

ABSTRACT
Amyloids are protein aggregates consisting of fibrils rich in β-sheets. Growth of amyloid fibrils occurs by the addition of protein molecules to the tip of an aggregate with a concurrent change of a conformation. Thus, amyloids are self-propagating protein conformations. In certain cases these conformations are transmissible / infectious; they are known as prions. Initially, amyloids were discovered as pathological extracellular deposits occurring in different tissues and organs. To date, amyloids and prions have been associated with over 30 incurable diseases in humans and animals. However, a number of recent studies demonstrate that amyloids are also functionally involved in a variety of biological processes, from biofilm formation by bacteria, to long-term memory in animals. Interestingly, amyloid-forming proteins are highly overrepresented among cellular factors engaged in all stages of mRNA life cycle: from transcription and translation, to storage and degradation. Here we review rapidly accumulating data on functional and pathogenic amyloids associated with mRNA processing, and discuss possible significance of prion and amyloid networks in the modulation of key cellular functions.
KEYWORDS: Amyloid, CPEB, Prion, Pub1, Sup35, S. cerevisiae, Tia1, yeast
INTRODUCTION
Pathological extracellular deposits, which were most likely amyloidoses, were described in liver, kidney and other human organs as early as in the 17th century.1,2 The term amyloid was coined in 1854, after the first chemical tests of extracellular waxy deposits in the brain and spinal cord: Rudolf Virchow detected their staining with iodine and postulated that they were composed of cellulose or starch.3 In just five years the prevailingly polysaccharide nature of such deposits was refuted. Based on their high nitrogen content, Friedreich and Kekule concluded that the deposits were primarily proteinaceous,4 but chose not to argue with the name. The iodine staining was eventually explained by the presence of polysaccharides in the form of glycosaminoglycans in most amyloids.5 Present-day evidence upholds that proteins determine the formation of amyloid deposits and remain their key components, and current chemical tests for amyloids are based on dyes that bind to amyloid-specific protein structures. These tests include apple-green birefringence in polarized light following staining with the Congo Red dye,6 or a shift of fluorescence spectrum upon binding of a fluorescent dye Thioflavine-T.7-9 These notable effects occur due to the presence of highly ordered amyloid fibrils where protein molecules form β-strands running perpendicular to the lateral axis of the fibril.10,11 Such architecture of amyloid fibrils also results in their characteristic “cross-β” pattern in X-ray diffraction,10 as well as an unusually high resistance to treatment with ionic detergents and,12-14 in some cases, with proteinases.15 The detailed structure of the fibers, and amino acids involved in the formation of β-strands, are known only for some amyloids, which were prepared in vitro from ectopically expressed and purified proteins. However, considering the frequent occurrence and fundamental similarity of amyloids, the term “amyloid” is also used, and the abovementioned structure is presumed, for a broad range of extracellular deposits in tissues, intracellular inclusions and protein aggregates satisfying at least one criterion of amyloids, as long as there is no evidence for an alternative structure.
To date, at least 32 proteins are known to form pathological amyloids, which have been associated with dozens of incurable diseases in humans and animals.16,17 However, the other side of the coin was revealed in 2000, when protective envelope of the egg of silk moth Bombyx mori18 and hydrophobins of basidiomycete Schizophyllum commune19 were proven to have amyloid properties. These studies demonstrated that amyloids can be not only pathogenic, but also functional, and were soon followed by a number of reports describing the biological importance of other amyloids. Eventually, functional amyloids were discovered in all domains of life. They participate in the formation of biofilms in Archaea,20 as well as in different species of Bacteria, for which at least six groups of functional amyloids have been identified.17,21 In Eukarya, amyloids possess a broad spectrum of functions including, in addition to the aforementioned, deposition of hormones,22 regulation of melanin polymerization,23 modulation of long-term memory,24 anti-viral response,25 and programmed necrosis.26
Unexpectedly, the path of amyloid research intersected with studies of infectious protein conformations, or prions. Extreme resistance of PrPSc to protease digestion was reported right upon the identification of this protein as the only component consistently co-purifying with the infectivity of transmissible spongiform encephalopathies.15,27 Furthermore, histopathological studies of brains of scrapie-infected animals and Creutzfeldt-Jakob disease patients revealed the accumulation of PrPSc in congophilic amyloid plaques.28,29 Finally, in vitro, PrP formed rod-like aggregates that exhibited apple-green birefringence upon binding Congo Red.30 With these characteristics being major hallmarks of amyloids, experimental evidence indicated that PrP was an amyloidogenic protein. Strikingly, while the infectious PrPSc was clearly associated with the aggregated amyloid state of the PrP protein, the non-prion form of PrP, PrPC, was soluble and non-amyloid. The explanation how amyloid structure could be so efficiently utilized by an infectious protein conformation was provided by the nucleation-polymerization model. This model postulates that prions appear through a nucleation event involving a conformational conversion and oligomerization, grow through the addition of protein monomers to the nucleus, with concurrent conformational conversion, and infect through the transmission of fragments of prion aggregates called seeds.31,32
Thus, once other prions were later discovered in Saccharomyces cerevisiae, Podospora anserina and, eventually, in higher Eukaryotes, the amyloid structure of prion conformations did not come as a surprise. The highest number of prions has been identified in fungi, and particularly in baker's yeast S. cerevisiae. While opinions still differ on whether the majority of yeast prions are functional epigenetic modifiers, egoistic elements or diseases, most of them do not have a significant negative effect on fitness and can be maintained in yeast populations for a long time, or even indefinitely.33,34 To date there are nine known amyloid-based yeast prions, [PSI+]35 (for which structural protein is Sup35),36 [URE3]37 (Ure2),36 [PIN+] (Rnq1),38,39 [SWI+] (Swi1),40 [OCT+] (Cyc8),41 [MOT3+] (Mot3),14 [ISP+] (Sfp1),42 [NUP100+] (Nup100),43 [MOD+] (Mod5),44 as well as two functional prion-like amyloids: [PUB1] (Pub1) and [PUB1 / SUP35] (Pub1 and Sup35).45 Also, the [HET-s] prion is an amyloid controlling heterokaryon incompatibility in filamentous fungus Podospora anserina.46,47 The only known prions that do not possess amyloid properties are [β]48 and [GAR+]49,50 from S. cerevisiae. For [β] the principle of self-propagation is completely different: this is a protease that propagates through covalent autoactivation. As for [GAR+], while the Pma1 and Std1 proteins have been associated with the formation of this prion, the involvement of other proteins and an amyloid structure cannot be excluded. Thus, it is currently unclear to what extent prions and amyloids should be considered as two overlapping sets, or if all prions not functioning through covalent automodification are amyloids.
The β-sheet-based structure of amyloid fibrils engenders great diversity both in pathogenic and functional aspects. It should be acknowledged that it is still not completely clear what makes a protein amyloidogenic, but some conclusions can already be drawn from studies of amyloidogenic sequences. The amyloid-forming proteins identified to date possess regions with “unusual” amino acid composition. Such regions, depending on the method used for their prediction, are called “compositionally biased regions” (CBRs)51 or “low complexity regions” (LCRs).52 In essence, the term CBR implies that such region is rich or poor in particular residues compared to the average occurrence frequency of these residues in the proteome.51 So far two types of CBRs have been described for amyloid-forming proteins: (i) sequences rich in glutamine (Q) and/or asparagine (N);51,53 and (ii) sequences rich in hydrophobic and non-polar residues such as I, W, F, Y, L and V.54,55 The term LCR implies that a region contains little diversity in its amino acid composition.52 To a certain extent, low complexity is an expected consequence of significant enrichment with one or several amino acids that reduces the representation of other amino acids. The following classification of LCRs can be suggested in connection with amyloid formation: (i) LCRs lacking, or exhibiting only loose clustering of overrepresented amino acids; (ii) LCRs with extended interrupted or uninterrupted runs of homo-amino acid repeat tracts; (iii) LCRs with periodic sequences, such as tandem oligopeptide repeats. CBRs / LCRs affect amyloid formation in two different ways: (i) upon initial protein folding they maintain the region of the protein as intrinsically disordered and thus available for a conformational switch, and (ii) they promote amyloid formation through the formation of intermolecular β-strands. The first is best achieved by CBRs / LCRs rich in polar and charged residues, which increase the solubility of the protein. Amino acid composition of such regions appears to be more critical than the exact amino acid sequence.56 The second is facilitated by monotonous and repetitious sequences, including those rich in hydrophobic residues,57 and in this case the position of each residue is very important: the same residue may either promote, or block the formation of amyloid, depending on the context.58 Notably, analysis of amyloidogenic proteins reveals that some of them carry both types of CBRs / LCRs.59
Computational prediction of amyloid properties is efficient for short peptides. The false positive rate for full-length proteins is much higher,60 although some recently developed algorithms provide up to 70% of true positive predictions for full-length proteins.61 The difference in the efficiency of predictions is because in real proteins amyloidogenic regions are interspersed by non-amyloidogenic ones in primary sequences. Currently, there are no reliable algorithms that analyze the interplay of amyloidogenic sequences with other regions of a protein and determine the ability or inability of an amyloidogenic region to drive aggregation under physiological conditions.
Noteworthy, amyloidogenic regions can exhibit complex organization, where one protein may contain not one, but numerous amyloidogenic determinants. For example, Rnq1, a structural protein for the [PIN+] prion,38 has four Q/N-rich regions. Each of these regions alone can promote the aggregation of Rnq1 in vitro, and one common Q/N region is sufficient for the transmission of the prion state between Rnq1 fragments, though the overall conformation of the [PIN+] prion is determined by the cooperative action of all four determinants.62 It has also been suggested that Rnq1 encompasses non-Q/N-rich amyloidogenic regions, both interspersing the Q/N-rich determinants and located outside of the prion domain, and that these regions differentially affect the maintenance of [PIN+] variants.63 Analogously, the prion domain of the CPEB3 protein carries two aggregation domains with non-identical roles in amyloid formation, which are separated by a module regulating aggregation by affecting CPEB3 interaction with the actin cytoskeleton.59
One intriguing feature of the amyloid-forming proteins identified to date is a significant overrepresentation of mRNA-processing proteins. This peculiarity was first noted for Q/N-rich proteins of the yeast Saccharomyces cerevisiae,53 and was later upheld for confirmed prions. Indeed, nine prion- and amyloid-forming proteins of S. cerevisiae and at least 13 amyloid-forming proteins of higher eukaryotes are known to possess mRNA-processing functions. In some cases, the amyloid state of these proteins is neutral or functional, in others – pathogenic. In this review we analyze biological diversity of amyloids formed by proteins involved in mRNA processing, summarize their functional and pathological roles, and discuss a possible significance of prion and amyloid networks for the modulation of key cellular processes. Data on biological diversity of such amyloids, their functions, and processes, in which they are involved, are summarized in Table 1 and Figure 1. Although many amyloid-forming proteins affect mRNA processing in many ways, for the purpose of this review we tried to group them according to the stages of the mRNA life cycle in which they are mostly involved: (i) transcription, (ii) mRNA turnover, and (iii) translation.
TABLE 1.
RNA-modulating prions, amyloids, and amyloidogenic proteins
| Gene / Protein | Amyloid (Prion)* | Functions of the protein** | Effects of prion or amyloid state | Organism | Compositionally biased regions*** |
|---|---|---|---|---|---|
| URE2 / Ure2 | [URE3] | Transcriptional regulator of the nitrogen catabolism | Blocks interaction of Ure2 with Gln380 that causes preference of poor nitrogen sources | S. cerevisiae | N, Q/N |
| SWI1 / Swi1 | [SWI+] | Transcriptional regulator, subunit of SWI/SNF chromatin remodeling complex | Inhibits growth on the media with non-fermentable carbon sources,40 provides resistance to benomyl14 | S. cerevisiae | A, N, Q, Q/N, T |
| SUP35 / Sup35 | [PSI+] | Translation release factor eRF3, mRNA decay | Reduces efficiency of translation termination178 | S. cerevisiae | E, G, K, Q, Q/N, Y |
| MOT3 / Mot3 | [MOT3+] | Transcription factor modulating mating, carbon metabolism, and stress response | Regulates flocculation; induction by ethanol and elimination by hypoxia85 | S. cerevisiae | A, H, N, P, Q, Q/N |
| CYC8 / Cyc8 | [OCT+] | Transcriptional repressor and activator, subunit of Cyc8-(Ssn6)-Tup1 complex | Inhibits growth of the strains bearing cyc1 deletion on the media with non-fermentable carbon source lactate41 | S. cerevisiae | A, E, P, Q/N, Q |
| SFP1 / Sfp1 | [ISP+] | Transcription regulator of genes encoding ribosomal proteins; regulates response to nutrients and stress | Antisuppression of nonsense mutations;87 enhances resistance to cycloheximide and paromomycin42 | S. cerevisiae | A, D, H, N, Q, Q/N, T |
| PUB1 / Pub1 | [PUB1] | Stress granule assembly, 3′UTR mRNA-binding | Unclear; aggregates co-localize with P-bodies / stress granules45 | S. cerevisiae | M, N, Q, Q/N |
| PUB1 and SUP35 / Pub1 and Sup35 | [PUB1 / SUP35] | Stress granule assembly / Translation release factor eRF3 | Formation of microtubule-associated cytoskeleton-associated complex carrying translational machinery and important for maintaining the integrity of the microtubular cytoskelton45 | S. cerevisiae | E, G, K, M, N, Q, Q/N, Y |
| TIA1 / TIA1 | [TIA1] | Stress granule assembly, 3′UTR mRNA-binding | Amyloid-like fibrils involved in stress granule assembly111 | M. musculus | Q |
| NUP100 / Nup100 | [NUP100+] | Subunit of nuclear pore complex; nucleocytoplasmic transport | Unknown, phenotypic manifestation is absent43 | S. cerevisiae | G, N, Q, Q/N, S, T |
| TP53 / p53 | p53, mutant | Transcriptional repressor | Amyloid-like oligomers and fibrils associated with carcinogenesis104,105 | H. sapiens | A, P |
| HTT / huntingtin | huntingtin, mutant | Transcriptional regulator | Pathological amyloid-like inclusions associated with Huntington's disease91 | H. sapiens | D, L, P, Q, Q/N, V |
| ATN / atrophin-1 | atrophin-1, mutant | Transcriptional repressor | Pathological amyloid-like inclusions associated with dentatorubral pallidoluysian atrophy94 | H. sapiens | D, E, G, H, P, Q, Q/N, R, S |
| TNRC6A / TNRC6A | TNRC6A | RNA-binding, component of P-bodies, involved in the regulation of post-transcriptional gene silencing through the RNA interference | Fibrils in P-bodies, function is unclear120 | H. sapiens | G, K, N, P, Q, Q/N, S, T, W, |
| DDX6 / DDX6 | DDX6 | RNA helicase found in P-bodies and stress granules, involved in translation suppression and mRNA degradation | Fibrils in P-bodies, function is unclear121 | H. sapiens | Q |
| TDP43 / TDP43 | TDP43, mutant | Transcriptional repressor, component of stress-granules | Pathological inclusions associated with frontotemporal dementia and amyotrophic lateral sclerosis127-129 | H. sapiens | G, N |
| FUS / FUS | FUS, mutant | RNA-binding, component of hnRNP-complex | Pathological inclusions associated with amyotrophic lateral sclerosis and polyglutamine diseases131,132 | H. sapiens | G, Q, R, S, Y |
| HNRNPA1 / hnRNPA1 | hnRNPA1, mutant | RNA-binding, involved in the packaging of pre-mRNA into hnRNP particles, transport of poly-A mRNA from the nucleus to the cytoplasm | Pathological, multisystem proteinopathy-associated intracellular amyloid-like inclusions136 | H. sapiens | G |
| HNRNPA2B1 / hnRNPA2B1 | hnRNPA2B1, mutant | RNA-binding, associated with pre-mRNAs in the nucleus | Pathological, multisystem proteinopathy-associated intracellular amyloid-like inclusions136 | H. sapiens | G, Y |
| CPEB / CPEB | CPEB | RNA-binding, binds U-rich sequence located in the 3′ untranslated regions of mRNAs, thus either promoting, or inhibiting translation | Functional amyloid involved in the control of long-term memory; induction by 5HT24,159 | A. californica | P, Q, Q/N, S |
| orb2 / Orb2A | Orb2A | see above (CPEB) | see above (CPEB); induction by dopamine, octopamine, or tyramine161,169 | D. melanogaster | G, H, M, Q, Q/N, S |
| CPEB3 / CPEB3 | CPEB3 | see above (CPEB) | see above (CPEB); induction by dopamine, glutamate, or glycine59,162,163 | M. musculus | A, P, Q, Q/N |
Column indicates the special designation of amyloid (or prion, in the case of yeast prions).
Based on data from the NCBI Gene database (http://www.ncbi.nlm.nih.gov/gene/), Saccharomyces Genome Database (http://www.yeastgenome.org/), and cited studies.
Overrepresented amino acids are shown. CBRs were predicted by SARP algorithm,179 and the probability thresholds were set to 10−6 for single residue CBRs and 10−12 for multiple residues CBRs to reduce false positives. CBRs for multiple residue were predicted for the Q/N pair only.
FIGURE 1.
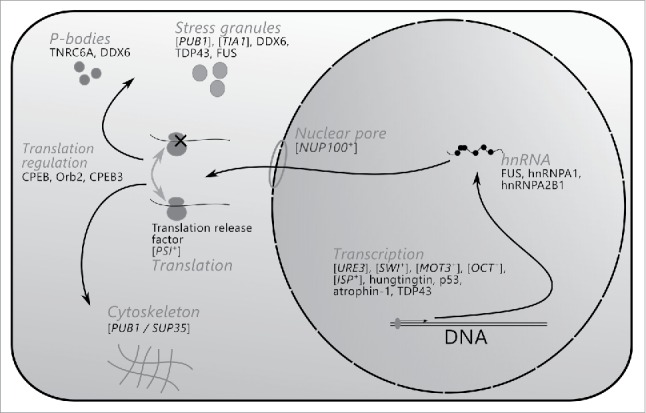
RNA-modulating prions and amyloids, and cellular processes in which they are implicated. Shown are cellular processes or protein complexes, which are associated with corresponding prions and amyloids. Arrows connect consequent stages of the mRNA life cycle.
I. Prions, Amyloids, and The Modulation of Transcription
Considerable data on the modulation of transcription by amyloid-forming proteins has been obtained for prions of the yeast S. cerevisiae. Currently, the list of prions for which prion-forming proteins possess an activity of transcriptional regulators includes [URE3], [SWI+], [MOT3+], [OCT+], and [ISP+]. In addition, the [NSI+] prion-like determinant,64,65 for which the prion-forming protein is unknown, was shown to slightly modulate the levels of mRNAs of several genes.66,67 Prion-forming proteins of the corresponding prions serve different roles in transcriptional regulation. Ure2 is a component of a system regulating nitrogen catabolism through the modulation of the localization of the GATA transcriptional activator Gln3.68 Swi1 is a subunit of SWI/SNF chromatin remodeling complex, which is required for the transcription of many genes including those controlling metabolism of sugars and mating type switching.69 Mot3 is a transcriptional repressor and activator involved in the regulation of mating, carbon metabolism, stress response, and controls a complex cell wall remodeling program during the adaptation to anaerobiosis.70 Cyc8, the prion-forming protein of [OCT+], is a component of Cyc8/Tup1 complex that represses the transcription of over 150 genes.71,72 Interestingly, while Mot3 contributes to the recruitment of the Cyc8/Tup1 complex to repress transcription, Cyc8/Tup1 is involved in the recruitment of the SWI/SNF complex to activate transcription.73 Finally, Sfp1 is a transcriptional activator of the genes coding for ribosomal proteins.74
All prions within the aforementioned group form aggregates with amyloid properties. However, an extensive characterization of amyloid fibrils including the Congo Red assay, electron microscopy and solid state NMR structural analysis of in vitro-made amyloid has been completed for Ure2 only.75-77 Swi1 and Mot3 were shown to form SDS-resistant aggregates in the yeast cytosol,14 and amyloid fibers assembled in vitro from the ectopically expressed protein fragments corresponding to prion-forming domains of these proteins caused a shift in fluorescence spectrum when stained with Thioflavin-T.14,78 For Cyc8 fused to YFP, fluorescent microscopy revealed formation of aggregates in [OCT+] cells.41 Currently, there is no evidence to suggest that they are detergent-resistant,41 although the Q/N-rich region of Cyc8 forms SDS-resistant aggregates.14 Sfp1 forms aggregates in the nuclei of [ISP+] cells. Such localization is unique among known yeast prions.42 Intriguingly, the Q/N rich region of Gln3, a protein interacting with Ure2 and playing the central role in the [URE3] manifestation, forms amyloid aggregates,14 although this propensity was not tested for the full-length protein.
Prions were originally described as deleterious factors.79 Accordingly, when prions were discovered in yeast, it was postulated that proteins in the prion conformation are inactivated, and the phenotype of prion state corresponds to a deletion or an inactivating mutation in the gene encoding the prion-forming protein.36 Initial experimental evidence for [URE3] was consistent with this notion. However, further in-depth studies of [URE3] and recent studies of [SWI+], [MOT3+], and [ISP+] indicate that the situation is not that simple. Formation of [URE3] is known to prevent the interaction of Ure2 with Gln3,80 which, through a cascade of transcriptional factors,81,82 makes yeast to catabolize poor nitrogen sources, such as ureidosuccinate, in the presence of rich nitrogen sources. The original phenotypic assay for [URE3] was based on this disruption of Gln3 regulation; and thus, the formation of [URE3] could be interpreted as inactivation of Ure2.37 However, in addition to transcriptional regulation, Ure2 possesses glutathione S-transferase activity,83 and this function persists in both [URE3] and [ure-0] states.84 Analogously, the appearance of [SWI+] leads to the inability to grow on non-fermentable carbon sources, which is characteristic of SWI/SNF inactivation. However, unlike the swi / snf mutants, yeast bearing the [SWI+] prion are not sensitive to 0.3 M LiCl or 1 M NaCl.40 [MOT3+] causes several phenotypes, including flocculation, that are not observed upon the deletion of the corresponding gene.85 Most strikingly, Sfp1 preserves its function of the transcriptional regulator in [ISP+] strains.86 Furthermore, while the SFP1 deletion leads to slow growth and smaller cell size, prionization of Sfp1 does not.42 [ISP+] even has the opposite effect on fitness: it increases growth rate relative to the [isp−] strain.87 So, prionization of transcriptional regulators can not only inhibit, but also modulate their functional activities.
Some of the prions discussed in this section also provide a beneficial resistance to various drugs and compounds: [SWI+] - to benomyl, [MOT3+] - to calcofluor white and Congo Red,14 and [ISP+] - to cycloheximide and paromomycin, but it is unclear whether these benefits are ever used by wild yeast.42
So far, of all these prions, the presence in natural yeast isolates has only been reported for [MOT3+]. 43 Notably, a specific mechanism of induction and elimination driven by changes in environmental conditions has been described for this prion: increasing ethanol concentration promotes [MOT3+] formation, while hypoxia eliminates it by repressing the expression of the MOT3 gene.85 Such changes in environmental conditions do occur during fermentation, and, taking into consideration that the appearance of [MOT3+] causes FLO11-mediated flocculation, which is important for survival, this prion is apparently beneficial, although, in fact, this has not been confirmed outside of the laboratory.85
[URE3] does not provide any apparent advantages to yeast, and some strains or conformational variants of this prion significantly inhibit vegetative growth.88 Also, [SWI+] manifests not only beneficial, but also harmful phenotypes: it inhibits vegetative growth on media containing non-fermentable carbon sources.40 This might mean that these prions are egoistic elements or even that they are harmful. However, we cannot exclude the possibility that these prions provide a survival advantage under certain conditions that occur rarely and which we do not yet know, and thus act as bet hedging prions.33
In mammals, mutant huntingtin and p53 are examples of potentially harmful amyloids formed by proteins involved in transcription. Mutant huntingtin protein containing an expanded poly-Q tract in its first exon89 forms intranuclear and cytoplasmic inclusions90 that have some amyloid characteristics91 and causes a lethal neurodegenerative disorder known as Huntington's disease.92,93 A hallmark of Huntington's disease and a set of disorders caused by the expansion of poly-Q tracts in several other proteins, such as ATN (atrophin-1), is a significant change in the transcription of different genes that is presumably associated with pathogenesis.94 Currently, the molecular mechanism, through which these changes occur, is unclear, but several interesting hypotheses are worth mentioning. One is based on the finding that inclusions of huntingtin and atrophin-1 sequester some poly-Q-containing transcription factors:95-97 it was suggested that such sequestration might interfere with the functional activity of the corresponding transcription factors.94 This model is still under consideration, although incomplete recruitment of the transcription factors by the inclusions98 is viewed as an argument against the sequestration hypothesis. Other explanations do not implicate inclusions formed by proteins with poly-Q expansions. For example, mutant and wild-type huntingtin have been shown to have direct DNA-binding sites, and these sites are different.99 Therefore, mutant huntingtin could perturb normal transcription by preventing or modulating the binding of normal transcription factors to DNA. Alternatively, there is evidence that transcriptional deregulation could be due to the presence of mRNAs carrying the expansions of CAG codons coding for the poly-Q stretches.100
The tumor suppressor p53, in its normal form, is a tetrameric transcription factor that blocks proliferation by inducing cell-cycle arrest.101 Mutations in TP53, the p53-encoding gene, often result in tumor progression,102 which occurs either due to the functional inactivation of p53, or the sequestration of the wild type protein by its dominant-negative mutant form.103 In the latter case, such a dominant-negative form can be manifested by amyloids of mutant p53.104 The exact role of p53-containing amyloids in carcinogenesis is currently unclear, though these amyloids are detected in samples of breast cancer105 and co-localize with wild-type p53 in several cell lines.104 In summary, there is evidence that proteins involved in transcriptional regulation form amyloids in mammals. Amyloidogenicity of these proteins is due to mutations in the corresponding protein-encoding genes, and amyloids are hypothesized to be pathogenic and cause toxicity via the loss-of-function effect, which can be either direct or mediated by sequestration of other proteins.
II. Prions, Amyloids, and mRNA Turnover
Several recent studies find that amyloidogenic proteins are widespread in the ribonucleoprotein (RNP) granules found in eukaryotic cells, from yeast to mammals.106-109 These granules contain non-translating mRNAs and are involved in the control of all stages of mRNA turnover, including storage and degradation. The structure of these granules is highly dynamic, as their protein and RNA composition undergoes significant changes depending on the particular needs of the cell. Two major types of RNP granules, stress granules and processing bodies (P-bodies), are formed in most cells. Stress granules are absent in normal physiological conditions, but assemble rapidly upon the inhibition of translation initiation, which usually occurs in response to various stresses. After the termination of a corresponding stressful condition, stress granules disassemble (for a review see refs.109,110). Stress granules comprise a number of proteins, many of which contain CBRs / LCRs and are able to form hydrogels containing amyloid-like aggregates in a cell-free system.108 One of these proteins, Tia1, carries three RNA-binding domains in the N-terminal part and a Q-rich CBR in the C-terminal part. The RNA-binding domains of Tia1 recognize the U- and A-U-rich motifs in the 3′UTRs of mRNAs recruited into stress granules,78 and the Q-rich region is required for the recruitment of Tia1 into stress granules, and alone forms protease-resistant polymers in the Hsp70-dependent manner in vivo.111,112 In vitro, Tia1 forms fibers that, according to Congo Red and Thioflavine-T tests, as well as EM and X-ray analysis, have amyloid structure.45,113 Moreover, a recent study in yeast demonstrated that full-length mouse Tia1 forms heritable SDS-resistant prion-like cytoplasmic aggregates that co-localize with P-bodies / stress-granules.45 Pub1, a yeast ortholog of Tia1, also forms detergent-resistant oligomers and visible prion-like aggregates that co-localize with P-bodies / stress-granules and, like most yeast prions, require the Hsp104 chaperone for their formation.45,114
The second type of ubiquitous RNP granules is processing bodies (P-bodies). P-bodies interact with stress granules and promote their assembly.115,116 In contrast to stress granules, P-bodies are constitutively present in cells, although their number and size are increased in response to stress.115 P-bodies encompass untranslated mRNAs and a complex of enzymes involved in mRNA decapping, deadenylation and degradation. In addition to mRNA degradation, P-bodies act as dynamic sites for different stages of mRNA processing, including translation repression and storage.117 In yeast, several P-body proteins contain Q/N-rich regions that can aggregate in vivo.118,119 In mammals, P-bodies contain fibrils of TNRC6A (GW182)120 and DDX6 (RCK, p54)121 proteins; both of which also have CBRs / LCRs.120,121
Even though the presence of multiple amyloidogenic proteins with CBRs / LCRs in P-bodies and stress granules led to the hypothesis that functional amyloids are implicated in the biogenesis of these RNPs,45,111,118,119 so far there is no proof that these proteins are present in RNPs in the amyloid state.122 Indeed, it appears that initial formation of these RNPs is enthalpy driven, depends on multivalent interactions involving both CBRs/LCRs and RNA-binding domains of proteins and RNAs, and leads to the formation of large RNP complexes in extremely dynamic phase-separated liquid-like droplets. Only at subsequent stages these droplets maturate into more stable structures that are more likely to incorporate functional amyloid.122-124 In the framework of this model formation of amyloid might also manifest an alternative, strictly pathogenic pathway.122,123 Consequently, the lack of negative selection toward the formation of amyloid by wild type or single-mutation proteins is explained either by the functionality of the amyloid state in the granules or exclusively by the need to maintain CBRs / LCRs prone for multivalent interactions.
A variety of aggregation-prone proteins associated with RNP-granules have been shown to form amyloid aggregates as a result of mutations. In humans these aggregates are hallmarks of multiple diseases. For example, a mutation in the Tia1 protein has recently been linked to Welander distal myopathy.125 Also, for TDP-43, which is involved in different stages of mRNA metabolism and reversibly incorporates into stress granules via direct interactions with some RNAs and proteins including Tia1,126 mutations, most of which are located in the Q/N-rich LCR, lead to the formation of irreversible intracellular inclusions.127 These inclusions are associated with several neurodegenerative disorders, such as frontotemporal lobar dementia and amyotrophic lateral sclerosis128 and, in some cases, possess amyloid-like properties.129,130 Also, wild type TDP-43 is a major component of inclusions in sporadic amyotrophic lateral sclerosis. Another example of pathologic inclusions associated with a stress granule-associated protein are formed by the RNA-binding protein FUS. Mutations in the FUS-encoding gene lead to the formation of FUS inclusions in amyotrophic lateral sclerosis131 and are also observed in diseases caused by the expansion of glutamine-encoding repeats in other proteins.132 Whether these inclusions are related to amyloidogenesis in vivo is still unclear,133 although FUS is capable of forming amyloid-like polymers in vitro.108 One of interesting hypotheses for both TDP-43- and FUS-associated amyotrophic lateral sclerosis proposes that stress granules are the nucleation sites for the pathologic aggregation of these proteins.134,135 Alternatively, pathologic aggregation of TDP-43 and FUS may represent off-pathway events in the formation of stress granules, essentially a loss of both nuclear and cytoplasmic functions for TDP-43 and FUS.135 Yet another example of disease-associated aggregation involves the mutations in hnRNPA2B1 and hnRNPA1 RNA-binding proteins that lead to formation of multisystem proteinopathy-associated inclusions with fibrillary properties.136 Disease-linked mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 make possible the formation of a steric zipper, which produces self-complementary β-sheets that comprise the spine of amyloid fibrils accumulating in non-RNP inclusions. This diverts these proteins from physiological folding trajectories connected with the RNP granule assembly and accelerates hnRNPA1 and hnRNPA2/B1 misfolding.136,137 A low-complexity region of Nab3, a yeast hnRNP homolog, is prone to form amyloid filaments in vitro.138 Overall, the RNP granules are highly dynamic structures comprising a repertoire of proteins with low-complexity domains that may either participate in the life cycle of these granules through promiscuous interactions or amyloid formation, or form pathogenic mutation-linked amyloid-like inclusions.
III. Translation-Coupled Prions and Amyloids
Undoubtedly, the best-studied prion in S. cerevisiae is [PSI+], which is formed by the translation release factor eRF3 (Sup35). Initially discovered as the non-chromosomal allosuppressor of super-suppressor tRNAs,35 this determinant was proposed36 and proven to be a prion form of Sup35 in a series of studies (for reviews see refs.139-141). Currently, Sup35 is known to be implicated in a number of functions,140 some of which are prion-associated. The C-terminal domain of Sup35 acts as a translational release factor eRF3,142-144 while the N-terminal domain is prion-forming145-148 and capable of assembling into bona fide amyloid fibrils.149,150 Interestingly, the Q/N-rich N-terminal prion-forming domain of Sup35 modulates mRNA decay via the regulation of deadenylation,151 although the influence of the [PSI+] prion on the degradation of mRNA was not shown. [PSI+] not only causes translational readthrough, but also acts as frameshift suppressor, thereby modulating the cellular content of polyamines.152 The appearance of [PSI+] is enhanced by different stressful conditions,153 and by the presence of another yeast prion, [PIN+],38,154 which is relatively widespread in yeast populations.155,156 Interestingly, [PSI+] exists in a number of dynamically changing variants,157 among which there are beneficial,156 neutral, and harmful88 in particular conditions. Moreover, the composition and the size of the [PSI+] aggregates, the characteristic feature of [PSI+] variants, determines to what extent [PSI+]-associated Sup35 retains its functionality.158
Also, Sup35 interacts with Pub1, an mRNA-binding protein, and this interaction occurs via the Q/N-rich LCRs present in both proteins.45 The tubulin-associated protein complex containing Sup35 and Pub1 in oligomeric SDS-resistant prion-like states normally exists in yeast cells and is involved in the maintenance of the integrity of the microtubular cytoskeleton. This complex contains tubulin mRNA and components of the translation machinery, which suggests that it is likely implicated in local cytoskeleton-associated protein synthesis.45 Thus, Sup35 can exist in the cell in at least three different states: as a functional monomeric translation termination factor eRF3, monoprotein prion [PSI+], which apparently acts as an epigenetic phenotypic modulator through a bet-hedging mechanism, and a functional two-protein [PUB1 / SUP35] prion-like assemblysome. Furthermore, Pub1 is also associated with two self-propagating structures, the one associated with P-bodies / stress granules and not involving Sup35, and the [PUB1 / SUP35] structure.
Remarkable examples of the translation-coupled functional amyloids in multicellular organisms are neuron-specific forms of the cytoplasmic polyadenylation element binding protein (CPEB), ApCPEB, Orb2, and CPEB3 in the mollusk Aplysia californica,24,159,160 fruit fly Drosophila melanogaster,161 and mouse,59,162,163 correspondingly. CPEB represents a family of RNA-binding proteins that bind U-rich sequences called CPE elements, which are located in the 3′ untranslated regions of a number of cellular mRNAs. Such binding either promotes, or inhibits translation.164 Neuron-specific isoforms of CPEB have been hypothesized to provide local protein synthesis in the synapses, which is important for the formation of the so-called “synaptic tag” that stabilizes long-term functional and structural changes in the synapse.24,159,165-167
Current models for ApCPEB postulate that in a naïve synapse it persists in a monomeric form and acts as a repressor of translation. Stimulation of the neurons with serotonin leads to the formation of multimers of ApCPEB. Such induction of the formation of the prion state in response to a physiologically relevant stimulus represents a key feature of a functional prion. Furthermore, in neurons, multimers of ApCPEB self-propagate and exist in the same physiological conditions as the monomers. The ApCPEB multimers possess amyloid properties and are, in fact, an active form of ApCPEB. Indeed, their binding with multimer-specific antibodies destabilizes the maintenance of long-term facilitation.24 Moreover, studies in yeast revealed that an amyloid isoform of ApCPEB possesses multiple hallmarks of a bona fide prion.160,168
Like ApCPEB, Orb2 exists in two states in the brain of D. melanogaster, monomeric and amyloid-like oligomeric. Multimerization of Orb2 is induced following stimulation with dopamine, octopamine, or tyramine, and persists up to 24 h.161 The Orb2 locus encodes six proteins, only two of which, Orb2A and Orb2B, are CPEB orthologs containing the same prion domain. Orb2A is shorter and, being fused to GFP, forms fluorescent foci, while Orb2B does not form aggregates alone. Endogenous oligomers consist of both, Orb2A and Orb2B, although Orb2A is critical for the involvement of Orb2B in the aggregated state. Mutational inactivation of Orb2A oligomerization leads to the impairment of long-term memory.161 Moreover, Orb2A stability and oligomerization was observed to be controlled by the protein network consisting of Lim kinase, protein phosphatase 2A, and Tob transcription regulator.169
Finally, three recent manuscripts analyzed aggregation and self-perpetuation of the mouse ortholog of ApCPEB, CPEB3.59,162,163 Studies of an ectopically expressed purified protein confirmed the ability of CPEB3 to form typical amyloid fibers with a characteristic birefringence upon Congo Red staining. Studies in yeast established that CPEB3 can act as a bona fide prion, i.e. a heritable protein conformation. Also, studies in yeast and mice uncovered some details of CPEB3 prionization: both CPEB3 expression and its aggregation are promoted by synaptic stimulation and occur upon de-SUMOylation of the CPEB3 protein and through its interaction with the actin cytoskeleton. Noteworthy, in agreement with the hypothesis that the CPEB3 prion acts as a synaptic tag in the maintenance of long-term memory persistence, CPEB3 aggregation not only self-perpetuates, but also supports the translation of proteins essential for the functioning of the synapse, such as GluR receptors, as well as proteins essential for CPEB3 regulation, such as actin and SUMO.59,162,163 Overall, CPEB proteins form functional amyloids with prion-like properties that control long-term memory and possess a complex regulatory network consisting of chemical and protein regulators.
FROM INDIVIDUAL AMYLOIDS TO FUNCTIONAL AMYLOID NETWORKS: PIECES OF A PUZZLE
The diversity of amyloids formed by RNA-binding proteins and proteins involved in mRNA processing and regulation identified to date suggests that amyloid formation is an important component of the key cellular processes related to all stages of mRNA metabolism. In the field of RNA regulation, amyloid-forming proteins participate in transcription, pre-mRNA splicing, mRNA transportation, storage, translation and degradation (Fig. 1). The feature that unites all RNA-modulating amyloids is the presence of CBRs / LCRs. Indeed, all proteins listed in Table 1 have such regions, and in 19 of 22 listed proteins they are rich in Q and/or N residues (Table 1, right column). This is not surprising, as the fact that Q/N-rich CBRs / LCRs contribute to amyloid formation is known for a long time.51,53 The most representative group illustrating this compositional bias are yeast prions, almost all of which are Q/N-rich. In addition, the Q/N domains of approximately 50 yeast proteins form detergent-resistant aggregates in vivo when overproduced, while 17 of them demonstrate prion-like properties.14
A number of studies also demonstrated that Q/N-enrichment is a common feature of amyloids in other organisms, including humans. The first group of such amyloids is represented by pathological amyloids, which are formed due to mutations. Such mutations can either be single-residue substitutions (as in the case of FUS, TDP-43, and HNRNPA) or occur through recombination-based mechanisms leading to the expansion of poly-Q-encoding repeats (huntingtin, atropnin-1, etc.). The second group includes functional amyloids, for which aggregation is unrelated to mutations and begins in response to specific stimuli (e.g. synaptic stimulation), or is associated with the assembly of specific intracellular structures (e.g., microtubular cytoskeleton or, possibly, stress granules). Most proteins forming the Q/N-rich amyloids and, in the case of functional amyloids, the amyloids themselves, are functionally related to the metabolism of RNA. Moreover, analysis of the functions of the proteomic subset of Q/N-rich proteins in S. cerevisiae demonstrates that functional groups overrepresented in this subset in comparison with the entire proteome, are related to transcription and RNA-binding (Table 2). This poses an important question: why does Q/N-enrichment strongly correlate with RNA-modulating activities?
TABLE 2.
Functional categories* overrepresented in the subset of Q/N-rich proteins** in comparison with the entire proteome of S. cerevisiae
| Function | Total number of proteins in category | Number of Q/N-rich proteins | The level of significance |
|---|---|---|---|
| GO:0004674 protein serine threonine kinase activity | 128 | 29 | 0.05 |
| GO:0004713 protein tyrosine kinase activity | 8 | 4 | 0.028455 |
| GO:0016251 general RNA polymerase II transcription factor activity | 72 | 20 | 0.05 |
| GO:0005515 protein binding | 486 | 62 | 0.04775 |
| GO:0003702 RNA polymerase II transcription factor activity | 147 | 50 | 0.05 |
| GO:0030276 clathrin binding | 13 | 6 | 0.047577 |
| GO:0016563 transcription activator activity | 50 | 26 | 0.05 |
| GO:0016564 transcription repressor activity | 39 | 16 | 0.05 |
| GO:0004672 protein kinase activity | 134 | 30 | 0.05 |
| GO:0003723 RNA binding | 442 | 56 | 0.04131 |
| GO:0046914 transition metal ion binding | 398 | 62 | 0.05 |
| GO:0019899 enzyme binding | 38 | 9 | 0.026086 |
| GO:0016773 phosphotransferase activity alcohol group as acceptor | 196 | 34 | 0.05 |
| GO:0016566 specific transcriptional repressor activity | 21 | 7 | 0.0416 |
| GO:0001071 nucleic acid binding transcription factor activity | 137 | 50 | 0.05 |
| GO:0019208 phosphatase regulator activity | 26 | 10 | 0.05 |
| GO:0019888 protein phosphatase regulator activity | 26 | 10 | 0.05 |
| GO:0016301 kinase activity | 222 | 34 | 0.047667 |
| GO:0003704 specific RNA polymerase II transcription factor activity | 56 | 24 | 0.05 |
| GO:0019789 SUMO ligase activity | 5 | 3 | 0.013763 |
| GO:0030234 enzyme regulator activity | 256 | 41 | 0.05 |
| GO:0035091 phosphoinositide binding | 66 | 16 | 0.048875 |
| GO:0004535 poly(A)-specific ribonuclease activity | 5 | 3 | 0.013763 |
| GO:0008289 lipid binding | 95 | 23 | 0.05 |
| GO:0030528 transcription regulator activity | 279 | 88 | 0.05 |
| GO:0043565 sequence-specific DNA binding | 261 | 77 | 0.05 |
| GO:0008143 poly(A) RNA binding | 7 | 4 | 0.038469 |
| GO:0008270 zinc ion binding | 313 | 56 | 0.05 |
| GO:0060589 nucleoside-triphosphatase regulator activity | 131 | 20 | 0.006615 |
| GO:0070717 poly-purine tract binding | 7 | 4 | 0.038469 |
| GO:0008601 protein phosphatase type 2A regulator activity | 5 | 3 | 0.013763 |
| GO:0003729 mRNA binding | 52 | 22 | 0.05 |
| GO:0003727 single-stranded RNA binding | 15 | 7 | 0.0482 |
| GO:0016455 RNA polymerase II transcription mediator activity | 30 | 8 | 0.029088 |
| GO:0003700 sequence-specific DNA binding transcription factor activity | 137 | 50 | 0.05 |
| GO:0005543 phospholipid binding | 84 | 21 | 0.05 |
Functional analysis of the subset was performed in the “Gene Ontology” Database (http://www.geneontology.org/); fractions of proteins were compared by “GoMiner” software.
The selection of Q/N-rich proteins from the S. cerevisiae proteome was obtained using the SARP algorithm.179 The probability thresholds were set to 10−6 for single residue CBRs and 10−12 for multiple residue CBRs; CBRs were detected for the N and Q residues, as well as for the Q/N pair.
First, poly-Q tracts have their own transcriptional activities. The incorporation of poly-Q stretches into the sequences of transcription factors stimulates their activity in different systems.170 Correlatively, the expression of poly-Q tracts in yeast causes changes in the transcriptome, and, although these changes depend on both the length of poly-Q tract and presence of the nuclear localization signal (NLS), they occur even in the case of relatively short stretches of 23Qs without NLS in a protein, which is unable to aggregate.171 The same is partly true for poly-N, since 104N fused with GFP and directed by NLS causes repression of transcription from PHO84 and HSP104 promoters.172 Additionally, analysis of the 104N aggregates172 supports the hypothesis that poly-Q (or poly-N) aggregates sequester Q/N-rich transcription factors thus modulating the transcriptome profile (see section I).94,173 Although this hypothesis has been experimentally proven,94,173 the real contribution of such sequestration to the transcriptomic effects of Q/N-rich amyloids remains unclear.
The recruitment of transcription factors by poly-Q aggregates manifests the influence of Q/N-rich regions on protein-protein interactions. In general, the presence of CBRs / LCRs in protein sequences might affect the probability of their interactions.52 Computational analysis of the Q/N-rich proteins of S. cerevisiae revealed that Q/N-rich proteins generally tend not to interact with each other.174 However, known yeast prion-forming proteins conversely demonstrate a significant tendency to interact with other Q/N-rich proteins.174 We propose that such selection against interaction of Q/N-rich proteins might have occurred through evolution to prevent non-specific binding between Q/N-rich regions, which is likely to lead to the co-aggregation of proteins and loss-of-function, which occurs in the case of various amyloid-associated diseases.
Simultaneously, similar Q/N-rich regions are widespread in subunits of different protein complexes. While the proteome of S. cerevisiae contains more than 400 protein complexes,175 approximately 40 of them possess one, and 10 – several Q/N-rich subunits (Fig. 2). All complexes with more than one Q/N-rich subunit are involved in mRNA metabolism (transcription, storage, and nucleocytoplasmic transport). Such enrichment with multiple Q/N-rich subunits in protein complexes suggests that Q/N-rich regions could play a role in the assembly of these complexes. Interestingly, the prion-forming proteins Cyc8, Swi1, Sup35, Pub1 and Nup100 are the components of large protein complexes. Currently, except for Pub1 / Sup35, for which interaction through the Q/N-rich regions is essential for the formation of a tubulin associate complex implicated in the maintenance of microtubule integrity,45 for most of these proteins it is unclear whether or not their prionization destabilizes or promotes the corresponding protein complexes. However, with the exception of a subunit of nuclear pore complex Nup100, the prionization of these proteins has specific phenotypes. In the case of Nup100, prion can be detected only biochemically or using the Nup100-YFP reporter and has no phenotypic manifestation, although [NUP100+] aggregates sequester other Q/N rich Nup proteins (at least, Nup116 and Nup145).43
FIGURE 2.
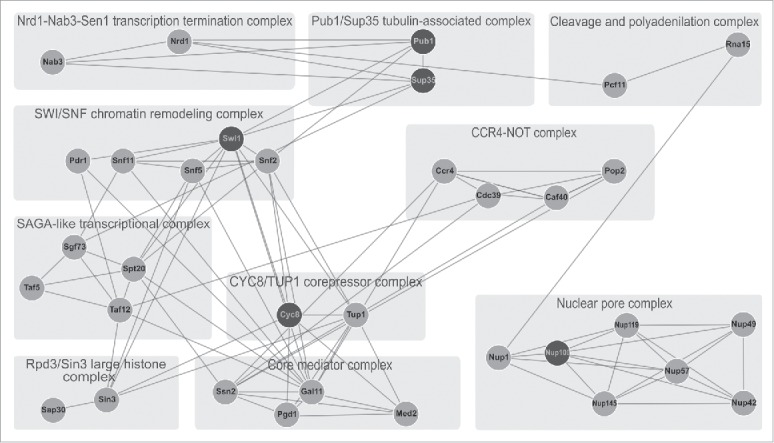
Q/N-rich subunits of protein complexes in S. cerevisiae and their interactions. Shown is the interactome of Q/N-rich subunits of protein complexes containing more than one Q/N-rich subunit. Lines indicate physical protein-protein interactions (according to data from “String” database, http://string-db.org/). Experimentally proven prion and amyloid-forming proteins are indicated by black circles, while other Q/N-rich proteins are indicated by gray circles. Light gray rectangles indicate corresponding protein complexes (including their names, as indicated).
Combining the data from the aforementioned studies, we propose that the interactome of prion- and amyloid-forming proteins includes specific groups of interactions occurring due to the presence of CBRs / LCRs in the amino acid sequences of these proteins. We may highlight 3 groups of such specific interactions: (i) prion or amyloid with other prions and amyloids, (ii) subunits of protein complex with a prion- or amyloid-forming protein in the case that this protein is a subunit of a complex, and (iii) interaction between corresponding prion or amyloid with LCR-containing proteins. Combined, this network of interactions is likely to make a significant contribution to the manifestation of the corresponding amyloid. Unfortunately, current data on proteomics of amyloids (or amyloidomics) is very poor, and verification of this hypothesis will only be possible in the future studies. However, several methods have been recently developed allowing to screen proteomes for amyloids. Such methods, TAPI176 and PSIA,177 facilitate rapid identification of amyloid proteins. PSIA uses two-dimensional difference gel electrophoresis (2D-DIGE) for the separation of proteins. The limitation of this method is that it does not allow the identification of proteins with extreme isoelectric points.177 TAPI employs very efficient high-performance liquid chromatography to separate proteins, but utilizes resistance to sodium dodecyl sulfate, to which not all amyloids are resistant, as a screening criterion.114,176 Despite the limitations, these methods were validated by detection of a wide spectrum of known amyloids and are promising for the identification of novel amyloid-forming and amyloid-associated proteins in different organisms at the proteomic level.
To conclude, RNA-modulating amyloids exhibit an extremely wide diversity of biological roles, including transcription, translation, storage and degradation of RNA, as well as pathogenesis. The ability of a protein to form amyloids and its RNA-modulating activity are closely linked in certain cases, as they are determined by the same sequences rich in Q and N. Such Q/N-rich sequences possess multiple activities: (i) the formation of amyloids, (ii) DNA- and RNA- binding, (iii) the participation in the protein-protein interactions, and likely (iv) the formation of protein complexes. Such a diversity of activities engenders diversity of functions and manifestations of Q/N rich amyloids. Simultaneously, the question is: what is the real number of amyloids in proteomes of different organisms? The amyloids identified to date are likely to be only pieces of a puzzle, since they were identified only by studies addressing particular proteins, and their real prevalence in proteomes could be considerably higher. Further proteomic studies of amyloids should highlight these issues and explain the biological significance and roles of amyloid structure.
ABBREVIATIONS
- CBR
compositionally biased region in protein sequence
- eRF
eukaryotic translation release factor
- hnRNP
heterogeneous nuclear ribonucleoprotein
- Hsp
heat shock protein
- LCR
low complexity region in protein sequence
- NLS
nuclear localization signal
- PSIA
proteomic screening and identification of amyloids
- RNP
ribonucleoprotein particle
- TAPI
technique for amyloid purification and identification
- YFP and GFP
yellow and green fluorescent proteins, respectively
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Institutes of Health (grant 7 R01 GM070934-06 to ILD), by the grant of the President of the Russian Federation (MK-4854.2015.4 to AAN and KSA), by Russian Foundation for Basic Research (grants 16-34-60153 to AAN and 16-34-00582 to SAB, KSA, and AAN), and St. Petersburg Government (to AAN and KSA). The authors acknowledge Saint-Petersburg University for a research grants 1.50.2543.2013 (to AAN and SGI), 15.61.2218.2013 (to SAB), and 1.37.291.2015 (to KSA, SAB, and AAN).
REFERENCES
- [1].Kyle RA. Amyloidosis: a convoluted story. Br J Haematol 2001; 114:529-38; PMID:11552976; http://dx.doi.org/ 10.1046/j.1365-2141.2001.02999.x [DOI] [PubMed] [Google Scholar]
- [2].Buxbaum JN, Linke RP. A molecular history of the amyloidoses. J Mol Biol 2012; 421:142-59; PMID:22321796; http://dx.doi.org/ 10.1016/j.jmb.2012.01.024 [DOI] [PubMed] [Google Scholar]
- [3].Virchow R. Ueber eine im Gehirn und Ruckenmark des Menschen aufgefunde Substanz mit der chemishen Reaction der Cellulose. Virchows Arch Path Anat Physiol 1854; 6:135-8; http://dx.doi.org/ 10.1007/BF01930815 [DOI] [Google Scholar]
- [4].Friedreich N, Kekule FA. Zur Amyloidfrage. Virchows Arch Path Anat Physiol 1859; 16:50-65; http://dx.doi.org/ 10.1007/BF01945246 [DOI] [Google Scholar]
- [5].Snow AD, Willmer J, Kisilevsky R. Sulfated glycosaminoglycans: a common constituent of all amyloids? Lab Invest 1987; 56:120-3; PMID:2432352 [PubMed] [Google Scholar]
- [6].Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Structural Biol 2000; 130:88-98; http://dx.doi.org/ 10.1006/jsbi.2000.4221 [DOI] [PubMed] [Google Scholar]
- [7].Vassar PS, Culling CF. Fluorescent stains, with special reference to amyloid and connective tissues. Arch Pathol 1959; 68:487-98; PMID:13841452 [PubMed] [Google Scholar]
- [8].Hobbs JR, Morgan AD. Fluorescence Microscopy with Thioflavine-T in the Diagnosis of Amyloid. J Pathol Bacteriol 1963; 86:437-42; PMID:14068952; http://dx.doi.org/ 10.1002/path.1700860218 [DOI] [PubMed] [Google Scholar]
- [9].LeVine H, 3rd. Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol 1999; 309:274-84; PMID:10507030; http://dx.doi.org/ 10.1016/S0076-6879(99)09020-5 [DOI] [PubMed] [Google Scholar]
- [10].Eanes ED, Glenner GG. X-ray diffraction studies on amyloid filaments. J Histochem Cytochem 1968; 16:673-7; PMID:5723775; http://dx.doi.org/ 10.1177/16.11.673 [DOI] [PubMed] [Google Scholar]
- [11].Tycko R, Wickner RB. Molecular structures of amyloid and prion fibrils: consensus versus controversy. Accounts Chem Res 2013; 46:1487-96; http://dx.doi.org/ 10.1021/ar300282r [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Meyer RK, McKinley MP, Bowman KA, Braunfeld MB, Barry RA, Prusiner SB. Separation and properties of cellular and scrapie prion proteins. Proc Natl Acad Sci U S A 1986; 83:2310-4; PMID:3085093; http://dx.doi.org/ 10.1073/pnas.83.8.2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem 2003; 278:49636-43; PMID:14507919; http://dx.doi.org/ 10.1074/jbc.M307996200 [DOI] [PubMed] [Google Scholar]
- [14].Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009; 137:146-58; PMID:19345193; http://dx.doi.org/ 10.1016/j.cell.2009.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science 1982; 218:1309-11; PMID:6815801; http://dx.doi.org/ 10.1126/science.6815801 [DOI] [PubMed] [Google Scholar]
- [16].Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJ, Westermark P. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014; 21:221-4; PMID:25263598; http://dx.doi.org/ 10.3109/13506129.2014.964858 [DOI] [PubMed] [Google Scholar]
- [17].Nizhnikov AA, Antonets KS, Inge-Vechtomov SG. Amyloids: from pathogenesis to function. Biochem Biokhimiia 2015; 80:1127-44; http://dx.doi.org/ 10.1134/S0006297915090047 [DOI] [PubMed] [Google Scholar]
- [18].Iconomidou VA, Vriend G, Hamodrakas SJ. Amyloids protect the silkmoth oocyte and embryo. FEBS Letters 2000; 479:141-5; PMID:10981723; http://dx.doi.org/ 10.1016/S0014-5793(00)01888-3 [DOI] [PubMed] [Google Scholar]
- [19].de Vocht ML, Reviakine I, Wosten HA, Brisson A, Wessels JG, Robillard GT. Structural and functional role of the disulfide bridges in the hydrophobin SC3. J Biol Chem 2000; 275:28428-32; PMID:10829014; http://dx.doi.org/ 10.1074/jbc.M000691200 [DOI] [PubMed] [Google Scholar]
- [20].Chimileski S, Franklin MJ, Papke RT. Biofilms formed by the archaeon Haloferax volcanii exhibit cellular differentiation and social motility, and facilitate horizontal gene transfer. BMC Biol 2014; 12:65; PMID:25124934; http://dx.doi.org/ 10.1186/s12915-014-0065-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Syed AK, Boles BR. Fold modulating function: bacterial toxins to functional amyloids. Front Microbiol 2014; 5:401; PMID:25136340; http://dx.doi.org/ 10.3389/fmicb.2014.00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KP, Simon R, Schubert D, et al.. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009; 325:328-32; PMID:19541956; http://dx.doi.org/ 10.1126/science.1173155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. Functional amyloid formation within mammalian tissue. PLoS Biol 2006; 4:e6; PMID:16300414; http://dx.doi.org/ 10.1371/journal.pbio.0040006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Si K, Choi YB, White-Grindley E, Majumdar A, Kandel ER. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 2010; 140:421-35; PMID:20144764; http://dx.doi.org/ 10.1016/j.cell.2010.01.008 [DOI] [PubMed] [Google Scholar]
- [25].Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, Chen ZJ. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014; 156:1207-22; PMID:24630723; http://dx.doi.org/ 10.1016/j.cell.2014.01.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al.. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012; 150:339-50; PMID:22817896; http://dx.doi.org/ 10.1016/j.cell.2012.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell 1983; 35:57-62; PMID:6414721; http://dx.doi.org/ 10.1016/0092-8674(83)90207-6 [DOI] [PubMed] [Google Scholar]
- [28].DeArmond SJ, McKinley MP, Barry RA, Braunfeld MB, McColloch JR, Prusiner SB. Identification of prion amyloid filaments in scrapie-infected brain. Cell 1985; 41:221-35; PMID:3922627; http://dx.doi.org/ 10.1016/0092-8674(85)90076-5 [DOI] [PubMed] [Google Scholar]
- [29].Kitamoto T, Tateishi J, Tashima T, Takeshita I, Barry RA, DeArmond SJ, Prusiner SB. Amyloid plaques in Creutzfeldt-Jakob disease stain with prion protein antibodies. Annals Neurol 1986; 20:204-8; http://dx.doi.org/ 10.1002/ana.410200205 [DOI] [PubMed] [Google Scholar]
- [30].Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 1983; 35:349-58; PMID:6418385; http://dx.doi.org/ 10.1016/0092-8674(83)90168-X [DOI] [PubMed] [Google Scholar]
- [31].Come JH, Fraser PE, Lansbury PT Jr.. A kinetic model for amyloid formation in the prion diseases: importance of seeding. Proc Natl Acad Sci U S A 1993; 90:5959-63; PMID:8327467; http://dx.doi.org/ 10.1073/pnas.90.13.5959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jarrett JT, Lansbury PT Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer disease and scrapie? Cell 1993; 73:1055-8; PMID:8513491; http://dx.doi.org/ 10.1016/0092-8674(93)90635-4 [DOI] [PubMed] [Google Scholar]
- [33].Newby GA, Lindquist S. Blessings in disguise: biological benefits of prion-like mechanisms. Trends Cell biol 2013; 23:251-9; PMID:23485338; http://dx.doi.org/ 10.1016/j.tcb.2013.01.007 [DOI] [PubMed] [Google Scholar]
- [34].Wickner RB, Edskes HK, Bateman DA, Kelly AC, Gorkovskiy A, Dayani Y, Zhou A. Amyloid diseases of yeast: prions are proteins acting as genes. Essays Biochem 2014; 56:193-205; PMID:25131596; http://dx.doi.org/ 10.1042/bse0560193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cox B. Psi, a cytoplasmic supperssor of supersuppressors in yeast. Heredity 1965; 20:505-21; http://dx.doi.org/ 10.1038/hdy.1965.65 [DOI] [Google Scholar]
- [36].Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 1994; 264:566-9; PMID:7909170; http://dx.doi.org/ 10.1126/science.7909170 [DOI] [PubMed] [Google Scholar]
- [37].Lacroute F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J Bacteriol 1971; 106:519-22; PMID:5573734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN(+)]. Cell 2001; 106:171-82; PMID:11511345; http://dx.doi.org/ 10.1016/S0092-8674(01)00427-5 [DOI] [PubMed] [Google Scholar]
- [39].Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell 2000; 5:163-72; PMID:10678178; http://dx.doi.org/ 10.1016/S1097-2765(00)80412-8 [DOI] [PubMed] [Google Scholar]
- [40].Du Z, Park KW, Yu H, Fan Q, Li L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat Genetics 2008; 40:460-5; PMID:18362884; http://dx.doi.org/ 10.1038/ng.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Patel BK, Gavin-Smyth J, Liebman SW. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat Cell Biol 2009; 11:344-9; PMID:19219034; http://dx.doi.org/ 10.1038/ncb1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rogoza T, Goginashvili A, Rodionova S, Ivanov M, Viktorovskaya O, Rubel A, Volkov K, Mironova L. Non-Mendelian determinant [ISP+] in yeast is a nuclear-residing prion form of the global transcriptional regulator Sfp1. Proc Natl Acad Sci U S A 2010; 107:10573-7; PMID:20498075; http://dx.doi.org/ 10.1073/pnas.1005949107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Halfmann R, Wright JR, Alberti S, Lindquist S, Rexach M. Prion formation by a yeast GLFG nucleoporin. Prion 2012; 6:391-9; PMID:22561191; http://dx.doi.org/ 10.4161/pri.20199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Suzuki G, Shimazu N, Tanaka M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 2012; 336:355-9; PMID:22517861; http://dx.doi.org/ 10.1126/science.1219491 [DOI] [PubMed] [Google Scholar]
- [45].Li X, Rayman JB, Kandel ER, Derkatch IL. Functional role of Tia1/Pub1 and Sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton. Mol Cell 2014; 55:305-18; PMID:24981173; http://dx.doi.org/ 10.1016/j.molcel.2014.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci U S A 1997; 94:9773-8; PMID:9275200; http://dx.doi.org/ 10.1073/pnas.94.18.9773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci U S A 2002; 99:7402-7; PMID:12032295; http://dx.doi.org/ 10.1073/pnas.072199199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Roberts BT, Wickner RB. Heritable activity: a prion that propagates by covalent autoactivation. Genes Dev 2003; 17:2083-7; PMID:12923060; http://dx.doi.org/ 10.1101/gad.1115803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brown JC, Lindquist S. A heritable switch in carbon source utilization driven by an unusual yeast prion. Genes Dev 2009; 23:2320-32; PMID:19797769; http://dx.doi.org/ 10.1101/gad.1839109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jarosz DF, Brown JC, Walker GA, Datta MS, Ung WL, Lancaster AK, Rotem A, Chang A, Newby GA, Weitz DA, et al.. Cross-kingdom chemical communication drives a heritable, mutually beneficial prion-based transformation of metabolism. Cell 2014; 158:1083-93; PMID:25171409; http://dx.doi.org/ 10.1016/j.cell.2014.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Harrison PM, Gerstein M. A method to assess compositional bias in biological sequences and its application to prion-like glutamine/asparagine-rich domains in eukaryotic proteomes. Genome Biol 2003; 4:R40; PMID:12801414; http://dx.doi.org/ 10.1186/gb-2003-4-6-r40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Coletta A, Pinney JW, Solis DY, Marsh J, Pettifer SR, Attwood TK. Low-complexity regions within protein sequences have position-dependent roles. BMC Systems Biol 2010; 4:43; http://dx.doi.org/ 10.1186/1752-0509-4-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Michelitsch MD, Weissman JS. A census of glutamine/asparagine-rich regions: implications for their conserved function and the prediction of novel prions. Proc Natl Acad Sci U S A 2000; 97:11910-5; PMID:11050225; http://dx.doi.org/ 10.1073/pnas.97.22.11910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Garbuzynskiy SO, Lobanov MY, Galzitskaya OV. FoldAmyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2010; 26:326-32; PMID:20019059; http://dx.doi.org/ 10.1093/bioinformatics/btp691 [DOI] [PubMed] [Google Scholar]
- [55].Conchillo-Sole O, de Groot NS, Aviles FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics 2007; 8:65; PMID:17324296; http://dx.doi.org/ 10.1186/1471-2105-8-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc Natl Acad Sci U S A 2005; 102:12825-30; PMID:16123127; http://dx.doi.org/ 10.1073/pnas.0506136102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Das S, Pal U, Das S, Bagga K, Roy A, Mrigwani A, Maiti NC. Sequence complexity of amyloidogenic regions in intrinsically disordered human proteins. PloS One 2014; 9:e89781; PMID:24594841; http://dx.doi.org/ 10.1371/journal.pone.0089781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, et al.. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Meth 2010; 7:237-42; http://dx.doi.org/ 10.1038/nmeth.1432 [DOI] [PubMed] [Google Scholar]
- [59].Stephan JS, Fioriti L, Lamba N, Colnaghi L, Karl K, Derkatch IL, Kandel ER. The CPEB3 Protein Is a Functional Prion that Interacts with the Actin Cytoskeleton. Cell Reports 2015; 11:1772-85; PMID:26074072; http://dx.doi.org/ 10.1016/j.celrep.2015.04.060 [DOI] [PubMed] [Google Scholar]
- [60].Ahmed AB, Kajava AV. Breaking the amyloidogenicity code: methods to predict amyloids from amino acid sequence. FEBS Lett 2013; 587:1089-95; PMID:23262221; http://dx.doi.org/ 10.1016/j.febslet.2012.12.006 [DOI] [PubMed] [Google Scholar]
- [61].Ahmed AB, Znassi N, Chateau MT, Kajava AV. A structure-based approach to predict predisposition to amyloidosis. Alzheimer Dementia 2015; 11:681-90; PMID:25150734; http://dx.doi.org/ 10.1016/j.jalz.2014.06.007 [DOI] [PubMed] [Google Scholar]
- [62].Kadnar ML, Articov G, Derkatch IL. Distinct type of transmission barrier revealed by study of multiple prion determinants of Rnq1. PLoS Genetics 2010; 6:e1000824; PMID:20107602; http://dx.doi.org/ 10.1371/journal.pgen.1000824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Stein KC, True HL. Extensive diversity of prion strains is defined by differential chaperone interactions and distinct amyloidogenic regions. PLoS Genetics 2014; 10:e1004337; PMID:24811344; http://dx.doi.org/ 10.1371/journal.pgen.1004337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Saifitdinova AF, Nizhnikov AA, Lada AG, Rubel AA, Magomedova ZM, Ignatova VV, Inge-Vechtomov SG, Galkin AP. [NSI (+)]: a novel non-Mendelian nonsense suppressor determinant in Saccharomyces cerevisiae. Curr Genetics 2010; 56:467-78; http://dx.doi.org/ 10.1007/s00294-010-0314-2 [DOI] [PubMed] [Google Scholar]
- [65].Nizhnikov AA, Magomedova ZM, Rubel AA, Kondrashkina AM, Inge-Vechtomov SG, Galkin AP. [NSI+] determinant has a pleiotropic phenotypic manifestation that is modulated by SUP35, SUP45, and VTS1 genes. Curr Genetics 2012; 58:35-47; http://dx.doi.org/ 10.1007/s00294-011-0363-1 [DOI] [PubMed] [Google Scholar]
- [66].Nizhnikov AA, Kondrashkina AM, Galkin AP. Interactions of [NSI+] prion-like determinant with SUP35 and VTS1 genes in Saccharomyces cerevisiae. Russian J Genetics 2013; 49:1004-12; http://dx.doi.org/ 10.1134/S1022795413100074 [DOI] [PubMed] [Google Scholar]
- [67].Kondrashkina AM, Antonets KS, Galkin AP, Nizhnikov AA. Prion-Like Determinant [NSI+] Decreases the Expression of the SUP45 Gene in Saccharomyces cerevisiae. Mol Biol 2014; 48:688-93; http://dx.doi.org/ 10.1134/S0026893314050069 [DOI] [PubMed] [Google Scholar]
- [68].Blinder D, Coschigano PW, Magasanik B. Interaction of the GATA factor Gln3p with the nitrogen regulator Ure2p in Saccharomyces cerevisiae. J Bacteriol 1996; 178:4734-6; PMID:8755910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Saha A, Wittmeyer J, Cairns BR. Chromatin remodelling: the industrial revolution of DNA around histones. Nat Rev Mol Cell Biol 2006; 7:437-47; PMID:16723979; http://dx.doi.org/ 10.1038/nrm1945 [DOI] [PubMed] [Google Scholar]
- [70].Grishin AV, Rothenberg M, Downs MA, Blumer KJ. Mot3, a Zn finger transcription factor that modulates gene expression and attenuates mating pheromone signaling in Saccharomyces cerevisiae. Genetics 1998; 149:879-92; PMID:9611199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 1997; 278:680-6; PMID:9381177; http://dx.doi.org/ 10.1126/science.278.5338.680 [DOI] [PubMed] [Google Scholar]
- [72].Smith RL, Johnson AD. Turning genes off by Ssn6-Tup1: a conserved system of transcriptional repression in eukaryotes. Trends Biochem Sci 2000; 25:325-30; PMID:10871883; http://dx.doi.org/ 10.1016/S0968-0004(00)01592-9 [DOI] [PubMed] [Google Scholar]
- [73].Proft M, Struhl K. Hog1 kinase converts the Sko1-Cyc8-Tup1 repressor complex into an activator that recruits SAGA and SWI/SNF in response to osmotic stress. Mol Cell 2002; 9:1307-17; PMID:12086627; http://dx.doi.org/ 10.1016/S1097-2765(02)00557-9 [DOI] [PubMed] [Google Scholar]
- [74].Marion RM, Regev A, Segal E, Barash Y, Koller D, Friedman N, O'Shea EK. Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc Natl Acad Sci U S A 2004; 101:14315-22; PMID:15353587; http://dx.doi.org/ 10.1073/pnas.0405353101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 1999; 283:1339-43; PMID:10037606; http://dx.doi.org/ 10.1126/science.283.5406.1339 [DOI] [PubMed] [Google Scholar]
- [76].Baxa U, Wickner RB, Steven AC, Anderson DE, Marekov LN, Yau WM, Tycko R. Characterization of β-sheet structure in Ure2p1-89 yeast prion fibrils by solid-state nuclear magnetic resonance. Biochem 2007; 46:13149-62 [DOI] [PubMed] [Google Scholar]
- [77].Kryndushkin DS, Wickner RB, Tycko R. The core of Ure2p prion fibrils is formed by the N-terminal segment in a parallel cross-β structure: evidence from solid-state NMR. J Mol Biol 2011; 409:263-77; PMID:21497604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Du Z, Crow ET, Kang HS, Li L. Distinct subregions of Swi1 manifest striking differences in prion transmission and SWI/SNF function. Mol Cell Biol 2010; 30:4644-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Prusiner SB. Prions and neurodegenerative diseases. N Eng J Med 1987; 317:1571-81 [DOI] [PubMed] [Google Scholar]
- [80].Baxa U, Speransky V, Steven AC, Wickner RB. Mechanism of inactivation on prion conversion of the Saccharomyces cerevisiae Ure2 protein. Proc Natl Acad Sci U S A 2002; 99:5253-60; PMID:11959975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Mitchell AP, Magasanik B. Regulation of glutamine-repressible gene products by the GLN3 function in Saccharomyces cerevisiae. Mol Cell Biol 1984; 4:2758-66; PMID:6152012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Rai R, Genbauffe F, Lea HZ, Cooper TG. Transcriptional regulation of the DAL5 gene in Saccharomyces cerevisiae. J Bacteriol 1987; 169:3521-4; PMID:3301804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Coschigano PW, Magasanik B. The URE2 gene product of Saccharomyces cerevisiae plays an important role in the cellular response to the nitrogen source and has homology to glutathione s-transferases. Mol Cell Biol 1991; 11:822-32; PMID:1990286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bai M, Zhou JM, Perrett S. The yeast prion protein Ure2 shows glutathione peroxidase activity in both native and fibrillar forms. J Biol Chem 2004; 279:50025-30; PMID:15371425 [DOI] [PubMed] [Google Scholar]
- [85].Holmes DL, Lancaster AK, Lindquist S, Halfmann R. Heritable remodeling of yeast multicellularity by an environmentally responsive prion. Cell 2013; 153:153-65; PMID:23540696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Drozdova P, Rogoza T, Radchenko E, Lipaeva P, Mironova L. Transcriptional response to the [ISP(+) ] prion of Saccharomyces cerevisiae differs from that induced by the deletion of its structural gene, SFP1. FEMS Yeast Res 2014; 14:1160-70; PMID:25227157 [DOI] [PubMed] [Google Scholar]
- [87].Volkov KV, Aksenova AY, Soom MJ, Osipov KV, Svitin AV, Kurischko C, Shkundina IS, Ter-Avanesyan MD, Inge-Vechtomov SG, Mironova LN. Novel non-Mendelian determinant involved in the control of translation accuracy in Saccharomyces cerevisiae. Genetics 2002; 160:25-36; PMID:11805042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].McGlinchey RP, Kryndushkin D, Wickner RB. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A 2011; 108:5337-41; PMID:21402947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes . The Huntington's Disease Collaborative Research Group. Cell 1993; 72:971-83; PMID:8458085 [DOI] [PubMed] [Google Scholar]
- [90].DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997; 277:1990-3; PMID:9302293 [DOI] [PubMed] [Google Scholar]
- [91].McGowan DP, van Roon-Mom W, Holloway H, Bates GP, Mangiarini L, Cooper GJ, Faull RL, Snell RG. Amyloid-like inclusions in Huntington's disease. Neuro Sci 2000; 100:677-80 [DOI] [PubMed] [Google Scholar]
- [92].Valor LM. Transcription, epigenetics and ameliorative strategies in Huntington's Disease: a genome-wide perspective. Mol Neurobiol 2015; 51:406-23; PMID:24788684; http://dx.doi.org/ 10.1007/s12035-014-8715-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Bithell A, Johnson R, Buckley NJ. Transcriptional dysregulation of coding and non-coding genes in cellular models of Huntington's disease. Biochem Society Transactions 2009; 37:1270-5; http://dx.doi.org/ 10.1042/BST0371270 [DOI] [PubMed] [Google Scholar]
- [94].Nucifora FC Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, et al.. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001; 291:2423-8; PMID:11264541; http://dx.doi.org/ 10.1126/science.1056784 [DOI] [PubMed] [Google Scholar]
- [95].Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A 2000; 97:6763-8; PMID:10823891; http://dx.doi.org/ 10.1073/pnas.100110097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Huang CC, Faber PW, Persichetti F, Mittal V, Vonsattel JP, MacDonald ME, Gusella JF. Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somatic Cell Mol Genetics 1998; 24:217-33; http://dx.doi.org/ 10.1023/B:SCAM.0000007124.19463.e5 [DOI] [PubMed] [Google Scholar]
- [97].Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, Kimura T, Koide R, Nozaki K, Sano Y, et al.. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genetics 2000; 26:29-36; PMID:10973244; http://dx.doi.org/ 10.1038/79139 [DOI] [PubMed] [Google Scholar]
- [98].Yu ZX, Li SH, Nguyen HP, Li XJ. Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genetics 2002; 11:905-14; http://dx.doi.org/ 10.1093/hmg/11.8.905 [DOI] [PubMed] [Google Scholar]
- [99].Benn CL, Sun T, Sadri-Vakili G, McFarland KN, DiRocco DP, Yohrling GJ, Clark TW, Bouzou B, Cha JH. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J Neurosci 2008; 28:10720-33; PMID:18923047; http://dx.doi.org/ 10.1523/JNEUROSCI.2126-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chan HY. RNA-mediated pathogenic mechanisms in polyglutamine diseases and amyotrophic lateral sclerosis. Front Cell Neurosci 2014; 8:431; PMID:25565965; http://dx.doi.org/ 10.3389/fncel.2014.00431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Silva JL, Rangel LP, Costa DC, Cordeiro Y, De Moura Gallo CV. Expanding the prion concept to cancer biology: dominant-negative effect of aggregates of mutant p53 tumour suppressor. Biosci Reports 2013; 33:e00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Olivier M, Langerod A, Carrieri P, Bergh J, Klaar S, Eyfjord J, Theillet C, Rodriguez C, Lidereau R, Bieche I, et al.. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin Cancer Res 2006; 12:1157-67; PMID:16489069; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-1029 [DOI] [PubMed] [Google Scholar]
- [103].Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol 2013; 15:2-8; PMID:23263379; http://dx.doi.org/ 10.1038/ncb2641 [DOI] [PubMed] [Google Scholar]
- [104].Ano Bom AP, Rangel LP, Costa DC, de Oliveira GA, Sanches D, Braga CA, Gava LM, Ramos CH, Cepeda AO, Stumbo AC, et al.. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J Biol Chem 2012; 287:28152-62; PMID:22715097; http://dx.doi.org/ 10.1074/jbc.M112.340638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Levy CB, Stumbo AC, Ano Bom AP, Portari EA, Cordeiro Y, Silva JL, De Moura-Gallo CV. Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int J Biochem Cell Biol 2011; 43:60-4; PMID:21056685; http://dx.doi.org/ 10.1016/j.biocel.2010.10.017 [DOI] [PubMed] [Google Scholar]
- [106].King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res 2012; 1462:61-80; PMID:22445064; http://dx.doi.org/ 10.1016/j.brainres.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, et al.. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 2012; 149:768-79; PMID:22579282; http://dx.doi.org/ 10.1016/j.cell.2012.04.016 [DOI] [PubMed] [Google Scholar]
- [108].Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, et al.. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012; 149:753-67; PMID:22579281; http://dx.doi.org/ 10.1016/j.cell.2012.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Buchan JR. mRNP granules. Assembly, function, and connections with disease. RNA Biol 2014; 11:1019-30; PMID:25531407; http://dx.doi.org/ 10.4161/15476286.2014.972208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci 2008; 33:141-50; PMID:18291657; http://dx.doi.org/ 10.1016/j.tibs.2007.12.003 [DOI] [PubMed] [Google Scholar]
- [111].Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell 2004; 15:5383-98; PMID:15371533; http://dx.doi.org/ 10.1091/mbc.E04-08-0715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 α to the assembly of mammalian stress granules. J Cell Biol 1999; 147:1431-42; PMID:10613902; http://dx.doi.org/ 10.1083/jcb.147.7.1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Furukawa Y, Kaneko K, Matsumoto G, Kurosawa M, Nukina N. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J Neurosci 2009; 29:5153-62; PMID:19386911; http://dx.doi.org/ 10.1523/JNEUROSCI.0783-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Urakov VN, Vishnevskaya AB, Alexandrov IM, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Interdependence of amyloid formation in yeast: implications for polyglutamine disorders and biological functions. Prion 2010; 4:45-52; PMID:20118659; http://dx.doi.org/ 10.4161/pri.4.1.11074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell 2007; 25:635-46; PMID:17349952; http://dx.doi.org/ 10.1016/j.molcel.2007.02.011 [DOI] [PubMed] [Google Scholar]
- [116].Buchan JR, Yoon JH, Parker R. Stress-specific composition, assembly and kinetics of stress granules in Saccharomyces cerevisiae. J Cell Sci 2011; 124:228-39; PMID:21172806; http://dx.doi.org/ 10.1242/jcs.078444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Buchan JR, Nissan T, Parker R. Analyzing P-bodies and stress granules in Saccharomyces cerevisiae. Methods Enzymol 2010; 470:619-40; PMID:20946828; http://dx.doi.org/ 10.1016/S0076-6879(10)70025-2 [DOI] [PubMed] [Google Scholar]
- [118].Reijns MA, Alexander RD, Spiller MP, Beggs JD. A role for Q/N-rich aggregation-prone regions in P-body localization. J Cell Sci 2008; 121:2463-72; PMID:18611963; http://dx.doi.org/ 10.1242/jcs.024976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Decker CJ, Teixeira D, Parker R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol 2007; 179:437-49; PMID:17984320; http://dx.doi.org/ 10.1083/jcb.200704147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Yang Z, Jakymiw A, Wood MR, Eystathioy T, Rubin RL, Fritzler MJ, Chan EK. GW182 is critical for the stability of GW bodies expressed during the cell cycle and cell proliferation. J Cell Sci 2004; 117:5567-78; PMID:15494374; http://dx.doi.org/ 10.1242/jcs.01477 [DOI] [PubMed] [Google Scholar]
- [121].Souquere S, Mollet S, Kress M, Dautry F, Pierron G, Weil D. Unravelling the ultrastructure of stress granules and associated P-bodies in human cells. J Cell Sci 2009; 122:3619-26; PMID:19812307; http://dx.doi.org/ 10.1242/jcs.054437 [DOI] [PubMed] [Google Scholar]
- [122].Kroschwald S, Maharana S, Mateju D, Malinovska L, Nuske E, Poser I, Richter D, Alberti S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. Elife 2015; 4:e06807; PMID:26238190; http://dx.doi.org/ 10.7554/eLife.06807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015; 163:123-33; PMID:26406374; http://dx.doi.org/ 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Lin Y, Protter DS, Rosen MK, Parker R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol Cell 2015; 60:208-19; PMID:26412307; http://dx.doi.org/ 10.1016/j.molcel.2015.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Klar J, Sobol M, Melberg A, Mabert K, Ameur A, Johansson AC, Feuk L, Entesarian M, Orlen H, Casar-Borota O, et al.. Welander distal myopathy caused by an ancient founder mutation in TIA1 associated with perturbed splicing. Hum Mutation 2013; 34:572-7 [DOI] [PubMed] [Google Scholar]
- [126].Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, et al.. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PloS One 2010; 5:e13250; PMID:20948999; http://dx.doi.org/ 10.1371/journal.pone.0013250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 2009; 284:20329-39; PMID:19465477; http://dx.doi.org/ 10.1074/jbc.M109.010264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al.. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314:130-3; PMID:17023659; http://dx.doi.org/ 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- [129].Robinson JL, Geser F, Stieber A, Umoh M, Kwong LK, Van Deerlin VM, Lee VM, Trojanowski JQ. TDP-43 skeins show properties of amyloid in a subset of ALS cases. Acta Neuropathologica 2013; 125:121-31; PMID:23124365; http://dx.doi.org/ 10.1007/s00401-012-1055-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Udan M, Baloh RH. Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion 2011; 5:1-5; PMID:21135580; http://dx.doi.org/ 10.4161/pri.5.1.14265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al.. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323:1208-11; PMID:19251628; http://dx.doi.org/ 10.1126/science.1165942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Doi H, Koyano S, Suzuki Y, Nukina N, Kuroiwa Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci Res 2010; 66:131-3; PMID:19833157; http://dx.doi.org/ 10.1016/j.neures.2009.10.004 [DOI] [PubMed] [Google Scholar]
- [133].Lashley T, Rohrer JD, Bandopadhyay R, Fry C, Ahmed Z, Isaacs AM, Brelstaff JH, Borroni B, Warren JD, Troakes C, et al.. A comparative clinical, pathological, biochemical and genetic study of fused in sarcoma proteinopathies. Brain 2011; 134:2548-64; PMID:21752791; http://dx.doi.org/ 10.1093/brain/awr160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Maniecka Z, Polymenidou M. From nucleation to widespread propagation: A prion-like concept for ALS. Virus Res 2015; 207:94-105; PMID:25656065; http://dx.doi.org/ 10.1016/j.virusres.2014.12.032 [DOI] [PubMed] [Google Scholar]
- [135].Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol 2013; 201:361-72; PMID:23629963; http://dx.doi.org/ 10.1083/jcb.201302044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al.. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013; 495:467-73; PMID:23455423; http://dx.doi.org/ 10.1038/nature11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Shorter J, Taylor JP. Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis 2013; 1:e25200; PMID:25002999; http://dx.doi.org/ 10.4161/rdis.25200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].O'Rourke TW, Loya TJ, Head PE, Horton JR, Reines D. Amyloid-like assembly of the low complexity domain of yeast Nab3. Prion 2015:9(1):34-47; PMID:25635624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Tuite MF, Serio TR. The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol 2010; 11:823-33; PMID:21081963; http://dx.doi.org/ 10.1038/nrm3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Nizhnikov AA, Antonets KS, Inge-Vechtomov SG, Derkatch IL. Modulation of efficiency of translation termination in Saccharomyces cerevisiae. Prion 2014; 8:247-60; PMID:25486049; http://dx.doi.org/ 10.4161/pri.29851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Liebman SW, Chernoff YO. Prions in yeast. Genetics 2012; 191:1041-72; PMID:22879407; http://dx.doi.org/ 10.1534/genetics.111.137760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Frolova L, Le Goff X, Rasmussen HH, Cheperegin S, Drugeon G, Kress M, Arman I, Haenni AL, Celis JE, Philippe M, et al.. A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature 1994; 372:701-3; PMID:7990965; http://dx.doi.org/ 10.1038/372701a0 [DOI] [PubMed] [Google Scholar]
- [143].Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov S, Kisselev L, Philippe M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J 1995; 14:4065-72; PMID:7664746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, Paushkin SV, Nierras CR, Cox BS, Ter-Avanesyan MD, Tuite MF. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J 1995; 14:4365-73; PMID:7556078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 1994; 137:671-6; PMID:8088512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J 1996; 15:3127-34; PMID:8670813 [PMC free article] [PubMed] [Google Scholar]
- [147].Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 1996; 144:1375-86; PMID:8978027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 1996; 273:622-6; PMID:8662547; http://dx.doi.org/ 10.1126/science.273.5275.622 [DOI] [PubMed] [Google Scholar]
- [149].King CY, Tittmann P, Gross H, Gebert R, Aebi M, Wuthrich K. Prion-inducing domain 2–114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci U S A 1997; 94:6618-22; PMID:9192614; http://dx.doi.org/ 10.1073/pnas.94.13.6618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997; 89:811-9; PMID:9182769; http://dx.doi.org/ 10.1016/S0092-8674(00)80264-0 [DOI] [PubMed] [Google Scholar]
- [151].Hosoda N, Kobayashi T, Uchida N, Funakoshi Y, Kikuchi Y, Hoshino S, Katada T. Translation termination factor eRF3 mediates mRNA decay through the regulation of deadenylation. J Biol Chem 2003; 278:38287-91; PMID:12923185; http://dx.doi.org/ 10.1074/jbc.C300300200 [DOI] [PubMed] [Google Scholar]
- [152].Namy O, Galopier A, Martini C, Matsufuji S, Fabret C, Rousset JP. Epigenetic control of polyamines by the prion [PSI+]. Nat Cell Biol 2008; 10:1069-75; PMID:19160487; http://dx.doi.org/ 10.1038/ncb1766 [DOI] [PubMed] [Google Scholar]
- [153].Tyedmers J, Madariaga ML, Lindquist S. Prion switching in response to environmental stress. PLoS Biol 2008; 6:e294; PMID:19067491; http://dx.doi.org/ 10.1371/journal.pbio.0060294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics 1997; 147:507-19; PMID:9335589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci U S A 2005; 102:10575-80; PMID:16024723; http://dx.doi.org/ 10.1073/pnas.0504882102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 2012; 482:363-8; PMID:22337056; http://dx.doi.org/ 10.1038/nature10875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Bateman DA, Wickner RB. The [PSI+] prion exists as a dynamic cloud of variants. PLoS Genetics 2013; 9:e1003257; PMID:23382698; http://dx.doi.org/ 10.1371/journal.pgen.1003257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Pezza JA, Villali J, Sindi SS, Serio TR. Amyloid-associated activity contributes to the severity and toxicity of a prion phenotype. Nat Commun 2014; 5:4384; PMID:25023996; http://dx.doi.org/ 10.1038/ncomms5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim JH, Zhu H, Kandel ER. A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in aplysia. Cell 2003; 115:893-904; PMID:14697206; http://dx.doi.org/ 10.1016/S0092-8674(03)01021-3 [DOI] [PubMed] [Google Scholar]
- [160].Si K, Lindquist S, Kandel ER. A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 2003; 115:879-91; PMID:14697205; http://dx.doi.org/ 10.1016/S0092-8674(03)01020-1 [DOI] [PubMed] [Google Scholar]
- [161].Majumdar A, Cesario WC, White-Grindley E, Jiang H, Ren F, Khan MR, Li L, Choi EM, Kannan K, Guo F, et al.. Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell 2012; 148:515-29; PMID:22284910; http://dx.doi.org/ 10.1016/j.cell.2012.01.004 [DOI] [PubMed] [Google Scholar]
- [162].Fioriti L, Myers C, Huang YY, Li X, Stephan JS, Trifilieff P, Colnaghi L, Kosmidis S, Drisaldi B, Pavlopoulos E, et al.. The Persistence of Hippocampal-Based Memory Requires Protein Synthesis Mediated by the Prion-like Protein CPEB3. Neuron 2015; 86:1433-48; PMID:26074003; http://dx.doi.org/ 10.1016/j.neuron.2015.05.021 [DOI] [PubMed] [Google Scholar]
- [163].Drisaldi B, Colnaghi L, Fioriti L, Rao N, Myers C, Snyder AM, Metzger DJ, Tarasoff J, Konstantinov E, Fraser PE, et al.. SUMOylation Is an Inhibitory Constraint that Regulates the Prion-like Aggregation and Activity of CPEB3. Cell Reports 2015; 11:1694-702; PMID:26074071; http://dx.doi.org/ 10.1016/j.celrep.2015.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Richter JD. CPEB: a life in translation. Trends Biochem Sci 2007; 32:279-85; PMID:17481902; http://dx.doi.org/ 10.1016/j.tibs.2007.04.004 [DOI] [PubMed] [Google Scholar]
- [165].Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 2012; 5:14; PMID:22583753; http://dx.doi.org/ 10.1186/1756-6606-5-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Miniaci MC, Kim JH, Puthanveettil SV, Si K, Zhu H, Kandel ER, Bailey CH. Sustained CPEB-dependent local protein synthesis is required to stabilize synaptic growth for persistence of long-term facilitation in Aplysia. Neuron 2008; 59:1024-36; PMID:18817739; http://dx.doi.org/ 10.1016/j.neuron.2008.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Kandel E, Derkatch I, Pavlopoulos E. The Role of Functional Prions in the Persistence of Memory Storage In: Jucker M, Christen Y, eds. Proteopathic Seeds and Neurodegenerative Diseases: Springer; Berlin Heidelberg, 2013:131-52 [Google Scholar]
- [168].Heinrich SU, Lindquist S. Protein-only mechanism induces self-perpetuating changes in the activity of neuronal Aplysia cytoplasmic polyadenylation element binding protein (CPEB). Proc Natl Acad Sci USA 2011; 108:2999-3004; PMID:21270333; http://dx.doi.org/ 10.1073/pnas.1019368108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].White-Grindley E, Li L, Mohammad Khan R, Ren F, Saraf A, Florens L, Si K. Contribution of Orb2A stability in regulated amyloid-like oligomerization of Drosophila Orb2. PLoS Biol 2014; 12:e1001786; PMID:24523662; http://dx.doi.org/ 10.1371/journal.pbio.1001786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Atanesyan L, Gunther V, Dichtl B, Georgiev O, Schaffner W. Polyglutamine tracts as modulators of transcriptional activation from yeast to mammals. Biol Chem 2012; 393:63-70; PMID:22628299; http://dx.doi.org/ 10.1515/BC-2011-252 [DOI] [PubMed] [Google Scholar]
- [171].Hughes RE, Lo RS, Davis C, Strand AD, Neal CL, Olson JM, Fields S. Altered transcription in yeast expressing expanded polyglutamine. Proc Natl Acad Sci U S A 2001; 98:13201-6; PMID:11687606; http://dx.doi.org/ 10.1073/pnas.191498198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Peters TW, Huang M. Protein aggregation and polyasparagine-mediated cellular toxicity in Saccharomyces cerevisiae. Prion 2007; 1:144-53; PMID:19164913; http://dx.doi.org/ 10.4161/pri.1.2.4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, Sakahira H, Siegers K, Hayer-Hartl M, Hartl FU. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell 2004; 15:95-105; PMID:15225551; http://dx.doi.org/ 10.1016/j.molcel.2004.06.029 [DOI] [PubMed] [Google Scholar]
- [174].Harbi D, Harrison PM. Interaction networks of prion, prionogenic and prion-like proteins in budding yeast, and their role in gene regulation. PloS One 2014; 9:e100615; PMID:24972093; http://dx.doi.org/ 10.1371/journal.pone.0100615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Pu S, Wong J, Turner B, Cho E, Wodak SJ. Up-to-date catalogues of yeast protein complexes. Nucleic Acids Res 2009; 37:825-31; PMID:19095691; http://dx.doi.org/ 10.1093/nar/gkn1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Kryndushkin D, Pripuzova N, Burnett BG, Shewmaker F. Non-targeted identification of prions and amyloid-forming proteins from yeast and mammalian cells. J Biol Chem 2013; 288:27100-11; PMID:23926098; http://dx.doi.org/ 10.1074/jbc.M113.485359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Nizhnikov AA, Alexandrov AI, Ryzhova TA, Mitkevich OV, Dergalev AA, Ter-Avanesyan MD, Galkin AP. Proteomic screening for amyloid proteins. PloS One 2014; 9:e116003; PMID:25549323; http://dx.doi.org/ 10.1371/journal.pone.0116003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Wickner RB, Masison DC, Edskes HK. [PSI] and [URE3] as yeast prions. Yeast 1995; 11:1671-85; PMID:8720070; http://dx.doi.org/ 10.1002/yea.320111609 [DOI] [PubMed] [Google Scholar]
- [179].Antonets KS, Nizhnikov AA. SARP: A Novel Algorithm to Assess Compositional Biases in Protein Sequences. Evolutionary Bioinformatics Online 2013; 9:263-73; PMID:23919085 [DOI] [PMC free article] [PubMed] [Google Scholar]