

Summary
Haem‐oxygenase‐1 (HO‐1) is an enzyme responsible for the degradation of haem that can suppress inflammation, through the production of carbon monoxide (CO). It has been shown in several experimental models that genetic and pharmacological induction of HO‐1, as well as non‐toxic administration of CO, can reduce inflammatory diseases, such as endotoxic shock, type 1 diabetes and graft rejection. Recently, it was shown that the HO‐1/CO system can alter the function of antigen‐presenting cells (APCs) and reduce T‐cell priming, which can be beneficial during immune‐driven inflammatory diseases. The molecular mechanisms by which the HO‐1 and CO reduce both APC‐ and T‐cell‐driven immunity are just beginning to be elucidated. In this article we discuss recent findings related to the immune regulatory capacity of HO‐1 and CO at the level of recognition of pathogen‐associated molecular patterns and T‐cell priming by APCs. Finally, we propose a possible regulatory role for HO‐1 and CO over the recently described mitochondria‐dependent immunity. These concepts could contribute to the design of new therapeutic tools for inflammation‐based diseases.
Keywords: antigen presentation, carbon monoxide, cytokine, dendritic cells, haem‐oxygenase 1
Biological function of HO‐1 activity
Several proteins, such as myoglobins, cytochromes and haemoblogins use the haem group as a cofactor.1 As is the case for most cellular components, these proteins are degraded after being damaged or aged.2, 3 Because haem is a pro‐oxidant molecule that can participate in the formation of oxidative radicals, leading to oxidative‐toxic injury that results in cell death, degradation of haem is required after the turnover of haem‐containing protein.4 Therefore, animal cells contain a specific set of haem‐degrading enzymes, known as haem‐oxygenases (HOs).2, 5, 6, 7 Depending on the tissue, three different HO isoforms can be expressed (HO‐1, HO‐2 and HO‐3). HO‐1 is mainly expressed in hepatic,8 endothelial,9 myeloid10, 11 and respiratory epithelial cells.12 HO‐2 is expressed in testis, brain and vascular system.7, 13, 14, 15 Although HO‐3 is constitutively expressed, it has no catalytic activity, and genetic studies in rats have shown that the Hmox3 gene is an HO‐2‐derived pseudogene.16
The HOs degrade the haem group into Fe3+, biliverdin and carbon monoxide (CO). Although Fe3+ and CO are conserved and employed as second physiological signals, biliverdin is rapidly converted into bilirubin by the biliverdin reductase system.2, 7 Due to their functions, HOs are known as shock‐stress‐protecting enzymes.4
As a consequence of the high biological impact recently described for HO enzymatic activity, HO‐1 function has been the most studied and characterized HO enzyme. An HO‐1 deficiency leads to haem accumulation, causing several health burdens to the host.17, 18 The first patient suffering from HO‐1 deficiency was reported in 1999.19 In addition to all the metabolic, vascular and endothelial alterations, this patient suffered from an acute inflammatory state.18, 19 Lymph node swelling and leukocytosis were observed in this patient, which was in agreement with an advanced health deterioration and subsequent death.19 Accordingly, HO‐1 knockout (KO) mice display similar alterations, such as splenomegaly, lymph node swelling, altered CD4+ T‐cell numbers and an enhanced T‐cell activation state.18, 20 Furthermore, HO‐1 KO mice showed an unexpected increased susceptibility to lipopolysaccharide (LPS) ‐endotoxic shock.21 Splenocytes from HO‐1 KO mice also showed an augmented secretion of pro‐inflammatory cytokines, such as interleukin‐1 (IL‐1), IL‐6 and tumour necrosis factor‐α. Consistent with these observations, monocytes from patients suffering from the autoimmune disease systemic lupus erythematosus, which manifests as exacerbated general inflammation, showed a reduced expression of HO‐1.22 Similar results were seen in patients with multiple sclerosis, who displayed reduced levels of HO‐1 in peripheral blood mononuclear cells during disease exacerbation.23 These results suggest that HO‐1, in addition to a pro‐homeostatic function, can contribute to modulating the inflammatory response in the host.
Because antigen‐presenting cells (APCs), such as dendritic cells (DCs), monocytes and macrophages express high levels of HO‐1, the function and anti‐inflammatory capacity of this molecule have been extensively studied.24, 25, 26 A better understanding of the biology and function for HO‐1 within these APCs could contribute to the design of improved anti‐inflammatory therapies. In this article, we review recent findings for the role of HO‐1 in the modulation of immunity.
Regulation of HO‐1 gene expression
Regulation of HO‐1 gene expression (Hmox1) is driven by pro‐inflammatory and pro‐oxidant molecules, such as pathogen‐associated molecular pattern (PAMPs) and damage‐associated molecular pattern (DAMPs, e.g. haem group). These PAMPs and DAMPs activate signal transduction pathways that can modify intracellular equilibrium causing cell stress by activation of stress response genes.27, 28, 29 These pathways include mitogen‐activated protein kinases (MAPK) and the c‐Jun N‐terminal kinases (JNK).30, 31 HO‐1 expression is regulated by the Keap1/Nrf2 and the Bach‐1/Maf systems. During cellular stress or an inflammatory response,32, 33 nuclear erythroid 2‐related factor‐2 (Nrf2) dissociates from kelch‐like erythroid cell‐derived protein with CNC homology (ECH)‐associated protein 1 (Keap1), the molecule that retains Nrf2 at the cytoplasm (Fig. 1a). Activated kinases from the MAPK and JNK pathways catalyse the phosphorylation of Nrf2, allowing translocation to the nucleus and binding to antioxidant response elements (ARE) in the Hmox1 promoter site34 (Fig. 1b, c). In addition to pro‐inflammatory stimuli, the anti‐inflammatory cytokine IL‐10 can also induce the transcription of the Hmox1 gene through the p38–MAPK pathway to suppress the PAMP‐mediated and pro‐oxidant molecule‐mediated inflammatory responses.35, 36 Because HO‐1 expression also induces IL‐10 production, it is likely that a positive feedback loop takes place between IL‐10 and HO‐1 expression in the responding cells. On the other hand, the haem‐binding protein Bach‐1 has been shown to form a heterodimer with small Maf proteins and represses Hmox1 transcription by competing with Nrf2 for the binding to AREs37, 38 (Fig. 1). Only during stress responses, Bach‐1 dissociates from V‐maf musculoaponeurotic fibrosarcoma oncogene homologues (Mafs), allowing Nrf2 to heterodimerize with these molecules (Fig. 1b, c). Therefore, there is a constant competition between Nrf2 and Bach‐1 for the binding to small Maf proteins at the ARE. Recently, it was shown that IL‐10 and other anti‐inflammatory molecules can regulate the Bach‐1/Maf system by reducing the expression of miR‐155, a Bach‐1 repressor molecule.39, 40 Hence, it is thought that micro RNAs can also contribute to the balance between cellular homeostasis and inflammatory response by modulating the access of Nrf2 to the Hmox1 gene promoter. Consistently with this notion, Nrf2 KO mice develop several pathological manifestations including an enhanced proliferative response of CD4+ T cells and a lupus‐like syndrome with the presence of antinuclear antibodies, intravascular deposition of immune complexes, glomerulonephritis and decreased survival rates, showing a similar phenotype to HO‐1 KO mice.41
Figure 1.
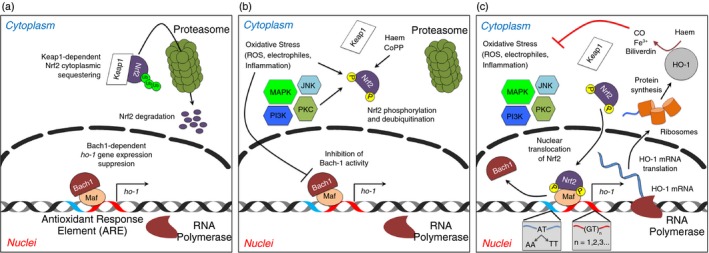
Hmox1 gene expression and its regulation. (a) In resting state, Nrf2 remains as bound to Keap1 in the cytoplasm. Nrf2 is constantly being ubiquitinated and targeted for proteasomal degradation. During this process, Bach‐1 heterodimerizes with small Mafs proteins at Antioxidant Response Elements (ARE) in the Hmox1 gene promoter site. (b) During cell stress caused either by pathogen‐associated molecular pattern (PAMPs) and/or pro‐oxidant molecules, Nrf2 is released from Keap1, increasing its stability and reducing its proteasome‐dependent degradation. Different kinases access to phosphorylate Nrf2 and activate its translocation to the nucleus. The binding of Bach‐1 to small Mafs is compromised by the same pro‐inflammatory and pro‐oxidants signals. (c) Phosphorylated Nrf2 migrates to the nucleus and displaces Bach‐1. Heterodimers Nrf2/Maf induce the activation of the Hmox1 gene promoter site and the recruitment of the RNA polymerase. The Hmox1 gene transcription begins. Depending on the (GT)n (n = number of repetitions) and the 413A > T (AT ‐> AA; AT ‐> TT) polymorphisms present in the promoter site, the amount of mRNA haem oxygenase 1 can vary.
Recent studies have shown that the expression of the Hmox1 gene can be modulated by the presence of certain microsatellites located in the gene promoter (GT)n.42 Individuals with short GT repetitions have been shown to have a reduced risk of suffering rheumatoid arthritis43 or chronic pulmonary emphysema,44 with a favourable outcome to sepsis,45 as well as other diseases,42 due to an increased HO‐1 promoter activity.29 In addition, the single nucleotide polymorphism 413 A > T in the Hmox1 gene promoter has been associated with increased transcription levels of HO‐1 transcription.46, 47, 48 The presence of this polymorphism correlated with an augmented incidence of hypertension in women.46 Interestingly, individuals harboring this genetic alteration manifested lower incidence of acute kidney injury48 and ischaemic heart disease,47 suggesting a differential protective capacity for the 413 A > T polymorphism. Hence, although the activities of Nrf2 and Bach‐1 are essential to regulate the transcription of Hmox1 gene and protein quantity, specific DNA modifications in the promoter region also contribute to regulating these processes.
Pharmacological modulation of HO‐1 expression using metalloporphyrins is an interesting experimental approach to study the role of this enzyme in several biological processes.49 Cobalt protoporphyrin IX (CoPP) is a haem group homologue that induces the up‐regulation of Hmox1 gene expression by promoting the degradation of Bach1 protein and decreasing degradation of Nrf2 protein.50 Contrary to CoPP, Tin protoporphyrin IX (SnPP), a metalloporphyrin formed by a chelate of tin with the porphyrin ring, is one of the most efficient inhibitors of HO‐1 activity at the catalytic site.51, 52 This molecule works as a competitive substrate for the haem group, although it enhances the synthesis of new enzyme without catalytic activity.51
Blockade of pro‐inflammatory receptors by HO‐1: the case of TLR4/MD2
As described above, HO‐1 is up‐regulated after PAMP recognition in APCs.29, 32, 33 Because PAMPs mainly signal through Toll‐like receptors (TLRs) and nucleotide‐binding oligomerization domain receptors (NODs), it has been suggested that these recognition pathways can play a central role in the induction of HO‐1 expression.32, 53 Therefore, the contribution of HO‐1 activity to the function of immune cells expressing TLR and NOD receptors, such as DCs and macrophages,53, 54, 55, 56 has been extensively studied. It has been shown that HO‐1 over‐expression inhibits the secretion of inflammatory cytokines after an LPS challenge in DCs.11, 27, 35, 57, 58 These studies underscored CO as the most important product responsible for the immune suppressive capacity of HO‐1.27, 59 CO can block the interferon regulatory factor 3/inducible protein 1 and the JNK/ inducible protein 1 inflammatory pathways in DCs27 and macrophages,10 respectively. Reduced signalling led to impaired de novo expression of different acute inflammatory cytokines, such as IL‐6.60 However, the molecular mechanisms used by CO to modify these pathways still remain unknown. Recent studies have provided insight as to how this gas produced by HO‐1 can block pro‐inflammatory pathways.21, 27, 57, 61 Two different groups have shown that CO directly interferes with the normal surface expression of the LPS‐recognizing receptor in DCs,62 neutrophils62 and macrophages.63 This receptor consists of a complex formed by the TLR4 and the myeloid differentiation factor 2 (hereafter TLR4/MD2). These studies suggested that CO modifies the native conformation of the TLR4/MD2 complex without altering the surface expression of either individual TLR4 or MD2. As a result, CO could impair a key step required for the proper conformational assembly of the TLR4/MD2 complex on the surface of APCs. This notion was further supported by the observation that CO exposure reduced the MD2‐dependent glycosylation of TLR4 induced by LPS and inhibited both the transport and surface expression of these two molecules in hepatic cells.64 Hence, a decrease in the expression of the TLR4/MD2 complex can reduce the sensitivity to LPS stimulation, reducing and dampening inflammation. Although DCs and macrophages did not up‐regulate TLR4 after LPS stimulation, reduced TLR4 glycosylation can explain the capacity of CO to impair subsequent LPS stimulation in monocytes (Fig. 2). Hence, by reducing the MD2‐dependent TLR4 glycosylation, CO would cause an absence of functionally assembled TLR4/MD2 on the surface of the monocyte without altering the total amount of individual TLR4 and MD2 over time. However, additional studies are required to demonstrate this hypothesis.
Figure 2.

Carbon monoxide (CO) reduces the expression of the Toll like receptor 4/ myeloid differentiation factor 2 (TLR4/MD2) complex receptor in the surface of myeloid cells. TLR4 and MD2 form a glycosylation‐dependent complex in the Golgi apparatus, which is then transported to the cell surface. Once in the surface, this complex can recognize lipopolysaccharide (LPS) and trigger an intracellular inflammatory cascade, which will lead to the activation of innate immune responses. After haem oxygenase 1 (HO‐1) expression and CO production, a blockade in the glycosylation is produced in the Golgi apparatus and the standard generation and assembly of the TLR4/MD2 complex is compromised. Then, a non‐well assembled complex is transported to the cell surface. Normal levels of TLR4 and MD2 are placed in the cell surface but its geometric association lack of effectiveness to recognize LPS. The innate immune response is compromised and reduced.
Due to the contribution of TLR4/MD2‐dependent inflammation to LPS‐mediated septic shock, a potential protective capacity for HO‐1 and CO has been explored.62 CO‐treated animals displayed reduced sensitivity to LPS‐induced shock.35, 62, 64 Several explanations are possible, but most of them rely on the ability of CO to reduce the secretion of pro‐inflammatory molecules and to increase the production of immune suppressive cytokines, such as IL‐10.11, 35 The role of IL‐10 has been widely studied during inflammatory diseases because, in addition to inducing HO‐1 expression as mentioned previously, this cytokine can efficiently suppress both innate and adaptive immunity.35 Hence, the HO‐1–CO system can reduce inflammation in vivo by, for instance, promoting the secretion of IL‐10.35, 65 CO‐mediated protection has been observed in some inflammatory pathologies, such as acute pancreatitis,63 haemorragic shock,66 Alzheimer's amyloid‐β1‐42‐induced toxicity,67 ischaemia/reperfusion,68 autoimmunity20 and graft rejection.69 Because these pathologies are mainly mediated by innate or adaptive immune responses, it remains unknown whether CO can ameliorate disease progression by blocking the same or different inflammatory pathways in either innate or adaptive immune cells. Because the innate and adaptive immune responses can be linked by professional APCs that express HO‐1 (DCs and macrophages), this enzyme's activity could be considered as a mechanism to down‐modulate APC function and reduce detrimental innate and adaptive immune responses.
The HO‐1–CO system as a modulator of antigen presentation by DCs
Dendritic cells are professional APCs that reside strategically in tissues that are normally exposed to foreign antigens and infectious agents.70 PAMPs induce the maturation of DCs and, in conjunction with the capture of surrounding antigens, promote the migration of mature DCs to secondary lymphoid organs where they prime antigen‐specific T cells (naive and memory).71 Because of the key role that DCs play during the innate and adaptive immune responses, a DC deficiency can cause significant immune suppression and an increased susceptibility to infections.72, 73
Haem‐oxygenase 1 is constitutively expressed by DCs and can be up‐regulated after PAMPs stimulation.11, 74 However, HO‐1 over‐expression is not observed at early times during maturation. Hence, it seems that this enzyme can contribute to the recovery of DCs after a long period of inflammatory stress. Recent studies have shown that HO‐1 induction by a haem homologue, CoPP, reduces maturation in human and rat DCs, by decreasing the expression of surface maturation markers as well as the secretion of inflammatory cytokines.11, 27 Furthermore, LPS‐mediated reactive oxygen species (ROS) production also was blocked by HO‐1 activity, which is consistent with an early defined role for this enzyme during oxidative stress. In agreement with these results, inhibition of the basal HO‐1 activity by SnPP enhanced maturation of murine DCs after stimulation of the p38–MAPK, cAMP‐responsive element binding protein and the activating transcription factor 1 pathways.75 Reduced DC maturation due to CoPP‐induced HO‐1 expression abolished activation of allogeneic T cells.11, 27, 76 These observations were consistent with data showing an augmented priming of antigen‐specific T cells by DCs in which HO‐1 was inhibited by SnPP.75 Similar results have been obtained with the HO‐1 inhibitory molecule Tin mesoporphyrin, which increased the capacity of cytomegaloviruspp65‐peptide‐pulsed peripheral blood mononuclear cells to prime virus‐specific naive T cells.77 Consistently with the ability of HO‐1 to down‐modulate the capacity of DCs to prime T cells, improved graft acceptance and reduced leucocyte infiltration were observed in an allogeneic aorta rat transplantation model after virus‐mediated Hmox1 gene transfer.59 Notably, both reduced leucocyte recruitment and tissue acceptance were reproduced in CO‐treated animals,59, 78 suggesting that this gas was the molecule responsible for the HO‐1‐mediated inhibition of T‐cell activation. The immune suppressive role proposed for CO has been corroborated by other studies showing that HO‐1 expression and CO production reduce MHC‐II expression in DCs. In addition, a deficiency of this enzyme increased the susceptibility to neuro‐inflammation in a murine experimental autoimmune encephalomyelitis model by inducing both increased accumulation of effector T cells and central nervous system damage.79
Either CoPP‐treated or CO‐treated human and murine DCs retained their capacity to secrete IL‐10, despite losing their ability to produce IL‐12p70,11, 76 a finding that was reproduced in murine cells.27, 62 However, although human DCs show reduced surface expression of maturation markers after CoPP/CO incubation, these molecules were not altered in murine DCs. These data suggest that the effect of HO‐1 activity could vary among species.27, 80 However, the precise explanation as to why the HO‐1–CO system shows different pattern of responses between human/rat and mouse cells remains unknown.
Because DCs are professional APCs, the capacity of HO‐1 to regulate antigen presentation has been an intensive area of research.80 In the murine system, it was shown that both the CoPP‐mediated induction of HO‐1 and CO were able to reduce the presentation of foreign antigens to naive CD4+ or CD8+ T cells (Fig. 3a). Furthermore, the effect of CO relied on a reduced capacity of both mature DCs and macrophages to target soluble extracellular antigens to intracellular lysosomal compartments. Conversely, HO‐1 activity and CO treatment had no effect on the processing and presentation of larger sized antigens, as those contained in 3‐μm latex beads.80
Figure 3.

Carbon monoxide (CO) impairs the endosome‐to‐lysosome pathway to soluble antigens in myeloid cells. (a) After the extracellular antigen is captured, it fuses with Rab5+ early endosomes. After that, these vesicles can fuse with proteasome/MHC‐I/TAP‐containing endosomes, which drive cross‐presentation. In parallel, antigen‐containing Rab5+ vesicles can fuse with Rab7+ endosomes to form late endosomes and then, sequentially, they can fuse with lysosomes (Lamp1+). These lysosomes harbour a full repertoire of MHC molecules that receive and present the small peptides obtained after the antigen is processed by lysosomal proteases. Once haem oxygenase 1 (HO‐1) is over‐expressed and CO is produced, there is an interference in the fusion between antigen‐containing late endosomes and lysosomes so compromising the correct antigen processing and antigen presentation to T cells. No effect of CO over cross‐presentation has been observed. (b) (i) Under local presence of pathogen‐associated molecular pattern (PAMPs); either by soluble molecules or presence of pathogens, dendritic cells (DCs) become activated. After binding the Toll like receptor 4/ myeloid differentiation factor 2 (TLR4/MD2) complex, LPS induces DC maturation by up‐regulation of co‐stimulatory molecules and secretion of cytokines. In addition, PAMPs cause local tissue damage and release of self‐ and non‐self‐antigens. (ii) Resident DCs capture soluble antigens presenting them to local T cells (something also observed in autoimmunity and graft rejection). Antigen‐containing mature DCs can travel to secondary lymphoid organs and activate antigen‐specific naive T cells. (iii) After PAMPs exposure [or treatment with cobalt protoporphyrin IX (CoPP), for example], DCs over‐express HO‐1, degrade haem‐group and produce CO. This process will modulate the immunogenicity of DCs recovering their initial homeostasis. (iv) CO‐producing mature DCs will lose their capacity to process antigens through the endosome‐to‐lysosome pathway. In addition, DCs reduce their secretion of cytokines. (v) Finally, mature DC‐dependent innate and adaptive immune inflammation is suppressed. Tissue homeostasis is recovered and pathologies caused by PAMPs and either foreign or self‐antigens are restricted.
These data support a model in which the HO‐1–CO system can discriminate between small and large antigens, by selectively inhibiting intracellular processing routes for the small soluble antigens.76 Along these lines, it seems that size is a crucial parameter for defining the intracellular processing pathway for an antigen. This notion is supported by recent studies showing that when extracellular large‐volume antigens make contact with cells, an endoplasmic reticulum (ER) ‐assisted phagocytosis occurs.81, 82 As part of this process, the ER supports phagosome formation by providing membrane fragments that form a structure known as the ERgosome (ER + phagosome). This compartment does not seem to require to be transported to the perinuclear zone for antigen‐processing because, at early times, it is enriched with lysosomal markers, proteasomal machinery and MHC molecules, all derived from cytoplasmic vesicles.83, 84 Hence, presentation on MHC‐II and cross‐presentation on MHC‐I of antigens attached to large bodies occurred regardless of CO treatment. In agreement with this observation is the fact that CO was unable to inhibit proteasome‐dependent cross‐presentation of intracellular soluble antigens, which suggested that only the endosome‐to‐lysosome route for extracellular soluble antigens can be targeted by CO.80
Despite the HO‐1–CO system only inhibiting the endosome‐to‐lysosome route, this pathway contributes to the onset of several antigen‐specific T‐cell‐dependent pathologies. Hence, CO‐mediated blockade of this pathway could be evaluated to interfere with the onset and progression of several inflammatory detrimental responses. Moreover, understanding how HO‐1 controls antigen‐dependent, as well as antigen‐independent induction of immunity by DCs can contribute to designing new therapies to prevent and treat inflammatory diseases.
Regulation of mitochondrial function by CO: implications for immunity
Recent studies have shown that mitochondria play a key role in the initiation of the antiviral innate immune response.85, 86 It has been observed that this organelle participates as a signalling platform for the activation of mitochondrial antiviral signalling protein, which ends mainly in the priming of the interferon response.86, 87 Furthermore, mitochondria can contribute to the processing of intracellular bacteria by enhancing ROS production after TLR signalling in macrophages.88 Thus, innate immune cells, such as APCs, employ mitochondria to exert some of their immune functions.
Because APCs require internal regulators to control inflammatory pathways and to restore homeostasis, it is possible that mitochondria could decrease immunity. Traditionally, mitochondria have been considered as cellular organelles associated with energetic, metabolic and genetic roles, whose oxidative function is carried out mainly by cytochrome‐containing complexes.89, 90, 91 Cytochromes are proteins that use Fe2+‐to‐Fe3+ porphyritic groups as co‐factors, which facilitate the transit of mobile electrons (high oxide‐redox potential). Based on their chemical properties, the porphyritic group and the Fe2+ also show high affinity for exposed electrons from diatomic gases, such as O2, NO and CO.92, 93 Hence, these gases in mitochondria can control both cytochrome‐dependent oxidative phosphorylation and ROS production. Experiments using soluble cell‐free/purified mitochondria have shown that CO binds to complex I and III in the respiratory chain.94, 95 CO affinity for these molecules prevents electron transport, reducing the mitochondrial membrane potential (ΔΨ), ATP and mitochondrial ROS generation (mROS, O2 −). Importantly, non‐toxic doses of CO have been used in these in vitro experiments to avoid mitochondrial disorganization and destruction.95 Because HOs are the only enzymes in mammal cells that produce CO at non‐toxic levels, it is likely that they could play a relevant role at modulating both mitochondrial function and at controlling immune cells, such as APCs. However, this hypothesis remains to be evaluated.
Because the HO‐1‐CO system interferes with the initiation of the APC‐dependent adaptive immune response, it is likely that a CO‐dependent mitochondrial blockade could reduce T‐cell priming.96 Consistent with this notion, it was shown that Kupffer cells require both a functional respiratory chain and high levels of mROS to prime antigen‐specific CD4+ T cells.97 These data agree with a recent study showing that mitochondrial stability is required to process and present soluble antigens by B cells to T cells in an ATP‐dependent manner.98 However, it remains unclear how and where mitochondria could be regulating antigen‐dependent immunity and whether endogenously produced CO can regulate these processes. Recent data from our laboratory propose a role for mitochondria in the transport and processing of antigen‐containing vesicles.99 These observations suggest that the HO‐1–CO system inhibits this pathway by dropping down mitochondrial ATP production without impairing the glycolysis‐dependent DC maturation.99 As a consequence, CO impairs mitochondrial function in DCs up to a point of down‐modulating antigen‐specific T‐cell priming. Because DCs link the innate and the adaptive immune responses, their important function could be modulated by targeting the inflammatory activity of mitochondria by using molecules that regulate ROS and ATP production. It is likely that HO‐1 activity and CO can act as natural regulators of the mitochondria‐dependent inflammatory pathway in APCs by blocking the maturation of antigen‐containing endosomes.
Regarding the interaction between mitochondria and HO‐1, studies performed in human alveolar and bronchial epithelial cells have shown the translocation of HO‐1 to the mitochondrial compartment after the exposure to cigarette smoke, LPS and haemin.100 The localization of HO‐1 at mitochondria in vivo has also been reported using the model of gastric mucosal tissue injury induced by non‐steroidal anti‐inflammatory drugs.101 This phenomenon resulted in the prevention of non‐steroidal anti‐inflammatory drug‐induced mitochondrial dysfunction and oxidative stress, gastric mucosal cell apoptosis and gastric mucosal injury. The proposed mechanism is the stabilization of complex I‐driven mitochondrial respiratory control and the transmembrane potential. These processes have been recently proposed as a novel cytoprotective effect of HO‐1.101 Mitochondrial HO‐1 translocation was also observed in macrophage RAW‐264.7 cells after the treatment with CoCl2 or exposure to hypoxia. However, in this case the in‐organ localization of HO‐1 caused mitochondrial dysfunction.102
Taken together, these findings suggest that the anti‐inflammatory effects of HO‐1 could be in part explained by the suppression of the antigen‐dependent immunity, which is directly associated with mitochondrial function. In addition, HO‐1‐mediated mitochondrial protection after the translocation of this enzyme to that organelle reduces cytotoxicity and massive cell death, reducing DAMPs release. These findings also uncover new therapeutic targets to control the HO‐1–CO system as a manner to approach diseases caused by the adaptive immune response.
The HO‐1–CO system reduces pathologies caused by the immune response
HO‐1 reduces innate immunity‐mediated inflammatory diseases
Several pathologies are associated with the effector function of the innate immune system.103 Fast and acute inflammatory conditions, such as fever, organ swelling and septic shock are mainly mediated by the rapid recruitment of monocytes and neutrophils to the site of infection.104 Cellular recruitment is associated with a gradient of inflammatory cytokines that are secreted either at the infection site or at damaged tissues (Fig. 3b(i)–(ii)).105, 106, 107 Hence, interference with the secretion of these pro‐inflammatory cytokines is likely to reduce the pathology. Expression of HO‐1 and the subsequent CO production can contribute to controlling several innate immunity‐driven inflammatory pathologies.108 For instance, CO can protect from the permeability induced by LPS in epithelial tissues by reducing inflammatory cytokine secretion and preventing the down‐regulation of tight junction proteins, such as ZO‐1 and occludin.109 Furthermore, it has been recently shown that an IL‐10‐dependent HO‐1 induction decreases both the recruitment of innate inflammatory cells and the secretion of inflammatory cytokines in a septic shock animal model.35 Hence, blockade of myeloid cell‐derived inflammatory cytokines by the HO‐1–CO system can work as a mechanism to reduce the sensitivity to stimulation by PAMPs (Fig. 3b(iii)–(iv)).
The HO‐1–CO system reduces adaptive immunity‐dependent inflammation. Implications in tolerance during transplantation and pregnancy
Activated T cells can contribute to several immune‐based pathologies. T cells become activated after recognizing antigens as peptide–MHC complexes (pMHC) on the surface of APCs. The pMHCs are generated by APCs as a result of the processing and presentation of internalized antigens, either foreign or self, as is the case of pathogen infections or autoimmune disorders, respectively.
Furthermore, allospecific T cells are primed by APCs expressing MHC molecules at variant from the host.110, 111, 112 Such an allogeneic recognition can lead to organ rejection in patients who have received a transplant to treat illnesses, such as kidney failure. During transplant rejection, DCs stimulate T cells through direct, indirect or semi‐direct pathways of allorecognition.111, 112 In the direct pathway, donor DCs can migrate out of the grafted tissue and present intact donor MHC/peptide complexes to allospecific T cells (which recognize non‐self antigens).111, 113 In the indirect pathway, recipient DCs process donor alloantigens (foreign/non‐self antigens) and present them to autologous reactive T cells.111, 113 Finally, through the semi‐direct pathway of allorecognition, recipient T cells recognize intact donor MHC/peptide complexes that have been transferred to the surface of recipient DCs by a process known as ‘nibbling’.114 From these three pathways, the direct route is associated with early graft rejection and has been classified as the most powerful mechanism of rejection.115 However, this mechanism decreases with time because donor DCs mainly die by senescence. On the other hand, recipient DCs arise as the most potent factor able to activate host T cells during the time. Hence, the indirect pathway arises as the major cause of chronic graft rejection after presentation and recognition of alloantigens.112, 113, 115
These mechanisms of allo‐recognition pose an important challenge to improve graft acceptance, so new specific approaches are required to prevent T‐cell activation by donor or recipient DCs after transplantation. Graft acceptance could be promoted either by suppressing de novo activation of T cells or inducing T‐cell antigen‐unresponsiveness. A promising approach to achieve these goals is the generation of antigen‐specific regulatory T cells.116, 117, 118 This technique contributes to specific protection and graft survival. Although regulatory T cells show antigen‐specificity, these cells are efficient at suppressing locally reactive effector T cells. Whether the regulatory T‐cell approach can produce as a side effect local immune suppression leading to pathogen spread remains to be defined. A combination of different strategies, for example regulatory T cells together with DCs that induce T‐cell unresponsiveness, might be a solution. However, these alternatives must be evaluated, both in animal models and in clinical studies.
It was recently shown that in vivo Hmox1 gene transfer using adenoviral vectors improves long‐term heart graft survival in a myeloid cell (DC) ‐dependent fashion.59, 78 Also, in another model of graft survival, injection of pigs with a lentiviral vector encoding for Hmox1 gene ameliorated the outcome of a Duchenne muscular dystrophy therapy with myogenic cell precursors.58 Similar results were obtained in diabetes,119, 120 renal transplantation121, 122 and human cardiac stem cell transference123 in which all the over‐expression of HO‐1 was induced with CoPP. This notion was further supported by the observation that regulatory T‐cell‐dependent suppression of activated T cells required the expression of HO‐1 in APCs.124 Similar observations were made in mouse models for inflammatory diseases, such as in lactobacillus‐mediated infection where HO‐1 activity was required to efficiently produce mesenteric Foxp3+ CD25+ CD4+ T cells.125 Moreover, transfer of wild‐type regulatory T cells into HO‐1 heterozygous mice restored the ratios between regulatory and effector T cells and reduced inflammation in a model of necrotizing enterocolitis, supporting a possible direct role for HO‐1 in the generation and function of regulatory T cells.126 Hence, the HO‐1–CO system arises as a powerful candidate to restrict antigen presentation to T cells in vivo and also to impair the activation of lymphocytes in transplantation and autoimmunity. Furthermore, the generation of regulatory T cells during bacteria‐dependent inflammation and graft acceptance seems to depend on the HO‐1 activity.
Because these pathologies are mainly mediated by the presentation of extracellular antigens that have been processed by the endosome‐to‐lysosome pathway, the mechanism by which CO could be reducing the activation of T cells can be associated with a blockade of this route (Fig. 3b(v)). Moreover, as seen in rat and human models of organ transplantation, reduced DC maturation by HO‐1 can impair the expression of both MHCs and co‐stimulatory molecules. As a result, the priming of MHC mismatched T cells would be reduced. Hence, due to its impact on antigen presentation, modulation of the HO‐1–CO system in vitro either by pharmacological or gene therapy could work as efficient strategies to induce antigen‐specific tolerogenic DCs that are useful for cell transfer‐based therapy during autoimmune diseases.
In addition, the HO‐1–CO system has also been characterized as protective during pregnancy.127, 128 During embryo development, new cells and antigens are produced. To protect the developing fetus from being attacked by the adaptive immune response of the mother, a tolerant immune state must be established in the fetal–maternal interface. Consistent with this notion, it has been shown that HO‐1 expression can prevent natural abortion in a well‐established mouse model.129 HO‐1 up‐regulation induces BCL‐associated athanogene‐1 (Bag‐1) and neuropilin‐1, two markers associated with the development of regulatory T cells and the induction of tolerance.130 Furthermore, recent studies showed that HO‐1 regulates regulatory T‐cell‐mediated protection against abortion in mice because this enzyme reduced DC maturation and regulatory T‐cell expansion.131 It is thought that reduced DC maturation avoided the priming of effector T cells in the mother, which protected embryo from the immune response. These mechanisms were supported by recent data suggesting that progesterone regulates both the expansion of regulatory CD8+ CD122+ T cells and the establishment of fetal tolerance after inducing expression of HO‐1 in the placenta.132 In addition, the progesterone–HO‐1 system reduced the expansion of cytotoxic CD8+ T cells that recognize non‐self antigens expressed by the embryo.132 Consistently, a correlation was shown for pregnant women between current miscarriage and a reduced amount of regulatory T cells, despite having regular numbers of circulating DCs.133 These data suggest that HO‐1 regulates DC activity and the capacity to induce T‐cell‐mediated tolerance.
Because DCs are crucial for the establishment of peripheral tolerance, their contributions to tolerance during pregnancy and to protecting the new embryo from immune recognition have been extensively studied. It has been shown that uterine DCs display an immature/IL‐10‐secreting phenotype during pregnancy and that maturation and IL‐12 secretion are directly associated with abortion in mice.134, 135 Indeed, absence of IL‐10 during pregnancy leads to an altered phenotype for DCs, macrophages, effector and regulatory T cells, which contribute to LPS‐mediated abortion.136 These data suggest that production of IL‐10 by APCs controls T‐cell responses after recognition/presentation of embryo‐derived antigens. In addition, uterine DCs might be crucial for embryo implantation because they accumulate before pregnancy during the oestrous cycle137 and their remotion definitely affects the success of implantation.138
Because mature uterine DCs release several inflammatory mediators, activation of these cells by PAMPs could promote inflammation in mother tissues and impair embryo development. Hence, it has been shown that bacterial infection is an important risk factor that can trigger abortion because, in addition to the inflammatory condition, pathogens down‐regulate HO‐1 expression in the placenta.139, 140 Up‐regulation of HO‐1 by CoPP increased fetus development and augmented cell survival of the placental tissue.128 This process is likely to be promoted by the capacity of HO‐1/IL‐10‐expressing DCs to induce regulatory T‐cell expansion after antigen presentation, which contributes to placental stability. It has also been shown that HO‐1 regulates placental development.141 Consistently with this notion, the amount of apoptotic placental cells was reduced when HO‐1 expression was promoted by transduction with an adenoviral Hmox1 gene transfer system.129 Another study showed decreased placenta size and weight, as well as impaired cell viability in mice heterozygous (Hmox1 +/−) for HO‐1.141, 142 Along these lines, it has been shown that both Hmox1 +/− and Hmox1 −/− mice show an impairment in uterine natural killer (uNK) ‐dependent maternal spiral arteries remodeling during implantation because of reduced numbers of uNK.143, 144 Interestingly, treatement of Hmox1 +/− mice with low doses of CO improved both uNK proliferation, spiral arteries remodelling and also stabilized blood presure, suggesting that this gas plays a key role during uNK‐dependent intrauterine growth.143, 144 Similar data were obtained in a trophoblastic stem cell line that resembles the development of trophoblasts into giant cells, which showed that HO‐1 inhibition compromises cell viability and their ability to differentiate.145 In agreement, trophoblastic tissue from women suffering spontaneous abortion displayed reduced HO‐1 expression levels compared with healthy pregnancies.142 It is noteworthy that CO administration during implantation and the early placentation window improved cell survival and increased Bag‐1 expression in the same manner as shown by the expression of HO‐1, suggesting that the main mediator for this enzyme in placenta stability is CO.146 Hence, the HO‐1–CO system not only plays an important role in the regulation and tolerance associated with the immune system, but also implies a direct function in the placental tissue development that will protect the embryo during the interaction with tissues of the mother.
Altogether, these results suggest that the HO‐1–CO system can prevent and reduce the outcome of inflammatory pathologies related to innate and adaptive immunity (Fig. 3b). In the case of innate immunity, both CO‐mediated cytokine secretion suppression and reduced cell recruitment to the site of PAMPs accumulation arise as the most important regulatory mechanism used by HO‐1. In the case of adaptive immunity, CO‐mediated suppression of both fusion between antigen‐containing endosomes and lysosomes, antigen surface presentation and impairment of DC maturation is associated with reduced T‐cell priming, lower tissue destruction and an establishment of tolerance.
Concluding remarks
HO‐1 is expressed in APCs, such as DCs and macrophages. The production of CO by this enzyme mediates the suppression of different inflammatory pathways. HO‐1 and CO reduce the capacity of APCs to recognize PAMPs and suppress both pro‐inflammatory cytokine secretion and antigen presentation. These modulatory processes uncouple the whole immune network by blocking the activation of either antigen‐specific or allogeneic T cells. HO‐1–CO‐mediated reduction of immunity controls the onset and progression of several diseases, such as sepsis, organ rejection and autoimmunity. The exploitation of the molecular targets of the HO‐1–CO system, such as mitochondria, arises as a promising alternative to create anti‐inflammatory therapies. More research is needed to evaluate whether by inhibiting mitochondria until non‐toxic levels, as CO does it, can lead to improving life quality in patients suffering from inflammatory ailments.
Disclosures
The authors have declared that no conflict of interest exists.
Ackowledgements
This study was supported by the following grants: FONDO NACIONAL DE CIENCIA Y TECNOLOGIA DE CHILE (FONDECYT numbers 1110397, 1131012, 1140010 and 1110604), Millennium Institute on Immunology and Immunotherapy P09/016‐F and Grant ‘Nouvelles Equipes‐nouvelles thématiques’ from the La Région Pays De La Loire. SAR and JAE are supported by the Comisión Nacional de Investigación Científica y Tecnológica (CONICYT). LJC is a Latin American Pew Fellow. AMK is a Chaire De La Région Pays De La Loire, Chercheur Étranger D'excellence, France.
References
- 1. Li T, Bonkovsky HL, Guo JT. Structural analysis of heme proteins: implications for design and prediction. BMC Struct Biol 2011; 11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maines MD. Overview of heme degradation pathway. Curr Protoc Toxicol 2001. DOI: 10.1002/0471140856.tx0901s00. [DOI] [PubMed] [Google Scholar]
- 3. Ryter SW, Tyrrell RM. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro‐ and antioxidant properties. Free Radic Biol Med 2000; 28:289–309. [DOI] [PubMed] [Google Scholar]
- 4. Dennery PA. Signaling function of heme oxygenase proteins. Antioxid Redox Signal 2014; 20:1743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J 1988; 2:2557–68. [PubMed] [Google Scholar]
- 6. Abraham NG, Cao J, Sacerdoti D, Li X, Drummond G. Heme oxygenase: the key to renal function regulation. Am J Physiol Renal Physiol 2009; 297:F1137–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kikuchi G, Yoshida T, Noguchi M. Heme oxygenase and heme degradation. Biochem Biophys Res Commun 2005; 338:558–67. [DOI] [PubMed] [Google Scholar]
- 8. Bauer M, Bauer I. Heme oxygenase‐1: redox regulation and role in the hepatic response to oxidative stress. Antioxid Redox Signal 2002; 4:749–58. [DOI] [PubMed] [Google Scholar]
- 9. Zhang Y, Jiang G, Sauler M, Lee PJ. Lung endothelial HO‐1 targeting in vivo using lentiviral miRNA regulates apoptosis and autophagy during oxidant injury. FASEB J 2013; 27:4041–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morse D, Pischke SE, Zhou Z, Davis RJ, Flavell RA, Loop T et al Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP‐1. J Biol Chem 2003; 278:36993–8. [DOI] [PubMed] [Google Scholar]
- 11. Chauveau C, Remy S, Royer PJ, Hill M, Tanguy‐Royer S, Hubert FX et al Heme oxygenase‐1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL‐10 expression. Blood 2005; 106:1694–702. [DOI] [PubMed] [Google Scholar]
- 12. Morse D. The role of heme oxygenase‐1 in pulmonary fibrosis. Am J Respir Cell Mol Biol 2003; 29:S82–6. [PubMed] [Google Scholar]
- 13. Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem 1986; 261:411–9. [PubMed] [Google Scholar]
- 14. Trakshel GM, Kutty RK, Maines MD. Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. J Biol Chem 1986; 261:11131–7. [PubMed] [Google Scholar]
- 15. Stout JM, Gousset MU, Drummond HA, Gray W 3rd, Pruett BE, Stec DE. Sex‐specific effects of heme oxygenase‐2 deficiency on renovascular hypertension. J Am Soc Hypertens 2013; 7:328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y et al Characterization of rat heme oxygenase‐3 gene. Implication of processed pseudogenes derived from heme oxygenase‐2 gene. Gene 2004; 336:241–50. [DOI] [PubMed] [Google Scholar]
- 17. Radhakrishnan N, Yadav SP, Sachdeva A, Pruthi PK, Sawhney S, Piplani T et al Human heme oxygenase‐1 deficiency presenting with hemolysis, nephritis, and asplenia. J Pediatr Hematol Oncol 2011; 33:74–8. [DOI] [PubMed] [Google Scholar]
- 18. Poss KD, Tonegawa S. Reduced stress defense in heme oxygenase 1‐deficient cells. Proc Natl Acad Sci USA 1997; 94:10925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T et al Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase‐1 deficiency. J Clin Invest 1999; 103:129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mackern‐Oberti JP, Riquelme SA, Llanos C, Schmidt CB, Simon T, Anegon I et al Heme oxygenase‐1 as a target for the design of gene and pharmaceutical therapies for autoimmune diseases. Curr Gene Ther 2014; 14:218–35. [DOI] [PubMed] [Google Scholar]
- 21. Kapturczak MH, Wasserfall C, Brusko T, Campbell‐Thompson M, Ellis TM, Atkinson MA et al Heme oxygenase‐1 modulates early inflammatory responses: evidence from the heme oxygenase‐1‐deficient mouse. Am J Pathol 2004; 165:1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Herrada AA, Llanos C, Mackern‐Oberti JP, Carreno LJ, Henriquez C, Gomez RS et al Haem oxygenase 1 expression is altered in monocytes from patients with systemic lupus erythematosus. Immunology 2012; 136:414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fagone P, Patti F, Mangano K, Mammana S, Coco M, Touil‐Boukoffa C et al Heme oxygenase‐1 expression in peripheral blood mononuclear cells correlates with disease activity in multiple sclerosis. J Neuroimmunol 2013; 261:82–6. [DOI] [PubMed] [Google Scholar]
- 24. Ryu EY, Park SY, Kim SG, da Park J, Kang JS, Kim YH et al Anti‐inflammatory effect of heme oxygenase‐1 toward Porphyromonas gingivalis lipopolysaccharide in macrophages exposed to gomisins A, G, and J. J Med Food 2011; 14:1519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hull TD, Agarwal A, George JF. The mononuclear phagocyte system in homeostasis and disease: a role for heme oxygenase‐1. Antioxid Redox Signal 2014; 20:1770–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen HG, Xie KL, Han HZ, Wang WN, Liu DQ, Wang GL et al Heme oxygenase‐1 mediates the anti‐inflammatory effect of molecular hydrogen in LPS‐stimulated RAW 264.7 macrophages. Int J Surg 2013; 11:1060–6. [DOI] [PubMed] [Google Scholar]
- 27. Remy S, Blancou P, Tesson L, Tardif V, Brion R, Royer PJ et al Carbon monoxide inhibits TLR‐induced dendritic cell immunogenicity. J Immunol 2009; 182:1877–84. [DOI] [PubMed] [Google Scholar]
- 28. De Nardo D. Toll‐like receptors: activation, signalling and transcriptional modulation. Cytokine 2015; 74:181–9. [DOI] [PubMed] [Google Scholar]
- 29. Wegiel B, Hauser CJ, Otterbein LE. Heme as a danger molecule in pathogen recognition. Free Radic Biol Med 2015; 89:651–61. [DOI] [PubMed] [Google Scholar]
- 30. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK‐activated protein kinases. Microbiol Mol Biol Rev 2011; 75:50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davies C, Tournier C. Exploring the function of the JNK (c‐Jun N‐terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans 2012; 40:85–9. [DOI] [PubMed] [Google Scholar]
- 32. Rushworth SA, Chen X‐L, Mackman N, Ogborne RM, O'Connell MA. Lipopolysaccharide‐induced heme oxygenase‐1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J Immunol 2005; 175:4408–15. [DOI] [PubMed] [Google Scholar]
- 33. Srisook K, Cha YN. Super‐induction of HO‐1 in macrophages stimulated with lipopolysaccharide by prior depletion of glutathione decreases iNOS expression and NO production. Nitric Oxide 2005; 12:70–9. [DOI] [PubMed] [Google Scholar]
- 34. Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser‐40 by protein kinase C regulates antioxidant response element‐mediated transcription. J Biol Chem 2002; 277:42769–74. [DOI] [PubMed] [Google Scholar]
- 35. Lee TS, Chau LY. Heme oxygenase‐1 mediates the anti‐inflammatory effect of interleukin‐10 in mice. Nat Med 2002; 8:240–6. [DOI] [PubMed] [Google Scholar]
- 36. Al‐Huseini LM, Aw Yeang HX, Sethu S, Alhumeed N, Hamdam JM, Tingle Y et al Nuclear factor‐erythroid 2 (NF‐E2) p45‐related factor‐2 (Nrf2) modulates dendritic cell immune function through regulation of p38 MAPK‐cAMP‐responsive element binding protein/activating transcription factor 1 signaling. J Biol Chem 2013; 288:22281–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Igarashi K, Sun J. The heme‐Bach1 pathway in the regulation of oxidative stress response and erythroid differentiation. Antioxid Redox Signal 2006; 8:107–18. [DOI] [PubMed] [Google Scholar]
- 38. Sun Jang J, Piao S, Cha YN, Kim C. Taurine chloramine activates Nrf2, increases HO‐1 expression and protects cells from death caused by hydrogen peroxide. J Clin Biochem Nutr 2009; 45:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pulkkinen KH, Yla‐Herttuala S, Levonen AL. Heme oxygenase 1 is induced by miR‐155 via reduced BACH1 translation in endothelial cells. Free Radic Biol Med 2011; 51:2124–31. [DOI] [PubMed] [Google Scholar]
- 40. McCoy CE, Sheedy FJ, Qualls JE, Doyle SL, Quinn SR, Murray PJ et al IL‐10 inhibits miR‐155 induction by toll‐like receptors. J Biol Chem 2010; 285:20492–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma Q, Battelli L, Hubbs AF. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant‐activated transcription factor Nrf2. Am J Pathol 2006; 168:1960–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Exner M, Minar E, Wagner O, Schillinger M. The role of heme oxygenase‐1 promoter polymorphisms in human disease. Free Radic Biol Med 2004; 37:1097–104. [DOI] [PubMed] [Google Scholar]
- 43. Rueda B, Oliver J, Robledo G, Lopez‐Nevot MA, Balsa A, Pascual‐Salcedo D et al HO‐1 promoter polymorphism associated with rheumatoid arthritis. Arthritis Rheum 2007; 56:3953–8. [DOI] [PubMed] [Google Scholar]
- 44. Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S et al Microsatellite polymorphism in the heme oxygenase‐1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet 2000; 66:187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sponholz C, Huse K, Kramer M, Giamarellos‐Bourboulis EJ, Claus RA, Kern A et al Gene polymorphisms in the heme degradation pathway and outcome of severe human sepsis. Shock 2012; 38:459–65. [DOI] [PubMed] [Google Scholar]
- 46. Ono K, Mannami T, Iwai N. Association of a promoter variant of the haeme oxygenase‐1 gene with hypertension in women. J Hypertens 2003; 21:1497–503. [DOI] [PubMed] [Google Scholar]
- 47. Ono K, Goto Y, Takagi S, Baba S, Tago N, Nonogi H et al A promoter variant of the heme oxygenase‐1 gene may reduce the incidence of ischemic heart disease in Japanese. Atherosclerosis 2004; 173:315–9. [DOI] [PubMed] [Google Scholar]
- 48. Askenazi DJ, Halloran B, Patil N, Keeling S, Saeidi B, Koralkar R et al Genetic polymorphisms of heme‐oxygenase 1 (HO‐1) may impact on acute kidney injury, bronchopulmonary dysplasia, and mortality in premature infants. Pediatr Res 2015; 77:793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hajdena‐Dawson M, Zhang W, Contag PR, Wong RJ, Vreman HJ, Stevenson DK et al Effects of metalloporphyrins on heme oxygenase‐1 transcription: correlative cell culture assays guide in vivo imaging. Mol Imaging 2003; 2:138–49. [DOI] [PubMed] [Google Scholar]
- 50. Shan Y, Lambrecht RW, Donohue SE, Bonkovsky HL. Role of Bach1 and Nrf2 in up‐regulation of the heme oxygenase‐1 gene by cobalt protoporphyrin. FASEB J 2006; 20:2651–3. [DOI] [PubMed] [Google Scholar]
- 51. Sardana MK, Kappas A. Dual control mechanism for heme oxygenase: tin(IV)‐protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc Natl Acad Sci USA 1987; 84:2464–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Anderson KE, Simionatto CS, Drummond GS, Kappas A. Tissue distribution and disposition of tin‐protoporphyrin, a potent competitive inhibitor of heme oxygenase. J Pharmacol Exp Ther 1984; 228:327–33. [PubMed] [Google Scholar]
- 53. Hung CC, Liu X, Kwon MY, Kang YH, Chung SW, Perrella MA. Regulation of heme oxygenase‐1 gene by peptidoglycan involves the interaction of Elk‐1 and C/EBPα to increase expression. Am J Physiol Lung Cell Mol Physiol 2010; 298:L870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Akira S, Takeda K. Toll‐like receptor signalling. Nat Rev Immunol 2004; 4:499–511. [DOI] [PubMed] [Google Scholar]
- 55. Akira S, Yamamoto M, Takeda K. Role of adapters in Toll‐like receptor signalling. Biochem Soc Trans 2003; 31:637–42. [DOI] [PubMed] [Google Scholar]
- 56. O'Neill LA, Golenbock D, Bowie AG. The history of Toll‐like receptors ‐ redefining innate immunity. Nat Rev Immunol 2013; 13:453–60. [DOI] [PubMed] [Google Scholar]
- 57. Ferenbach DA, Ramdas V, Spencer N, Marson L, Anegon I, Hughes J et al Macrophages expressing heme oxygenase‐1 improve renal function in ischemia/reperfusion injury. Mol Ther 2010; 18:1706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roach JP, Moore EE, Partrick DA, Damle SS, Silliman CC, McIntyre RC Jr et al Heme oxygenase‐1 induction in macrophages by a hemoglobin‐based oxygen carrier reduces endotoxin‐stimulated cytokine secretion. Shock 2009; 31:251–7. [DOI] [PubMed] [Google Scholar]
- 59. Chauveau C, Bouchet D, Roussel JC, Mathieu P, Braudeau C, Renaudin K et al Gene transfer of heme oxygenase‐1 and carbon monoxide delivery inhibit chronic rejection. Am J Transplant 2002; 2:581–92. [DOI] [PubMed] [Google Scholar]
- 60. Tzima S, Victoratos P, Kranidioti K, Alexiou M, Kollias G. Myeloid heme oxygenase‐1 regulates innate immunity and autoimmunity by modulating IFN‐β production. J Exp Med 2009; 206:1167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laumonier T, Yang S, Konig S, Chauveau C, Anegon I, Hoffmeyer P et al Lentivirus mediated HO‐1 gene transfer enhances myogenic precursor cell survival after autologous transplantation in pig. Mol Ther 2008; 16:404–10. [DOI] [PubMed] [Google Scholar]
- 62. Riquelme SA, Bueno SM, Kalergis AM. Carbon monoxide down‐modulates TLR4/MD2 expression on innate immune cells and reduces endotoxic shock susceptibility. Immunology 2014; 144:321–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xue J, Habtezion A. Carbon monoxide‐based therapy ameliorates acute pancreatitis via TLR4 inhibition. J Clin Invest 2014; 124:437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rocuts F, Ma Y, Zhang X, Gao W, Yue Y, Vartanian T et al Carbon monoxide suppresses membrane expression of TLR4 via myeloid differentiation factor‐2 in βTC3 cells. J Immunol 2010; 185:2134–9. [DOI] [PubMed] [Google Scholar]
- 65. Gomez‐Hurtado I, Zapater P, Bellot P, Pascual S, Perez‐Mateo M, Such J et al Interleukin‐10‐mediated heme oxygenase 1‐induced underlying mechanism in inflammatory down‐regulation by norfloxacin in cirrhosis. Hepatology 2011; 53:935–44. [DOI] [PubMed] [Google Scholar]
- 66. Nassour I, Kautza B, Rubin M, Escobar D, Luciano J, Loughran P et al Carbon monoxide protects against hemorrhagic shock and resuscitation‐induced microcirculatory injury and tissue injury. Shock 2015; 43:166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hettiarachchi N, Dallas M, Al‐Owais M, Griffiths H, Hooper N, Scragg J et al Heme oxygenase‐1 protects against Alzheimer's amyloid‐β1‐42‐induced toxicity via carbon monoxide production. Cell Death Dis 2014; 5:e1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ruan Y, Wang L, Zhao Y, Yao Y, Chen S, Li J et al Carbon monoxide potently prevents ischemia‐induced high‐mobility group box 1 translocation and release and protects against lethal renal ischemia‐reperfusion injury. Kidney Int 2014; 86:525–37. [DOI] [PubMed] [Google Scholar]
- 69. Nakao A, Toyoda Y. Application of carbon monoxide for transplantation. Curr Pharm Biotechnol 2012; 13:827–36. [DOI] [PubMed] [Google Scholar]
- 70. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature 1998; 392:245–52. [DOI] [PubMed] [Google Scholar]
- 71. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ et al Immunobiology of dendritic cells. Annu Rev Immunol 2000; 18:767–811. [DOI] [PubMed] [Google Scholar]
- 72. Kim SJ, Diamond B. Modulation of tolerogenic dendritic cells and autoimmunity. Semin Cell Dev Biol 2014; 41:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol 2003; 21:685–711. [DOI] [PubMed] [Google Scholar]
- 74. Mackern‐Oberti JP, Llanos C, Carreno LJ, Riquelme SA, Jacobelli SH, Anegon I et al Carbon monoxide exposure improves immune function in lupus‐prone mice. Immunology 2013; 140:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Al‐Huseini LM, Aw Yeang HX, Hamdam JM, Sethu S, Alhumeed N, Wong W et al Heme oxygenase‐1 regulates dendritic cell function through modulation of p38 MAPK‐CREB/ATF1 signaling. J Biol Chem 2014; 289:16442–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Listopad J, Asadullah K, Sievers C, Ritter T, Meisel C, Sabat R et al Heme oxygenase‐1 inhibits T cell‐dependent skin inflammation and differentiation and function of antigen‐presenting cells. Exp Dermatol 2007; 16:661–70. [DOI] [PubMed] [Google Scholar]
- 77. Bunse CE, Fortmeier V, Tischer S, Zilian E, Figueiredo C, Witte T et al Modulation of heme oxygenase‐1 by metalloporphyrins increases anti‐viral T cell responses. Clin Exp Immunol 2015; 179:265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Braudeau C, Bouchet D, Tesson L, Iyer S, Remy S, Buelow R et al Induction of long‐term cardiac allograft survival by heme oxygenase‐1 gene transfer. Gene Ther 2004; 11:701–10. [DOI] [PubMed] [Google Scholar]
- 79. Chora AA, Fontoura P, Cunha A, Pais TF, Cardoso S, Ho PP et al Heme oxygenase‐1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest 2007; 117:438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tardif V, Riquelme SA, Remy S, Carreno LJ, Cortes CM, Simon T et al Carbon monoxide decreases endosome‐lysosome fusion and inhibits soluble antigen presentation by dendritic cells to T cells. Eur J Immunol 2013; 43:2832–44. [DOI] [PubMed] [Google Scholar]
- 81. Gagnon E, Bergeron JJ, Desjardins M. ER‐mediated phagocytosis: myth or reality? J Leukoc Biol 2005; 77:843–5. [DOI] [PubMed] [Google Scholar]
- 82. Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele‐Mortimer O et al Endoplasmic reticulum‐mediated phagocytosis is a mechanism of entry into macrophages. Cell 2002; 110:119–31. [DOI] [PubMed] [Google Scholar]
- 83. Guermonprez P, Amigorena S. Pathways for antigen cross presentation. Springer Semin Immunopathol 2005; 26:257–71. [DOI] [PubMed] [Google Scholar]
- 84. Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A et al Phagosomes are competent organelles for antigen cross‐presentation. Nature 2003; 425:402–6. [DOI] [PubMed] [Google Scholar]
- 85. Zemirli N, Arnoult D. Mitochondrial anti‐viral immunity. Int J Biochem Cell Biol 2012; 44:1473–6. [DOI] [PubMed] [Google Scholar]
- 86. Arnoult D, Soares F, Tattoli I, Girardin SE. Mitochondria in innate immunity. EMBO Rep 2011; 12:901–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Haller O, Kochs G, Weber F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 2006; 344:119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument‐Bromage H, Tempst P et al TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011; 472:476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Piantadosi CA, Suliman HB. Redox regulation of mitochondrial biogenesis. Free Radic Biol Med 2012; 53:2043–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Roy M, Reddy PH, Iijima M, Sesaki H. Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol 2015; 33:111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Turnbull DM, Rustin P. Genetic and biochemical intricacy shapes mitochondrial cytopathies. Neurobiol Dis 2015; 15:00023–6. [DOI] [PubMed] [Google Scholar]
- 92. Winter MB, Herzik MA Jr, Kuriyan J, Marletta MA. Tunnels modulate ligand flux in a heme nitric oxide/oxygen binding (H‐NOX) domain. Proc Natl Acad Sci USA 2011; 108:E881–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Weber C, Cole DJ, O'Regan DD, Payne MC. Renormalization of myoglobin‐ligand binding energetics by quantum many‐body effects. Proc Natl Acad Sci USA 2014; 111:5790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Desmard M, Boczkowski J, Poderoso J, Motterlini R. Mitochondrial and cellular heme‐dependent proteins as targets for the bioactive function of the heme oxygenase/carbon monoxide system. Antioxid Redox Signal 2007; 9:2139–55. [DOI] [PubMed] [Google Scholar]
- 95. Long R, Salouage I, Berdeaux A, Motterlini R, Morin D. CORM‐3, a water soluble CO‐releasing molecule, uncouples mitochondrial respiration via interaction with the phosphate carrier. Biochim Biophys Acta 2014; 1837:201–9. [DOI] [PubMed] [Google Scholar]
- 96. Almeida AS, Figueiredo‐Pereira C, Vieira HL. Carbon monoxide and mitochondria‐modulation of cell metabolism, redox response and cell death. Front Physiol 2015; 6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Maemura K, Zheng Q, Wada T, Ozaki M, Takao S, Aikou T et al Reactive oxygen species are essential mediators in antigen presentation by Kupffer cells. Immunol Cell Biol 2005; 83:336–43. [DOI] [PubMed] [Google Scholar]
- 98. Bonifaz L, Cervantes‐Silva M, Ontiveros‐Dotor E, Lopez‐Villegas E, Sanchez‐Garcia F. A role for mitochondria in antigen processing and presentation. Immunology 2014; 144:461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Riquelme SA, Pogu J, Anegon I, Bueno SM, Kalergis AM. Carbon monoxide impairs mitochondria‐dependent endosomal maturation and antigen presentation in dendritic cells. Eur J Immunol 2015; 45:3269–88. [DOI] [PubMed] [Google Scholar]
- 100. Slebos DJ, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ et al Mitochondrial localization and function of heme oxygenase‐1 in cigarette smoke‐induced cell death. Am J Respir Cell Mol Biol 2007; 36:409–17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101. Bindu S, Pal C, Dey S, Goyal M, Alam A, Iqbal MS et al Translocation of heme oxygenase‐1 to mitochondria is a novel cytoprotective mechanism against non‐steroidal anti‐inflammatory drug‐induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J Biol Chem 2011; 286:39387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bansal S, Biswas G, Avadhani NG. Mitochondria‐targeted heme oxygenase‐1 induces oxidative stress and mitochondrial dysfunction in macrophages, kidney fibroblasts and in chronic alcohol hepatotoxicity. Redox Biol 2014; 2:273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Beutler B. Innate immunity: an overview. Mol Immunol 2004; 40:845–59. [DOI] [PubMed] [Google Scholar]
- 104. Delneste Y, Beauvillain C, Jeannin P. Innate immunity: structure and function of TLRs. Med Sci (Paris) 2007; 23:67–73. [DOI] [PubMed] [Google Scholar]
- 105. Brown KA, Brain SD, Pearson JD, Edgeworth JD, Lewis SM, Treacher DF. Neutrophils in development of multiple organ failure in sepsis. Lancet 2006; 368:157–69. [DOI] [PubMed] [Google Scholar]
- 106. Efron P, Moldawer LL. Sepsis and the dendritic cell. Shock 2003; 20:386–401. [DOI] [PubMed] [Google Scholar]
- 107. Nautiyal KM, McKellar H, Silverman AJ, Silver R. Mast cells are necessary for the hypothermic response to LPS‐induced sepsis. Am J Physiol Regul Integr Comp Physiol 2009; 296:R595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hoetzel A, Dolinay T, Schmidt R, Choi AM, Ryter SW. Carbon monoxide in sepsis. Antioxid Redox Signal 2007; 9:2013–26. [DOI] [PubMed] [Google Scholar]
- 109. Mu X, Pan C, Zheng S, Alhamdi Y, Sun B, Shi Q et al Protective effects of carbon monoxide‐releasing molecule‐2 on the barrier function of intestinal epithelial cells. PLoS One 2014; 9:e104032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Warren EH, Zhang XC, Li S, Fan W, Storer BE, Chien JW et al Effect of MHC and non‐MHC donor/recipient genetic disparity on the outcome of allogeneic HCT. Blood 2012; 120:2796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol 2007; 7:610–21. [DOI] [PubMed] [Google Scholar]
- 112. Gould DS, Auchincloss H Jr. Direct and indirect recognition: the role of MHC antigens in graft rejection. Immunol Today 1999; 20:77–82. [DOI] [PubMed] [Google Scholar]
- 113. Ali JM, Bolton EM, Bradley JA, Pettigrew GJ. Allorecognition pathways in transplant rejection and tolerance. Transplantation 2013; 96:681–8. [DOI] [PubMed] [Google Scholar]
- 114. Harshyne LA, Watkins SC, Gambotto A, Barratt‐Boyes SM. Dendritic cells acquire antigens from live cells for cross‐presentation to CTL. J Immunol 2001; 166:3717–23. [DOI] [PubMed] [Google Scholar]
- 115. Benichou G, Thomson AW. Direct versus indirect allorecognition pathways: on the right track. Am J Transplant 2009; 9:655–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Schliesser U, Streitz M, Sawitzki B. Tregs: application for solid‐organ transplantation. Curr Opin Organ Transplant 2012; 17:34–41. [DOI] [PubMed] [Google Scholar]
- 117. Guillonneau C, Picarda E, Anegon I. CD8+ regulatory T cells in solid organ transplantation. Curr Opin Organ Transplant 2010; 15:751–6. [DOI] [PubMed] [Google Scholar]
- 118. Picarda E, Bezie S, Venturi V, Echasserieau K, Merieau E, Delhumeau A et al MHC‐derived allopeptide activates TCR‐biased CD8+ Tregs and suppresses organ rejection. J Clin Invest 2014; 124:2497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Pileggi A, Molano RD, Berney T, Cattan P, Vizzardelli C, Oliver R et al Heme oxygenase‐1 induction in islet cells results in protection from apoptosis and improved in vivo function after transplantation. Diabetes 2001; 50:1983–91. [DOI] [PubMed] [Google Scholar]
- 120. Hu CM, Lin HH, Chiang MT, Chang PF, Chau LY. Systemic expression of heme oxygenase‐1 ameliorates type 1 diabetes in NOD mice. Diabetes 2007; 56:1240–7. [DOI] [PubMed] [Google Scholar]
- 121. Kupiec‐Weglinski JW, Busuttil RW. Ischemia and reperfusion injury in liver transplantation. Transplant Proc 2005; 37:1653–6. [DOI] [PubMed] [Google Scholar]
- 122. Baan C, Peeters A, Lemos F, Uitterlinden A, Doxiadis I, Claas F et al Fundamental role for HO‐1 in the self‐protection of renal allografts. Am J Transplant 2004; 4:811–8. [DOI] [PubMed] [Google Scholar]
- 123. Cai C, Teng L, Vu D, He JQ, Guo Y, Li Q et al The heme oxygenase 1 inducer (CoPP) protects human cardiac stem cells against apoptosis through activation of the extracellular signal‐regulated kinase (ERK)/NRF2 signaling pathway and cytokine release. J Biol Chem 2012; 287:33720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. George JF, Braun A, Brusko TM, Joseph R, Bolisetty S, Wasserfall CH et al Suppression by CD4+CD25+ regulatory T cells is dependent on expression of heme oxygenase‐1 in antigen‐presenting cells. Am J Pathol 2008; 173:154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Karimi K, Kandiah N, Chau J, Bienenstock J, Forsythe P. A Lactobacillus rhamnosus strain induces a heme oxygenase dependent increase in Foxp3+ regulatory T cells. PLoS One 2012; 7:e47556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Schulz S, Chisholm KM, Zhao H, Kalish F, Yang Y, Wong RJ et al Heme oxygenase‐1 confers protection and alters T‐cell populations in a mouse model of neonatal intestinal inflammation. Pediatr Res 2015; 77:640–8. [DOI] [PubMed] [Google Scholar]
- 127. Schumacher A, Zenclussen AC. Effects of heme oxygenase‐1 on innate and adaptive immune responses promoting pregnancy success and allograft tolerance. Front Pharmacol 2014; 5:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ozen M, Zhao H, Lewis DB, Wong RJ, Stevenson DK. Heme oxygenase and the immune system in normal and pathological pregnancies. Front Pharmacol 2015; 6:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zenclussen ML, Anegon I, Bertoja AZ, Chauveau C, Vogt K, Gerlof K et al Over‐expression of heme oxygenase‐1 by adenoviral gene transfer improves pregnancy outcome in a murine model of abortion. J Reprod Immunol 2006; 69:35–52. [DOI] [PubMed] [Google Scholar]
- 130. Sollwedel A, Bertoja AZ, Zenclussen ML, Gerlof K, Lisewski U, Wafula P et al Protection from abortion by heme oxygenase‐1 up‐regulation is associated with increased levels of Bag‐1 and neuropilin‐1 at the fetal‐maternal interface. J Immunol 2005; 175:4875–85. [DOI] [PubMed] [Google Scholar]
- 131. Schumacher A, Wafula PO, Teles A, El‐Mousleh T, Linzke N, Zenclussen ML et al Blockage of heme oxygenase‐1 abrogates the protective effect of regulatory T cells on murine pregnancy and promotes the maturation of dendritic cells. PLoS One 2012; 7:e42301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Solano ME, Kowal MK, O'Rourke GE, Horst AK, Modest K, Plosch T et al Progesterone and HMOX‐1 promote fetal growth by CD8+ T cell modulation. J Clin Invest 2015; 125:1726–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kwiatek M, Geca T, Krzyzanowski A, Malec A, Kwasniewska A. Peripheral dendritic cells and CD4+CD25+Foxp3+ regulatory T cells in the first trimester of normal pregnancy and in women with recurrent miscarriage. PLoS One 2015; 10:e0124747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kammerer U, Eggert AO, Kapp M, McLellan AD, Geijtenbeek TB, Dietl J et al Unique appearance of proliferating antigen‐presenting cells expressing DC‐SIGN (CD209) in the decidua of early human pregnancy. Am J Pathol 2003; 162:887–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Blois SM, Alba Soto CD, Tometten M, Klapp BF, Margni RA, Arck PC. Lineage, maturity, and phenotype of uterine murine dendritic cells throughout gestation indicate a protective role in maintaining pregnancy. Biol Reprod 2004; 70:1018–23. [DOI] [PubMed] [Google Scholar]
- 136. Prins JR, Zhang B, Schjenken JE, Guerin LR, Barry SC, Robertson SA. Unstable Foxp3+ regulatory T cells and altered dendritic cells are associated with lipopolysaccharide‐induced fetal loss in pregnant interleukin 10‐deficient mice. Biol Reprod 2015; 93:95. [DOI] [PubMed] [Google Scholar]
- 137. Zenclussen AC, Olivieri DN, Dustin ML, Tadokoro CE. In vivo multiphoton microscopy technique to reveal the physiology of the mouse uterus. Am J Reprod Immunol 2013; 69:281–9. [DOI] [PubMed] [Google Scholar]
- 138. Plaks V, Birnberg T, Berkutzki T, Sela S, BenYashar A, Kalchenko V et al Uterine DCs are crucial for decidua formation during embryo implantation in mice. J Clin Invest 2008; 118:3954–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Tachibana M, Hashino M, Nishida T, Shimizu T, Watarai M. Protective role of heme oxygenase‐1 in Listeria monocytogenes‐induced abortion. PLoS One 2011; 6:e25046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tachibana M, Watanabe K, Yamasaki Y, Suzuki H, Watarai M. Expression of heme oxygenase‐1 is associated with abortion caused by Brucella abortus infection in pregnant mice. Microb Pathog 2008; 45:105–9. [DOI] [PubMed] [Google Scholar]
- 141. Zhao H, Wong RJ, Kalish FS, Nayak NR, Stevenson DK. Effect of heme oxygenase‐1 deficiency on placental development. Placenta 2009; 30:861–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kahlo K, Fill Malfertheiner S, Ignatov T, Jensen F, Costa SD, Schumacher A et al HO‐1 as modulator of the innate immune response in pregnancy. Am J Reprod Immunol 2013; 70:24–30. [DOI] [PubMed] [Google Scholar]
- 143. Linzke N, Schumacher A, Woidacki K, Croy BA, Zenclussen AC. Carbon monoxide promotes proliferation of uterine natural killer cells and remodeling of spiral arteries in pregnant hypertensive heme oxygenase‐1 mutant mice. Hypertension 2014; 63:580–8. [DOI] [PubMed] [Google Scholar]
- 144. Zenclussen ML, Linzke N, Schumacher A, Fest S, Meyer N, Casalis PA et al Heme oxygenase‐1 is critically involved in placentation, spiral artery remodeling, and blood pressure regulation during murine pregnancy. Front Pharmacol 2014; 5:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Zenclussen ML, Casalis PA, El‐Mousleh T, Rebelo S, Langwisch S, Linzke N et al Haem oxygenase‐1 dictates intrauterine fetal survival in mice via carbon monoxide. J Pathol 2011; 225:293–304. [DOI] [PubMed] [Google Scholar]
- 146. El‐Mousleh T, Casalis PA, Wollenberg I, Zenclussen ML, Volk HD, Langwisch S et al Exploring the potential of low doses carbon monoxide as therapy in pregnancy complications. Med Gas Res 2012; 2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]