

Abstract
The emergence and spread of carbapenem-resistant Gram-negative pathogens is a global public health problem. The acquisition of metallo-β-lactamases (MBLs) such as NDM-1 is a principle contributor to the emergence of carbapenem-resistant Gram-negative pathogens that threatens the use of penicillin, cephalosporin, and carbapenem antibiotics to treat infections. So far a clinical inhibitor of MBLs that could reverse resistance and re-sensitize resistant Gram-negative pathogens to carbapenems does not exist. Here we have identified a fungal natural product, aspergillomarasmine A (AMA) that is a rapid and potent inhibitor of the NDM-1 enzyme and another clinically relevant MBL, VIM-2. AMA also fully restored the activity of meropenem against Enterobacteriaceae, Acinetobacter spp. and Pseudomonas spp. possessing either VIM or NDM-type alleles. In mice infected with NDM-1-expressing Klebsiella pneumoniae, AMA efficiently restored meropenem activity, demonstrating that a combination of AMA and a carbapenem antibiotic has therapeutic potential to address the clinical challenge of MBL positive carbapenem-resistant Gram-negative pathogens.
The β-lactams (penicillins, cephalosporins, carbapenems & monobactams) are one of the most important and frequently used classes of antibiotics in medicine and are essential in the treatment of serious Gram-negative infections. Since the clinical introduction of penicillins and cephalosporins over 60 years ago, the emergence of β-lactamases, enzymes that hydrolyse the β-lactam ring that is essential for their cell-killing activity, has been an ongoing clinical problem 1. Antibiotic resistance has intensified medicinal chemistry efforts to broaden antibacterial spectrum while shielding the core β-lactam scaffold from β-lactamase-catalyzed hydrolysis. The result has been multiple generations of β-lactams with improved efficacy and tolerance to existing β-lactamases. However, pathogenic bacteria have in turn evolved further resistance mechanisms primarily by acquiring new or modified β-lactamases. This is typified by the emergence of extended spectrum β-lactamases that inactivate many of the latest generation cephalosporins and penicillins (but not carbapenems)2. Consequently, the past two decades have seen significant increases in the utilization of carbapenems such as imipenem and meropenem. Predictably, this increase in carbapenem consumption has been accompanied by the emergence of CRGNP 3,4. In particular, carbapenem-resistant Enterobacteriaceae (CRE) is a growing crisis across the globe 5 as witnessed by recent outbreaks in Chicago 6 and British Columbia 7.
Carbapenemases, β-lactamases that inactivate carbapenems, can be divided into two categories based on their mechanism of β-lactam ring hydrolysis. The first deploy an active site Serine residue that covalently attacks the β-lactam ring e.g. KPC and OXA-48 types 8. The second are MBLs that use Zn2+ atoms to activate a nucleophilic water molecule that opens the ring e.g. VIM and NDM types 9. Several inhibitors of Ser β-lactamases are clinically available as co-drugs where the inhibitor is formulated with a β-lactam antibiotic in order to overcome resistance (e.g. clavulanic acid-amoxicillin, tazobactam-piperacillin, sulbactam-ampicillin and the more recent Ser β-lactamase inhibitor avibactam, which is in phase III clinical trials paired with various cephalosporins) 10. Despite ongoing efforts11,12,, there are no equivalent inhibitors for MBLs in the clinic for practical and technical reasons. First, until recently, MBL-derived CRE was not thought to be a major clinical problem and its rapid increase has outpaced MBL-inhibitor development. Second, the development of a single inhibitor to neutralise key clinically important MBLs (VIM and NDM) has been deemed too technically challenging, and overcoming in vivo toxicity associated with cross reactivity with human metallo-enzymes has been a concern. With the recent emergence of MBLs as a significant clinical threat, a potent and safe inhibitor of MBLs particularly against VIM and NDM would greatly benefit infectious disease management.
We initiated a cell-based screen for inhibitors of the NDM-1 MBL using our in-house collection of DMSO-dissolved natural product extracts derived from environmental microorganisms. To increase the sensitivity of the screen to MBL inhibitor discovery we generated a test strain of Escherichia coli BW25113 in which the bamB and tolC genes were independently deleted (E. coli BW25113ΔbamBΔtolC). BamB is essential to outer membrane porin assembly and disruption results in increased permeability to small molecules13 and TolC is a key component of tripartite small molecule efflux systems such as AcrA-AcrB-TolC that serve to actively eliminate small molecules from the cell 14. Therefore, E. coli BW25113ΔbamBΔtolC increases the sensitivity of the screen to discovery of “hits”. This strain was further modified by integrating the blaNDM-1β-lactamase gene under control of the pLac promoter into the chromosome [E. coli BW25113ΔbamBΔtolCΔaraDAB::pLac(blaNDM-1)]. We screened this strain in the presence of a sub-lethal concentration of meropenem (¼ of the minimum inhibitory concentration 0.125 μg/mL) in combination with ~500 natural product extracts.
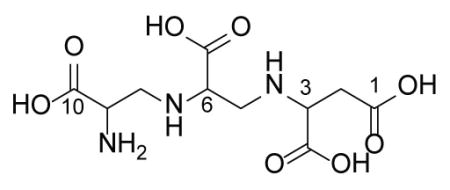
The screen generated one reproducible hit from an extract of a strain of Aspergillus versicolor (as identified by 18S rRNA gene sequence) with excellent ability to restore meropenem antibiotic activity against the E. coli screening strain. The selectivity of the extract to neutralise NDM-1 activity in vitro was confirmed using purified enzyme and the colorimetric β-lactamase substrate nitrocefin and with the carbapenem drug meropenem. Activity-guided purification of the active compound from the fermentation broth of A. versicolor and subsequent detailed chemical characterization identified the MBL inhibitor as aspergillomarasmine A (AMA; Fig. 1a, Extended Data Table 1 and Fig. 1–5), a fungus-derived molecule that was discovered and reported in the early 1960s for its wilting and necrotic activity on plant leaves 15. This molecule was re-evaluated in the 1980s as an inhibitor of angiotensin-converting enzyme (ACE) 16 and in the early 1990s as a pre-clinical candidate for the inhibition of activation of human endothelin 17, a peptide that modulates blood vessel muscle contraction. The activation of this vasoconstricting peptide requires endothelin-converting enzyme, which, like angiotensin-converting enzyme, is a metalloproteinase that shares some mechanistic similarities with MBLs. This previous work demonstrated that AMA was well-tolerated and had low toxicity in mice (LD50 159.8 mg/kg, i.v. compared to EDTA at 28.5 mg/kg) and had no effect on mean atrial blood pressure 18.
Fig. 1. AMA inactivates MBLs.

(a) Chemical structure of AMA. (b) AMA inhibits NDM-1 (●) (IC50 4.0 ± 1.0 μM) and VIM-2 (○) (IC50 of 9.6 ± 2.4 μM). Activity of OXA-48 (■) was unaffected by AMA. (c) Removal of AMA via PD10 column does not restore NDM-1 activity, confirming irreversible inactivation. (d) Addition of excess ZnSO4 restores activity post5 inactivation. (e) The rate of inactivation of NDM-1 and VIM-2 is saturable with [AMA]. (f) ICP-MS confirms depletion of Zn from NDM-1. For all experiments error bars denote standard deviation of three technical replicates.
AMA showed potent in vitro dose-dependent inhibition of NDM-1 and the related MBL VIM-2 (Fig. 1b), with weaker activity against the IMP-7 MBL. AMA had no effect on the Serine β-lactamases TEM-1 and CTX-M-15 as well as the Serine-carbapenemases KPC-2 and OXA-48 (Fig. 1b and Extended Data Fig. 6B). The fact that AMA was identified as an inhibitor of unrelated mammalian metalloproteinases in the past suggested that the compound interacts with the MBL metal centres. Inhibition of NDM-1 was shown to be irreversible after removal of AMA by gel filtration (Fig. 1c), but enzymatic activity could be restored by addition of excess ZnSO4 consistent with a metal depletion mechanism (Fig. 1d). The inhibition of mammalian metalloenyzmes could be seen as a potential side effect however AMA was only able to reduce the activity of rabbit lung ACE by ~ 35% in concentration-response assays (Extended Data Fig. 6a). Extended incubation of the metalloenzymes NDM-1, VIM-2, IMP-7, and ACE at high concentrations of AMA (0.5 mM) in Zn2+-depleted buffer prepared as previously described 19 led to complete inactivation of NDM-1 and VIM-2, and ~ 70% and ~50% inhibition of activity in IMP-7 and ACE, respectively demonstrating selectivity toward NDM and VIM MBLs (Extended Data Table 2). Time-dependent inactivation was shown to be saturable for NDM-1 (Ki = 11 nM, k+2 = 0.0062 s−1) and VIM-2 (Ki = 7 nM, k+2 = 0.0065 s−1) (Fig. 1e), consistent with an inactivation mechanism whereby AMA removes Zn2+ ions similar to known in vitro chelators that interact with subclass B1 MBLs 20. This mechanism of action was confirmed by inductively coupled mass spectrometry that showed a loss of ~ 1.8 Zn equivalents in NDM-1 inactivated by AMA (Fig. 1f).
Systematic titration of AMA and meropenem concentrations against our engineered E. coli and a panel of clinical CRE strains demonstrated that AMA restored meropenem activity consistent with NDM inhibition. Checkerboard MIC studies confirmed the expected synergy between meropenem and AMA only in NDM-1 expressing CRE and not in carbapenem sensitive strains (Fig. 2a, b). Fractional inhibitory concentration (FIC) index values were determined to be < 0.1 for a panel of 16 clinical CRE isolates tested against meropenem and AMA combinations (FIC values of ≤ 0.5 are defined as synergistic 21) (Extended Data Table 3). Potentiation of AMA (8 μg/ml) with meropenem was further investigated using 229 MBL positive (SPM-1, IMP, NDM, AIM and VIM) non-clonal clinical isolates (Enterobacteriaceae, Acinetobacter spp. and Pseudomonas spp.) (Fig. 2c). 76 isolates were also tested that possessed serine carbapenemases, or MBLs and serine carbapenemases. Strains were amassed over a 10 year period as part of a global MBL collection including isolates from Russia, India, Pakistan, Australia, North Africa, and South America. AMA restored meropenem sensitivity (2 μg/ml) in 88% of NDM positives isolates and 90% of VIM positive isolates. Importantly, AMA was active in Pseudomonas spp. (mainly Pseudomonas aeruginosa), which are viewed as a highly challenging model for new antibiotics. AMA showed very little potentiation with SPM-1, IMP and AIM but these MBLs are less numerous than the “global” VIM and NDM MBLs and therefore deemed to be less clinically relevant. The lack of potentiation with IMP-expressing strains correlates well with biochemical data showing less potent inactivation of purified IMP-7 compared to NDM-1 or VIM-2 (Extended Data Table 2).
Fig. 2. AMA potentiates the activity of meropenem against CRGNP.

(a, b) Microdilution checkerboard analysis showing the combined effect of AMA and meropenem selectively against CRE (a, K. pneumoniae N11-2218 MIC meropenem = 32 μg/ml) but not a carbapenem sensitive strain (b, E. coli BW25113 MIC = 0.008–0.016 μg/ml). Heat plots shows the average of two technical replicates. (c) VIM- and NDM-expressing Gram negative pathogens were highly susceptible to meropenem/AMA combination (respectively 2μg/ml and 8 μg/ml) while AIM-, IMP-, and SPM-1-expressing isolates remained resistant.
The resistance profile of NDM-1-positive clinical CRE and the efficacy with which AMA potentiated meropenem activity against NDM-1-positive clinical CRE suggested that AMA would reverse NDM-1-mediated resistance to meropenem in vivo and restore clinical efficacy of this antibiotic. To test this, CD1 mice were infected intraperitoneally with a lethal dose of NDM-1-positive K. pneumoniae N11-2218 to initiate a lethal systemic infection and the effects of meropenem or AMA monotherapy or antibiotic-inhibitor combination therapy was evaluated. Preliminary dosing experiments determined empirically that the bacterial load of NDM-1-positive K. pneumoniae in tissues was unaffected by treatment with AMA alone, and that this strain was resistant to meropenem monotherapy at doses below 50 mg/kg, leading to lethal infection (Extended Data Fig. 7). However, combination therapy with AMA and meropenem significantly reduced the bacterial load in the spleen (Fig. 3a) and to a lesser extent in the liver (Fig. 3b) after a single intraperitoneal dose. Remarkably, while meropenem or AMA alone were unable to prevent lethal infection by NDM-1-positive K. pneumoniae, a single dose of combination therapy led to >95% survival at 5 days following infection (Fig. 3c).
Fig. 3. AMA rescues meropenem activity in vivo.

CD-1 mice were given a sub-lethal dose of K. pneumoniae N11-2218 (meropenem MIC 32 μg/mL) by i.p. injection. (a, b) Groups of mice were treated with a single dose of meropenem (10 mg/kg), a combination of meropenem (10 mg/kg) + AMA (10 mg/kg), or PBS by s.c. injection. Bacterial load in the spleen (a) and liver (b) was determined by selective plating. Data are the means with standard error from two separate experiments (n=7 per group). (c) For survival experiments, CD-1 mice were given a lethal dose of K. pneumoniae N11-2218, and treated with a single dose of meropenem (10 mg/kg), a combination of meropenem (10 mg/kg) + AMA (30 mg/kg), AMA alone (30 mg/kg), or PBS by s.c. injection. Data are the means with standard error from four separate experiments (n=12 per group, except AMA only treatment where n=13). Groups were analyzed using a non-parametric Mann-Whitney t-test. P-values <0.05 were considered statistically significant.
AMA presents a non-toxic candidate for an antibiotic adjuvant that can overcome resistance mediated by NDM and VIM MBLs and re-sensitize carbapenem-resistant Gram-negative pathogens to carbapenems. Active drug/inhibitor combinations continue to be highly successful in the clinic with inhibitors targeted to Ser-β-lactamases 22. AMA presents for the first time, in vitro and in vivo, complementary activity against key MBLs that have become rapidly global and result in significant human morbidity particularly in developing countries. In combination with a β-lactam antibiotic such as meropenem as we show here, resistance can be overcome and antibiotic activity fully restored. AMA, or semi-synthetic derivatives, is therefore an excellent lead for an antibiotic adjuvant co-therapy to address the recent emergence of MBLs in the clinic.
METHODS
Reagents
All enzymes and chemicals of analytical grade were purchased from Sigma-Aldrich, unless otherwise stated. Proton and carbon NMR spectra were recorded on a Bruker 700 MHz spectrometer. Nitrocefin was synthesized as reported previously23
DNA manipulations and plasmid construction
Plasmid DNA purification and gel extraction were performed using the PureLink Quick plasmid miniprep and PureLink Quick gel extraction kits (Invitrogen), respectively. Restriction enzymes were purchased from Fermentas. Primers for PCR DNA amplification were purchased from IDT (Coralville, Iowa). PCR was performed using Phusion High-Fidelity DNA polymerase (Thermo Scientific) using reaction conditions specified by the manufacturer. All ligation reactions were performed using T4 DNA ligase (Thermo Scientific) according to manufacturer’s instructions. All β-lactamase overexpression constructs were generated without the leader peptide in a pET-28b plasmid containing a N-terminal His Tag. Leader peptide sequences were determined using SignalP 4.0 25 All vectors were transformed in E. coli TOP10 chemically competent cells.
Protein purification
VIM-2, IMP-7, CTX-M-15, TEM-1, and OXA-48: An E. coli BL21(DE3) colony transformed with its respective β-lactamase construct was inoculated into LB medium containing 50 μg/mL kanamycin and grown at 37°C. Protein expression was induced with 1 mM IPTG at OD600 0.7 and cultures were incubated overnight at 16°C. Cells were harvested by centrifugation and cell paste from 1 L of culture expressing β-lactamase was washed with 8 mL 0.85 % NaCl, resuspended in buffer containing 20 mM HEPES pH 7.5, 500 mM NaCl, 20 mM imidazole and 20 μM ZnSO4 (for metalloenzymes) then lysed by sonication. Lysate was centrifuged using a Beckman JA 25.50 rotor at 20 000 RPM (48 254 × g) for 45 min at 4°C. The supernatant was applied to a 5-mL HiTrap Ni-NTA column (GE Lifesciences) at a constant flow rate of 3 mL/min. The column was washed with 5 column volumes of the same buffer and step gradients of increasing imidazole were used for wash and elution steps. Fractions containing purified β-lactamase, based on SDS-PAGE, were pooled and dialyzed overnight at 4°C in buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 20% glycerol and 20 μM ZnSO4 for metalloenzymes. NDM-1 was purified as above with the addition of 84 μg SUMO protease. Protease and uncleaved NDM-1 were removed by applying dialyzed solution to a 5-mL HiTrap Ni-NTA column and collecting the flow through fractions. All purified enzymes were verified to be >95% pure as assessed by SDS-PAGE and stored at −20°C.
Cell-based screen
~500 natural product extracts were screened against E. coli BW25113ΔbamBΔtolCΔaraDAB::pLac(blaNDM-1) in combination with 0.125 μg/mL meropenem. Screen was conducted in 96-well plates in duplicate using cation-adjusted Mueller-Hinton broth (CAMHB). Low growth control was 2X MIC meropenem (1 μg/mL) and growth control was ¼ MIC meropenem (0.125 μg/mL). Z′ was determined to be 0.77, indicating an excellent screening window 26.
Purification of AMA
WAC-138 (Aspergillus versicolor) (4L) was evaporated under reduced pressure with 2% (W/V) HP-20 resin (Diaion) to give a residue, WAC138-E. The crude mixture was applied on a HP-20 (100g) column eluting with H2O (1L), 10% MeOH (1L), 25% MeOH (1L), 60% MeOH (1L), and 100% MeOH (1L) to yield five fractions, WAC138-E-1~5. The active fraction WAC138-E-1 was applied to reverse-phase CombiFlash ISCO (RediSep Rf C18, Teledyne) and eluted with a Water-Acetonitrile linear gradient system (0–100% Acetonitrile) to give 65 fractions WAC138-E-1-1~65. The active subfractions WAC138-E-1-5 were passed through a Sephadex LH-20 column (100 ml), eluting with 25% MeOH, to yield 12 subfractions. The active subfractions WAC138-E-1-5-6~8 were combined and recrystallized in 5 ml 1% acetic acid (V/V) to give AMA as white crystals. 1 L of culture yielded ~ 200 mg AMA. (previously reported value = −48°)15. Predicted m/z = 308.1094. Exact m/z = 308.1094. Optical rotations were determined on a Perkin-Elmer 241 polarimeter. Mass spectrometry experiments were conducted using a Bruker Maxis 4G Q/TOF, ESI MS Direct infusion (3 μL/min) in positive ion mode.
IC50 enzyme inhibition assays
Enzyme (NDM-1, 5 nM; VIM-2, 500 pM; CTX-M-15, 500 pM; KPC-2, 5 nM; OXA-48, 1 nM; TEM-1, 100 pM; ACE, 50 nM) was mixed with 30 μM nitrocefin (100 μM nitrocefin for TEM-1; 250 μM furanacryloyl-L-phenylalanylglycylglycine [FAPGG] for ACE24) after a 5–10 minute preincubation with AMA and linear portions of curves were used to analyze data. Metallo-β-lactamases were supplemented with 10 μM ZnSO4. Assays were read in 96-well microplate format at 490 nm using a Spectramax reader (Molecular Devices) at 30–37°C.
Incubation of AMA with metalloenzymes
For Extended Data Table 2, Enzyme (500 nM) was incubated with AMA (500 μM) for 10 minutes. 20 μL of the above was diluted with 180 μL nitrocefin or FAPGG substrate for the following final concentrations: Enzyme (50 nM), FAPGG (50 μM)/nitrocefin (20 μM), AMA (50 μM). Buffer (50 mM HEPES pH 7.5, 300 mM NaCl) was stirred overnight with 2 g/100 mL Chelex-100 (Biorad; Richmond, CA). Assays were read in 96-well microplate format at 490 nm using a Spectramax reader (Molecular Devices) at 37°C.
Reversibility assays
5 mL NDM-1 (500 nM) was incubated either with AMA (100 μM) or without on ice for 1 hour. No enzyme control was buffer alone (Chelex-treated 50 mM HEPES pH 7.5). 2.5 mL was passed through a PD-10 spin column (GE healthcare) following column equilibration with buffer and centrifuged at 2,000×g for 2 minutes. Pre-PD-10 (+ AMA, − AMA, - NDM-1), Post-PD-10 ((+ AMA, − AMA, - NDM-1), and nitrocefin were equilibrated to 30°C. 20 μL of the enzyme solution was added to 180 μL nitrocefin for final enzyme concentration of 50 nM, and nitrocefin of 100 μM and AMA of 10μM. Assays were read in 96-well microplate format at 490 nm using a Spectramax reader (Molecular Devices) for 1 hr at 30°C.
Zn2+ restoration assays
NDM-1 (5 nM) supplemented with 10 μM ZnSO4 was incubated with 20 μM AMA for 15 minutes at 30°C. Nitrocefin (30 μM) and ZnSO4 from 500 nM-40 μM were added to a final volume of 100 μL and absorbance at 490 nm was monitored using a Spectramax reader (Molecular Devices) for 30 minutes at 30°C. Percent residual activity was calculated from no AMA control. Slightly negative percent residual activity was reported as 0.
Inactivation kinetics
NDM-1 (50 nM) was added to 20 μM nitrocefin containing AMA in serial ½ dilutions from 8 μM. Assay was performed in 50 mM HEPES pH 7.5, 200 μL final volume. Assay was read in 96-well microplate format at 490 nm using a Spectramax reader (Molecular Devices) for 10 minutes at 37°C. VIM-2 (10 nM) was added to 20 μM nitrocefin containing AMA in serial ½ dilutions from 16 μM. Assay was performed in 50 mM HEPES pH 7.5, 200 μL final volume. Assay was read in 96-well microplate format at 490 nm using a Spectramax reader (Molecular Devices) for 10 minutes at 37°C. For all assays experiments the offset between reaction initiation and the first read was ~ 6s.
Rate constants characterizing the inactivation of enzyme were calculated based on the dependence of the pseudo-first order rate constant, ki, upon AMA concentration according to the following model:
Where E•Zn, A, E•Zn•A, and Zn•A are the active metalloenzyme, AMA, the ternary metalloenzyme-AMA complex, and AMA-metal complex, respectively. Ki represents the dissociation constant of the ternary complex and k+2 is the rate constant for dissociation of ternary complex into inactivated enzyme and AMA-Zn complex. Steady-state progress curves were fit to the integrated equation:
| (1) |
Where v0 is the initial rate of reporter substrate turnover and ki is the pseudo-first-order inactivation rate constant. The individual values of Ki and k+2 were determined by fitting the value of ki to equation 2 as described previously 18:
| (2) |
Where [A] is the concentration of AMA and [S] and KM were the concentration and KM of the reporter substrate, respectively.
ICP Mass Spectrometry
Inductively coupled mass spectrometry (ICP-MS) was used to analyze the ability of AMA to chelate Zn66 from purified NDM-1 (27–270). The NDM-1 protein was purified as previously described 27 and freshly exchanged using a 15 kDa cutoff dialysis tubing into ICP-MS buffer (20 mM HEPES, 100 mM NaCl, pH 7.5) overnight at 4°C in order to remove any contaminating metals. The protein was concentrated to ~5 mg/mL and varying concentrations of AMA were incubated with the protein samples in triplicate for 3 hours at room temperature with gentle shaking. The protein-AMA samples were again dialyzed overnight at 4°C into ICP-MS buffer using 12–14 kDa cutoff D-tube dialyzer mini (EMD biosciences) microdialysis cassettes. The final protein was diluted to 1 mg/mL in ICP-MS buffer, followed by a 1/40 dilution in an internal standard (10 ug/L Sc45, 1% nitric acid, Inorganic Ventures). Prior to sample analysis, the ICP MS was calibrated using a standard solution containing the metal isotopes of interest (Inorganic Ventures). The protein sample was then transferred by nebulization into a NexION 3000 ICP mass spectrometer (Perkin Elmer). Quantitative analysis was performed in triplicate for each sample with 60 sweeps per reading using the peak-hopping mode with a 50 ms/AMU dwell time for each element. Instrument settings were: rf power (1600 W), integration time (35s), collision gas (Ar40), RPQ voltage (25V) and sample flow rate (4 rpm). Isotope abundance was determined by integrating peak areas using the NexION software program, and the data was represented graphically using Microsoft Excel.
FIC index determination
FIC values were determined by standard methods 20 setting up checkerboards with 8 concentrations of each meropenem and AMA in serial ½ dilutions. Experiments were done in duplicate and the mean used for calculation. The MIC for each compound was the lowest [compound] showing no growth. The FIC for each compound was calculated as the [compound in the presence of co-compound] for a well showing no growth, divided by the MIC for that compound. The FIC index is the sum of the two FICs.
Clinical isolate screening
Various concentrations of AMA were chosen in combination with 2 mg/L of meropenem which is the EUCAST breakpoint for resistance 28 Synergistic properties of the two compounds were examined in a micro-titre tray using BHI as the growth medium (Oxford Science Park, England) and an inoculum of 0.5 MacFarland. All plates contained the control strains Escherichia coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853. Overall, 226 non-clonal clinical isolates (Enterobacteriaceae, P. aeruginosa and Acinetobacter spp.) were challenged containing one of the following MBLs: SPM-1 (n = 17), AIM-1 (n = 8), NDM-1 (n = 67), VIM-type (n = 114) or IMP-type (n = 20). Three E. coli and 5 K. pneumoniae carrying VIM-1 also carried the serine carbapenemase KPC; and 4 E. coli and one K. pneumoniae carrying NDM-1 also possessed the carbapenemase OXA-181. Plates were incubated at 37C and read after 18 hrs. A sub-set of strains (#48) were repeated to examine reproducibility and showed no deviation from the original data.
Animal studies
All animals were housed in specific pathogen-free unit in the Central Animal Facility at McMaster University. All experimental protocols were approved by, and performed in accordance with the McMaster Animal Research Ethics Board. Female, 7–9 week old CD1 mice were purchased from Charles River. Animals were randomized to cages of 5 per group for each experiment. Sample sizes were chosen based on empirical data from pilot experiments and to maximize statistical power. The preparation of treatments, animal infections, and tissue processing was performed by two independent investigators to maximize blinding.
Bacterial Infections
Experiments were based on previously established murine models of infection with β-lactamase-expressing Enterobacteriaceae. 29, 30 Mice were infected intraperitoneally (ip) with a dose of 2×106 colony forming units (cfu) of Klebsiella pneumoniae N11-2218 for all organ bacterial load experiments, or with a dose of 5×107 cfu for all survival experiments. For all organ bacterial load experiments, mice were euthanized 48 hours post infection, and spleen and liver were harvested. Organs were placed into 1 mL sterile PBS on ice, and then homogenized (Mixer Mill 400; Retsch). Organ homogenates were then serially diluted in PBS, and plated on Brilliant Green agar (Oxoid) for cfu enumeration. For survival curves, mice were monitored for endpoint until day 4 post infection. For all experiments, mice were treated 30 minutes post infection with a specified subcutaneous dose of either PBS, meropenem, AMA inhibitor, or a combination of both antibiotic and inhibitor.
Extended Data
Extended Data Figure 1.

1H NMR spectrum of AMA in D2O
Extended Data Figure 2.

13C NMR spectrum of AMA in D2O
Extended Data Figure 3.

1H-1H COSY NMR spectrum of AMA in D2O
Extended Data Figure 4.

1H-13C HSQC NMR spectrum of AMA in D2O
Extended Data Figure 5.

1H-13C HMBC NMR spectrum of AMA in D2O
Extended Data Figure 6. IC50 inhibition profiles for select SBLs and ACE.

Experiments were done as in Fig. 1b for (a) ACE and (b) CTX-M-15 (●), KPC-2 (○), and TEM-1 (■). Error bars denote standard deviation of at least two replicates.
Extended Data Figure 7. Effects of meropenem dosage on spleen burden.

CD-1 mice were infected with K. pneumoniae N11-2218 by i.p. injection. Mice were treated with either PBS (n=6), or various doses of meropenem (n=3 per group) by s.c. injection. Mice were euthanized 48 h after infection, and the bacterial load in the spleen was determined by selective plating. Data are the means with standard error.
Extended Data Table 1.
| 1H and 13C NMR Data of Aspergillomarasmine in D2O
| ||
|---|---|---|
| No. | δH | δC |
| 1 | 176.6 | |
| 2 | 2.73 (dd, J = 17.9, 3.6 Hz) | 35.0 |
| 2.61 (dd, J = 17.9, 9.1 Hz) | ||
| 3 | 3.75 (dd, J = 9.1, 3.6 Hz) | 58.7 |
| 5 | 3.21 (dd, J = 12.8, 4.5 Hz) | 47.2 |
| 3.06 (dd, J = 12.8, 9.5 Hz) | ||
| 6 | 3.35 (dd, J = 9.5, 4.5 Hz) | 59.0 |
| 8 | 3.19 (dd, J = 13.5, 5.7 Hz) | 46.4 |
| 2.80 (dd, J = 13.5, 4.1 Hz) | ||
| 9 | 3.74 (dd, J = 5.7, 4.1 Hz) | 53.8 |
| 10 | 176.2 | |
| 3-COOH | 172.5 | |
| 6-COOH | 172.4 | |
Extended Data Table 2.
Percent Residual Activity following metalloenzyme incubation with AMA.
Initial rates of reporter substrate hydrolysis following incubation of enzyme with AMA over time. % residual activity values are calculated by using a no inhibitor and no enzyme sample as high and low controls, respectively. Error values are the standard deviations of 3 technical replicates
| Enzyme | % Residual Activity (0 min) | % Residual Activity (60 min) |
|---|---|---|
| NDM-1 | 84.3 ± 5.5 | 0.3 ± 0.2 |
| VIM-2 | 59.3 ± 3.6 | 1.0 ± 0.2 |
| IMP-7 | 87.4 ± 5.1 | 35.6 ± 2.0 |
| ACE | 53.1 ± 8.4 | 46.9 ± 8.9 |
Extended Data Table 3.
FIC indices against select clinical isolates of CRE.
Values are from single assay according to CLSI protocols.
| Strain | FIC index |
|---|---|
| M. morganii N10-3295 | 0.09 |
| K. pneumoniae N11-0306 | 0.08 |
| K. pneumoniae N11-0459 | 0.07 |
| K. pneumoniae N11-2218 | 0.07 |
| K. pneumoniae GN529 | 0.05 |
| P. rettgeri GN570 | 0.06 |
| E. cloacae GN574 | 0.06 |
| M. morganii GN575 | 0.06 |
| P. stuartii GN576 | 0.09 |
| E. cloacae GN579 | 0.09 |
| E. coli GN610 | 0.08 |
| K. pneumoniae GN629 | 0.08 |
| E. cloacae GN687 | 0.06 |
| E. coli GN688 | 0.06 |
| K. oxytoca GN942 | 0.05 |
| C. freundii GN978 | 0.09 |
Acknowledgments
We thank Dr Michael Mulvey (Public Health Agency of Canada) and Dr Roberto Melano (Public Health Ontario) for clinical strains. We thank Laura Rossi for her work in constructing the screening strain. AMA inhibition activity on clinical strains by T.R.W. was funded by the UK Medical Research Council (G1100135). This research was funded by a Canadian Institutes of Health Research Grant (MT-13536), Natural Sciences and Engineering Research Council Grant (237480), and by a Canada Research Chair in Infectious Disease Pathogenesis (to B.K.C.) and Antibiotic Biochemistry (to G.D.W.).
Footnotes
Author contributions AMK, GDW, SAR, BKC, TRW, and NCS designed experiments, GDP and AMK designed and engineered the E. coli strains and screened extracts, AMK synthesized nitrocefin, AMK cloned constructs, AMK and DTK purified enzymes, AMK performed enzyme kinetics, WW and AMK fermented WAC-138 and purified AMA, WW elucidated AMA structure, AMK performed FIC experiments, DTK performed ICP-MS, SAR and BKC designed the animal studies, SAR and AMK performed animal experiments, TRW performed clinical isolate screen, AMK and GDW principally wrote the manuscript with input from all.
References
- 1.Frère JM. Beta-Lactamases. Nova Science Publishers; New York: 2011. [Google Scholar]
- 2.Pitout JD, Laupland KB. Extended-spectrum beta-lactamase-producing Enterobacteriaceae: an emerging public-health concern. The Lancet infectious diseases. 2008;8:159–166. doi: 10.1016/S1473-3099(08)70041-0. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States. Atlanta, GA: 2013. [Google Scholar]
- 4.Edelstein MV, et al. Spread of extensively resistant VIM-2-positive ST235 Pseudomonas aeruginosa in Belarus, Kazakhstan, and Russia: a longitudinal epidemiological and clinical study. The Lancet infectious diseases. 2013;13:867–876. doi: 10.1016/S1473-3099(13)70168-3. [DOI] [PubMed] [Google Scholar]
- 5.Patel G, Bonomo RA. “Stormy waters ahead”: global emergence of carbapenemases. Frontiers in microbiology. 2013;4:48. doi: 10.3389/fmicb.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frias M, et al. New Delhi Metallo-β-Lactamase–Producing Escherichia coli Associated with Endoscopic Retrograde Cholangiopancreatography — Illinois, 2013. MMWR. 2014;62:1051. [PMC free article] [PubMed] [Google Scholar]
- 7.Chang Y. Laboratory Trends. BC Public Health Microbiology & Reference Laboratory; Vancouver, BC: Dec 17, 2013. p. 8. [Google Scholar]
- 8.Yigit H, et al. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrobial agents and chemotherapy. 2001;45:1151–1161. doi: 10.1128/AAC.45.4.1151-1161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bush K. Proliferation and significance of clinically relevant beta-lactamases. Annals of the New York Academy of Sciences. 2013;1277:84–90. doi: 10.1111/nyas.12023. [DOI] [PubMed] [Google Scholar]
- 10.Drawz SM, Bonomo RA. Three decades of beta-lactamase inhibitors. Clinical microbiology reviews. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fast W, Sutton LD. Metallo-beta-lactamase: inhibitors and reporter substrates. Biochimica et biophysica acta. 2013;1834:1648–1659. doi: 10.1016/j.bbapap.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 12.Buynak JD. beta-Lactamase inhibitors: a review of the patent literature (2010–2013) Expert Opin Ther Pat. 2013;23:1469–1481. doi: 10.1517/13543776.2013.831071. [DOI] [PubMed] [Google Scholar]
- 13.Ricci DP, Silhavy TJ. The Bam machine: a molecular cooper. Biochimica et biophysica acta. 2012;1818:1067–1084. doi: 10.1016/j.bbamem.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blair JM, Piddock LJ. Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: an update. Current opinion in microbiology. 2009;12:512–519. doi: 10.1016/j.mib.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Haenni AL, et al. Structure chimique des aspergillomarasmines A et B. Helvetica Chimica Acta. 1965;48:729–750. doi: 10.1002/hlca.19650480409. [DOI] [PubMed] [Google Scholar]
- 16.Mikami Y, Suzuki T. Novel microbial inhibitors of angiotensin-converting enzyme, aspergillomarasmines A and B. Agric Biol Chem. 1983;47:2693–2695. [Google Scholar]
- 17.Arai K, et al. Aspergillomarasmine A and B, potent microbial inhibitors of endothelin-converting enzyme. Biosci Biotech Biochem. 1993;57:1944. [Google Scholar]
- 18.Matsuura A, et al. Pharmacological profiles of aspergillomarasmines as endothelin converting enzyme inhibitors. Japanese journal of pharmacology. 1993;63:187–193. doi: 10.1254/jjp.63.187. [DOI] [PubMed] [Google Scholar]
- 19.Hernandez Valladares M, et al. Zn(II) dependence of the Aeromonas hydrophila AE036 metallo-beta-lactamase activity and stability. Biochemistry. 1997;36:11534–11541. doi: 10.1021/bi971056h. [DOI] [PubMed] [Google Scholar]
- 20.Docquier JD, et al. On functional and structural heterogeneity of VIM-type metallo-beta-lactamases. The Journal of antimicrobial chemotherapy. 2003;51:257–266. doi: 10.1093/jac/dkg067. [DOI] [PubMed] [Google Scholar]
- 21.Pillai SK, Moellering RC, Jr, Eliopoulos GM. In: Antibiotics in Laboratory Medicine. Lorian V, editor. Williams & Wilkins; Philadelphia: 2005. pp. 365–440. [Google Scholar]
- 22.Shlaes DM. New beta-lactam-beta-lactamase inhibitor combinations in clinical development. Annals of the New York Academy of Sciences. 2013;1277:105–114. doi: 10.1111/nyas.12010. [DOI] [PubMed] [Google Scholar]
- 23.Holmquist B, Bunning P, Riordan JF. A continuous spectrophotometric assay for angiotensin converting enzyme. Anal Biochem. 1979;95:540–548. doi: 10.1016/0003-2697(79)90769-3. [DOI] [PubMed] [Google Scholar]
- 24.Lee M, Hesek D, Mobashery S. A Practical Synthesis of Nitrocefin. J Org Chem. 2005;70:367–369. doi: 10.1021/jo0487395. [DOI] [PubMed] [Google Scholar]
- 25.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 26.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 27.King DT, Worrall LJ, Gruninger R, Strynadka NC. New Delhi metallo-beta-lactamase: structural insights into beta-lactam recognition and inhibition. J Am Chem Soc. 2012;134:11362–11365. doi: 10.1021/ja303579d. [DOI] [PubMed] [Google Scholar]
- 28.EUCAST. Breakpoint tables for interpretation of MICs and zone diameters. Version 4.0, 2014. 2014. [Google Scholar]
- 29.Marra A, et al. Effect of linezolid on the 50% lethal dose and 50% protective dose in treatment of infections by Gram-negative pathogens in naive and immunosuppressed mice and on the efficacy of ciprofloxacin in an acute murine model of septicemia. Antimicrob Agents Chemother. 2012;56:4671–4675. doi: 10.1128/AAC.00276-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Endimiani A, et al. Evaluation of Ceftazidime and NXL104 in Two Murine Models of Infection Due to KPC-Producing Klebsiella pneumoniae. Antimicrobial Agents and Chemotherapy. 2011;55:82–85. doi: 10.1128/AAC.01198-10. [DOI] [PMC free article] [PubMed] [Google Scholar]