

Abstract
Two series of 4,6-diaryl-2-imino-1,2-dihydropyridine-3-carbonitriles and their isosteric 4,6-diaryl-2- oxo-1,2-dihydropyridine-3-carbonitriles were synthesized through a combinatorial approach. The prepared analogues were evaluated for their in vitro capacity to inhibit PDE3A and the growth of the human HT-29 colon adenocarcinoma tumor cell line. Compound 6-(4-bromophenyl)-4-(2-ethoxyphenyl)-2- imino-1,2-dihydropyridine-3-carbonitrile (Id) exhibited the strongest PDE3 inhibition when cGMP but not cAMP is the substrate with a IC50of 27 μM, which indicates a highly selective mechanism of enzyme inhibition. On the other hand, compound 6-(1,3-benzodioxol-5-yl)-4-(2-ethoxyphenyl)-2-imino-1,2- dihydropyridine-3-carbonitrile (Ii) was the most active in inhibiting colon tumor cell growth with a IC50 of 3 μM. The electronic effects, steric effects and conformational aspects of Id seem to be the most crucial for the PDE3 inhibition. Meanwhile, steric factors and the H-bonding capability seem to be the most important factors for tumor cell growth inhibitory activity. Conversely, there is no direct correlation between PDE3 inhibition and anticancer activity for the prepared compounds. An in silico docking experiment indicates the potential involvement of other potential molecular targets such as PIM-1 kinase to explain its tumor cell growth inhibitory activity.
Keywords: Combinatorial chemistry, 3-Cyano-2-pyridone, 3-Cyano-2-iminopyridine, PDE3 inhibitor, Growth inhibition
1. Introduction
Cheney et al., reported novel 4, 6-diaryl-2-oxo-1,2-dihydropyridine- 3-carbonitriles (e.g., Fig. 1, compound 1), as inhibitors of the oncogenic serine/threonine kinase PIM-1, which plays a role in cancer cell survival, differentiation and proliferation. PIM-1 kinase has been shown to be over expressed in a variety of cancer cell lines.1 Wendt et al., showed that several compounds with the same general formula as above but with higher lipophilic properties (e.g., Fig. 1, compound 2) can inhibit survivin which is a member of the inhibitor of apoptosis family (IAP).2 The level of expression of survivin in tumor cells is often associated with poor prognosis and shorter patient survival rates. Survivin is highly expressed in most human tumors and fetal tissue but undetectable in most terminally differentiated adult tissues. This fact therefore makes survivin an ideal target for cancer therapy.3,4
Figure 1.

Various 3-cyano-2-oxopyridine derivatives with potential growth inhibitory and/or antiangiogenic actions through PIM-1 kinase inhibition (compound 1), survivin inhibition (compound 2) or PDE3 inhibition (compound 3).
Milrinone (Fig. 1, compound 3) is a 3-cyano-2-oxopyridine derivative that has been introduced to the clinic for the treatment of congestive heart failure. Its mechanism of action involves PDE3 inhibition, leading to high levels of cAMP and consequent inotropic effect. Recent studies showed that PDE3, PDE4 and PDE5 are over expressed in cancerous cells compared with normal cells. In addition, cross inhibition of PDE3 together with other PDEs may lead to inhibition of tumor cell growth and angiogenesis.5–9
In this work, we report upon the synthesis of novel derivatives of the formula 4,6-diaryl-2-oxo-1,2-dihydropyridine-3-carbonitriles and their 2-imino isosteres. The final compounds were evaluated for their PDE3A inhibitory effect, in addition to their tumor cell growth inhibitory using the human HT-29 colon adenocarcinoma tumor cell line. Structure–activity relationships were studied.
2. Results and discussion
2.1. Chemistry
The general synthesis of 4,6-diaryl-2-imino-1,2-dihydropyridine- 3-carbonitriles (Ia–j) and 4,6-diaryl-2-oxo-1,2-dihydropyridine- 3-carbonitriles (IIa–j) is illustrated in Schemes 1 and 2. A combinatorial approach was adopted utilizing in-solution phase MCRs. Briefly, 4-bromoacetophenone or 1-(1,3-benzodioxol-5-yl) ethanone were reacted with the appropriate aldehyde, namely 2- methoxybenzaldehyde, 2-ethoxybenzaldehyde, 4-ethoxybenzaldehyde, thiophene-2-carboxaldehyde and thiophene-3-carboxaldehyde in the presence of malononitrile or ethyl cyanoacetate and ammonium acetate. All of the products derived from 1-(1,3-benzodioxol- 5-yl)ethanone showed a methylene protons as a deshielded singlet peak at the downfield region δ ≈ 6.1 due to their flanking between 2 oxygen atoms. Mass spectrometry to all products derived from 4-bromoacetophenone showed molecular ion peaks atM+ and M++2 due to the isotopic nature of bromine atom. In addition, the molecular ion peaks were also the base peaks signifying their stable character. The infrared (IR) spectra of all derivativesshowedbands at a stretching frequency (ν) around 3400 cm−1 corresponding for the N–H showed relatively lower values of the carbonyl stretching at ν around 1650 cm−1 than the typical carbonyl stretching at a stretching frequency around 1700 cm−1. Thismaybe due to the single-bond character of the tautomeric enol form, leading to lower absorption frequency.
Scheme 1.
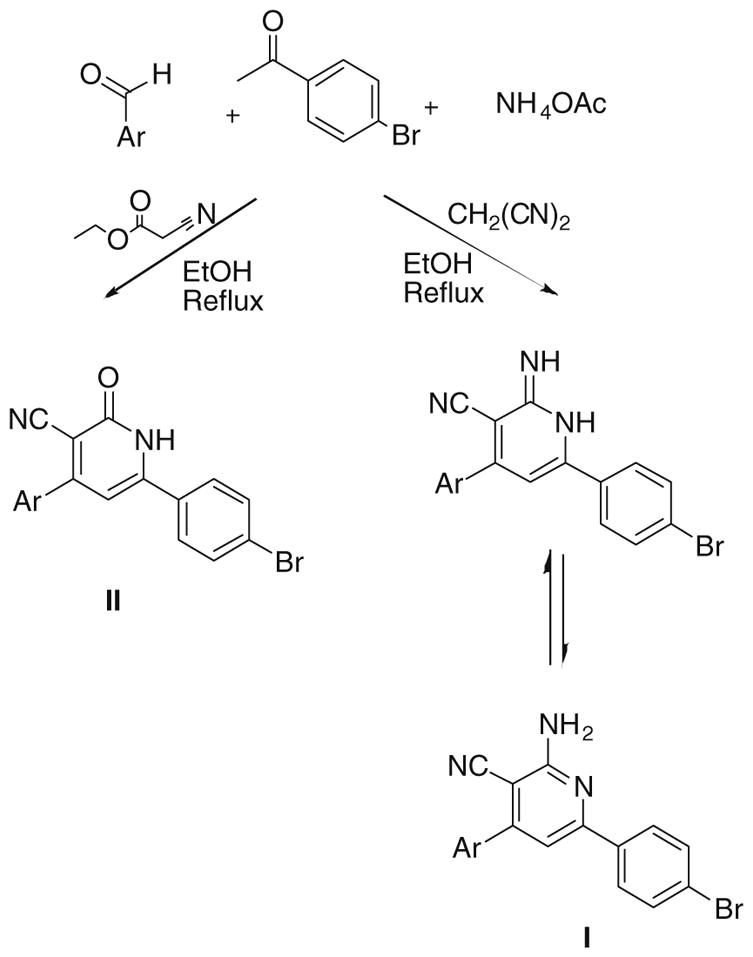
For Ar see Table 1 and Experimental.
Scheme 2.
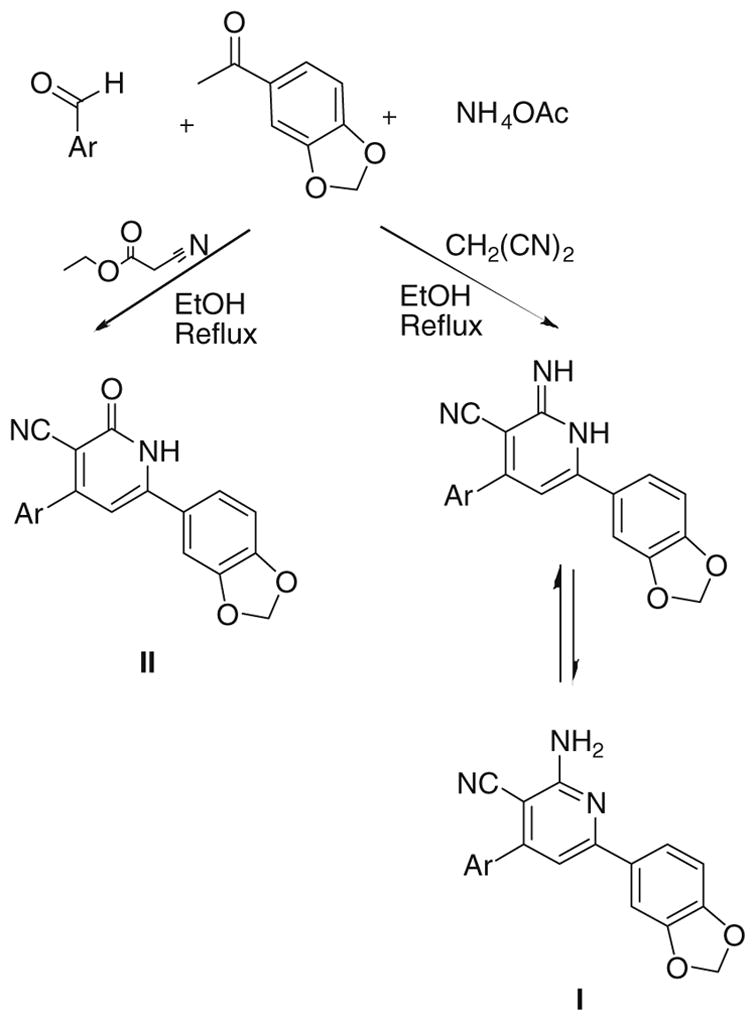
For Ar see Table 1 and Experimental.
2.2. Biology
All the synthesized compounds were tested for their in vitro ability to inhibit the growth of human HT-29 colon adenocarcinoma tumor cells and to inhibit recombinant human PDE3A. Initially, all compounds were screened at a dose of 50 μM in triplicate, followed by a full dose–response to calculate the exact IC50 value. Compounds displaying percentage of inhibition >70% was determined by testing a range of 10 concentrations with at least two replicates per concentration.
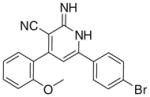
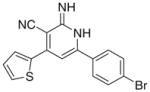
The previous biological results showed only one compound (Id) active as PDE3A inhibitor when cGMP was used as a substrate and seven compounds (Ia, Ib, Id, Ie, If, Ii, Ij) displayed tumor cell growth inhibitory activity as summarized in, Table 1.
Table 1.
Inhibitory effect of the synthesized compounds on HT-29 cells and PDE3
| Compd # | Chemical structure | % Growth inhibition (at 50 μm) | Growth inhibition IC50 (μM) | % PDE3 inhibition (at 50 μm)
|
PDE3 inhibition IC50 (μM)
|
||
|---|---|---|---|---|---|---|---|
| cAMP | cGMP | cAMP | cGMP | ||||
| Ia |
|
75.03 | 50 | 16.32 | −15.46 | ND | |
| Ib |
|
98.68 | 35 | 3.57 | −19.78 | ND | |
| Ic |
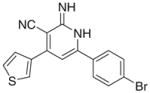
|
−5 | NDa | 15 | 42 | ND | |
| Id |
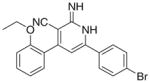
|
99.78 | 13 | 37.92 | 125.53 | ND | 27 |
| Ie |
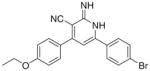
|
75 | 14.19 | 30 | 50 | ND | |
| If |
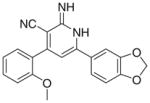
|
95 | 4.17 | 45 | 35 | ND | |
| Ig |
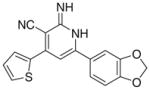
|
−20 | ND | 5 | 10 | ND | |
| Ih |
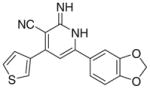
|
45 | ND | 18 | 3 | ND | |
| Ii |
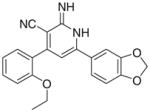
|
91.25 | 3 | 19.04 | 26.25 | ND | |
| Ij |
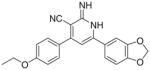
|
87.68 | 12 | 45.91 | −8.99 | ND | |
| IIa |
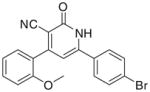
|
13.59 | ND | 6.12 | 15.10 | ND | |
| IIb |
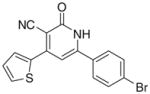
|
−20 | ND | 3 | 4 | ND | |
| IIc |
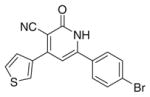
|
−25 | ND | 2 | −5 | ND | |
| IId |
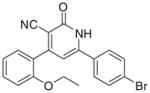
|
2.81 | ND | 13.09 | 35.25 | ND | |
| IIe |
|
10.65 | ND | 4.76 | 10.79 | ND | |
| IIf |
|
0.00 | ND | 30 | 30 | ND | |
| IIg |
|
6.14 | ND | 32.31 | 49.64 | ND | |
| IIh |
|
−25 | ND | 8 | 20 | ND | |
| IIi |
|
8.28 | ND | 7.65 | 14.38 | ND | |
| IIj |
|
3.84 | ND | 10.54 | 22.30 | ND | |
| Milrinone |
|
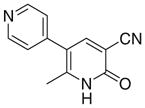
0 | >50 | 77 | 95 | 11.4 | 3.6 |
ND: Not determined.
For PDE3A inhibition, the activity of (Id) with 2-ethoxyphenyl at position 4 and 4-bromophenyl at position 6 of the lactam ring, relative to the inactivity of Ia, Ie showed the importance of the size of the substituent (steric factors) and positional properties upon activity, respectively. The presence of the o-ethoxy group (Id) in the phenyl ring leads to the non-coplanarity between the o-ethoxyphenyl and the substituted 1,2-dihydropyridine group in the ligand–protein complex (Fig. 2). This non-coplanarity possibly permits the optimum interaction with the binding site of PDE3A. Comparing the active Id to its inactive congener Ii indicates the relevance of the nature of the substituent at position 6 of the lactam. The molecular electrostatic potential surface of compound (Id) showed 2 intense red (negative) regions similar to amrinone which is a standard PDE3A inhibitor; due to the –CO and CN functions., By comparison, the inactive congener Ii showed 4 intense red regions with extra negative areas that may lead to non-favorable interaction with the receptor (Fig. 3).
Figure 2.

Docking of PDE3A with compound (Id) in 2D diagram (a), and a MEP on a Gaussian contact surface of the binding pocket (b).
Figure 3.

Molecular electrostatic potential on a Connolly surface of compound (Id) (a); amrinone (b); and compound (Ii) (c). A comparable electrostatic distribution of compound (Id) and amrinone is shown. Red is negative and blue is positive.
In silico docking experiment showed Id mostly interacts with PDE3A through H bonding of His25 with the cyano, Thr97 with the imino N and Glu55 with the 2NHs (Fig. 2). It worthy to mention that the –CO isostere (IId) of Id was inactive as PDE3A inhibitor, which may be due to the lack of the proton able to afford hydrogen bonding with Glu55. It should be noted that Id has not been able to inhibit PDE3A utilizing cAMP as a substrate and opens the horizons towards substrate specific inhibition of dual PDEs for example, PDE3.
As per the in vitro tumor cell growth inhibitory activity, the 2-imino-1,2-dihydropyridine derivatives, namely Ia, Ib, Id, Ie, If, Ii and Ij were more active than their 2-pyridone congeners IIa, IIb, IId, IIe, IIe and IIj, which indicates the potential involvement of proton of the imino in hydrogen bonding with the receptor responsible for the anticancer activity. For the top three growth inhibitory compounds viz Ii > If > Ij with IC50 = 3, 4.17, 12 μM, respectively, highlights the importance of a bulky ortho substituent upon non coplanarity. This is confirmed from the higher activity of Id versus Ia, IC50 = 50 and 13 μM, respectively. Comparing Ic (active) versus Ib (inactive) showed the in vitro anticancer activity increases when the electronegative S atom is at 2 position rather than 3 position of thiophenyl group.
Only compound Id showed dual cancer-PDE3 inhibitory activity with IC50 = 13 and 27 μM, for anticancer and PDE3 inhibition (when cGMP is the substrate), respectively; while the other active compounds possess only anticancer activity. From these experiments we conclude that PDE3 inhibition is not responsible for the tumor cell growth inhibitory activity of these milrinone analogs.
Docking of compound (Ii) with other potential targets, namely PIM-1 kinase showed potential H-bonding network. The apparent H-bonding network resulted from the interaction of the 2-imino group and 1-NH group with the conserved water molecule that interacts with the PIM-1 kinase catalytic residues Asp186. Additionally, the 2-imino and 3-cyano groups are making H-bonding interactions with PIM-1 kinase catalytic residue Lys67; Figure 4. Obviously, the docking of the most potent compound (Ii) shows comparable interactions with the catalytic residues as compound 1 does; therefore, the docking results suggested that PIM-1 kinase may be a potential target that mediates the tumor cell growth inhibitory effect. On the other hand, docking of (Ii) with survivin shows non-specific interactions (data not shown).
Figure 4.

Docking of PIM-1 kinase with compound (Ii) in 2D diagram (a) and overlay of the reference compound 1 (green) and Ii (red) in the binding pocket of PIM1 kinase (b).
3. Experimental
3.1. Chemistry
All reactions were performed with commercially available reagents and they were used without further purification. Solvents were dried by standard methods and stored over molecular sieves. All reactions were monitored by thin-layer chromatography (TLC) carried out on precoated silica gel plates (ALUGRAM SIL G/UV254) and detection of the components was made by short and long UV light. Melting points were determined in open capillaries using a Buchi Melting Point B-540 apparatus and are uncorrected. 1H NMR spectra were recorded on Varian spectrometer at 300 MHz using tetramethylsilane (TMS) as internal reference. Chemical shift values are given in ppm at room temperature using DMSO-d6 as a solvent; chemical shifts (δ) were reported in parts per million (ppm) downfield from TMS; multiplicities are abbreviated as: s: singlet; d: doublet; q: quartet; m: multiplet; dd: doublet of doublet; br s: broad singlet. Yields are not optimized. Elemental analyses were performed by Institute of Organic Chemistry, Jena University; and the Microanalytical Unit, Faculty of Science, Cairo University; found values were within ±0.4% of the theoretical ones, unless otherwise indicated.
3.2. General procedure for the preparation of 4-aryl-6-(4- bromophenyl)-2-imino-1,2-dihydropyridines-3-carbonitriles and 4-Aryl-6(1,3-benzodioxol-5-yl)-2-imino-1,2- dihydropyridines-3-carbonitriles (I a–j)
A mixture of p-bromoacetophenone or 3′,4′-methylenedioxy-acetophenone (2.5 mmol), appropriate aldehyde (2.5 mmol), malononitrile (0.16 g, 2.5 mmol) and ammonium acetate (1.54 g, 20 mmol) in ethanol (50 ml) was heated under reflux for 18– 24 h. The reaction mixture was cooled and the formed precipitate was filtered, washed with ethanol, then washed successively with water, dried and crystallized from DMF/ethanol 1:2, respectively.
3.2.1. 6-(4-Bromophenyl)-2-imino-4-(2-methoxyphenyl)-1,2- dihydropyridine-3-carbonitrile (Ia)
Yield 50%; mp 255–257 °C; IR (cm−1): 3446, 2201; 1H NMR (DMSO-d6): 4.20 (s, 3H, –OCH3), 6.93–7.81 (m, 11H, aromatic + – NHs); Anal. Calcd for C19H14BrN3O: C, 60.02; H, 3.71; N, 11.05. Found: C, 60.15; H, 3.82; N, 11.12.
3.2.2. 6-(4-Bromophenyl)-2-imino-4-(thiophen-2-yl)-1,2- dihydropyridine-3-carbonitrile (Ib)
Yield 60%; mp 243–245 °C; IR (cm−1): 3355, 2213. 1H NMR (DMSO-d6): 6.97–7.77 (m, 10H, aromatic + –NHs); Anal. Calcd for C16H10BrN3S: C, 53.94; H, 2.83; N, 11.80; S, 9.00. Found: C, 54.14; H, 2.92; N, 11.73; S, 9.23.
3.2.3. 6-(4-Bromophenyl)-2-imino-4-(thiophen-3-yl)-1,2- dihydropyridine-3-carbonitrile (Ic)
Yield 65%; mp 204–206 °C; IR (cm−1): 3357, 2209; 1H NMR (DMSO-d6): 6.91–7.75 (m, 10H, aromatic + –NHs); Anal. Calcd for C16H10BrN3S: C, 53.94; H, 2.83; N, 11.80; S, 9.00. Found: C, 54.09; H, 2.82; N, 11.91; S, 9.17.
3.2.4. 6-(4-Bromophenyl)-4-(2-ethoxyphenyl)-2-imino-1,2- dihydropyridine-3-carbonitrile (Id)
Yield 60%; mp 281–283 °C; IR (cm−1): 3448, 2202; 1H NMR (DMSO-d6): 1.41–1.46 (t, 3H, –CH2–CH3), 4.48–4.55 (q, 2H, –CH2– CH3), 6.85–8.06 (m, 11H, aromatic + –NHs); Anal. Calcd for C20H16BrN3O: C, 60.93; H, 4.09; N, 10.66. Found: C, 61.11; H, 4.10; N, 10.61.
3.2.5. 6-(4-Bromophenyl)-4-(4-ethoxyphenyl)-2-imino-1,2- dihydropyridine-3-carbonitrile (Ie)
Yield 50%; mp 173–175 °C; IR (cm−1): 3435, 2206; 1H NMR (DMSO-d6): 1.33–1.36 (t, 3H, –CH2–CH3), 4.06–4.09 (q, 2H, –CH2– CH3) 6.85–8.30 (m, 11H, aromatic + –NHs); Anal. Calcd for C20H16BrN3O: C, 60.93; H, 4.09; N, 10.66. Found: C, 60.76; H, 3.92; N, 11.03.
3.2.6. 6-(1,3-Benzodioxol-5-yl)-2-imino-4-(2-methoxyphenyl)- 1,2-dihydropyridine-3-carbonitrile (If)
Yield 58%; mp 223–225 °C; IR (cm−1): 3354, 2208; 1H NMR (DMSO-d6): 3.80 (s, 3H, –OCH3), 6.08 (s, 2H, –O–CH2–O–), 6.98– 7.68 (m, 10H, aromatic + –NHs); Anal. Calcd for C20H15N3O3: C, 69.56; H, 4.38; N, 12.17. Found: C, 69.10; H, 4.09; N, 12.18.
3.2.7. 6-(1,3-Benzodioxol-5-yl)-2-imino-4-(thiophen-2-yl)-1,2- dihydropyridine-3-carbonitrile (Ig)
Yield 45%; mp 172–174 °C; IR (cm−1): 3380, 2209; 1H NMR (DMSO-d6): 6.14 (s, 2H, –O–CH2–O–), 6.95–7.58 (m, 9H, aromatic + –NHs); Anal. Calcd for C17H11N3O2S: C, 63.54; H, 3.45; N, 13.08; S, 9.97. Found: C, 63.64; H, 3.71; N,13.43; S, 9.94.
3.2.8. 6-(Benzodioxol-5-yl)-2-imino-4-(thiophen-3-yl)-1,2- dihydropyridine-3-carbonitrile (Ih)
Yield 40%, buff crystals; mp 178–180 °C; IR (cm−1): 3391, 2204; 1H NMR (DMSO-d6): 6.1 (s, 2H, –O–CH2–O–), 6.80–8.16 (m, 9H, aromatic + –NHs); Anal. Calcd for C17H11N3O2S: C, 63.54; H, 3.45; N, 13.08; S, 9.98. Found: C, 63.39; H, 3.44; N, 12.67; S, 9.66.
3.2.9. 6-(1,3-Benzodioxol-5-yl)-4-(2-ethoxyphenyl)-2-imino- 1,2-dihydropyridine-3-carbonitrile (Ii)
Yield 60%; mp 182–184 °C; IR (cm−1): 3347, 2212; 1H NMR (DMSO-d6): 1.25–1.30 (t, 3H, –CH2–CH3), 4.07–4.10 (q, 2H, –CH2– CH3), 6.08 (s, 2H, –O–CH2–O–), 6.78–7.66 (m, 10H, aromatic + – NHs); Anal. Calcd For C21H17N3O3·0.25H2O: C, 69.32; H, 4.85; N, 11.55. Found: C, 69.59; H, 4.86; N, 11.21.
3.2.10. 6-(1,3-Benzodioxol-5-yl)-4-(4-ethoxyphenyl)-2-imino- 1,2-dihydropyridine-3-carbonitrile (Ij)
Yield 70%; mp 197–200 °C; IR (cm−1): 3356, 2217; 1H NMR (DMSO-d6): 1.32–1.37 (t, 3H, –CH2–CH3), 4.06–4.14 (q, 2H, –CH2– CH3), 6.08 (s, 2H, –O–CH2–O–), 6.85–7.72 (m, 10H, aromatic + – NHs); Anal. Calcd for C21H17N3O3: C, 70.18; H, 4.77; N, 11.69. Found: C, 70.05; H, 4.91; N, 11.52.
3.3. General procedure for the preparation of 6-(4- bromophenyl)-4-aryl-2-oxo-1,2-dihydropyridin-3-carbonitriles and 6-(1,3-benzodioxol-5-yl)-4-aryl-2-oxo-1,2-dihydropyridin- 3-carbonitriles (IIa–j)
A mixture of p-bromoacetophenone or 3′,4′-methylenedioxy-acetophenone (2.5 mmol), appropriate aldehyde (2.5 mmol), ethyl cyanoacetate (0.28 g, 2.5 mmol) and ammonium acetate (1.54 g, 20 mmol) in ethanol (50 ml) was heated under reflux for 10– 20 h. the reaction mixture was cooled and the formed precipitate was filtered, washed with ethanol, then washed successively with water, dried and crystallized from DMF/ethanol 1:2, respectively.
3.3.1. 6-(4-Bromophenyl)-4-(2-methoxyphenyl)-2-oxo-1,2- dihydropyridin-3-carbonitrile (IIa)
Yield 55%; mp 310–312 °C; IR (cm−1): 3390, 2220, 1647; 1H NMR (DMSO-d6): 3.81 (s, 3H, –OCH3), 7.07–7.83 (m, 10H, aromatic + –NH–), Anal. Calcd for C19H13BrN2O2: C, 59.86; H, 3.44; N, 7.35. Found: C, 59.73; H, 3.40; N, 7.34.
3.3.2. 6-(4-Bromophenyl)-4-(thiophen-2-yl)-2-oxo-1,2- dihydropyridin-3-carbonitrile (IIb)
Yield 40%; mp 330–332 °C; IR (cm−1): 3446, 2220, 1653; 1H NMR (DMSO-d6): 7.30–8.06 (m, 9H, aromatic + –NH–); Anal. Calcd for C16H9BrN2OS: C, 53.80; H, 2.54; N, 7.84; S, 8.98. Found: C, 53.66; H, 2.65; N, 7.96; S, 8.90.
3.3.3. 6-(4-Bromophenyl)-4-(thiophen-3-yl)-2-oxo-1,2- dihydropyridin-3-carbonitrile (IIc)
Yield 65%; mp 347–349 °C; IR (cm−1): 2222, 1653; 1H NMR (DMSO-d6): 6.98–8.35 (m, 9H, aromatic + –NH–); MS (EI); m/z 356 (M+, 100%), m/z 358 (M++2, 99.5%); Anal. Calcd for C16H9BrN2OS: C, 53.80; H, 2.54; N, 7.84; S, 8.98. Found: C, 53.60; H, 2.75; N, 7.64; S, 8.71.
3.3.4. 6-(4-Bromophenyl)-4-(2-ethoxyphenyl)-2-oxo-1,2- dihydropyridin-3-carbonitrile (IId)
Yield 70%; mp 281–283 °C; IR (cm−1): 3442, 2221, 1647; 1H NMR (DMSO-d6): 1.40–1.44 (t, 3H, –CH2–CH3), 4.15–4.17 (q, 2H, –CH2–CH3), 6.70–7.71 (m, 10H, aromatic + –NH–); MS (EI): m/z 394 (M+, 100%), m/z 396 (M+2+, 83%); Anal. Calcd for C20H15BrN2O2: C, 60.78; H, 3.83; N, 7.09. Found: C, 60.71; H, 3.73; N, 6.96.
3.3.5. 6-(4-Bromophenyl)-4-(4-ethoxyphenyl)-2-oxo-1,2- dihydropyridin-3-carbonitrile (IIe)
Yield 60%; mp 269–271 °C; IR (cm−1): 3446, 2220, 1656; 1H NMR (DMSO-d6): 1.45–2.50 (t, 3H, –CH2–CH3), 4.10–4.17 (q, 2H, –CH2–CH3), 6.71–7.80 (m, 10H, aromatic + –NH–); MS (EI): m/z 396 (M+, 100%); Anal. Calcd for C20H15BrN2O2: C, 60.78; H, 3.83; N, 7.09. Found: C, 60.48; H, 3.56; N, 6.58.
3.3.6. 6-(1,3-Benzodioxol-5-yl)-4-(2-methoxyphenyl)-2-oxo- 1,2-dihydropyridin-3-carbonitrile (IIf)
Yield 50%; mp 285–287 °C; IR (cm−1): 3432, 2227, 1680; 1H NMR (DMSO-d6): 3.79 (s, 3H, –O–CH3), 6.03 (s, 2H, –O–CH2–O–), 6.12–7.49 (m, 9H, aromatic + –NH); MS (EI): m/z 346 (M+, 100%); Anal. Calcd for C17H10N2O3·0.25H2O: C, 68.41; H, 3.99; N, 7.98. Found: C, 68.36; H, 4.33; N, 8.07.
3.3.7. 6-(1,3-Benzodioxol-5-yl)-4-(thiophen-2-yl)-2-oxo1,2- dihydropyridin-3-carbonitrile (IIg)
Yield 70%; mp 312–314 °C; IR (cm−1): 3440, 2213, 1637; 1H NMR (DMSO-d6): 6.13 (s, 2H, –O–CH2–O–), 6.88–8.05 (m, 8H, aromatic +–NH); MS (EI): m/z 322 (M+, 100%); Anal. Calcd for C17H10N2O3S·0.25H2O: C, 62.41; H, 3.06; N, 8.58; S, 9.78. Found: C, 62.39; H, 3.23; N, 8.59; S, 9.75.
3.3.8. 6-(1,3-Benzodioxol-5-yl)-4-(thiophen-3-yl)-2-oxo1,2- dihydropyridin-3-carbonitrile (IIh)
Yield 70%; mp 303–305 °C; IR (cm−1): 3420, 2218, 1655; 1H NMR (DMSO-d6): 6.12 (s, 2H, –O–CH2–O–), 6.88–8.34 (m, 8H, aromatic + –NH); MS (EI): m/z 322 (M+, 100%); Anal. Calcd for C17H10N2O3S·0.25H2O: C, 62.41; H, 3.06; N, 8.56; S, 9.78. Found: C, 62.66; H, 3.42; N, 8.55; S, 9.58.
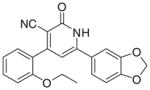
3.3.9. 6-(1,3-Benzodioxol-5-yl)-4-(2-ethoxyphenyl)-2-oxo1,2- dihydropyridin-3-carbonitrile (IIi)
Yield 40%; mp 274–276 °C; IR (cm−1): 3444, 2220, 1652; 1H NMR (DMSO-d6): 1.27–1.31 (t, 3H, –CH2–CH3), 4.09–4.12 (q, 2H, –CH2–CH3), 6.12 (s, 2H, –OCH2O–), 7.03–7.47 (m, 9H, aromatic +–NH); MS (EI): m/z 360 (M+, 100%); Anal. Calcd for C21H16N2O4: C, 69.99; H, 4.48; N, 7.77. Found: C, 69.77; H, 4.23; N, 6.96.
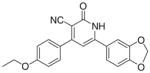
3.3.10. 6-(1,3-Benzodioxol-5-yl)-4-(4-ethoxyphenyl)-2-oxo1,2- dihydropyridin-3-carbonitrile (IIj)
Yield 60%; mp 288–290 °C; IR (cm−1): 3430, 2219, 1680; 1HNMR (DMSO-d6): 1.44–1.56 (t, 3H, –CH2–CH3), 4.11–4.13 (q, 2H, –CH2– CH3), 6.09 (s, 2H, –O–CH2–O–), 6.61–7.68 (m, 9H, aromatic +); MS (EI): m/z 360 (M+; 100%); Anal. Calcd for C21H16N2O4·0.5H2O: C, 68.23; H, 4.33; N, 7.58. Found: C, 68.54; H, 4.44; N, 7.56.
3.4. Biology
3.4.1. Cell cultures
HT-29 tumor cells were obtained from ATCC. They were grown under standard cell culture conditions at 37 °C in a humidified atmosphere with 5% CO2. Cells were grown in RPMI 1640 containing 5% fetal bovine serum. Cell count and viability was determined by Trypan blue staining followed by hemocytometry. Only cultures displaying >95% cell viability were used for experiments.
3.4.2. Growth assays
Tissue culture treated microtiter 96-well plates were seeded at a density of 5000 cells/well. The plates were incubated for 18–24 h prior to any treatment. Cell viability was measured 72 h after treatment by the Cell Titer Glo Assay (Promega), which is a luminescent assay that is an indicator of live cells as a function of metabolic activity and ATP content. The assay was performed according to manufacturer’s specifications.10
3.4.3. Phosphodiesterase inhibitory activity
PDE activity was measured using an adaptation of the IMAP® fluorescence polarization phosphodiesterase assay (Molecular Devices). PDE hydrolysis of the fluorescent-labeled substrate allows it to bind the IMAP® reagent, which increases fluorescence polarization (FP). The assay used fluorescein (Fl)-cAMP and tetramethylrhodamine (TAMRA)-cGMP as substrates. The different excitation and emission spectra of the substrates (485–530 nm for Fl and 530–590 nm for TAMRA) allowed for simultaneous measurement of cAMP and cGMP hydrolysis in the same well. The assays were performed in 96-well microtiter plates using a reaction buffer containing 10 mMTris–HCl (pH 7.2) 10 mMMgCl2, 0.05% NaN3 and 0.1% phosphate-free BSA as the carrier. Each well contained 20 μl of recombinant enzyme (BPS Biosciences, San Diego, CA) and 10 μl inhibitor. The reaction was initiated by the addition of 10 μl of a substrate solution containing 50 nM Fl-cAMP and/or TAMRA-cGMP. After incubating at room temperature for 60 min, the reaction was terminated by adding 120 μl of binding solution. FP was measured with a BioTek Synergy 4.
3.4.4. Experimental design and data analysis
Drug effects on tumor cell growth and PDE activity were measured and potency was expressed by an IC50 value (50% inhibitory concentration). For growth assays, the IC50 value was determined by testing a range of 8 concentrations with at least four replicates per concentration. For enzyme assays, the IC50 value was determined by testing a range of 10 concentrations with at least two replicates per concentration. Dose response curves were analyzed using Prism™ 4 software (GraphPad) to calculate IC50 values using a four parameter logistic equation.
3.5. Molecular modeling
The compounds with the correct stereochemistry and subjected to energy minimization using Force Field-MMFF94x by Molecular Operating Environment (MOE) software.11
3.5.1. Source of target proteins
The Crystal structure of Human PIM-1 kinase complexed with its inhibitor compound 1 (PDB ID code: 20BJ),1 survivin (PDB ID code: 1F3H),12 and PDE3A a theoretical homology model (PDB ID code: 1LRC) were downloaded from the Protein data bank and opened with MOE software. The complexed inhibitor-if any-was removed when using the synthesized compounds as ligands for docking. MOE was also used to calculate the best score between the ligands and the enzymes’ binding sites as given in the software manual. MOE–Dock is used to search for favorable binding configurations between the ligand and the macromolecular target.
3.5.2. Energy minimization procedure
Selected compounds for docking were drawn in mol format and all hydrogens are added, they are energy minimized using Hamiltonian- Force Field-MMFF94x and the partial charges are also calculated.
3.5.3. Docking procedure
Milrinone was drawn and energy minimized and docked to the PDE3A homology model (PDB ID code 1LRC) using the whole protein as the binding site to identify the binding pocket. For the 200 retained structures; the binding site was identified from the abundance of 30 top score conformers; the default docking procedure is done for milrinone as standards for validation in the identified binding site and then for the selected synthesized compounds.
Docking procedure of PIM-1 kinase was as following: (a) the cocrystallized compound 1 was identified; therefore, the binding site was identified with its residues. (2) Ligand interactions were computed for the X-ray co-crystallized compound 1 to reveal the different types of interaction as a validation for the coming docking procedure. (3) The co-crystallized compound 1 is then removed and the selected synthesized compounds are used instead. (4) The docking was done with the default settings of the MOE–DOCK as following: a. the option: Rotate Bonds was selected to give flexible ligand-rigid receptor docking; b. the scoring function was London dG with a replacement of Alpha Triangle; c. 30 conformers of the ligand were retained with highest and best score by default; d. the top score ligand-receptor docking was then demonstrated by 2D ligand-receptor interactions and by 3D MEP (Molecular Electrostatic Potential) on a Gaussian contact surface of the binding pocket.
Docking procedure of survivin was as following: (1) Survivin is an identical dimeric protein; therefore, the unnecessary chain was deleted. (2) ZnSO4 moieties of co-crystallization were deleted. (3) Then, the binding site was selected by selecting the residues L6, L96, L98, L102, L104, F86, F93, F101, V89 and K15. These residues were reported to form the binding site of survivin inhibitors of compound 1.2 (4) Then the docking procedure was done by the default settings of MOE–DOCK as previously mentioned.
Acknowledgments
The first author is grateful to the Alexander von Humboldt foundation, Germany for sponsoring a postdoctoral research stay in Germany. The second author is indebted to the Faculty of post-graduate studies, German University in Cairo, for partial finance of this research.
References and notes
- 1.Cheney IW, Yan S, Appleby T, Walker H, Vo T, Yao N, Hamatake R, Hong Z, Wu JZ. Bioorg Med Chem Lett. 2007;17:1679. doi: 10.1016/j.bmcl.2006.12.086. [DOI] [PubMed] [Google Scholar]
- 2.Wendt MD, Sun C, Kunzer A, Sauer D, Sarris K, Hoff E, Yu L, Nettesheim DG, Chen J, Jin S, Comess KM, Fan Y, Anderson SN, Isaac B. Bioorg Med Chem Lett. 2007;17:3122. doi: 10.1016/j.bmcl.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 3.Aqui NA, Vonderheide RH. Cancer Biol Ther. 2008;7:1888. doi: 10.4161/cbt.7.12.7219. [DOI] [PubMed] [Google Scholar]
- 4.Ambrosini G, Adida C, Altieri DC. Nat Med. 1997;3:917. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 5.Gary P, Soh JW, Mao Y, Kim MG, Pamukcu R, Li H, Thompson WJ, Weinstein IB. Clin Cancer Res. 2000;6:4136. [PubMed] [Google Scholar]
- 6.Cheng J, Grande JP. Exp Biol Med. 2007;232:38. [PubMed] [Google Scholar]
- 7.Murata T, Shimizu K, Narita M, Manganiello V, Tagawa T. Anticancer Res. 2002;22:3171. [PubMed] [Google Scholar]
- 8.Murata T, Sugatani T, Shimizu K, Manganiello V, Tagawa T. Anticancer Drugs. 2001;12:79. doi: 10.1097/00001813-200101000-00011. [DOI] [PubMed] [Google Scholar]
- 9.Moon E, Lee R, Near R, Weintraub L, Wolda S, Lerner A. Clin Cancer Res. 2002;8:589. [PubMed] [Google Scholar]
- 10.Liu L, Li H, Underwood T, Lloyd M, David M, Sperl G, Pamukcu R, Thompson WJ. J Pharmacol Exp Ther. 2001;299:583. [PubMed] [Google Scholar]
- 11.MOE. Chemical Computing Group Inc; Montreal: http://www.chemcomp/com. [Google Scholar]
- 12.Verdecia MA, Huang H, Dutil E, Kaiser DA, Hunter T, Noel JP. Nat Struct Biol. 2000;7:602. doi: 10.1038/76838. [DOI] [PubMed] [Google Scholar]