

Abstract
Uniquely among known natural ribozymes that cleave RNA sequence-specifically, the glmS ribozyme-riboswitch employs a small molecule, glucosamine-6-phosphate (GlcN6P) as a catalytic cofactor. In vitro selection was employed to search for coenzyme-independent variants of this ribozyme. In addition to shedding light on the catalytic mechanism of the ribozyme, such variants could resemble the evolutionary ancestors of the modern, GlcN6P-regulated ribozyme-riboswitch. A mutant pool was constructed such that the secondary structure elements, which define the triply-pseudoknotted global fold of the ribozyme, was preserved. A stringent selection scheme that relies on thiol-mercury affinity chromatography for separating active and inactive sequences ultimately yielded a triple mutant with a cleavage rate exceeding 3 min−1 that only requires divalent cations for activity. Mutational analysis demonstrated that a point reversion of the variant toward the wild-type sequence was sufficient to partially restore GlcN6P-dependence, suggesting that coenzyme dependence can be readily be acquired by RNAs that adopt the glmS ribozyme fold. The methods employed to perform this selection experiment are described in detail in this review.
1. Introduction
1.1 Background
The glmS ribozyme-riboswitch is a catalytic, gene-regulatory mRNA domain widespread in Gram-positive bacteria. It is the only natural RNA known to be both a riboswitch and a ribozyme (reviewed in [1, 2]). Riboswitches are mRNA elements that regulate gene expression in cis in response to direct binding to a cognate ligand [3–6]. The glmS gene encodes the protein enzyme glucosamine 6-phosphate (GlcN6P) synthetase, and the ribozyme-riboswitch lies in its 5′-untranslated region. The self-cleavage activity of the ribozyme-riboswitch is activated by binding to GlcN6P, and self-scission leads to degradation of the mRNA [7, 8]. In this manner, the glmS ribozyme performs negative-feedback regulation of the intracellular levels of GlcN6P, an essential precursor for the synthesis of the bacterial cell wall. Because disruption of glmS ribozyme function is deleterious in vivo in the model organism Bacillus subtilis, this catalytic RNA is a candidate antibiotic target, and its molecular mechanism of action has been studied intensively[2].
Activation of the glmS ribozyme by GlcN6P could result from either of two distinct mechanisms. The small molecule could function as an allosteric effector whose binding allows the RNA to achieve a catalytically competent conformation. Alternatively, GlcN6P could be a coenzyme, providing functional groups essential for chemical catalysis to an otherwise incomplete ribozyme active site. Biochemical and crystallographic studies support the latter mechanism. The amine of GlcN6P is positioned to protonate the 5′-OH leaving group of the internal transestrification through which the ribozyme cleaves RNA, thus functioning as a general acid catalyst [9, 10]. Binding to the ribozyme lowers the pKa of the amine group, thereby improving its catalytic efficiency [11–13]. Further arguing against an allosteric mechanism, the ribozyme has been shown readily to adopt its active conformation under physiological Mg2+ concentrations [14, 15], and its rigid, prefolded structure is not perturbed by binding of GlcN6P, or cleavage of the substrate strand [9, 16–19]. The catalytic mechanism of the glmS ribozyme, which relies on binding of a small molecule coenzyme, is unique among all known ribozymes that cleave RNA through internal transestrification, and it suggests the possible involvement of catalytic cofactors in a primordial RNA world [20].
1.2 In vitro selection of glmS ribozyme variants
Several studies have employed in vitro selection to isolate sequence variants of the glmS ribozyme [21, 22]. These studies aimed to investigate the coenzyme selectivity of the ribozyme, to discover variants that could efficiently employ molecules other than GlcN6P as catalytic cofactors, and to explore possible evolutionary origins of this gene-regulatory RNA. Because GlcN6P is a metabolite present in all kingdoms of life, it is difficult to selectively activate the glmS ribozyme in a heterologous context. Identification of a glmS ribozyme variant that employs a coenzyme other than GlcN6P, and which is not activated by GlcN6P itself, would provide a ribozyme and coenzyme set that is orthognal [23] to the natural ribozyme, and thus be useful for synthetic biology applications. Moreover, characterization of such an orthogonal system may also shed light on the molecular basis of coenzyme selectivity by the wild-type ribozyme-riboswitch.
It is thought that complex biological functions arise step-wise, with each step providing a selective advantage [24]. Thus, in the case of the glmS ribozyme-riboswich, it is conceivable that an ancestral molecule existed that was either a ribozyme or a riboswitch, but not both [22]. Phylogenetic analyses have thus far failed to uncover possible molecular ancestors of the glmS ribozyme-riboswitch, but because none of the other known natural ribozymes that cleave RNA through internal transesterification employ a coenzyme, and because other riboswitches that bind sugar derivatives are unknown, a “ribozyme-first” scenario is more likely. That is, the molecular ancestor of this gene-regulatory RNA could have been an RNA-cleaving ribozyme that only subsequently acquired dependence on GlcN6P, and thus the ability to regulate cellular levels of this metabolite.
We recently employed in vitro selection to search for coenzyme-independent variants of the wild-type glmS ribozyme (hereafter glmSWT) [22]. In order to preserve the overall structure of the ribozyme, only nucleotides in the catalytic core that do not participate in forming the characteristic triply-pseudoknotted glmS ribozyme fold were mutagenized. The resulting library of sequence variants was subjected to in vitro selection, allowing us to identify an RNA with three adenosine mutations (hereafter glmSAAA) that is active in cleaving RNA in the absence of GlcN6P. This mutant RNA achieved cleavage rates as high as 3 min−1 in the presence of the cations Ca2+ or Mg2+, but its activity was not enhanced by GlcN6P. SAXS and crystallographic analyses confirmed that glmSAAA adopts the same fold as the wild-type ribozyme. The three adenosine mutations surround the scissile phosphate and abrogate RNA functional groups that coordinate GlcN6P in the wild-type. Biochemical characterization of point mutants of glmSAAA demonstrated that a single nucleotide reversion to the wild-type sequence is sufficient to confer GlcN6P-dependence (neither glmSAAA nor the various revertants have yet been tested in vivo). Thus, in addition to pinpointing the molecular requirements for GlcN6P utilization by the glmS ribozyme, this work revealed a possible path for the evolution of the bacterial ribozyme-riboswitch from an unregulated RNA-cleaving ribozyme that only requires divalent cations for full activity. Because the wild-type glmS ribozyme is very active (close to 100 min−1) and can employ adventitious coenzymes (such as the buffer Tris) in the absence of GlcN6P [11], stringent conditions were needed for in vitro selection. The methods employed for the selection of glmSAAA could be useful for other in vitro ribozyme evolution experiments and are detailed in this article.
2. Methodology
2.1 Mutagenized pool construction
A mutagenized DNA pool based on glmSWT was designed to retain sequences forming the helical elements (secondary structure) of the RNA (Figure 1A). Nucleotides from the core of the ribozyme, along with those from loops and bulges (43 positions in total, Note 1), were each mutagenized by 30% relative to glmSWT (Figure 1A, [22, 25]). Flanking the pool sequences are constant regions. The 5′ constant region contains a bacteriophage T7 RNA polymerase promoter, followed by a 40-nt leader (“substrate”) sequence (optimal length, Note 2). Following transcription, this 40-nt RNA leader can be cleaved in cis if its downstream sequence encodes an active ribozyme. The 3′ constant region contains a primer-binding site for reverse transcription and PCR. For our selection, the 205-nt single-stranded DNA pool was synthesized at the one-micromole scale using standard cyanoethyl phosphoramidite chemistry. For the synthesis, a molar ratio of 0.28:0.23:0.27:0.22 (dA:dG:dC:dT) was used to compensate for differences in coupling efficiencies [26, 27]. Following synthesis, ~60 random isolates from the DNA pool were sequenced (Note 3) to provide an estimate of the actual sequence diversity and mutation rate per position.
Figure 1. In vitro selection of a coenzyme-independent glmS ribozyme variant, glmSAAA.
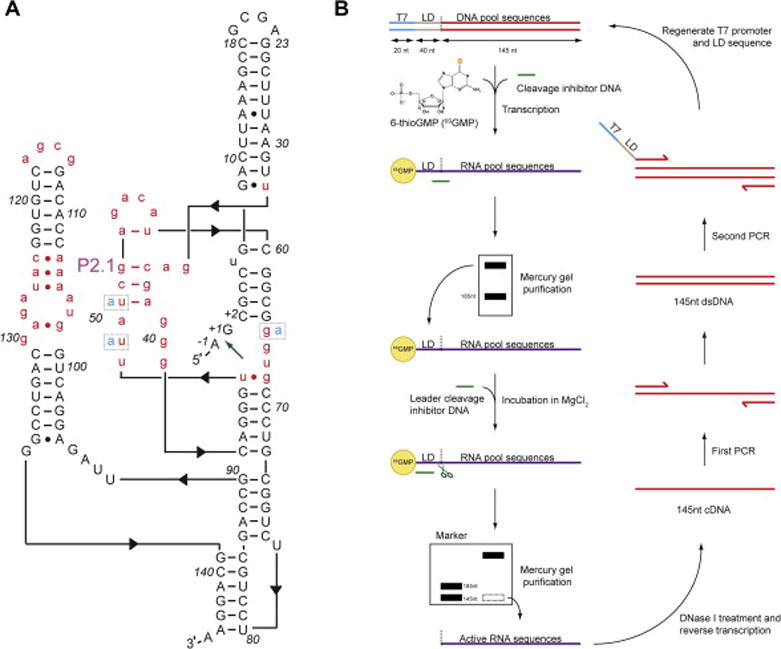
(A) Template design for the starting mutagenized pool based on the tertiary structure [9] of glmSWT. Nucleotides in black upper-case were kept constant, and those in red lower-case were mutagenized by 30% relative to glmSWT. The green arrow indicates position of the scissile phosphate of glmSWT. Positions 49, 51 and 65 (dotted boxes) were found to be mutated to adenosines (cyan) in glmSAAA. (B) In vitro selection scheme employing a thiol-mercury gel (APM-PAGE). Black dotted line, glmSWT cleavage site; LD, leader sequence; T7, T7 RNA polymerase promoter sequence.
2.2 Synthesis of double-stranded DNA template pool
The single-stranded DNA pool (~5 nanomoles, ~3 × 1015 unique sequences) was converted to double-stranded and amplified in a 5 mL PCR reaction containing 20 mM Tris-HCl pH 8.4, 50 mM KCl, 0.2 mM of each dNTP, 1.5 mM MgCl2, 2.5 μM of 5′ selection DNA primer (5′ – TTC TAA TAC GAC TCA CTA TAG GAA ACC ATC ATA ACA GTT CTT GCT ACA AAT CAT TTA TCA GGG CCT GGA CTT AAA GCC GCG AGG – 3′), 2.5 μM 3′ reverse selection primer (5′ – TTC CTG CCC GGA CTG – 3′), 0.03 U/μL Taq DNA polymerase (Invitrogen), using the following cycling program in a thermal cycler with a heated lid: (1) 72°C for 5 minutes (3′-primer extension); (2) 94°C for 4 minutes (denaturation); (3) 55°C for 5 minutes (annealing); (4) 72°C for 8 minutes (primer extension). Steps (2) to (4) were repeated for 4 cycles, which should produce 8 copies of each unique sequence in the starting population (2n−1, where n is the number of PCR cycles). The resulting DNA pool was purified by extraction with one volume of 50% (v/v) Tris-saturated phenol in chloroform, and concentrated by precipitation by addition of 2.5 volumes of ethanol.
2.3 Transcription and purification of RNA pools for selection
2.3.1 First selection round
Approximately 20 nanomoles of DNA were transcribed into RNA (50 mM HEPES-KCl pH 7.5, 25 mM MgCl2, 5 mM of each NTPs, 5 U/μL of T7 RNA polymerase, Figure 1B), in the presence of 200 μM of a 33-nt DNA oligonucleotide (5′ - GCT TTA AGT CCA GGC GCT GAT AAA TGA TTT GTA – 3′) that hybridizes to the RNA region encoding the potential cleavage site. This functions to inhibit ribozyme cleavage and retain full-length RNA constructs during the transcription process where millimolar concentrations of Mg2+ as well as other molecules that may serve as adventitious cofactors are present (Figure 3). Transcription was terminated after an incubation of 2 hrs at 37°C by adding one volume of loading buffer (90% formamide, 25 mM EDTA), and purified by electrophoresis through a denaturing 8% PAGE. The 185-nt RNA was visualized by UV shadowing, excised, and eluted overnight by electro-elution or passively with water. The eluted RNA was brought up to 100 mM NaCl and precipitated with 2.5 volumes of chilled ethanol.
Figure 3. Self-cleavage of glmSWT is blocked in the presence of a cleavage inhibitor DNA.
Autoradiogram of a PAGE analysis of the transcription of body-labeled cis-cleaving glmSWT in the presence or absence of a 33-nt DNA oligonucleotide complementary to sequences flanking the cleavage site.
2.3.2 Subsequent selection rounds
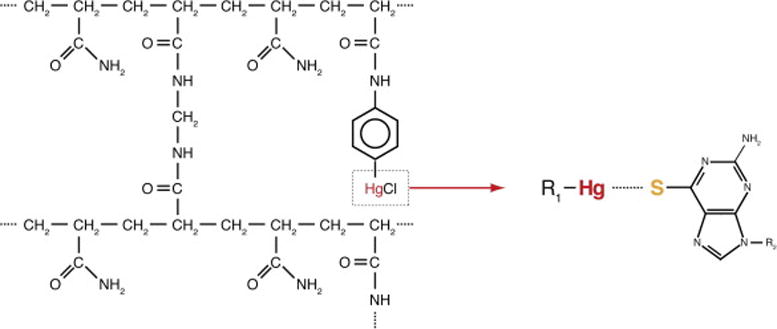
To increase the stringency of the selection, sulfur-mercury affinity PAGE was employed starting from the second selection round (Figure 1B). In each round, sulfur-mercury affinity PAGE was performed twice; first for purification of a 5′ thiol-tagged RNA pool, and then for the selection of active ribozyme sequences. Sulfur-mercury affinity PAGE exploits the strong interaction between thiol-tagged RNAs and mercury-containing compounds. N-acryloylaminophenyl-mercuric acetate (APM) can be used to incorporate mercury into polyacrylamide gels during polymerization (Figure 2, [22, 27]). Pool RNAs were 5′ thiol-tagged by including 6-thioguanosine monophosphate (6SGMP) during transcription (one of the tautomeric forms of 6SGMP has a free thiol). RNAs synthesized in this manner can have 6-thioguanosine only at their 5′ termini, since the monophosphate form of the nucleotide cannot serve as a substrate for transcription elongation. 100 μL reactions contained 1 nmol DNA template, 8 mM 6SGMP, and 2 mM NTPs (reduced from 2–5 mM in normal conditions; Note 4; [28, 29]). The reduced NTP concentration enhances incorporation of 6SGMP during transcription initiation. Following the reaction, the thiol-tagged RNAs were purified through an 8% denaturing PAGE containing 2 μg APM per mL of gel solution (otherwise comprised of acrylamide: bisacrylamide (29:1), urea, TBE buffer, and water). RNAs with a 5′ 6SGMP have a reduced electrophoretic mobility in the APM gel compared to unlabelled RNAs [27, 30]. Gel slices containing the labeled RNAs were recovered by elution overnight in 1 mM dithiothreitol (DTT), which competes for binding to mercury, helping release the thiol-tagged RNAs from the gel.
Figure 2. Chemical basis of thiol-mercury affinity PAGE.
(A) Structure of N-acryloylaminophenyl-mercuric acetate (APM) incorporated into the polyacrylamide gel matrix. The mercury derived from APM interacts strongly with thiol-containing molecules, enabling APM-PAGE separation between inactive RNAs that bear an intact 5′-terminal 6-thioguanosine and active RNAs that have cleaved their leader sequence.
2.4 Selection of active ribozyme sequences
2.4.1 Size selection in the first round
In the initial round, ~5 nanomoles of transcribed RNAs (at 5 μM) were incubated overnight at room temperature in 25 mM MgCl2 (50 mM HEPES, 200 mM KCl, pH 7.5) together with 40 μM of a cleavage inhibitor DNA (5′ - AAA TGA TTT GTA GCA AGA ACT GTT ATG ATG G – 3′), which hybridizes to the first 31 nucleotides of the RNA leader sequence. This DNA oligonucleotide was added in excess to the RNAs to prevent catalytic scission within the 5′ leader sequence, i.e., to enforce cleavage at the wild-type cleavage site. Following incubation, the reaction was stopped by the addition of 25 mM EDTA, desalted with water using a centrifugal microfilter (Amicon, 30 kDa nominal molecular weight cut-off), and electrophoresed through a 8% denaturing PAGE. Markers of unrelated RNAs with lengths 185-nt, 145-nt, and 40-nt were loaded in an adjacent lane to indicate the position of the active ribozymes (145-nt) after cis cleavage of the leader sequence at the wild-type locus. To enhance stringency, subsequent selection steps employed mercury-thiol affinity electrophoresis.
2.4.2 Thiol-mercury affinity PAGE selection in subsequent rounds
In subsequent rounds, purified RNA pool sequences with a 5′- thiol tag were incubated in Mg2+ buffer with a DNA that inhibits cleavage within the leader sequence, as described, and purified through a second APM-PAGE (Figure 1B and Figure 2). Active ribozymes with their leader sequence removed would migrate faster than the inactive RNAs that retained their 5′-thiol tag. The combined effect of the difference in length and the presence or absence of the 5′- 6SGMP tag between the inactive and active RNA sequences results in a large difference in electrophoretic mobility. As a size marker, a wild-type glmS ribozyme was incubated in parallel in 25 mM MgCl2 and 200 μM GlcN6P to induce self-cleavage, and electrophoresed in an adjacent lane on the APM gel (see Note 5). In the later selection rounds, the selection pressure was increased to promote the isolation of the fastest ribozymes by reducing the incubation time. In our selection, we halved the incubation time in each round starting from round 5 and continued for 4 additional rounds.
2.5 Amplification of active sequences
2.5.1 DNase I digestion and reverse transcription
Following passive overnight elution (into 1 mM DTT in the case of APM gels), RNAs were ethanol precipitated, and treated with 1 U/μL of DNase I (Roche) at 37°C for 30 minutes (400 mM Tris-HCl, 100 mM NaCl, 60 mM MgCl2, 10 mM CaCl2, pH 7.9). To inactivate the nuclease, the reaction was incubated at 75 °C for 20 minutes, followed by extraction with one volume of phenol and chloroform, and precipitation with 2.5 volumes of chilled ethanol. These steps ensure that DNAs present during the transcription and selection steps are removed and not enriched in the subsequent PCR amplification. Following DNase treatment, the active RNAs were reverse-transcribed to generate single-stranded cDNAs (50 mM Tris-HCl, 75 mM KCl, 3 mM MgCl2, 10 mM DTT, 560 μM of each dNTP, dNTP, pH 7.5, 10 U/μL Superscript II (Invitrogen), incubation at 48°C for 1 hr). The reaction was stopped by heating the sample at 90 °C for 20 minutes after adjusting to 100 mM KOH, in order to degrade the RNAs and denature the enzyme. This was followed by neutralization by adjusting to 250 mM Tris-HCl, pH 8.5.
2.5.2 PCR amplification and enrichment of active sequences
The reverse-transcribed cDNAs were amplified in two sequential PCR reactions. The first was performed under standard conditions in 100 μL volume using the 5′ internal primer, which corresponds to the first 20-nt of the RNA selection pool minus the 40-nt leader sequence (5′-GCG CCT GGA CTT AAA GCC GC -3′), and the 3′ selection primer. Amplification using the 5′ internal primer in this first PCR step is essential in the selection as it avoids amplifying 185-nt cDNAs generated from reverse-transcription of full-length, inactive RNAs. The 145-nt PCR products, after ~12 cycles of PCR (see Note 6), were analyzed and purified using a 1.5% agarose gel and recovered using an agarose gel purification kit (Qiagen).
In the second PCR reaction (500 μL), ~0.1 nmols of DNA recovered from the first PCR amplification were used as template and amplified using the 5′ selection and 3′ reverse-selection primers. This PCR reaction regenerates the T7 promoter and the 40-nt substrate sequences, both of which are needed for synthesis of full-length RNAs for the next selection round (Figure 1B). Starting from round 4, the leader sequence of the 5′ selection primer was changed every two rounds to avoid enrichment of ribozymes that are specific to particular leader sequences.
The PCR amplification step can also be used to increase sequence diversity, thereby enhancing the likelihood of finding active ribozymes that are close in sequence space to RNAs abundant in the selection pool. This technique (mutagenic PCR) uses skewed dNTP and metal ion concentrations (20 mM Tris-HCl pH 8.4, 50 mM KCl, 7 mM MgCl2, 0.5 mM MnCl2, 0.2 mM dATPs, 0.2 mM dGTPs, 1 mM dCTPs, 1 mM dTTPs) and a prolonged PCR cycling program that make the Taq DNA polymerase more prone to adding incorrect nucleotides [31]. A typical 30-cycle mutagenic PCR will introduce mutations at a frequency of ~0.7% per position.
2.6 Sequence analysis of the selection
Starting from round 4, the amplified DNA pool at the end of each round was cloned and ~30 isolates were sequenced as described (see Note 3). Sequences were aligned using the Clustal algorithm from MegAlign (DNASTAR) and compared with the glmSWT sequence. Duplicate sequences, or isolates that shared recognizable sequence features in the mutagenized regions, were not observed until round 6. At the end of the final selection round, ~70 isolates were sequenced. Of these, 26 retained the core secondary structure that characterizes the wild-type glmS ribozyme. Alignment of the sequences of these 26 isolates revealed 3 highly conserved mutations. The glmSAAA variant containing just these 3 mutations was synthesized and further characterized.
3. Concluding remarks
Pioneering experiments with the Tetrahymena group I intron demonstrated that one RNA, or close sequence variants, can catalyze a number of biochemically distinct reactions such as self-splicing [32], RNA cleavage [33], aminoacyl ester hydrolysis [34], template-dependent oligonucleotide polymerization [35] etc., Nonetheless, for many of those reactions, the ribozymes derived from the Tetrahymena intron exhibited only modest catalytic activity. Advances in the structural elucidation of RNAs with compact, stable folds [36] provide an opportunity to mutagenize ribozymes rationally, in ways that are likely to preserve their overall structural integrity, and to subject them to in vitro selection for new biochemical activities. Our discovery that three point mutations suffice to convert the GlcN6P-dependent glmS ribozyme into a highly active and strictly divalent-cation-dependent catalytic RNA that nonetheless retains the overall three-dimensional structure of the wild-type suggests that only a small number of nucleotides can be responsible for the specific reactivity of a given RNA (see also Ref. [22, 37]). Thus, in vitro selection applied to stable RNA scaffolds is likely to result in the discovery of families of closely related RNAs that catalyze several biochemically distinct transformations.
Acknowledgments
We thank S. Bachas, N. Baird, M. Chen, J. Hogg, C. Jones, J. Posakony, R. Trachman, K. Warner, and J. Zhang for discussions. M.W.L. was a Croucher Foundation Postdoctoral Fellow. This work was supported in part by the Intramural Program of the National Heart, Lung and Blood Institute, NIH.
Abbreviations
- APM
N-acryloylaminophenylmercuric acetate
- dNTPs
deoxyribonucleotide triphosphates
- DTT
dithiothreitol
- GlcN6P
glucosamine-6-phosphate
- glmSWT
wild-type glmS ribozyme
- 6SGMP
6-thioguanosine monophosphate
- glmSAAA
triple adenosine-mutant glmS ribozyme
- nt
nucleotides
- NTPs
ribonucleotide triphosphates
- PAGE
polyacrylamide gel electrophoresis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The 30% mutagenic rate was chosen because it provides sufficient sequence diversity (a starting pool of ~3 × 1015 unique sequences with an average of ~13 mutations per sequence), while biasing the sampling of sequence space in the proximity of the glmSWT ribozyme. Higher mutational frequencies (i.e., > 35%) can be used, but this increases the chance of generating sequences with alternative structural folds, leading to isolation of ribozymes that are unrelated to glmSWT [38–40]. For an in-depth review on random and doped selection pool design, see [25].
This 40-nt DNA sequence, after transcription, will serve as the RNA “substrate” or leader sequence, which will be removed by active cis-cleaving ribozymes. This sequence can vary in length, but should be > 30-nt to ensure that there is a sufficient separation between the cleaved and uncleaved RNAs during denaturing PAGE purification to minimize cross-contamination. Long leader sequences (i.e., > 100-nt) are best avoided to prevent their becoming targets of adventitious cleavage reactions.
The PCR-amplified pool can be directly cloned using a TA TOPO cloning kit (Invitrogen), transformed into competent bacterial cells, and individual colonies sequenced (alternatively, next-gen sequencing of an aliquot of the pool can be used to provide the same information [41]).
The optimal ratio between the concentrations of 6SGMP and the NTPs for maximizing both the transcription yield and 6SGMP incorporation efficiency will depend on the specifics of each RNA pool. In our experiments, ~40% of the transcribed RNAs initiated with 6SGMP.
The 185-nt cis-cleaving glmS ribozyme construct (40-nt leader sequence plus 145-nt Thermoanaerobacter tengcongensis glmS ribozyme, [9]) was used as a positive control in each round and subjected to the same selection process as the pool sequences. The glmS RNA served both, as a marker during APM gel analysis, and to ensure that each step in the selection cycle (i.e., tagging of RNAs with 5′- 6SGMP, cleavage of leader sequence in Mg2+ etc.) took place as planned.
In each selection round, an aliquot (~10%) of the reverse transcription reaction was removed prior to the addition of the Superscript II enzyme, and used as a negative control. For the selection pool (reverse transcriptase added), the subsequent PCR step generated an observable 145-nt product after ~13 cycles. For the negative control, on the other hand, a similar product was not detectable (ethidium bromide stained 1.5% agarose gel, UV detection) until at least after 20 rounds of PCR amplification. This difference in the number of cycles can be used to provide an estimate of the amount of contaminant DNAs that have been carried over during the selection, which affects the overall stringency of the selection experiment.
References
- 1.Ferré-D’Amaré AR, Scott WG. Cold Spring Harbor Persp Biol. 2010 doi: 10.1101/cshperspect.a003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferré-D’Amaré AR. Q Rev Biophys. 2010;43:423–447. doi: 10.1017/S0033583510000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grundy FJ, Henkin TM. Cell. 1993;74:475–482. doi: 10.1016/0092-8674(93)80049-k. [DOI] [PubMed] [Google Scholar]
- 4.Mironov AS, Gusarov I, Rafikov R, Lopez LE, Shatalin K, Kreneva RA, Perumov DA, Nudler E. Cell. 2002;111:747–756. doi: 10.1016/s0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 5.Winkler W, Nahvi A, Breaker RR. Nature. 2002;419:952–956. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Lau MW, Ferré-D’Amaré AR. Biochemistry. 2010;49:9123–9131. doi: 10.1021/bi1012645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Nature. 2004;428:281–286. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 8.Collins JA, Irnov I, Baker S, Winkler WC. Genes Dev. 2007;21:3356–3368. doi: 10.1101/gad.1605307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein DJ, Ferré-D’Amaré AR. Science. 2006;313:1752–1756. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- 10.Viladoms J, Fedor MJ. J Am Chem Soc. 2012;134:19043–19049. doi: 10.1021/ja307021f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCarthy TJ, Plog MA, Floy SA, Jansen JA, Soukup JK, Soukup GA. Chem Biol. 2005;12:1221–1226. doi: 10.1016/j.chembiol.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Davis JH, Dunican BF, Strobel SA. Biochemistry. 2011;50:7236–7242. doi: 10.1021/bi200471c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong B, Klein DJ, Ferré-D’Amaré AR, Carey PR. J Am Chem Soc. 2011;133:14188–14191. doi: 10.1021/ja205185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hampel KJ, Tinsley MM. Biochemistry. 2006;45:7861–7871. doi: 10.1021/bi060337z. [DOI] [PubMed] [Google Scholar]
- 15.Tinsley RA, Furchak JR, Walter NG. RNA. 2007;13:468–477. doi: 10.1261/rna.341807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein DJ, Wilkinson SR, Been MD, Ferré-D’Amaré AR. J Mol Biol. 2007;373:178–189. doi: 10.1016/j.jmb.2007.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klein DJ, Been MD, Ferré-D’Amaré AR. J Am Chem Soc. 2007;129:14858–14859. doi: 10.1021/ja0768441. [DOI] [PubMed] [Google Scholar]
- 18.Cochrane JC, Lipchock SV, Strobel SA. Chem Biol. 2007;14:97–105. doi: 10.1016/j.chembiol.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cochrane JC, Lipchock SV, Smith KD, Strobel SA. Biochemistry. 2009;48:3239–3246. doi: 10.1021/bi802069p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferré-D’Amaré AR. Phil Trans Royal Soc B. 2011;366:2942–2948. doi: 10.1098/rstb.2011.0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Link K, Guo L, Breaker R. Nucleic Acids Res. 2006;34:4968–4975. doi: 10.1093/nar/gkl643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lau MWL, Ferré-D’Amaré AR. Nat Chem Biol. 2013;9:805–810. doi: 10.1038/nchembio.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixon N, Duncan JN, Geerlings T, Dunstan MS, Mccarthy JEG, Leys D, Micklefield J. Proc Natl Acad Sci USA. 2010;107:2830–2835. doi: 10.1073/pnas.0911209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darwin C. On the Origin of Species. London: John Murray; 1872. [Google Scholar]
- 25.Hall B, Micheletti JM, Satya P, Ogle K, Pollard J, Ellington AD. Curr Prot Mol Biol. 2009:24.2.1–24.2.27. doi: 10.1002/0471142727.mb2402s88. [DOI] [PubMed] [Google Scholar]
- 26.Zaher HS, Unrau PJ. Methods Mol Biol. 2005;288:359–378. doi: 10.1385/1-59259-823-4:359. [DOI] [PubMed] [Google Scholar]
- 27.Lau MW, Cadieux KE, Unrau PJ. J Am Chem Soc. 2004;126:15686–15693. doi: 10.1021/ja045387a. [DOI] [PubMed] [Google Scholar]
- 28.Ferré-D’Amaré AR, Doudna JA. Nucleic Acids Res. 1996;24:977–978. doi: 10.1093/nar/24.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rupert PB, Ferré-D’Amaré AR. Methods Mol Biol. 2004;252:303–311. doi: 10.1385/1-59259-746-7:303. [DOI] [PubMed] [Google Scholar]
- 30.Lau MW, Unrau PJ. Chem Biol. 2009;16:815–825. doi: 10.1016/j.chembiol.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Cadwell RC, Joyce GF. Genome Res. 1994;3:S136–S140. doi: 10.1101/gr.3.6.s136. [DOI] [PubMed] [Google Scholar]
- 32.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 33.Zaug AJ, Cech TR. Science. 1986;231:470–475. doi: 10.1126/science.3941911. [DOI] [PubMed] [Google Scholar]
- 34.Piccirilli JA, McConnell TS, Zaug AJ, Noller HF, Cech TR. Science. 1992;256:1420–1424. doi: 10.1126/science.1604316. [DOI] [PubMed] [Google Scholar]
- 35.Doudna JA, Usman N, Szostak JW. Biochemistry. 1993;32:2111–2115. doi: 10.1021/bi00059a032. [DOI] [PubMed] [Google Scholar]
- 36.Ferré-D’Amaré AR, Doudna JA. Annu Rev Biophys Biomolec Struct. 1999;28:57–73. doi: 10.1146/annurev.biophys.28.1.57. [DOI] [PubMed] [Google Scholar]
- 37.Pitt JN, Ferré-D’Amaré AR. Science. 2010;330:376–379. doi: 10.1126/science.1192001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson DS, Szostak JW. Annu Rev Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- 39.Bartel DP, Szostak JW. Science. 1993;261:1411–1418. doi: 10.1126/science.7690155. [DOI] [PubMed] [Google Scholar]
- 40.Bartel DP, Zapp ML, Green MR, Szostak JW. Cell. 1991;67:529–536. doi: 10.1016/0092-8674(91)90527-6. [DOI] [PubMed] [Google Scholar]
- 41.Pitt JN, Rajapakse I, Ferré-D’Amaré AR. Nucleic Acids Res. 2010;38:7908–7915. doi: 10.1093/nar/gkq661. [DOI] [PMC free article] [PubMed] [Google Scholar]