

ABSTRACT
Disturbance of endoplasmic reticulum (ER) proteostasis is observed in Prion-related disorders (PrDs). The protein disulfide isomerase ERp57 is a stress-responsive ER chaperone up-regulated in the brain of Creutzfeldt-Jakob disease patients. However, the actual role of ERp57 in prion protein (PrP) biogenesis and the ER stress response remained poorly defined. We have recently addressed this question using gain- and loss-of-function approaches in vitro and animal models, observing that ERp57 regulates steady-state levels of PrP. Our results revealed that ERp57 modulates the biosynthesis and maturation of PrP but, surprisingly, does not contribute to the global cellular reaction against ER stress in neurons. Here we discuss the relevance of ERp57 as a possible therapeutic target in PrDs and other protein misfolding disorders.
KEYWORDS: ERp57, ER stress, Grp58, PDI, PDIA3, protein folding, PrP
Prion-related disorders (PrDs) are fatal neurodegenerative diseases characterized by spongiform degeneration of the brain and neurological dysfunction, where the most common forms are Creutzfeldt-Jakob disease (CJD) in humans and scrapie and bovine spongiform encephalopathy (or mad cow disease) in other animals. The accumulation of misfolded and protease resistant form of the prion protein (PrP), referred to as PrPSc for the scrapie isoform, is the hallmark of these diseases. According to ‘the protein-only’ hypothesis, misfolded PrP acts as a seed for the propagation of PrPSc mediated by a conformational change of the wild-type cellular prion protein (PrPC) through a physical interaction, enriching its structure with beta sheets. The etiology of PrDs is complex and involves 3 main variants, including familial forms, infectious, or of unknown origin (sporadic forms).1 During the pre-symptomatic phase of PrDs, PrP misfolds and propagates, altering several cellular functions including the homeostasis in the endoplasmic reticulum (ER). As the disease progresses, synaptic dysfunction leads to dendritic loss and spongiform degeneration, ending with extensive neuronal death and global cognitive and motor dysfunction.2 Although PrDs are rare diseases, they are becoming more relevant since the prion-like mechanism of protein misfolding propagation also operates in the disease spreading process of prevalent disorders like Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS).3
Despite the fact PrP misfolding has been extensively studied for more than 30 years, the molecular pathways controlling its folding, misfolding and clearance remain poorly understood. Early studies have demonstrated that cellular factors, in addition to intrinsic properties of PrP structure, are critical to the efficiency of PrP propagation.4 It is critical to gain insights into these processes in order to generate efficient therapeutic targets to prevent the initiation and propagation of the protein misfolding process in neurodegenerative diseases. PrPC is a highly glycosylated protein containing an intramolecular disulfide bond and a GPI anchor that targets the protein to lipid rafts at the plasma membrane.1 PrP synthesis and maturation occur in the secretory pathway, undergoing processing and quality control at the ER, to further glycosylation at the Golgi apparatus followed by its sorting to the plasma membrane.5,6 Around 10% of the newly synthesized PrPC is not correctly folded, and undergoes clearance through the ER-associated degradation (ERAD) pathway.7 The ER-resident chaperone Grp78/BiP has been implicated in the process of recognition of misfolded PrP for proteosomal degradation, playing a role in the maintenance of PrP quality control. In genetic forms of PrDs, the conformation changes triggered by PrP mutations are proposed to occur during its trafficking through the ER, leading to its retention at the ER or the Golgi apparatus, or a delay in its trafficking through the secretory pathway.2 Consequently, ER-resident chaperones may play a pivotal role in controlling the folding/refolding and quality control of PrP, thus emerging as novel therapeutic targets in PrDs.
Disturbance to ER proteostasis is a common feature of most protein misfolding disorders (PMDs) affecting the nervous system.8 The accumulation of misfolded proteins at the ER lumen causes stress.8 To restore ER proteostasis, cells activate the unfolded protein response (UPR), a signal transduction pathway that orchestrates adaptation to protein folding stress by boosting protein folding, quality control, and degradation mechanisms. However, under unresolvable ER stress, the UPR triggers apoptosis through a crosstalk with mitochondria.9 ER stress and UPR activation have been extensively described in models of infectious forms of PrDs (see examples in10–13). At the mechanistic level, alterations in ER calcium homeostasis may be one of the factors generating ER stress in PrDs.14 Interestingly, we and others have described that ER stress triggers the partial misfolding of PrP, enhancing its susceptibility to be converted into PrPSc, suggesting a vicious cycle between PrP propagation and the occurrence of ER stress.15,16 One of the main ER factors upregulated in PrDs is the protein disulfide isomerase (PDI) family member ERp57 (also known as PDIA3 or GRP58)10,11,17,18 (Fig. 1A). ERp57 is an ER-resident foldase that catalyzes disulfide bond formation of a subset of glycoproteins and represents a key component of the calnexin and calreticulin cycle (Fig. 1B).19 We previously reported that ERp57 expression protects cells against PrPSc toxicity,10 suggesting that chronic ER stress may be part of the mechanism mediating neuronal loss in PrDs. Interestingly, ER stress may also contribute to early neuronal dysfunction in PrDs by inducing the activation of the ER stress sensor PERK, which inhibits protein synthesis by phosphorylating the eukaryotic initiation factor 2α (eIF2α). This event was proposed to repress the translation of a cluster of synaptic proteins, generating neurological and behavioral impairment12,13
FIGURE 1.
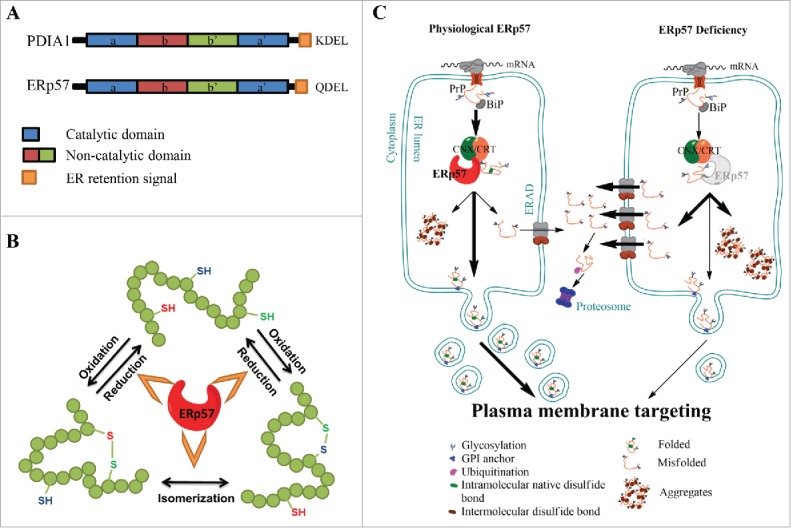
ERp57, the calnexin/calreticulin cycle and PrP biosynthesis. (A) ERp57/PDIA3/Grp58 and PDIA1 are members of the Protein Disulfide Isomerase (PDI) family. The domain structure is conserved in other members of the PDI family and is indicated using color code: in blue, catalytic domains a and a’, in red and green, the non-catalytic b and b’ domains, respectively and in orange, the ER retention signal. (B) Schematic representation of the protein folding activity of ERp57, comprising the oxidation, reduction and/or the isomerization of disulfide bonds in nascent glycoproteins. (C) Schematic representations of the biosynthesis of Prion protein (PrP) through the secretory pathway (left panel), emphasizing the impact of ERp57 deficiency (right panel). Left panel: Translation of PrP is directed toward the lumen of the endoplasmic reticulum (ER), where it receives a series of post-translational modifications, including glycosylation, intramolecular disulfide bond, and a GPI anchor that targets the protein to the plasma membrane. During its passing through the ER, PrP undergoes processing and quality control, to then mature at the Golgi apparatus for its sorting to the plasma membrane. Around 10% of the newly synthesized PrP is not correctly folded, and undergoes clearance through the ER-associated degradation (ERAD) pathway. Right panel: ERp57 deficiency alters PrP biosynthesis leading to alteration in the ratio of glycosylation, accumulation of large aggregates of the protein due to cross-links by aberrant intermolecular disulfide bonds, and reduction of steady-state levels of PrP protein related to decreased half-life of the protein, which lead to altered PrP trafficking.
A proteomic study identified ERp57 as the major protein up-regulated in sporadic CJD brain tissue and speculated that it may participate in the global cellular response to proteostasis impairment,17 a phenomenon that we confirmed in new variant CJD in addition to sporadic cases.11 PDIs are emerging as important stress factors in several PMDs in the nervous system, including PrDs, AD and ALS20 (Table 1). A physical interaction between PrP with ERp57 has been also demonstrated.10,21 Nevertheless, only few cell culture studies have explored the contribution of ERp57 to PrP misfolding.10
TABLE 1.
Role of Protein Disulfide Isomerases in neurodegenerative diseases.
| Disease | Effect | Reference |
|---|---|---|
| PrDs | • ERp57 and PDIA1 are upregulated in post mortem brain samples of CJD patients.• ERp57 levels correlate with disease progression in murine scrapie models.• ERp57 and PDIA1 expression protect against PrPSc toxicity in vitro.• PDIA1 is nitrosylated in PrDs models.• Inhibition of PDI activity reduces PrPSc replication in vitro. | 10,20,22 |
| AD | • PDIA1 inhibits Tau aggregation.• Pharmacological activation of ERp57 reduces clinical hallmarks of AD in mouse models.• ERp57 physically interacts with amyloid-β and prevents its aggregation.• Inhibition of ERp57 and PDIA1 protects against amyloid-β toxicity. | 20,27,33 |
| PD | • PDIA1 is upregulated in many toxicological models of PD.• PDIA1 is S-nitrosylated in brain tissue of PD patients. | 20 |
| ALS | • ERp57 and PDIA1 mutations are risk factors to develop ALS.• PDIA1 and ERp57 are upregulated in mouse models and post mortem tissue and CSF.• Upregulation of ERp57 in blood of patients is a possible biomarker of the disease• PDIA1 reduces mutant SOD1 aggregation in vitro.• PDIA1 co-localizes with SOD1, FUS and TDP-43 inclusion in ALS-derived tissue. | 20,29,31,32 |
| HD | • Inhibition of ERp57 or PDIA1 suppresses neuronal degeneration induced by mutant Huntingtin in vitro. | 20 |
A summary of selected literature related to protein disulfide isomerases in protein misfolding disorders (PMDs). Abbreviations used: PrDs, Prion related Disorders; AD, Alzheimer disease; PD, Parkinson disease; ALS, Amyotrophic Lateral Sclerosis; HD, Hungtington's disease. Most primary references can be found in.20
We have recently addressed this question using a combination of cell culture and mouse models, in addition to human post-mortem tissue derived from CJD patients from Chile.22 We demonstrated increased levels of ERp57 and PDIA1 in Purkinje cells of CJD cases, supporting previous reports.17 Using gain- and loss-of-function approaches in vitro and in vivo, we systematically studied the impact of ERp57 on the synthesis and folding of PrP, in addition to the susceptibility of cells to ER stress. Unexpectedly, steady state protein levels of PrP were directly dependent on ERp57 expression. These results were validated across several cell lines, involving an alteration in the half-life of the protein. Importantly, the impact of ERp57 on steady-state PrP levels was observed also on PrDs-linked mutants. Moreover, deficiency of ERp57 not only led to decreased levels of PrP, but also caused accumulation of large aggregates of the protein that were possibly cross-linked by aberrant intermolecular disulfide bonds. Notably, disulfide bonds have been previously suggested to mediate PrP misfolding and toxicity.23,24 These results correlated with a physical interaction between PrP and ERp57. We also validated these findings on a novel ERp57 transgenic mouse that we recently described25 in addition to a conditional knockout mouse for ERp57 in the central nervous system.22 Targeting ERp57 in vivo affected the ratio between glycosylated and non-glycosylated forms of PrP.
Despite initial predictions, neither ERp57 deficiency nor its overexpression altered the global susceptibility of cells to ER stress in vitro and in vivo on a mouse model. This is consistent with the fact that targeting XBP1 in the brain, a major UPR transcription factor, does not alter PrP replication or the neurodegenerative process.18 Moreover, the upregulation of PDIA1 and ERp57/Grp58 in scrapie prion-infected animals was not altered upon Xbp1 deletion.18 Our recent results may help reinterpreting previous findings in the puzzlingly relationship between ER stress and PrDs because up-regulation of ERp57 in patient tissue has been interpreted as part of the cellular mechanism to adapt cells to ER proteostasis disturbances. Rather, increased ERp57 levels may represent a specific response to tackle PrP folding and possibly enhance synthesis efficiency of a subset of ER client proteins that may be relevant to neuronal function.
Little is known about therapeutic targeting of PDIs in vivo in PrDs. Small molecules to inhibit PDI activity or gene therapy approaches may have specific applications depending on the disease. PDI inhibitors represent efficient anticancer compounds26 whereas they could reduce neurodegeneration in models of Alzheimer or Huntington´s disease.27,28 Five molecules where found that targeted ERp57 and PDIA1 and inhibited the canonical apoptosis pathway triggered by the PDIs. In vitro evidence indicated that the overexpression of ERp57 protects cells against the toxicity of misfolded PrP and pharmacological inhibition of PDIs increases PrPs replication rates.10 Monitoring PDI levels in body fluids may also serve as biomarkers in multiple neurodegenerative diseases associated with protein misfolding and ER stress as suggested in ALS.29,30 Thus, the consequences of targeting PDIs in PMDs may depend on the specific disease conditions. Given the rising relevance of PDIs in PMDs, and more specifically in PrDs, proof of concepts are needed using pharmacological or gene therapy approaches to validate PDIs as a possible therapeutic avenue for these disorders.
Together, our findings point toward a role of ERp57 in PrP biology (Fig. 1C). We propose that upregulation of ERp57 in the brain of CJD patients is a cellular response to buffer PrP maturation itself and not a reaction against global ER stress. Although PrP mutants do not seem to cause ER stress, at least in our hands, they can alter ER calcium content,14 which may translate into alterations to neuronal physiology. Interestingly, we recently reported that the overexpression of ERp57 in transgenic mice improves peripheral nerve regeneration after mechanical injury, but it did not affect the survival of dopaminergic neurons on a model of Parkinson´s disease,25 suggesting that ERp57 may underlay the differential vulnerability of specific cell types to disease-related perturbations. In addition, we recently reported that mutations in ERp57 and PDIA1 in ALS cases operates as a risk factors to develop the disease.31 Characterization of these PDIs variants uncovered a novel role of these foldases in promoting neuronal connectivity, a function that is lost in ALS contributing to the appearance of motor symptoms.32 Along these lines, some important questions arise: could ERp57 exert a protective role on a pathological context involving replication of misfolded PrP in vivo or other aggregation prone proteins? Or, could ERp57 directly rescue the toxic conformation of PrP? However it may be feasible that the upregulation ERp57 in CJD cases may enhance the accumulation of protease-resistant PrP due to increase availability of the PrPC substrate. The answer to these questions will reveal the potential of ERp57 as a possible therapeutic target to treat PrDs and other PMDs. Since ERp57 and other PDIs are also deregulated in many neurodegenerative diseases, the tools and animal models generated in our recent studies may represent unique instruments to determine the functional contribution of these ER foldases to PMDs including PD, ALS and AD.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
Funding
This work was funded by FONDECYT 11150579 (DBM), Millennium Institute No. P09-015-F, and FONDAP 15150012, the Frick Foundation, ALS Therapy Alliance 2014-F-059, Muscular Dystrophy Association 382453, CONICYT-USA2013-0003, Michael J Fox Foundation for Parkinson's Research, COPEC-UC Foundation, Ecos-Conicyt C13S02 and FONDECYT no. 1140549, Office of Naval Research-Global (ONR-G) N62909-16-1-2003 and CDMRP Amyotrophic Lateral Sclerosis Research Program (ALSRP) Therapeutic Idea Award AL150111 (C.H.). PR is supported by a CONICYT fellowship.
REFERENCES
- [1].Prusiner SB. Prions. Proc Natl Acad Sci 1998; 95(23):13363–83; http://dx.doi.org/ 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hetz C, Soto C. Stressing Out the ER: A Role of the Unfolded Protein Response in Prion-Related Disorders. Curr Mol Med 2006; 6(1):37–43; PMID:16472111; http://dx.doi.org/ 10.2174/156652406775574578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Soto C. Transmissible proteins: Expanding the prion heresy. Cell 2012; 149(5):968–77; PMID:22632966; http://dx.doi.org/ 10.1016/j.cell.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Abid K, Morales R, Soto C. Cellular factors implicated in Prion replication. FEBS Lett 2010; 584(11):2409–14; PMID:20412808; http://dx.doi.org/ 10.1016/j.febslet.2010.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Puig B, Altmeppen H, Glatzel M. The GPI-anchoring of PrP. Prion 2014; 8(1):11–8; PMID:24509692; http://dx.doi.org/ 10.4161/pri.27892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rudd PM, Merry AH, Wormald MR, Dwek RA. Glycosylation and prion protein. Curr Opin Struct Biol 2002; 12(5):578–86; PMID:12464308; http://dx.doi.org/ 10.1016/S0959-440X(02)00377-9 [DOI] [PubMed] [Google Scholar]
- [7].Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A. Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J 2001; 20(19):5383–91; PMID:11574470; http://dx.doi.org/ 10.1093/emboj/20.19.5383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 2014; 15(4):233–49; PMID:24619348; http://dx.doi.org/ 10.1038/nrn3689 [DOI] [PubMed] [Google Scholar]
- [9].Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol 2015; 17(7):1–10; PMID:25679028; http://dx.doi.org/ 10.1038/ncb3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hetz C, Russelakis-Carneiro M, Walchli S, Carboni S, Vial-Knecht E, Maundrell K, Castilla J, Soto C. The Disulfide Isomerase Grp58 Is a Protective Factor against Prion Neurotoxicity. J Neurosci 2005; 25(11):2793–802; PMID:15772339; http://dx.doi.org/ 10.1523/JNEUROSCI.4090-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J 2003; 22(20):5435–45; PMID:14532116; http://dx.doi.org/ 10.1093/emboj/cdg537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, et al.. Oral treatment targeting the unfolded protein response preventsneurodegeneration and clinical disease in prion-infected mice. 2013; 138(206):206ra138-206ra138; http://dx.doi.org/10.1126/scitranslmed.3006767 [DOI] [PubMed] [Google Scholar]
- [13].Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, et al.. Sustained translational repression by eIF2Į – P mediates prion neurodegeneration. Nature 2012; 485(7399):507–11; PMID:22622579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Torres M, Castillo K, Armisén R, Stutzin A, Soto C, Hetz C. Prion Protein Misfolding Affects Calcium Homeostasis and Sensitizes Cells to Endoplasmic Reticulum Stress. PLoS One 2010; 5(12):e15658; PMID:21209925; http://dx.doi.org/ 10.1371/journal.pone.0015658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hetz C, Castilla J, Soto C. Perturbation of Endoplasmic Reticulum Homeostasis Facilitates Prion Replication. J Biol Chem 2007; 282(17):12725–33; PMID:17329244; http://dx.doi.org/ 10.1074/jbc.M611909200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rane NS, Kang S, Chakrabarti O, Feigenbaum L, Hedge RS. Reduced Translocation of Nascent Prion Protein During ER Stress Contributes to Neurodegeneration. Dev Cell 2008; 15(3):359–70; PMID:18804434; http://dx.doi.org/ 10.1016/j.devcel.2008.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yoo BC, Krapfenbauer K, Cairns N, Belay G, Bajo M, Lubec G. Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt-Jakob disease. Neurosci Lett 2002; 334(3):196–200; PMID:12453628; http://dx.doi.org/ 10.1016/S0304-3940(02)01071-6 [DOI] [PubMed] [Google Scholar]
- [18].Hetz C, Lee A-H, Gonzalez-Romero D, et al.. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc Natl Acad Sci 2008; 105(2):757–62; http://dx.doi.org/ 10.1073/pnas.0711094105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Coe H, Michalak M. ERp57, a multifunctional endoplasmic reticulum resident oxidoreductase. Int J Biochem Cell Biol 2010; 42(6):796–9; PMID:20079872; http://dx.doi.org/ 10.1016/j.biocel.2010.01.009 [DOI] [PubMed] [Google Scholar]
- [20].Andreu CI, Woehlbier U, Torres M, Hetz C. Protein disulfide isomerases in neurodegeneration: From disease mechanisms to biomedical applications. FEBS Lett 2012; 586(18):2826–34; PMID:22828277; http://dx.doi.org/ 10.1016/j.febslet.2012.07.023 [DOI] [PubMed] [Google Scholar]
- [21].Watts JC, Huo H, Bai Y, Ehsani S, Jeon AH, Shi T, Daude N, Lau A, Young R, Xu L, et al.. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog 2009; 5(10):e1000608; PMID:19798432; http://dx.doi.org/ 10.1371/journal.ppat.1000608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Torres M, Medinas DB, Matamala JM, Woehlbier U, Cornejo VH, Solda T, Andreu C, Rozas P, Matus S, Muñoz N, et al.. The Protein-disulfide Isomerase ERp57 Regulates the Steady-state Levels of the Prion Protein. J Biol Chem 2015; 290(39):23631–45; PMID:26170458; http://dx.doi.org/ 10.1074/jbc.M114.635565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tompa P, Tusnády GE, Friedrich P, Simon I. The Role of Dimerization in Prion Replication. Biophys J 2002; 82(4):1711–8; PMID:11916832; http://dx.doi.org/ 10.1016/S0006-3495(02)75523-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yanai A, Meiner Z, Gahali I, Gabizon R, Taraboulos A. Subcellular trafficking abnormalities of a prion protein with a disrupted disulfide loop. FEBS Lett 1999; 460(1):11–6; PMID:10571052; http://dx.doi.org/ 10.1016/S0014-5793(99)01316-2 [DOI] [PubMed] [Google Scholar]
- [25].Castillo V, Oñate M, Woehlbier U, Rozas P, Andreu C, Medinas D, Valdés P, Osorio F, Mercado G, Vidal RL, et al.. Functional Role of the Disulfide Isomerase ERp57 in Axonal Regeneration. PLoS One 2015; 10(9):e0136620; PMID:26361352; http://dx.doi.org/ 10.1371/journal.pone.0136620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lovat PE, Corazzari M, Armstrong JL, et al.. Increasing melanoma cell death using inhibitors of protein disulphide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Research 2010; 68(13):5363–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tohda C, Urano T, Umezaki M, Nemere I, Kuboyama T. Diosgenin is an exogenous activator of 1,25D3-MARRS/Pdia3/ERp57 and improves Alzheimer's disease pathologies in 5XFAD mice. Sci Rep 2012; 2:1–11; http://dx.doi.org/ 10.1038/srep00535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hoffstrom BG, Kaplan A, Letso R, Schmid RS, Turmel GJ, Lo DC, Stockwell BR. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol 2010; 6(12):900–6; PMID:21079601; http://dx.doi.org/ 10.1038/nchembio.467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nardo G, Pozzi S, Pignataro M, Lauranzano E, Spano G, Garbelli S, Mantovani S, Marinou K, Papetti L, Monteforte M, et al.. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One 2011; 6(10):e25545; PMID:21998667; http://dx.doi.org/ 10.1371/journal.pone.0025545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis 2008; 30(3):400–7; PMID:18440237; http://dx.doi.org/ 10.1016/j.nbd.2008.02.009 [DOI] [PubMed] [Google Scholar]
- [31].Gonzalez-Perez P, Woehlbier U, Chian R-J, Sapp P, Rouleau GA, Leblond CS, Daoud H, Dion PA, Landers JE, Hetz C, et al.. Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene 2015; 566:158–65; PMID:25913742; http://dx.doi.org/ 10.1016/j.gene.2015.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Woehlbier U, Colombo A, Saaranen MJ, et al.. ALS-linked Protein Disulfide Isomerase Varians cause motor dysfunction. EMBO J, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Xu L-R, Liu X-L, Chen J, Liang Y. Protein Disulfide Isomerase Interacts with Tau Protein and Inhibits Its Fibrillization. PLoS One 2013; 8(10):e76657; PMID:24098548; http://dx.doi.org/ 10.1371/journal.pone.0076657 [DOI] [PMC free article] [PubMed] [Google Scholar]