

Abstract
Objective:
The objective of this study is to retrospectively collect and then describe the clinico-radiographical profile of confirmed cases of leukodystrophy who presented over a 5-year period to a tertiary care teaching hospital in North India.
Materials and Methods:
The case records of 80 confirmed cases of leukodystrophy were reviewed and the cases have been described in terms of their clinical presentation and neuroimaging findings.
Results:
The cases have been grouped into five categories: Hypomyelinating, demyelinating, disorders with vacuolization, cystic, and miscellaneous. The commonest leukodystrophies are megalencephalic leukoencephalopathy with subcortical cysts (MLC), Pelizaeus-Merzbacher disease (PMD), and metachromatic leukodystrophy (MLD). A notable proportion of hypomyelinating disorders were uncharacterized.
Conclusions:
Leukodystrophies at this point of time have no definite cure. They have a progressively downhill clinical course. Early diagnosis is imperative for appropriate genetic counseling. A simplified approach to diagnose common leukodystrophies has also been provided. It is important to develop a registry, which can provide valuable epidemiological data to prioritize research in this field, which has many unanswered questions.
Keywords: Clinico-radiological, cystic, genetic counselling, demyelination, hypomyelination, leukodystrophy
Introduction
Leukoencephalopathies are disorders, which selectively involve the cerebral white matter. It is an umbrella term and encompasses disorders with hereditary, inflammatory, autoimmune, vascular, infectious and neoplastic etiologies. The term “leukodystrophies” refer to the disorders with primary white matter involvement with demonstrable biochemical or molecular defect and usually with a progressive clinical course.
The clinical and radiological clues may be very helpful in guiding the investigations of a child with suspected leukodystrophy.[1,2,3,4] However, around half of the cases of leukodystrophies remain undiagnosed despite recent advances. This figure may be higher in India due to limited availability and cost of investigations and limited awareness among pediatricians and pediatric neurologists. This study aimed to describe the spectrum of leukodystrophies managed at a tertiary care and referral center in North India.
Materials and Methods
The medical records of children diagnosed with leukodystrophy admitted in the pediatric neurology ward or attending the pediatric neurology clinic at a tertiary care and a referral hospital in North India from January 2008 to December 2012 were retrospectively reviewed. The diagnosis was based on the clinical phenotype, suggestive neuroimaging, and definitive investigations wherever applicable and available. The data were extracted as per a pre-designed pro forma. The clinical and radiological data of each case were subsequently summarized and reported. The summary statistics were calculated where required. The study was approved by the ethics committee of the institute.
Results
During the study period, 1,314 children were seen. Out of these, 80 cases were diagnosed as leukodystrophy. Table 1 shows the spectrum of leukodystrophies in children seen during the study period. The summarized clinical and radiological data of these children are detailed below.
Table 1.
Spectrum of leukodystrophies in children seen during the study period
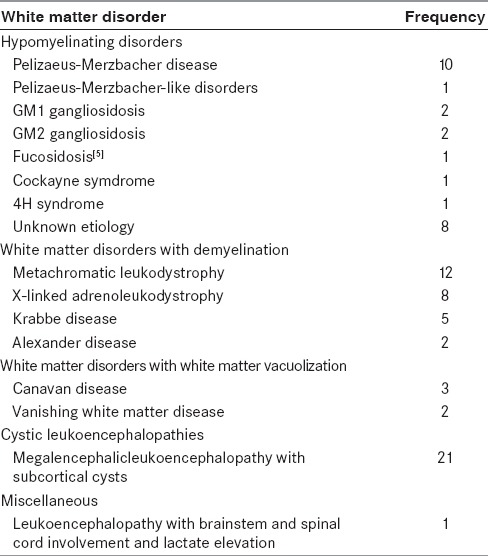
Hypomyelinating disorders
Twenty-six children were diagnosed with hypomyelinating disorders radiologically. The distribution is shown in Table 1.
Ten children had Pelizaeus-Merzbacher disease (PMD). The diagnosis was based on suggestive clinical phenotype and neuroimaging and genetic testing. The mean age at the time of presentation was 10.1 months (SD: –2.2, range: 6-14 months). All the children presented with global developmental delay, central hypotonia, and nystagmus. Other signs included ataxia (6), athetoid movements (2), and seizures. (1). Nerve conduction studies were available in two children and were normal. Magnetic resonance imaging (MRI) of the brain showed global hypomyelination in all the affected children. Mild cerebellar atrophy was seen in two children. Brainstem and basal ganglia were normal in all the children. All the children had proteolipid protein 1 (PLP1) gene duplication.
One 9-year-old girl was diagnosed with Pelizaeus-Merzbacher-like disorder radiologically [Figure 1]. She presented with intellectual disability, ataxia, mild dysarthria, and lower limb long tract signs. Her nerve conduction studies were normal. She had no mutation in PLP1 gene.
Figure 1.
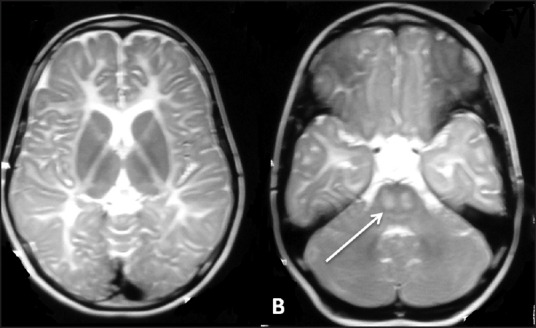
Magnetic resonance imaging of the brain in a Pelizaeus-Merzbacher-like disorder: axial T2-weighted images show hypomyelination (a) and image at the level of the pons (b) show T2hyperintensityin the pyramidal tracts (white arrows in B)
One child was diagnosed with Cockayne syndrome. He presented at 13 years of age with short stature, intellectual disability, frequent falls, cutaneous photosensitivity, small head, normal fundus, cerebellar signs, and lower limb long tract signs. Nerve conduction study revealed symmetric demyelinating sensori-motor polyneuropathy in the lower limbs. Computed tomography scan of the head showed bilateral lentiform calcifications. MRI of the brain showed hypomyelination with mild cerebellar atrophy.
One child was diagnosed with 4H syndrome. She was a 12-year-old girl who presented with progressive ataxia since 5 years of age. There was no history of impaired cognition, seizures, impaired vision or hearing, night blindness, malabsorption, headache, vomiting, or episodic deterioration. Examination revealed widely spaced poorly formed supernumerary upper central incisors, delayed sexual development (Tanner stage 0), lower limb spasticity, and truncal ataxia. There was no nystagmus or oculomotor ataxia and the fundus was normal. Nerve conduction studies were normal. MRI of the brain showed hypomyelination with cerebellar atrophy. Basal ganglia, brainstem, and thalamus were normal. Plasma follicle-stimulating hormone (FSH) and luteinizing hormone (LH) were markedly reduced. The clinico-radiological presentation was suggestive of 4H syndrome.
The below mentioned disorders are classically described as gray matter disorders. However, they have both gray and white matter abnormalities. Hypomyelination is present in all these disorders and hence, they have been included in the group of hypomyelination disorders.
Two children diagnosed with infantile GM1 gangliosidosis, based on clinical phenotype and deficient β-galactosidase in the leukocytes, had hypomyelination on neuroimaging. One child was a 2.5-year-old male who presented with global developmental delay, large head, coarse facies, increased Mongolian spots, hydrocele, hepatosplenomegaly, mild disc pallor on fundoscopy, and central hypotonia. His investigations revealed beaking of the lumbar vertebrae and suggestive neuroimaging [Figure 2]. The other child presented at 14 months of age with global developmental delay, large head, coarse facies, hepatosplenomegaly, cherry-red spot on fundoscopy, and central hypotonia. His skeletal survey showed diffuse osteopenia. Neuroimaging showed global hypomyelination with bilateral T2-weighted basal ganglia hyperintensities [Figure 2]. Brainstem and cerebellum were normal in both the cases.
Figure 2.
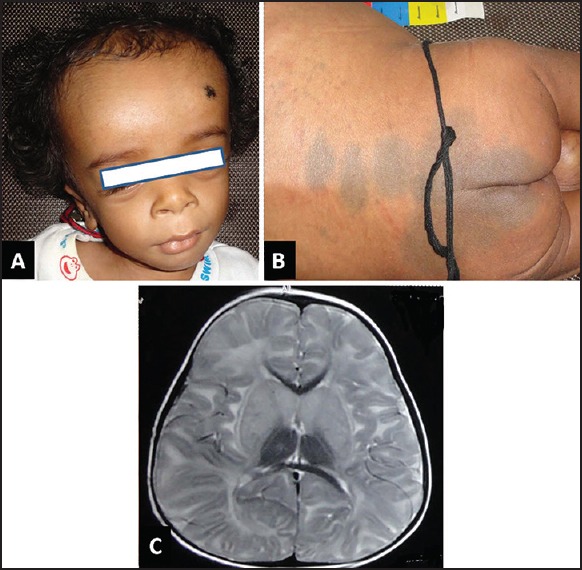
Child with GM1 gangliosidosis: (a) shows coarse facial facies (b) shows increased Mongolian spots (c) Magnetic resonance imaging of the brain, T2-weighted axial image, shows hypomyelination with bilateral caudate and lentiform hyperintensities
Two children diagnosed with classical Tay Sach disease, based on clinical phenotype and deficient hexosaminidase A in the leukocytes, had hypomyelination on neuroimaging. One boy presented at 22 months of age with initial normal development followed by global neuroregression since 15 months of age with vision impairment, increased startles, bilateral cherry-red spot on fundoscopy, and generalized spasticity. The visually -evoked potential was extinguished in both the eyes. Neuroimaging showed hypomyelination with bilateral T2-weighted basal ganglia hyperintensities with normal brainstem and cerebellum. The second case was a 2-year-old boy, born of a third-degree consanguineous couple, who had a similar clinical phenotype and radiology. Neuroregression started at 9 months of age following a febrile illness.
The child with fucosidosis was reported earlier.[5]
Eight children diagnosed with a hypomyelinating disorder, based on neuroimaging, compatible clinical phenotype, and exclusion of other causes, could not achieve final diagnosis.
White matter disorders with demyelination
Metachromatic leukodystrophy
Twelve children were diagnosed with metachromatic leukodystrophy (MLD) on the basis of suggestive clinical phenotype and neuroimaging and deficient arylsulfatase A activity in the peripheral leukocytes. The median age at the onset of symptoms was 11.5 months (range: 6-48 months). Only one child had juvenile variant of MLD and the rest had late infantile variants. Ten out of twelve children were males. Eight children were born to third-degree consanguineous couples.
All the children with late infantile variant MLD presented with global neuroregression, cognitive decline, and spasticity. Other observed features included optic atrophy (10/11), seizures (1/11), and microcephaly (3/11). The child with juvenile variant presented at 4 years of age with gait difficulties, cognitive decline, and ataxia. Nerve conduction studies were available for only three children. All studies were suggestive of demyelinating motor-sensory polyneuropathy.
MRI of the brain was available for all children. It showed bilateral nearly symmetrical confluent white matter T2- and fluid attenuated inversion recovery (FLAIR) hyperintensities with sparing of subcortical-U-fibers [Figure 3]. Four children had occipital predominant white matter changes and the rest of the children had diffuse involvement with no specific gradient. The “tigroid” pattern was appreciated in seven children. Posterior limb of the internal capsule and brainstem long tracts were involved in nine and two children, respectively. Basal ganglia and cerebellum were normal in all children.
Figure 3.
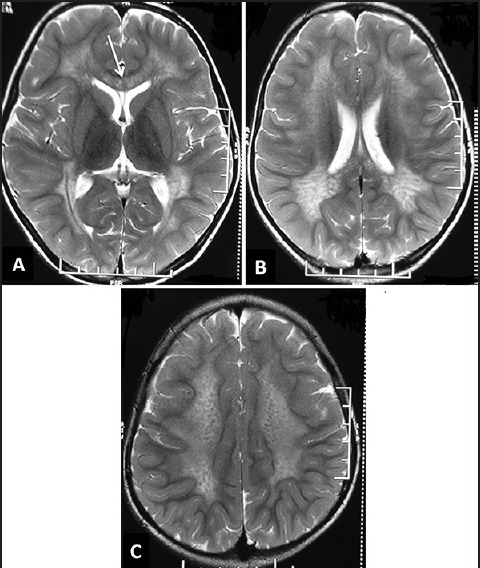
Magnetic resonance imaging of the brain in metachromatic leukodystrophy. T2-weighted axial images (a-c) of a child show bilateral confluent symmetrical occipital predominant white matter involvement with involvement of corpus callosum (white arrow in A). Also note the “tigroid” appearance in C
X-linked adrenoleukodystrophy
Eight boys were diagnosed with X-linked adrenoleukodystrophy based on suggestive clinical phenotype, neuroimaging and elevated very long chain fatty acids. The mean age at presentation was 7.8 years [standard deviation (SD): -1.7, range: 5-10]. The presenting complaints included gait difficulties (7), behavioral abnormalities (7), impaired vision (6), decreased hearing (6), poor school performance (6), seizures (2), and body hyperpigmentation (2). One boy presented with an episode of generalized tonic-clonic seizures following a trivial fall from the bicycle. His MRI of the brain was highly suggestive of X-linked adrenoleukodystrophy at that time. Within 2 months, he developed gait difficulties and became bedridden within 5 months of symptom onset. All children had a rapid course with loss of ambulation occurring after a mean of 4.5 months (SD: –1.3, range: 3-7). Family history was unremarkable in all the children.
All the three parameters, the concentration of C26:0 fatty acids, and ratios of C26:0/C22:0 and C24:0/C22:0 fatty acids were elevated in all the children. The screening of the siblings of the affected children with plasma very long chain fatty acids was negative in all cases. The screening for adrenal insufficiency was performed at the time of diagnosis by random blood sugar, serum electrolytes, and basal and post-adrenocorticotrophic hormone (ACTH) serum cortisol levels. It was normal in all cases. Nerve conduction studies were not available for any child.
MRI of the brain showed parieto-occipital predominant lesions classical of X-linked adrenoleukodystrophy in seven children [Figure 4]. One child showed corticospinal tract involvement from the cortex to internal capsule with mild parieto-occipital involvement and no contrast enhancement. He also showed a rapid course with loss of ambulation within 4 months of symptom onset. Cerebellum, brainstem, and basal ganglia were normal in all the children.
Figure 4.
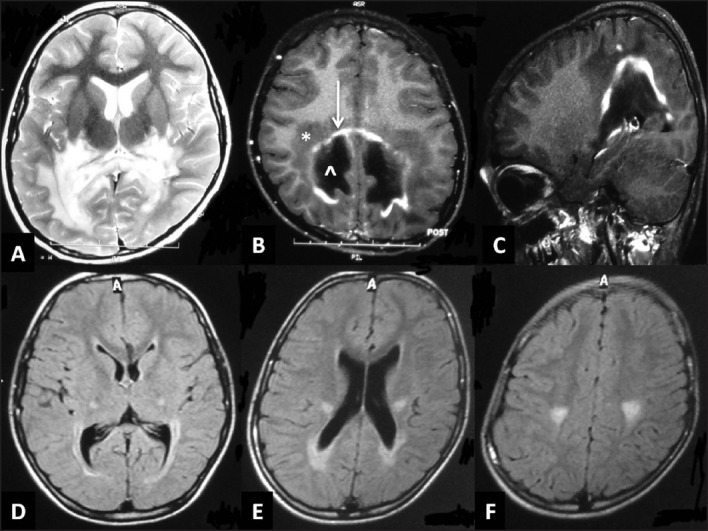
Magnetic resonance imaging of the brain in x-linked adrenoleukodystrophy: (a) T2-weighted axial image shows bilateral confluent symmetrical white matter hyperintensity in the parieto-occipital region with involvement of the splenium (b,c) T1-weighted axial and saggital image with contrast-enhancement shows the classical three zones: zone of active demyelination without inflammation (*), zone of prominent inflammation (white arrow) and zone of completely demyelinated/burnt-out areas (^). FLAIR axial images of another child (d-f) Shows selective corticospinal tract involvement from cortex to internal capsule with mild parieto-occipital involvement
None of the children could be treated with hematopoetic stem cell transplantation as all had advanced cerebral X-linked adrenoleukodystrophy.
Krabbe's disease
Five children were diagnosed with Krabbe's disease based on suggestive clinical phenotype and neuroimaging and deficient galactocerebrosidase activity in the leukocytes. Four children had infantile-onset and one child had juvenile-onset disease. The child with juvenile onset has been reported earlier.[6] The chief presenting features were neuroregression (5), irritability (4), impaired visison (3), seizures (2), exaggerated startle reaction (1), “sterile” pyrexia (1), microcephaly (2) and spasticity (5). All the children were bed-ridden at the time of diagnosis except the child with juvenile-onset disease.
MRI of the brain showed bilateral nearly symmetrical confluent white matter T2- and FLAIR hyperintensities with involvement of corpus callosum with sparing of subcortical-U-fibers in four children. It was parieto-occipital dominant in two children and diffuse in the other two. The child with juvenile-onset disease had isolated selective cortico-spinal tract involvement. The other findings included mild cerebral atrophy (2), cerebellar white matter involvement (1), and lentiform hyperintensities (1). Optic nerve enlargement was not seen in any case.
Alexander disease
Two cases were diagnosed with Alexander disease based on suggestive clinical phenotype, diagnostic neuroimaging,[7] and genetic testing (in one patient). The genetically confirmed child has been reported earlier.[8] The other child presented at 2 years of age with neuroregression, seizures, a large head, and spasticity. He was already bed-ridden at the time of presentation. MRI of the brain showed bilateral confluent symmetrical frontally dominant white matter hyperintenities with involvement of the caudate and putamen. There were cystic changes in the frontal areas with patchy contrast -enhancement in the centrum semiovale. There was no involvement of cerebellum, brainstem, spinal cord, and optic nerves.
White matter disorders with vacuolization
Canavan disease
Three children were diagnosed with infantile Canavan disease based on suggestive clinical phenotype and neuroimaging, high N-acetylaspartate resonance on proton MRI, and elevated urinary N-acetylaspartic acid. All three children were males with disease onset within 6 months of age. The presenting features included global developmental delay (3), neuroregression (1), seizures (2), impaired vision (2), feeding difficulties (3), macrocephaly (3), central hypotonia (3), and optic atrophy (2).
MRI of the brain revealed bilateral confluent symmetrical white matter hyperintenities (deep and subcortical) (3), globus pallidus involvement with sparing of the caudate and putamen (3), cerebellar white matter involvement (2), brainstem involvement (3), and bilateral temporal cysts (1).
Vanishing white matter disease
Two cases were diagnosed with vanishing white matter disease based on suggestive clinical phenotype and neuroimaging. One genetically confirmed case was reported earlier.[9]
The 2nd case presented at 4 years of age with global developmental delay, progressive ataxia, and two episodes of unprovoked generalized tonic-clonic seizures at 2 years of age. There was no episodic deterioration. Examination revealed optic disc pallor, cerebellar ataxia, and spasticity. MRI of the brain revealed bilateral symmetrical confluent diffuse white matter hyperintensities with “melting away” pattern with brainstem involvement. The cerebellum and basal ganglia were normal.
Cystic leukoencephalopathies Megalencephalic leukoencephalopathy with subcortical cysts
Twenty-one children were diagnosed with megalencephalic leukoencephalopathy with subcortical cysts (MLC) during the study period. The diagnosis was based on suggestive clinical phenotype and classical neuroimaging findings. The age at the onset of symptoms ranged from early infancy to 11 years with a median age at onset of 4 years [interquartile range (IQR): 2-7 years]. The male-to-female ratio was 2:1. The composition with regard to the ethnic community was as follows: Agarwal (19) and Khandelwal (2). None of the children had significant family history.
The most frequently reported symptoms included a large head (19), seizures (16), motor delay (13), and frequent falls (8). Out of 16 children with epilepsy, nine had impact seizures. All children with epilepsy had easily controlled seizures. None of the children had status epilepticus. Other signs observed were lower limb hyperreflexia (15), ataxia (6), mild dysarthria (5), and nystagmus (3).
One of the children had serious deterioration following trivial trauma with loss of all milestones and severe neurodevelopemntal sequelae. Out of 12 children of the school-going age, 11 were attending age-appropriate classes.
MRI of the brain was available for all children [Figure 5], which showed bilateral confluent white matter T2- and FLAIR hyperintensities with prominent subcortical involvement. The cerebellar white matter involvement was seen in three cases at the time of diagnosis. Bilateral temporal subcortical cysts were seen in all the children. Cysts were also seen in frontal (2) and parietal areas (1). Basal ganglia and brainstem were normal in all the children. Serial imaging was not available in none of the affected children.
Figure 5.
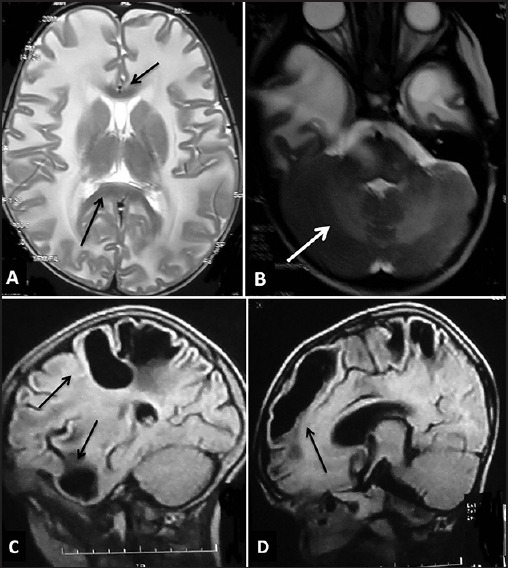
Magnetic resonance imaging of the brain in megalencephalic leukoencephalopathy with subcortical cysts. T2-weighted axial images (a and b) of a child show bilateral confluent symmetrical diffuse white matter involvement with swollen appearance with sparing of corpus callosum (black arrows in a) with temporal lobe cysts (b) and cerebellar white matter involvement (white arrow in B). Note was also made of cavum septi pellucidi and cavum vergae (a). The saggital FLAIR images of another child show subcortical cysts in temporal, frontal, and parietal areas (black arrows in C and D)
All children showed homozygous mutation in exon 2 of the MLC1 gene.
Miscellaneous
The child with leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation has been reported earlier.[10]
Discussion
The patients affected with leukodystrophy suffer high morbidity and mortality.[11] Further, due to lack of trained personnel in developing countries, their diagnosis, management, and genetic counseling are challenging issues. This study tried to explore the spectrum of leukodystrophies at a tertiary care center in North India.
The true burden of leukodystrophies is largely unknown. The population-based incidence of leukodystrophies has been estimated to be 1 in 7,663 live births in an American study.[12] MLD, PMD-, mitochondrial diseases, and adrenoleukodystrophy were the common leukodystrophies in this cohort. The Indian data in this regard is paltry. MLC was reported to be the commonest leukodystrophy in India.[13] In this study conducted at a tertiary care referral center, MLC, MLD, and PMD -were the commonest.
MRI — plays an important role in the diagnosis of leukodystrophies. Pattern recognition is important as it may provide a clue toward or clinch the diagnosis in many conditions.[2,3,4] This is especially important in developing countries such as India where the confirmatory tests for these disorders are either not available or are very expensive.
Schiffman and van der Knaap[2] reviewed the MRI-based approach in the diagnosis of white matter disorders. The first step is to differentiate a hypomyelinating disorder from other white matter pathologies. In case of a hypomyelinating disorder, the presence or absence of peripheral nerve involvement will further aid in diagnosis. The next step is to determine whether the white matter changes are confluent, patchy, or multifocal in other white matter pathologies. Patchy or multifocal signal abnormalities favor the diagnosis of an acquired white matter disorder although exceptions exist. If the white matter changes are confluent, it is important to note their predominant localization (frontal, parieto-occipital, diffuse, periventricular, or sub-cortical). Finally, the presence of additional features such as cysts, calcifications, microbleeds, and the involvement of other structures (gray matter, basal ganglia, spinal cord) may help in the diagnosis.[14] A simplified flow diagram based approach to leukodystrophies is being presented here [Figure 6].
Figure 6.

A simplified flow diagram-based approach to leukodystrophies
Further, the clinical phenotype of many leukodystrophies may be misdiagnosed as cerebral palsy. These neuroimaging clues may help us to avoid this misdiagnosis. “Red flags” for considering neurodegenerative disorders include the absence of definite perinatal insult, a pattern of disease inheritance or consanguinity, neuroregression or worsening symptoms, and isolated hypotonia, rigidity, or paraplegia.[15] Identification of these entities is vital with respect to therapy, prognosis, and genetic counseling.
The treatment options for leukodystrophies are unfortunately limited. Bone marrow transplant is only available for a few leukodystrophies (X-linked adrenoleukodystrophy, MLD, and Krabbe disease) and that too if done early in the disease.[16,17,18] Thus, supportive care and genetic counseling become pivotal.
This study reported the spectrum of leukodystrophies in children at a referral center in North India. With more availability of trained pediatric neurologists and radiologists, the ever -increasing availability of MRI brain, biochemical, and genetic testing in various centers across India, these disorders will be diagnosed more often. Population-based studies and studies in the different regions of the country may be required to truly estimate the prevalence of these rare disorders. The establishment of a pediatric white matter disorder registry will also be highly desirable. This would encourage much needed research in this field in terms of further diagnostics and therapeutics as well.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Renaud DL. Clinical approach to leukoencephalopathies. Semin Neurol. 2012;32:29–33. doi: 10.1055/s-0032-1306383. [DOI] [PubMed] [Google Scholar]
- 2.Schiffmann R, van der Knaap MS. Invited article: An MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750–9. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: Leukodystrophies and beyond. Pediatr Radiol. 2008;38:729–49. doi: 10.1007/s00247-008-0817-x. [DOI] [PubMed] [Google Scholar]
- 4.Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. 2010;133:2971–82. doi: 10.1093/brain/awq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain P, Ramesh K, Mohamed A, Kumar A, Gulati S. Teaching NeuroImages: Distinct neuroimaging features of fucosidosis. Neurology. 2012;78:e33. doi: 10.1212/WNL.0b013e3182452910. [DOI] [PubMed] [Google Scholar]
- 6.Sehgal R, Sharma S, Sankhyan N, Kumar A, Gulati S. Selective corticospinal tract involvement in late-onset Krabbe disease. Neurology. 2011;77:e20. doi: 10.1212/WNL.0b013e318225aaf5. [DOI] [PubMed] [Google Scholar]
- 7.van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, et al. Alexander disease: Diagnosis with MR imaging. AJNR Am J Neuroradiol. 2001;22:541–52. [PMC free article] [PubMed] [Google Scholar]
- 8.Ramesh K, Sharma S, Kumar A, Salomons GS, van der Knaap MS, Gulati S. Infantile-onset Alexander disease: A genetically proven case with mild clinical course in a 6-year-old Indian boy. J Child Neurol. 2013;28:396–8. doi: 10.1177/0883073812444313. [DOI] [PubMed] [Google Scholar]
- 9.Sharma S, Arya R, Raju KN, Kumar A, Scheper GC, van der Knaap MS, et al. Vanishing white matter disease associated with ptosis and myoclonic seizures. J Child Neurol. 2011;26:366–8. doi: 10.1177/0883073810381529. [DOI] [PubMed] [Google Scholar]
- 10.Sharma S, Sankhyan N, Kumar A, Scheper GC, van der Knaap MS, Gulati S. Leukoencephalopathy with brain stem and spinal cord involvement and high lactate: A genetically proven case without elevated white matter lactate. J Child Neurol. 2011;26:773–6. doi: 10.1177/0883073810390695. [DOI] [PubMed] [Google Scholar]
- 11.Brimley CJ, Lopez J, van Haren K, Wilkes J, Sheng X, Nelson C, et al. National variation in costs and mortality for leukodystrophy patients in US children's hospitals. Pediatr Neurol. 2013;49:156–62.e1. doi: 10.1016/j.pediatrneurol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonkowsky JL, Nelson C, Kingston JL, Filloux FM, Mundorff MB, Srivastava R. The burden of inherited leukodystrophies in children. Neurology. 2010;75:718–25. doi: 10.1212/WNL.0b013e3181eee46b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singhal BS. Leukodystrophies: Indian scenario. Indian J Pediatr. 2005;72:315–8. doi: 10.1007/BF02724013. [DOI] [PubMed] [Google Scholar]
- 14.Van der Knaap M, Valk J. Magnetic Resonance of Myelination and Myelin Disorders. 3rd ed. New York: Springer; 2005. [Google Scholar]
- 15.Lee RW, Poretti A, Cohen JS, Levey E, Gwynn H, Johnston MV, et al. A diagnostic approach for cerebral palsy in the genomic era. Neuromolecular Med. 2014;16:821–44. doi: 10.1007/s12017-014-8331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sevin C, Aubourg P, Cartier N. Enzyme, cell and gene-based therapies for metachromatic leukodystrophy. J Inherit Metab Dis. 2007;30:175–83. doi: 10.1007/s10545-007-0540-z. [DOI] [PubMed] [Google Scholar]
- 17.Orchard PJ, Tolar J. Transplant outcomes in leukodystrophies. Semin Hematol. 2010;47:70–8. doi: 10.1053/j.seminhematol.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Boelens JJ. Trends in haematopoietic cell transplantation for inborn errors of metabolism. J Inherit Metab Dis. 2006;29:413–20. doi: 10.1007/s10545-005-0258-8. [DOI] [PubMed] [Google Scholar]