets relevant to collagens,13, 35 the WNT pathway,33, 34, 35 bone development,31, 33, 34 and angiogenesis.13, 33
OVERLAP AND DIFFERENCES BETWEEN JOINTS
While most studies of knee or hip OA have focused exclusively on one joint, a few recent studies have considered the commonalities and differences between knee and hip OA (Supplementary Table S3).
Methylation
One study of cartilage looked for regions differentially methylated in a comparison between hip OA and controls, as well as in comparison between knee OA and the same control samples,13 and found that shared DMLs were enriched in genes involved in immune response, but constituted a limited proportion of DMLs individually found in hip and knee OA. In a comparison between hip and knee samples, DMLs were enriched in genes involved in development. In a different study, a clustering analysis of intact and damaged cartilage from knee and hip OA samples showed that knee and hip samples clustered separately.9 Here, the regions with differential methylation between joints were also enriched in genes involved in development, especially in homeodomain genes.
Gene Transcription
One study compared the differentially expressed genes identified in a cartilage case/control analysis for hip OA33 to the genes from an analogous study of knee OA.19 There was moderate overlap between the differentially expressed genes. However, 34 canonical pathways were significantly enriched in both studies, suggesting that different genes but similar pathways might play a role across joints in OA. In a different case/control study in blood, ascertained on the basis of OA in multiple joints (knee, hip, hand, or spine) or multiple joint sites in the hand,37 the differentially expressed genes were enriched among biological processes relating to protein transport and localisation, macromolecular complex subunit organisation, cell cycle, RNA processing, and apoptosis.
CONCLUSIONS
The last few years have witnessed a flurry of studies focussing on the functional genomics of OA. While there is limited concordance of study results even within the same joint, some gene sets have been implicated across multiple studies, notably those related to immune response, the extracellular matrix, angiogenesis, and WNT signalling. These associations may reflect causal processes as well as consequences of OA, especially when relatively intact tissue from individuals with OA serves as the control. Across the hip and knee OA joints, both similarities and differences have been observed, with higher overlap at the level of biological pathways than that of individual genes. Notably, the etiology of OA may be heterogeneous even within the same joint. This is supported by the clustering of OA samples into two distinct groups as observed in several studies. Further research will be required to explore this hypothesis.
Biological changes may be mediated by epidemiological OA risk factors, such as body mass index (BMI), joint morphology, and pain perception that impact the symptomatic aspects of OA. Detailed phenotypes at large scale will be required to address their interplay with molecular phenotypes. In addition, the etiology of OA may differ across ethnicities, as suggested by the different prevalence of lateral compared to medial OA between Chinese and European‐descent individuals.38 Consequently, studies of different ethnicities have the potential to provide insights into different genetic and environmental risk factors and their effect on molecular phenotypes.
Functional genomics studies carried out to date in OA have been small or modest in scale, and although necessary to demonstrate experimental feasibility and provide first insights into disease biology, they are restricted in terms of power. Using data from RNA sequencing in intact and affected cartilage from individuals with knee OA (m/s in review), we estimate that the genes identified as differentially expressed at 5% false‐discovery rate from 10 to 12 individuals represent less than 10% of the actually changed genes (Fig. 2). Using the same p‐value threshold and extrapolation from the pilot data, over 300 cases and 300 controls would be needed to identify at least 90% of the “true” differentially expressed genes (Fig. 2). Notably, none of the studies conducted to date approach these numbers. While these estimates are not exact and may depend on joint and cell type, they indicate the required level of magnitude. Importantly, replication is the gold standard for declaring robust findings, and to date, few of the functional genomics studies in OA have explicitly included a replication component. Further development of analytical methodology and standardized approaches to declaring significance in the presence of multiple—omics testing will additionally help utilize existing and future samples to their full potential.
Figure 2.
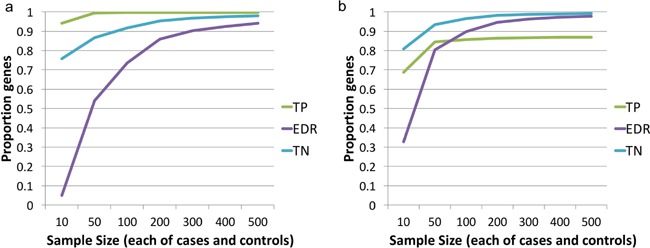
(a) At a sample size of 10 cases and 10 controls, an estimated less than 10% of the “true” differentially expressed genes are significant at 5% FDR. Sample sizes of about 300 cases and 300 controls are needed to detect over 90% of differentially expressed genes in OA (expected discovery rate, EDR). Higher sample sizes also increase true positive (TN) and true negative (TN) rates. The results are estimates obtained from PowerAtlas by extrapolation from the analysis of cartilage from 12 individuals, with intact and degraded cartilage from each individual, and significance defined at a p‐value threshold of p ≤ 0.001 which corresponds to 5% false‐discovery rate in the analysis of the given samples. (b) Defining significance at a nominal p‐value of p < 0.05 leads to lower rates of true positive (TP) results. Estimates obtained as in (a).
There are additional genomics approaches for which application to OA has been limited to date, including metabolomics (e.g., Zhang et al.39) and studies of non‐coding RNA (e.g., microRNAs40).
Genomics approaches can also help to clarify the mechanisms through which genetic risk variants exert their effect. For example, some OA risk variants were recently found to be correlated with DNA methylation levels of nearby probes.41 However, the mechanism of action is unknown for most variants. Some could regulate genes located far way—such long‐range interactions could be identified through chromosome conformation capture.42 For example, a range of genetic and genomic approaches was recently applied to elucidate that the obesity risk variant located in FTO 43 acts through affecting regulation of genes IRX3 and IRX5 which are over 500 kb away.44 To determine the causal variant among several in LD and verify their action, genome editing techniques such as CRISPR‐Cas945, 46 will be crucial.
The combination of different genomics data will also provide further insights. For example, a recent study has combined methylation and gene expression data from OA patients to identify genes whose expression was correlated with methylation of nearby probes, with differential expression of the genes and differential methylation of the probes in intact versus degraded cartilage.47 Some of these gene expression or probe methylation values were also correlated with nearby genetic variants.47
Finally, OA is a disease affecting multiple tissues, and interactions between tissues may be important for disease etiopathogenesis.48 Studies so far have only considered one tissue and have largely been focused on cartilage (Supplementary Tables S1–3), as it is both central to the disease and consists of only one cell type (chondrocytes). Building on this work, analyses taking into account several relevant tissues will be necessary to elucidate the corresponding biological mechanisms and to identify biomarkers. In particular, cross‐tissue analyses may help to resolve the role of inflammation in OA, which is currently still unclear and may depend on the tissue examined. Ultimately, integration across multiple tissues and different omics levels at large scale holds the promise of characterising OA processes in depth, thus leading to the development of new therapeutic interventions.
AUTHORS’ CONTRIBUTIONS
J.S. and E.Z. have designed and written the review. Both authors have read and approved the final submitted manuscript.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Table S1. Genome‐wide, hypothesis‐free knee OA functional genomics studies.
Table S2. Genome‐wide, hypothesis‐free hip OA functional genomics studies.
Table S3. Genome‐wide, hypothesis‐free functional genomics studies analysing data across multiple joints.
REFERENCES
- 1. Petersson IF. 1996. Occurrence of osteoarthritis of the peripheral joints in European populations. Ann Rheum Dis 55:659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glyn‐Jones S, Palmer AJR, Agricola R, et al. 2015. Osteoarthritis. Lancet 386:376–387. [DOI] [PubMed] [Google Scholar]
- 3. Spector TD, MacGregor AJ. 2004. Risk factors for osteoarthritis: genetics. Osteoarthritis Cartilage 12:39–44. [DOI] [PubMed] [Google Scholar]
- 4. Evangelou E, Kerkhof HJ, Styrkarsdottir U, et al. 2014. A meta‐analysis of genome‐wide association studies identifies novel variants associated with osteoarthritis of the hip. Ann Rheum Dis 73:2130–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Panoutsopoulou K, Zeggini E. 2013. Advances in osteoarthritis genetics. J Med Genet 50:715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Styrkarsdottir U, Thorleifsson G, Helgadottir HT, et al. 2014. Severe osteoarthritis of the hand associates with common variants within the ALDH1A2 gene and with rare variants at 1p31. Nat Genet 46:498–502. [DOI] [PubMed] [Google Scholar]
- 7. Lango Allen H, Estrada K, Lettre G, et al. 2010. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467:832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Speliotes EK, Willer CJ, Berndt SI, et al. 2010. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 42:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. den Hollander W, Ramos YFM, Bos SD, et al. 2014. Knee and hip articular cartilage have distinct epigenomic landscapes: implications for future cartilage regeneration approaches. Ann Rheum Dis 73:2208–2212. [DOI] [PubMed] [Google Scholar]
- 10. Fernández‐Tajes J, Soto‐Hermida A, Vázquez‐Mosquera ME, et al. 2014. Genome‐wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann Rheum Dis 73:668–677. [DOI] [PubMed] [Google Scholar]
- 11. Jeffries MA, Donica M, Baker LW, et al. 2014. Genome‐eide DNA methylation study identifies significant epigenomic changes in osteoarthritic cartilage. Arthritis Rheumatol 66:2804–2815. [DOI] [PubMed] [Google Scholar]
- 12. Moazedi‐Fuerst FC, Hofner M, Gruber G, et al. 2014. Epigenetic differences in human cartilage between mild and severe OA. J Orthop Res 32:1636–1645. [DOI] [PubMed] [Google Scholar]
- 13. Rushton MD, Reynard LN, Barter MJ, et al. 2014. Characterization of the cartilage DNA methylome in knee and hip osteoarthritis. Arthritis Rheumatol 66:2450–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taylor SEB, Smeriglio P, Dhulipala L, et al. 2014. A global increase in 5‐hydroxymethylcytosine levels marks osteoarthritic chondrocytes. Arthritis Rheumatol 66:90–100. [DOI] [PubMed] [Google Scholar]
- 15. Fang H, Beier F. 2014. Mouse models of osteoarthritis: modelling risk factors and assessing outcomes. Nat Rev Rheumatol 10:413–421. [DOI] [PubMed] [Google Scholar]
- 16. van der Kraan PM. 2013. Relevance of zebrafish as an OA research model. Osteoarthritis Cartilage 21:261–262. [DOI] [PubMed] [Google Scholar]
- 17. Chou CH, Lee MTM, Song IW, et al. 2015. Insights into osteoarthritis progression revealed by analyses of both knee tibiofemoral compartments. Osteoarthritis Cartilage 23:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Geyer M, Grässel S, Straub RH, et al. 2009. Differential transcriptome analysis of intraarticular lesional vs intact cartilage reveals new candidate genes in osteoarthritis pathophysiology. Osteoarthritis Cartilage 17:328–335. [DOI] [PubMed] [Google Scholar]
- 19. Karlsson C, Dehne T, Lindahl A, et al. 2010. Genome‐wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage 18:581–592. [DOI] [PubMed] [Google Scholar]
- 20. Tew SR, McDermott BT, Fentem RB, et al. 2014. Transcriptome‐wide analysis of messenger RNA decay in normal and osteoarthritic human articular chondrocytes. Arthritis Rheumatol 66:3052–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fu M, Huang G, Zhang Z, et al. 2015. Expression profile of long noncoding RNAs in cartilage from knee osteoarthritis patients. Osteoarthritis Cartilage 23:423–432. [DOI] [PubMed] [Google Scholar]
- 22. Liu Q, Zhang X, Dai L, et al. 2014. Long noncoding RNA related to cartilage injury promotes chondrocyte extracellular matrix degradation in osteoarthritis. Arthritis Rheumatol 66:969–978. [DOI] [PubMed] [Google Scholar]
- 23. Lambert C, Dubuc J‐E, Montell E, et al. 2014. Gene expression pattern of cells from inflamed and normal areas of osteoarthritis synovial membrane. Arthritis Rheumatol 66:960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chou CH, Wu CC, Song IW, et al. 2013. Genome‐wide expression profiles of subchondral bone in osteoarthritis. Arthritis Res Ther 15:R190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Attur M, Belitskaya‐Lévy I, Oh C, et al. 2011. Increased interleukin‐1β gene expression in peripheral blood leukocytes is associated with increased pain and predicts risk for progression of symptomatic knee osteoarthritis. Arthritis Rheumatol 63:1908–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ruiz‐Romero C, Carreira V, Rego I, et al. 2008. Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteomics 8:495–507. [DOI] [PubMed] [Google Scholar]
- 27. Lambrecht S, Verbruggen G, Verdonk PCM, et al. 2008. Differential proteome analysis of normal and osteoarthritic chondrocytes reveals distortion of vimentin network in osteoarthritis. Osteoarthritis Cartilage 16:163–173. [DOI] [PubMed] [Google Scholar]
- 28. Stenberg J, Rüetschi U, Skiöldebrand E, et al. 2013. Quantitative proteomics reveals regulatory differences in the chondrocyte secretome from human medial and lateral femoral condyles in osteoarthritic patients. Proteome Sci 11:43–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ritter SY, Subbaiah R, Bebek G, et al. 2013. Proteomic analysis of synovial fluid from the osteoarthritic knee: comparison with transcriptome analyses of joint tissues. Arthritis Rheumatol 65:981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gobezie R, Kho A, Krastins B, et al. 2007. High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res Ther 9:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Delgado‐Calle J, Fernández AF, Sainz J, et al. 2013. Genome‐wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheumatol 65:197–205. [DOI] [PubMed] [Google Scholar]
- 32. Welter D, MacArthur J, Morales J, et al. 2014. The NHGRI GWAS catalog, a curated resource of SNP‐trait associations. Nucl Acids Res 42:D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu Y, Barter MJ, Swan DC, et al. 2012. Identification of the pathogenic pathways in osteoarthritic hip cartilage: commonality and discord between hip and knee OA. Osteoarthritis Cartilage 20:1029–1038. [DOI] [PubMed] [Google Scholar]
- 34. Hopwood B, Tsykin A, Findlay DM, et al. 2007. Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor‐beta/bone morphogenic protein signalling. Arthritis Res Ther 9:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lamas JR, Rodríguez‐Rodríguez L, Vigo AG, et al. 2010. Large‐scale gene expression in bone marrow mesenchymal stem cells: a putative role for COL10A1 in osteoarthritis. Ann Rheum Dis 69:1880–1885. [DOI] [PubMed] [Google Scholar]
- 36. Rollín R, Marco F, Camafeita E, et al. 2008. Differential proteome of bone marrow mesenchymal stem cells from osteoarthritis patients. Osteoarthritis Cartilage 16:929–935. [DOI] [PubMed] [Google Scholar]
- 37. Ramos YFM, Bos SD, Lakenberg N, et al. 2014. Genes expressed in blood link osteoarthritis with apoptotic pathways. Ann Rheum Dis 73:1844–1853. [DOI] [PubMed] [Google Scholar]
- 38. Felson DT, Nevitt MC, Zhang Y, et al. 2002. High prevalence of lateral knee osteoarthritis in Beijing Chinese compared with Framingham Caucasian subjects. Arthritis Rheumatol 46:1217–1222. [DOI] [PubMed] [Google Scholar]
- 39. Zhang W, Sun G, Likhodii S, et al. 2016. Metabolomic analysis of human plasma reveals that arginine is depleted in knee osteoarthritis patients. Osteoarthritis Cartilage 24:827–834. [DOI] [PubMed] [Google Scholar]
- 40. Prasadam I, Batra J, Perry S, et al. 2016. Systematic identification, characterization and target gene analysis of microRNAs involved in osteoarthritis subchondral bone pathogenesis. Calcif Tissue Int. [DOI] [PubMed] [Google Scholar]
- 41. Rushton MD, Reynard LN, Young DA, et al. 2015. Methylation quantitative trait locus analysis of osteoarthritis links epigenetics with genetic risk. Hum Mol Genet 24:7432–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dekker J, Rippe K, Dekker M, et al. 2002. Capturing chromosome conformation. Science 295:1306–1311. [DOI] [PubMed] [Google Scholar]
- 43. Frayling TM, Timpson NJ, Weedon MN, et al. 2007. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Claussnitzer M, Dankel SN, Kim K‐H, et al. 2015. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med 373:895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jinek M, Chylinski K, Fonfara I, et al. 2012. A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sander JD, Joung JK. 2014. CRISPR‐Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. den Hollander W, Ramos YFM, Bomer N, et al. 2015. Transcriptional associations of osteoarthritis‐mediated loss of epigenetic control in articular cartilage. Arthritis Rheumatol 67:2108–2116. [DOI] [PubMed] [Google Scholar]
- 48. Mahjoub M, Berenbaum F, Houard X. 2012. Why subchondral bone in osteoarthritis? The importance of the cartilage bone interface in osteoarthritis. Osteoporos Int 23:841–846. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Table S1. Genome‐wide, hypothesis‐free knee OA functional genomics studies.
Table S2. Genome‐wide, hypothesis‐free hip OA functional genomics studies.
Table S3. Genome‐wide, hypothesis‐free functional genomics studies analysing data across multiple joints.