

Summary
Dysregulated lipid metabolism is a characteristic of malignancies. Sterol regulatory element-binding protein-1 (SREBP-1), a transcription factor playing a central role in lipid metabolism, is highly activated in malignancies. Here, we unraveled a previously unappreciated link between miR-29 and the SCAP/SREBP-1 pathway in glioblastoma (GBM) growth. EGFR signaling enhances miR-29 expression in GBM cells via upregulation of SCAP/SREBP-1, and SREBP-1 activates miR-29 expression via binding to specific sites in its promoter. In turn, miR-29 inhibits SCAP and SREBP-1 expression by interacting with their 3′-untranslated regions. miR-29 transfection suppressed lipid synthesis and GBM cell growth, which were rescued by addition of fatty acids or N-terminal SREBP-1 expression. Xenograft studies showed that miR-29 mimics significantly inhibit GBM growth and prolong the survival of GBM-bearing mice. Our study reveals a previously unrecognized negative feedback loop in SCAP/SREBP-1 signaling mediated by miR-29, and suggests that miR-29 treatment may represent an effective means to target GBM.
Keywords: miR-29, SCAP, SREBP-1, EGFR, lipid metabolism, Glioblastoma
eTOC Blurb
In this study, Peng et al. unravel a negative feedback loop mediated by miR-29 in SCAP/SREBP-1 signaling, advancing our understanding of lipid metabolism. The study also indicates that miR-29-mediated inhibition of SCAP/SREBP-1 may be a promising approach for targeting glioblastoma.
Introduction
Regulation of lipid homeostasis is critical for cell growth and function (Nohturfft and Zhang, 2009; van Meer et al., 2008). Sterol regulatory element-binding proteins (SREBPs), a family of endoplasmic reticulum (ER)-bound transcription factors, play a central role in the synthesis of fatty acids, phospholipids, and cholesterol (Goldstein et al., 2006; Jeon and Osborne, 2012; Shao and Espenshade, 2012). There are two SREBP genes, SREBF1 and SREBF2, in mammalians. SREBF1 encodes two isoforms, SREBP-1a and SREBP-1c, which differ in their first exon, and regulate fatty acid synthesis; SREBF2 encodes the SREBP-2 protein, which mainly controls cholesterol synthesis (Goldstein et al., 2006; Horton et al., 2002; Horton et al., 2003).
SREBPs are synthesized as inactive precursors and bind to SREBP-cleavage activating protein (SCAP) in the ER membrane (Goldstein et al., 2006). Large amount of evidence demonstrate that SREBP activation is regulated by a sterol-mediated negative feedback loop (Goldstein et al., 2006). High amounts of cholesterol bind to SCAP and enhance its association with ER-resident protein, insulin induced gene protein (Insig), which retains the SCAP/SREBP complex in the ER (Adams et al., 2004; Goldstein et al., 2006; Sun et al., 2007; Yang et al., 2002). Intriguingly, a study from Brown & Goldstein laboratory (Goldstein et al., 2006; Radhakrishnan et al., 2008; Sun et al., 2007) shows that as low as 5% reduction in ER cholesterol is sufficient to trigger the conformational change of SCAP and promote its dissociation from Insig, allowing the COPII complex-mediated vesicular transport of SCAP/SREBP from the ER to the Golgi for subsequent proteolytic SREBP activation (Goldstein et al., 2006; Radhakrishnan et al., 2008; Sun et al., 2007). This study raises the question of whether different regulatory factors might participate in SCAP/SREBP signaling in order to tightly control lipid homeostasis.
Glioblastoma (GBM) is the most deadly primary brain tumor (Bell and Guo, 2012; Guo et al., 2013; Guo et al., 2014; Ru et al., 2013; Wen and Kesari, 2008). About 50% of GBM patients express mutated or amplified EGFR (Brennan et al., 2013; Furnari et al., 2015). Our recent studies have demonstrated that oncogenic EGFR signaling activates SREBP-1 and its regulated lipid synthesis and uptake pathways via upregulation of SCAP, to promote rapid GBM growth (Cheng et al., 2015; Guo, 2016; Guo et al., 2009a; Guo et al., 2009b; Guo et al., 2011). However, the mechanisms by which cancer cells regulate lipid metabolism in the context of constitutively activated oncogenic signaling are still poorly understood.
Recently, several microRNAs (miR), the small noncoding RNAs that play an important role in the regulation of various cellular processes including apoptosis, differentiation and tumorigenesis (Schickel et al., 2008), have been reported to participate in the regulation of lipid metabolism (Fernandez-Hernando et al., 2011; Jeon et al., 2013). In particular, miR-29 has been shown to suppress GBM cell growth in vitro when transfected (Xu et al., 2015) and to connect with lipid metabolism pathways in liver and hepatoma cells (Kurtz et al., 2014; Xu et al., 2016). Thus, we asked the question whether miR-29 is involved in the EGFR signaling-regulated SCAP/SREBP-1 pathway and GBM tumor growth.
Results
EGFR signaling promotes miR-29 expression via upregulation of SCAP/SREBP-1
Our recent studies demonstrated that EGFR signaling upregulates SCAP and activates SREBP-1 (Cheng et al., 2015; Guo, 2016; Guo et al., 2009b; Guo et al., 2011). Since miR-29 has been reported to be involved in lipid metabolism (Kurtz et al., 2015; Kurtz et al., 2014; Xu et al., 2016), we performed a correlation analysis between SREBF1 expression and miR-29 levels based on genomic EGFR status in a large cohort of GBM patient samples (TCGA database) (Cerami et al., 2012; Gao et al., 2013). Interestingly, in GBM patients with altered EGFR (amplification and/or mutation), the data show that expression of the 3 members of the miR-29 family, miR-29a, -29b and -29c (Zhang et al., 2012), positively correlated with the expression of SREBF1 (Figures 1A and S1A). In contrast, no significant correlation was observed in GBM patients with normal EGFR (no amplification or mutation) (Figure S1B). These data suggest that miR-29 is possibly involved in oncogenic EGFR regulated-SREBP-1 signaling.
Figure 1. EGFR signaling promotes miR-29 expression via upregulation of SCAP/SREBP-1.

(A) Pearson correlation analysis of the expression of SREBF1 and miR-29 family (-29a, -29b and 29c) in a large cohort of GBM patient tissues with altered genomic EGFR (amplification and/or mutation, n=132) from the TCGA database.
(B, C) Real-time PCR analysis of the levels of pri-miR29a/b1, pri-miR29b2/c, and mature miR-29 (mean ± SD) (B) or western blot analysis of the indicated proteins (C) in U87/EGFR cells stimulated with EGF (50 ng/ml) at the indicated times after serum starvation for 24 hr. Statistical significance was determined by Student’s t-test (n=3); *p<0.001 compared with control cells without EGF stimulation (0 hr).
(D, E) Real-time PCR analysis of the levels of pri-miR29a/b1, pri-miR29b2/c, and mature miR-29 (mean ± SD) (D) or western blot analysis of the indicated proteins (E) in U87/EGFR cells stimulated with EGF (50 ng/ml) for 12 hr after pre-treatment with various kinase inhibitors, i.e., Erlotinib (Erloti, 10 μM) for EGFR, BKM120 (BKM, 10 μM) for PI3K, and MK2206 (MK, 10 μM) for Akt, for 1 hr. Statistical significance was determined by Student’s t-test (n=3); #p<0.05, +p<0.01 and *p<0.001.
(F, G) Western blot analysis of the indicated proteins (F) or real-time PCR analysis of pri-miR29a/b1, pri-miR29b2/c and mature miR-29a/b/c (mean ± SD) (G) in U87/EGFR cells stimulated with EGF (50 ng/ml) for 12 hr after knockdown of SCAP by specific siRNA. Statistical significance was determined by Student’s t-test (n=3). +p<0.01 and *p<0.001.
(H, I) Western blot analysis of the indicated proteins (H) or real-time PCR analysis of pri-miR29a/b1, pri-miR29b2/c and mature miR-29a/b/c (mean ± SD) (I) in U87/EGFR cells stimulated with EGF (50 ng/ml) for 12 hr after knockdown of SREBP-1 by specific siRNA. Statistical significance was determined by Student’s t-test (n=3); +p<0.01 and *p<0.001.
(J–M) Western blot analysis of lysates (J and L) or real-time PCR analysis of pri-miR-29/mature miR-29 (mean ± SD) (K and M) from human primary GBM83 cells after siRNA transfection (50 nM) against SCAP (J and K) or SREBP-1 (L and M) for 48 hr. Statistical significance was determined by Student’s t-test; *p<0.01 compared with siControl cells.
See also Figures S1.
We then determined the intrinsic connection between EGFR signaling, SCAP/SREBP-1 activation and miR-29 expression in GBM U87 cells that were stably expressing EGFR (U87/EGFR) (Guo et al., 2009b). The data show that activating EGFR/PI3K/Akt signaling with EGF significantly enhanced the expression of pri-miR-29a/b1, -29b2/c and mature miR-29a, -29b and -29c in a time-dependent manner (Figures 1B, 1C and S1C) (Zhang et al., 2012). As expected from our previous reports, EGF stimulation strongly upregulated the SCAP protein levels and activated SREBP-1 (as assessed by the increased N-terminal cleavage fragment of SREBP-1) (Figure 1C) (Cheng et al., 2015; Guo et al., 2009b; Guo et al., 2011). Inhibition of EGFR/PI3K/Akt signaling by their specific inhibitors significantly reduced the levels of pri-miR-29a/b1, -29b2/c and mature miR-29 (Figures 1D and 1E), and downregulated the EGF stimulation-enhanced SCAP protein levels and SREBP-1 cleavage (Figure 1E).
We further investigated whether miR-29 expression induced by EGFR signaling was mediated by SCAP/SREBP-1. The data show that knockdown of SCAP or SREBP-1 with specific siRNA significantly reduced EGF-enhanced expression of pri-miR-29a/b1, -29b2/c and mature miR-29 (Figures 1F–1I). Moreover, to confirm that SCAP/SREBP-1 regulates miR-29 expression in a physiological context, we examined the direct effects of knockdown of SCAP or SREBP-1 on miR-29 expression in low-passage primary GBM83 cells, which are able to form GBM-like tumors in mouse brains, making it a good orthotopic GBM xenograft model, as previously described (Mao et al., 2013). The data show that knockdown of SCAP or SREBP-1 significantly downregulated miR-29 expression in GBM83 cells (Figures 1J–1M). Taken together, these data demonstrate that SCAP/SREBP-1 regulates miR-29 expression.
SREBP-1 functions as a transcription factor to promote miR-29 expression
We wondered whether SREBP-1 regulates miR-29 expression via its activity as a transcription factor. First, we examined the effects of expression of adenovirus-mediated N-terminal active forms of SREBP-1a or -1c (nSREBP-1a or -1c) on miR-29 expression in U87 cells. The data show that overexpression of nSREBP-1a or -1c strongly upregulated the expression of its downstream targets, acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN) and stearoyl-CoA desaturase-1 (SCD1) (Figure 2A), and significantly promoted the expression of pri-miR-29 and mature miR-29 (Figure 2B). Moreover, our data also show that upregulation of SREBP-1 by the liver X receptor (LXR) agonist GW3965 (Guo et al., 2011) significantly enhanced the expression of miR-29 in U87 cells (Figure S2). These data demonstrate that SREBP-1 regulates miR-29 expression.
Figure 2. SREBP-1 functions as a transcription factor to promote miR-29 expression.

(A) Western blot analysis of U87 cells after infection with the adenovirus expressing the N-terminal SREBP-1a or -1c (nSREBP-1a or -1c) for 48 hr.
(B) Real-time PCR analysis of the expression of pri-miR29a/b1, pri-miR29b2/c (upper panel) and mature miR-29 (lower panel) in U87 cells after infection with the adenovirus expressing the N-terminal form of SREBP-1a or -1c (nSREBP-1a or -1c) for 48 hr (mean ± SD). Statistical significance was determined by Student’s t-test (n=3); *p<0.001.
(C, D) Schemas at the top show putative SREBP-1 binding sites (SREs) on miR-29a/b1 (C, upper panels) or miR-29b2/c promoter (D, upper panel). Agarose gel electrophoresis of PCR products after chromosomal immunoprecipitation (ChIP) assay using IgG or anti-SREBP-1 antibody in U87/EGFR cells with/without EGF (50 ng/ml) stimulation for 12 hr (C and D, middle panels). The PCR products of the ChIP assay shown on the agarose gel were quantified by ImageJ and normalized with the value of the input without EGF stimulation (C and D, lower panels) (mean ± SD). Statistical significance was determined by Student’s t-test (n=3); #p<0.05, +p<0.01 and *p<0.001.
(E, F) PCR analysis of SREBP-1 binding to the specific SRE motifs located in pri-miR-29a/b1 (E) or pri-miR-29b2/c promoter (F) in primary GBM83 cells. The PCR products of the ChIP assay shown on the agarose gel were quantified by the ImageJ software and normalized with the value of the input (mean ± SD). Statistical significance was determined by an unpaired Student’s t test (n=3). * p<0.01.
(G, H) Luciferase activity analysis of the promoters of miR-29a/b1 (G) or miR-29b2/c (H) with different deletions of SRE binding sites cloned in the pGL3-basic vector that were transfected into HEK293T cells together with Renilla and PC3.1, nSREBP-1a or -1c plasmids for 48 hr (mean ± SD). Statistical significance was determined by Student’s t-test (n=3); +p<0.01, +p<0.01, *p<0.001and N.S, no significance. See also Figures S2.
As SREBP-1 is a transcription factor, we speculated that it might bind to the promoter region of miR-29 to activate its expression. As previously reported, miR-29a and miR-29b1 are located on chromosome 7q32 and share the same promoter (miR-29a/b1), while miR-29b2 and miR-29c reside on chromosome 1q32 and have a different promoter (miR-29b2/c) (Figure S1C) (Zhang et al., 2012). We analyzed the promoter regions of miR-29a/b1 and miR-29b2/c using the Tfsearch online promoter analysis tool and referring to the previous reports of the DNA binding motif for SREBP-1 (Seo et al., 2009; Yang et al., 2014). Interestingly, four putative SREBP-1 binding sites, also named sterol regulatory-element binding sites (SREs) (Seo et al., 2009), were found in the promoter of miR-29a/b1 (Figure 2C, upper panel), and one in the promoter of miR-29b2/c (Figure 2D, upper panel). We then examined whether SREBP-1 directly binds to these SRE regions using a chromatin immunoprecipitation (ChIP) approach in GBM U87/EGFR cells. As shown in Figures 2C and 2D, SREBP-1 directly bound to SRE-1, SRE-2 and SRE-3/4 (a cross-linked region) on the promoter of miR-29a/b1 (Figure 2C, middle and lower panels), and to the single SRE region on the promoter of miR-29b2/c in U87/EGFR cells (Figure 2D, middle and lower panels). Moreover, EGF stimulation significantly enhanced the binding of SREBP-1 to all SRE motifs of the miR-29 promoters (Figures 2C and 2D), which is consistent with the enhanced miR-29 expression and increased nuclear form of SREBP-1 induced by EGF in U87/EGFR cells (Figures 1B and 1C). Moreover, the binding of SREBP-1 to the promoters of miR-29a/b1 and -29b2/c was confirmed by ChIP assay in the low-passage primary GBM83 cells (Figures 2E and 2F). Taken together, these data strongly support that SREBP-1 transcriptionally regulates miR-29 expression.
We then examined the transcriptional activity of SREBP-1 on miR-29 promoters using a promoter-driven luciferase (luc) reporter system. As shown in Figures 2E and 2F, we cloned the different fragments of the miR-29 promoters into the pGL3-luc expression vector, and transfected them into HEK293T cells together with active nSREBP-1a or -1c plasmids. Consistent with promoting miR-29 expression (Figure 2B), expression of nSREBP-1a or -1c markedly enhanced the luciferase activity driven by the full-length promoter of miR-29a/b1 or miR-29b2/c (including all SRE binding sites) (Figures 2G and 2H). Deletion of the SRE binding sites in the promoters of miR-29a/b1 or -29b2/c significantly decreased nSREBP-1-enhanced luc activity, which was reduced to the levels of the control vector (empty PC3.1 plasmid) transfection when all SRE binding sites were removed (Figures 2G and 2H), demonstrating that SREBP-1 directly activates miR-29 expression via binding to the specific SRE motifs in its promoters.
miR-29 reversely inhibits SCAP and SREBP-1 expression via binding to their 3′-UTR
To explore the function of miR-29 in GBM, we searched the potential targets of miR-29 by using microRNA.org and miRBase.org prediction resources. Interestingly, all members of the miR-29 family (-29a, -29b or -29c) have the same putative targeting site in the 3′-untranslated region (3′-UTR) of SCAP or SREBF1 mRNA (Figures 3A and 3B, upper panels). We cloned the 3′-UTR of SCAP or SREBF1 into a luciferase reporter vector and examined the effects of its interaction with miR-29 on luc activity in HEK293T cells. As shown in Figures 3A and 3B, miR-29 transfection significantly reduced the activity of luciferase that was linked with wild-type 3′-UTR of SCAP or SREBF1, and the reduction was completely abolished when the targeting site on the 3′-UTR was mutated, suggesting that miR-29 inhibits SCAP or SREBF1 expression via direct binding to their 3′-UTR.
Figure 3. miR-29 reversely inhibits SCAP and SREBP-1 expression through binding to their 3′-UTR.

(A, B) Schemas (upper panels) and luciferase activity analysis of putative miR-29 binding to the 3′-UTR of SCAP (A) or SREBF1 (B) in HEK293T cells transfected with pmirReport vector carrying the wild-type or mutant 3′-UTR together with miR-29 (50 nM) and renilla. The luciferase activity of each sample was normalized with the control transfected with wild-type 3-UTR vector (mean ± SD) (lower panels). Statistical significance was determined by Student’s t-test (n=3); #p<0.01; *p<0.001 compared with scramble control miRNA (Ctrl miR) transfection with wild-type SCAP or SREBF1 3′-UTR. WT, wild-type; MT, mutant.
(C, D) Western blot analysis of the indicated proteins (C) or real-time PCR analysis of gene expression (mean ± SD) (D) for GBM cells transfected with miR-29 (50 nM) for 48 hr. Statistical significance was determined by Student’s t-test (n=3); #p<0.01 or *p<0.001 compared with scramble control miRNA (Ctrl miR) transfection.
See also Figure S3.
We then examined the effects of miR-29 on SCAP and SREBP-1 expression by analyzing their protein and mRNA levels in various GBM cell lines and patient-derived cells (GBM83) after transfection with miR-29. Western blot analysis demonstrated that miR-29 transfection markedly reduced the protein levels of SCAP and SREBP-1 (Figure 3C). Moreover, real-time PCR analysis showed that miR-29 transfection significantly reduced the expression of SCAP, SREBF1, SREBP-1a and -1c (Figure S3), and downregulated the expression of their targets, ACACA, FASN, LDLR and SCD1 that control de novo fatty acid synthesis (Figure 3D).
Collectively, these data demonstrate that miR-29 serves as a negative regulator, inhibiting SCAP/SREBP-1 and lipid synthesis pathway in GBM cells.
miR-29 inhibits GBM growth in vitro and in vivo via suppressing SCAP/SREBP-1
Next, we examined whether miR-29 suppressed de novo lipid synthesis, in line with its inhibitory effects on SCAP/SREBP-1 signaling (Figure 3). Radiolabeled 14C-glucose was added to the culture medium of U87 cells after transfection with miR-29. As shown in Figure 4A, miR-29 significantly inhibited the production of 14C-labeled lipids generated from 14C-glucose. These data were confirmed by thin-layer chromatography analysis, which showed that miR-29 transfection significantly reduced the levels of fatty acids (FFA) and triglycerides (TG) in GBM cells compared to control cells transfected with scramble miRNA (Figure S4A and S4B).
Figure 4. miR-29 inhibits GBM growth via suppressing SCAP/SREBP-1.

(A) Analysis of radiolabeled lipid products by scintillation counting for U87 cells in culture with 14C-glucose for 2 hr after transfection with miR-29 or scramble control miRNA (50 nM) for 48 hr (mean ± SD). Statistical significance was determined by Student’s t-test (n=5); #p<0.05 or *p<0.001 compared with scramble control miRNA (Ctrl miR) transfection.
(B) Growth analysis of U87 cells transfected with miR-29 or scramble control miRNA (50 nM) overnight, then infected with the adenovirus constitutively expressing nSREBP-1a or -1c in 1% LPDS media for 3 days. Cell number was counted after trypan blue staining (mean ± SD). Statistical significance was determined by Student’s t-test (n=3); *p<0.001 for comparison with scramble control miRNA (Ctrl miR) transfection; #p<0.01 for the indicated comparison group.
(C) Growth analysis of GBM cells transfected with miR-29 or scramble control miRNA (50 nM) in the presence or absence of palmitate (PA, 10 μM) and oleic acid (OA, 10 μM) in 1% LPDS media for 3 days (U87, T98 and U251 cells) or neurobasal medium (GBM83 cells) for 2 days. Cells were counted after trypan blue staining (mean ± SD). Statistical significance was determined by Student’s t-test (n=3); *p<0.001 for comparison with scramble control miRNA (Ctrl miR) transfection; #p<0.001 for the indicated comparison group.
(D) Luminescence imaging of GBM tumor growth in mouse brains at day 14 post intracranial implantation of 1×105 U87/EGFRvIII-luc cells after transfection with miR-29 or scramble control miRNA (50 nM) for 48 hr (upper panel). Lower panel shows the quantification of the luminescence signal intensity from intracranial tumor on day 14 (mean ± SEM). Statistical significance was determined by Student’s t-test (n=7); *p<0.001 for comparison with scramble control miRNA (Ctrl miR) transfection.
(E) Immunohistochemistry analysis of SCAP and SREBP-1 protein levels in intracranial tumor tissues from U87/EGFRvIII-bearing mice transfected with miR-29 at day 20 or scramble control miRNA at day 15 (left panels) or from human primary GBM83-bearing mice transfected with miR-29 or control miRNA at day 14 (right panels). Lower panels show the relative protein levels in tumor tissues quantified by ImageJ and averaged from 5 separate areas in each tumor. The results were normalized with control miRNA tumors (mean ± SEM, n=5). Statistical significance was determined by Student’s t-test; *p<0.001 for comparison with scramble control miRNA transfection. Scale bar, 50 μm.
(F) Kaplan-Meier analysis of GBM-bearing mice implanted with 1×105 U87/EGFRvIII cells (left panel) or 2×104 GBM83 cells (right panel) after transfection with miR-29 or scramble control miRNA (50 nM) for 48 hr. Statistical significance was determined by log-rank test (n=7).
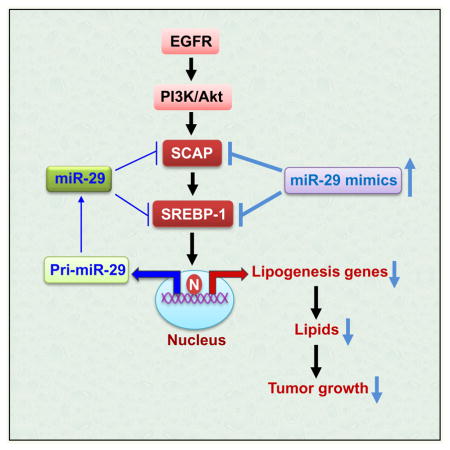
(G) Schematic model of the previously unrecognized negative feedback loop regulating lipid metabolism and associated tumor growth. EGFR signaling promotes miR-29 expression via upregulation of SCAP/SREBP-1, and SREBP-1 functions as a transcription factor to activate miR-29 transcription. In turn, miR-29 reversely inhibits SCAP and SREBP-1 expression via directly binding to their 3′-UTR to modulate tumor growth.
See also Figures S4.
Our previous studies have revealed that GBM growth is highly dependent on SCAP/SREBP-1 signaling (Cheng et al., 2015; Geng et al., 2016; Guo et al., 2009b; Guo et al., 2011). We asked whether miR-29 transfection was able to inhibit GBM cell growth through suppressing SCAP/SREBP-1-regulated lipogenesis. As shown in Figures 4B and S4C, transfection with miR-29 markedly inhibited GBM cell growth, which was significantly rescued by the overexpression of nSREBP-1a or -1c. Moreover, addition of the fatty acids, palmitate (PA) and oleic acid (OA), two major products of the fatty acid synthesis pathway regulated by SCAP/SREBP-1 (Horton et al., 2002), markedly rescued miR-29-mediated growth inhibition in various GBM cell lines and GBM patient-derived cells (GBM83) (Figures 4C and S4D). These data strongly demonstrate that miR-29 transfection inhibits GBM growth through suppressing SREBP-1 expression and downstream lipogenesis.
Next, we used a xenograft mouse model where U87/EGFRvIII-luc cells that stably expresses luciferase were stereotactically implanted into mouse brain in order to examine the in vivo antitumor effects of miR-29 using bioluminescence imaging (Cheng et al., 2015). The data show that transfecting miR-29 into U87/EGFRvIII-luc cells markedly inhibited GBM tumor growth in the mouse brains showing by the luminescent imaging (Figure 4D) and suppressed the expression of SCAP and SREBP-1 in the GBM tumor tissues from U87/EGFRvIII-luc-bearing mice (Figures 4E, left panels and S4E). Consistent with the intracranial tumor growth, Kaplan-Meier analysis showed that miR-29 transfection significantly prolonged the overall survival of U87/EGFRvIII-luc-bearing mice (Figure 4F, left panel). These data were further confirmed in the primary GBM83 cells-generated intracranial mouse model. The data show that miR-29 transfection significantly reduced the expression of SCAP and SREBP-1 in tumor tissues (Figures 4E, right panels, and S4E) and prolonged the overall survival of GBM83-bearing mice (Figure 4F, right panel).
Discussion
Lipids are critical cellular components, and their alteration leads to various metabolic diseases such as atherosclerosis and obesity (Nohturfft and Zhang, 2009; van Meer et al., 2008). Recent evidence shows that lipid metabolism is also reprogrammed in malignancies to support rapid tumor growth (Bell and Guo, 2012; Geng et al., 2016; Guo et al., 2013; Guo et al., 2014; Menendez and Lupu, 2007; Ru et al., 2013). In particular, our recent work has revealed that oncogenic EGFR signaling activates lipid metabolism via upregulation of SCAP/SREBP-1 signaling (Cheng et al., 2015; Guo, 2016; Guo et al., 2009b; Guo et al., 2011). Thus, a better understanding of how lipid metabolism is reprogrammed in cancer cells may lead to development of promising strategies to treat malignancies.
miR-29 has previously been reported to function as a tumor suppressor by suppressing different oncogenes involved in methylation, apoptosis and metastasis pathways (Fabbri et al., 2007; Ru et al., 2012; Schickel et al., 2008; Zhang et al., 2012). Our study does not exclude the possibility that these miR-29 antitumor functions play a role in GBM. Nevertheless, in this study, we unraveled a previously unappreciated critical function of miR-29 in the regulation of lipid metabolism, and identified a previously unrecognized negative feedback loop mediated by miR-29 in SCAP/SREBP-1 signaling (Figure 4G). We found that EGFR signaling enhanced miR-29 expression via upregulation of SCAP/SREBP-1 expression, with SREBP-1 transcriptionally activating specific sterol regulatory-element motifs in the promoter of miR-29. Interestingly, miR-29 inversely suppresses SCAP and SREBP-1 expression by binding to their 3′-UTR regions. These data demonstrate that a multilayer of regulatory mechanisms converge on the regulation of the SCAP/SREBP-1 pathway (Figure 4G).
Although the role of SCAP/SREBP-1 in lipogenesis is well established (Goldstein et al., 2006; Guo et al., 2014), its precise regulation in malignancies is still poorly understood. Our present study provides a molecular link between EGFR signaling, miR-29 and SCAP/SREBP-1, demonstrating an elaborate feedback loop operating in cancer cells to regulate lipid synthesis driven by oncogenic signaling. Addition of fatty acids or expression of active N-terminal SREBP-1 rescued miR-29-inhibited GBM cell growth, demonstrating that decreased lipogenesis underlies the mechanism of miR-29-mediated GBM growth suppression. Moreover, the increased survival in mice implanted with GBM cells transfected with miR-29 suggests that suppressing SCAP and SREBP-1 with miR-29 provides an effective means to target malignancies and other metabolic syndromes. Analyzing further the levels and distribution of miR-29 in normal brain tissues versus tumor tissues will be important to design a better strategy to treat GBM.
Although SREBP-1, a master transcription factor in lipid metabolism, was identified over 20 years ago (Wang et al., 1994), no targeting pharmaceutics have been developed so far. Using miR-29 mimics to inhibit this central pathway controlling lipid metabolism may be a promising strategy to suppress GBM growth. Further studies should also be dedicated to the development of effective methods to deliver the miR-29 mimics into brain tumor tissues.
EXPERIMENTAL PROCEDURES
Analysis of GBM Patient Tumor Tissues
The Cancer Genome Atlas (TCGA) mRNA (Affymetrix, u133a) and miRNA expression data (Agilent, 8×15k) for GBM patient tumor tissues were downloaded through the TCGA data portal (http://cancergenome.nih.gov/; updated on December 2014), and the patients’ EGFR status information (Alteration: amplification or mutation, n=132; Normal EGFR: no amplification or mutation, n=97) was obtained from cbioportal (http://www.cbioportal.org/) (Cerami et al., 2012; Gao et al., 2013). The association between the expression of SREBF1 and miR-29 was evaluated by the Pearson correlation method. Data analyses were performed using SAS 9.4 (SAS, Inc; Cary, NC).
Cell Culture and Vectors
Human GBM cell lines, i.e., U87, U87/EGFRvIII, U87/EGFRvIII-luc, U87/EGFR, U251, T98, and the HEK293T cell line were cultured in Dulbecco’s modified Eagle’s media (DMEM; Cellgro) supplemented with 5% FBS (Gemini Bio-Products) The GBM patient-derived cells, GBM83, which were previously molecularly characterized and described (Mao et al., 2013), were cultured in Neurobasal medium supplemented with B-27 (1x), Heparin (2 μg/ml), EGF (50 ng/ml) and FGF (50 ng/ml) in a humidified atmosphere of 5% CO2, 95% air at 37°C. U87/EGFRvIII cells were generated by stably expressing EGFRvIII, a constitutively active mutant form of EGFR, in U87 cells (Guo et al., 2009a). U87/EGFRvIII-luc cells stably express luciferase (Cheng et al., 2015). U87/EGFR cells were generated by retrovirus-mediated transduction of wild-type EGFR into U87 cells followed by selection of stable clones (Guo et al., 2009b). pcDNA3.1-2xFLAG-SREBP-1a and -1c (N-terminal fragment) were a gift from Dr. Timothy Osborne (Addgene plasmid # 26801 and # 26802) (Toth et al., 2004). Adenovirus containing N-terminal SREBP-1a or -1c was produced and amplified as previously described (Dif et al., 2006).
ChIP Assay
The details of the ChIP assay are described in the Supplemental Experimental Procedures.
Real-time RT-PCR
Details are described in the Supplemental Experimental Procedures.
3′-UTR Luciferase assay
Details are described in the Supplemental Experimental Procedures.
Promoter Luciferase Assay
Details are described in the Supplemental Experimental Procedures.
Intracranial Mouse Model and Survival
Female athymic nude mice (6–8 weeks of age obtained from the National Cancer Institute) were used for the intracranial xenograft models. Cells (1×105 U87/EGFRvIII-luc or 2×104 GBM83 cells in 4 μl of PBS) after transfected with miR-29 or mimic control for 48 hr were stereotactically implanted into mouse brain. Mice were then observed until they became moribund, at which point they were sacrificed. All animal procedures were approved by the Subcommittee on Research Animal Care at Ohio State University Medical Center.
Mice Bioluminescence Imaging
Analysis of mouse intracranial tumor by bioluminescence imaging was performed as previously described (Cheng et al., 2015). Imaging experiments were conducted at the OSU Small Animal Imaging Core.
Statistical Analysis
Statistical analysis was performed with Excel or GraphPad Prism5. The matched samples were compared using paired Student’s t test and samples between treatments were compared by using 2-sample t-tests. Kaplan-Meier plots were used for analysis of the mice overall survival (significance was analyzed by Log-rank test). P value < 0.05 was considered significant.
Supplementary Material
Highlights.
EGFR/PI3K signaling enhances miR-29 expression via upregulation of SCAP/SREBP-1.
SREBP-1 transcriptionally activates miR-29 expression via binding to its promoter.
miR-29 suppresses SCAP and SREBP-1 expression by interacting with their 3′-UTRs.
miR-29 transfection inhibits GBM growth in vivo via suppression of SCAP/SREBP-1.
Acknowledgments
We thank Dr. Martine Torres for her critical review and helpful comments of the manuscript. This work was supported by NIH/NINDS NS072838 (DG) and NS079701 (DG), American Cancer Society Research Scholar Grant RSG-14-228-01–CSM (DG), OSUCCC start-up funds (DG) and OSU Neuroscience Imaging Core pilot grant (DG).
Footnotes
CONFLICT OF INTEREST
The authors declare that there are no conflict of interest for this manuscript.
AUTHOR CONTRIBUTIONS
D.G. conceived the ideas. P.R. and D.G. designed the experiments. P.R., P.H., F.G., X.M., C.C., J.Y.Y., X.C., X.W., J.Y.G. and D.G. performed the experiments. P.R., P.H., F.G., B.K., A.C. and D.G. analyzed the data. I.N. and E.L. provided critical materials. P.R. and D.G. wrote the manuscript. D.G. supervised the study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, Goldstein JL. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. The Journal of biological chemistry. 2004;279:52772–52780. doi: 10.1074/jbc.M410302200. [DOI] [PubMed] [Google Scholar]
- Bell EH, Guo D. Biomarkers for malignant gliomas. Malignant Gliomas, Radiation Medicine Rounds. 2012;3:389–357. [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Ru P, Geng F, Liu J, Yoo JY, Wu X, Cheng X, Euthine V, Hu P, Guo JY, et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell. 2015;28:569–581. doi: 10.1016/j.ccell.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dif N, Euthine V, Gonnet E, Laville M, Vidal H, Lefai E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J. 2006;400:179–188. doi: 10.1042/BJ20060499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Hernando C, Suarez Y, Rayner KJ, Moore KJ. MicroRNAs in lipid metabolism. Curr Opin Lipidol. 2011;22:86–92. doi: 10.1097/MOL.0b013e3283428d9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. 2015;15:302–310. doi: 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, Mo X, Ru P, Hurwitz B, Kim S, et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-2973. pii: clincanres.2973.2015. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Guo D. SCAP links glucose to lipid metabolism in cancer cells. Mol Cell Oncol. 2016;3 doi: 10.1080/23723556.2015.1132120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Bell EH, Chakravarti A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncology. 2013;2:289–299. doi: 10.2217/cns.13.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des. 2014;20:2619–2626. doi: 10.2174/13816128113199990486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Hildebrandt IJ, Prins RM, Soto H, Mazzotta MM, Dang J, Czernin J, Shyy JY, Watson AD, Phelps M, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009a;106:12932–12937. doi: 10.1073/pnas.0906606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Prins RM, Dang J, Kuga D, Iwanami A, Soto H, Lin KY, Huang TT, Akhavan D, Hock MB, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal. 2009b;2:ra82. doi: 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S, et al. An LXR agonist promotes GBM cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1:442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon TI, Esquejo RM, Roqueta-Rivera M, Phelan PE, Moon YA, Govindarajan SS, Esau CC, Osborne TF. An SREBP-Responsive microRNA Operon Contributes to a Regulatory Loop for Intracellular Lipid Homeostasis. Cell metabolism. 2013;18:51–61. doi: 10.1016/j.cmet.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends in endocrinology and metabolism: TEM. 2012;23:65–72. doi: 10.1016/j.tem.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz CL, Peck BC, Fannin EE, Beysen C, Miao J, Landstreet SR, Ding S, Turaga V, Lund PK, Turner S, et al. MicroRNA-29 fine-tunes the expression of key FOXA2-activated lipid metabolism genes and is dysregulated in animal models of insulin resistance and diabetes. Diabetes. 2014;63:3141–3148. doi: 10.2337/db13-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A. 2013;110:8644–8649. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Nohturfft A, Zhang SC. Coordination of lipid metabolism in membrane biogenesis. Annual review of cell and developmental biology. 2009;25:539–566. doi: 10.1146/annurev.cellbio.24.110707.175344. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 2008;8:512–521. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru P, Steele R, Newhall P, Phillips NJ, Toth K, Ray RB. miRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol Cancer Ther. 2012;11:1166–1173. doi: 10.1158/1535-7163.MCT-12-0100. [DOI] [PubMed] [Google Scholar]
- Ru P, Williams TM, Chakravarti A, Guo D. Tumor metabolism of malignant gliomas. Cancers (Basel) 2013;5:1469–1484. doi: 10.3390/cancers5041469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27:5959–5974. doi: 10.1038/onc.2008.274. [DOI] [PubMed] [Google Scholar]
- Seo YK, Chong HK, Infante AM, Im SS, Xie X, Osborne TF. Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13765–13769. doi: 10.1073/pnas.0904246106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Espenshade PJ. Expanding Roles for SREBP in Metabolism. Cell metabolism. 2012;16:414–419. doi: 10.1016/j.cmet.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LP, Seemann J, Goldstein JL, Brown MS. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6519–6526. doi: 10.1073/pnas.0700907104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth JI, Datta S, Athanikar JN, Freedman LP, Osborne TF. Selective coactivator interactions in gene activation by SREBP-1a and -1c. Molecular and cellular biology. 2004;24:8288–8300. doi: 10.1128/MCB.24.18.8288-8300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nature reviews Molecular cell biology. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sato R, Brown MS, Hua X, Goldstein JL. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell. 1994;77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- Wen PY, Kesari S. Malignant gliomas in adults. The New England journal of medicine. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- Xu H, Sun J, Shi C, Sun C, Yu L, Wen Y, Zhao S, Liu J, Xu J, Li H, et al. miR-29s inhibit the malignant behavior of U87MG glioblastoma cell line by targeting DNMT3A and 3B. Neurosci Lett. 2015;590:40–46. doi: 10.1016/j.neulet.2015.01.060. [DOI] [PubMed] [Google Scholar]
- Xu P, Wu M, Chen H, Xu J, Li M, Qian F. Bioinformatics analysis of hepatitis C virus genotype 2a-induced human hepatocellular carcinoma in Huh7 cells. Onco Targets Ther. 2016;9:191–202. doi: 10.2147/OTT.S91748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Liu W, Pellicane C, Sahyoun C, Joseph BK, Gallo-Ebert C, Donigan M, Pandya D, Giordano C, Bata A, et al. Identification of miR-185 as a regulator of de novo cholesterol biosynthesis and low density lipoprotein uptake. J Lipid Res. 2014;55:226–238. doi: 10.1194/jlr.M041335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhao X, Fiskus W, Lin J, Lwin T, Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012;22:506–523. doi: 10.1016/j.ccr.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.