

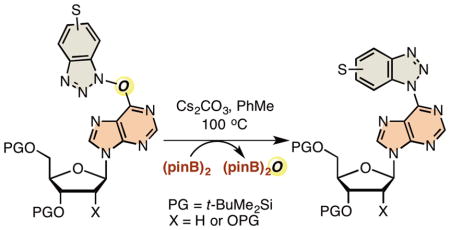
Abstract
Reaction of amide bonds in t-butyldimethylsilyl-protected inosine, 2′-deoxyinosine, guanosine, 2′-deoxyguanosine, and 2-phenylinosine with commercially available peptide-coupling agents (benzotriazol-1H-yloxy)tris(dimethylaminophosphonium) hexafluorophosphate (BOP), (6-chloro-benzotriazol-1H-yloxy)trispyrrolidinophosphonium hexafluorophosphate (PyClocK), and (7-azabenzotriazol-1H-yloxy)trispyrrolidinophosphonium hexafluorophospate (PyAOP) gave the corresponding O6-(benzotriazol-1-yl) nucleoside analogues containing a C–O–N bond. Upon exposure to bis(pinacolato)diboron and base, the O6-(benzotriazol-1-yl) and O6-(6-chlorobenzotriazol-1-yl) purine nucleoside derivatives obtained from BOP and PyClocK, respectively, underwent N–O bond reduction and C–N bond formation, leading to the corresponding C6 benzotriazolyl purine nucleoside analogues. In contrast, the 7-azabenzotriazolyloxy purine nucleoside derivatives did not undergo efficient deoxygenation, but gave unsymmetrical nucleoside dimers instead. This is consistent with a prior report on the slow reduction of 1-hydroxy-1H-4-aza and 1-hydroxy-1H-7-azabenzotriazoles. Because of the limited number of commercial benzotriazole-based peptide coupling agents, and to show applicability of the method when such coupling agents are unavailable, 1-hydroxy-1H-5,6-dichlorobenzotriazole was synthesized. Using this compound, silyl-protected inosine and 2′-deoxyinosine were converted to the O6-(5,6-dichlorobenzotriazol-1-yl) derivatives via in situ amide activation with PyBroP. The O6-(5,6-dichlorobenzotriazol-1-yl) purine nucleosides so obtained also underwent smooth reduction to afford the corresponding C6 5,6-dichlorobenzotriazolyl purine nucleoside derivatives. A total of 13 examples were studied with successful reactions occurring in 11 cases (the azabenzotriazole derivatives, mentioned above, being the only unreactive entities). To understand whether these reactions are intra or intermolecular processes, a crossover experiment was conducted. The results of this experiment as well as those from reactions conducted in the absence of bis(pinacolato)diboron and in the presence of water indicate that detachment of the benzotriazoloxy group from the nucleoside likely occurs, followed by reduction, and reattachment of the ensuing benzotriazole, leading to products.
Graphical Abstract
Introduction
Nucleosides are central to life processes and, therefore, it is not altogether unexpected that modified nucleosides continue to play a major role in biochemistry, biology, as probes of biological processes, and as pharmaceuticals.1–10 In the context of developing novel approaches to modified nucleosides, we and others have been involved with the development of SNAr reactions of nucleosides not involving conventional leaving groups such as halides and sulfonates.11–19 Specifically, our efforts have been directed towards the use of benzotriazole-based peptide-coupling agents for activation of the amide groups in purine nucleosides. Briefly, reactions of the purinyl amides with (benzotriazol-1-yloxy)tris(dimethylaminophosphonium) hexafluorophosphate (BOP) lead to the O6-(benzotriazol-1-yl)purine nucleosides. These compounds can either be isolated for subsequent SNAr reaction with nucleophiles,11–13,15,16,19 or amide activation can be done in situ followed by reaction with nucleophiles (Scheme 1).17
Scheme 1.

Formation of O6-(benzotriazol-1-yl)purine nucleoside derivatives and their conversion to other modified purine nucleosides.
In the course of these investigations we had attempted to use 3′,5′-O-disilyl protected O6-(benzotriazol-1-yl)-2′-deoxyinosine for Pd-catalyzed C–C bond formation.12 In this reaction only deoxygenation was observed, leading to the C6 benzotriazol-1-yl nucleoside analogue. This had prompted us to consider use of the oxophilic bis(pinacolato)diboron [(pinB)2] for the deoxygenation, and with this reagent the O6-(benzotriazol-1-yl) nucleoside was smoothly converted to the benzotriazol-1-yl derivative.12 This has now prompted us to query whether such a deoxygenation can be generally accomplished to access C6 benzotriazolyl nucleosides in a metal-free process. Notably, other than our previous results on the deoxygenation chemistry and two Pd-mediated reactions reported therein for structure elucidation purposes,12 only a solitary example exits on the synthesis of 6-(benzotriazol-1-yl)ribofuranosylpurine.20 In the latter work, 6-chloropurine riboside was reacted with 30 mol % excess of 1,2,3-benzotriazole, as a melt at 150 °C.20 Whereas this procedure was applicable to the synthesis of the ribonucleosides, its applicability to the synthesis of the more sensitive 2′-deoxyribonucleosides remains unknown. Further, high-temperature reactions of benzotriazoles can potentially be hazardous due to the possibility for their exothermic decomposition.21
Results and Discussion
At the outset we wanted to compare our proposed method for the synthesis of 6-benzotriazolyl purine nucleosides, which can also be considered N-aryl benzotriazoles, to other approaches. The relevant ones for this comparison are: (a) the single known example of a neat reaction between 6-chloropurine riboside and benzotriazole at 150 °C,20 (b) aryne-azide cycloaddition,22 and (c) metal-catalyzed C–N bond formation between benzotriazoles and an aryl halide.12,23 Between these, (a) and (b) are comparable to the proposed approach (d) because they are both metal-free transformations (Scheme 2).
Scheme 2.
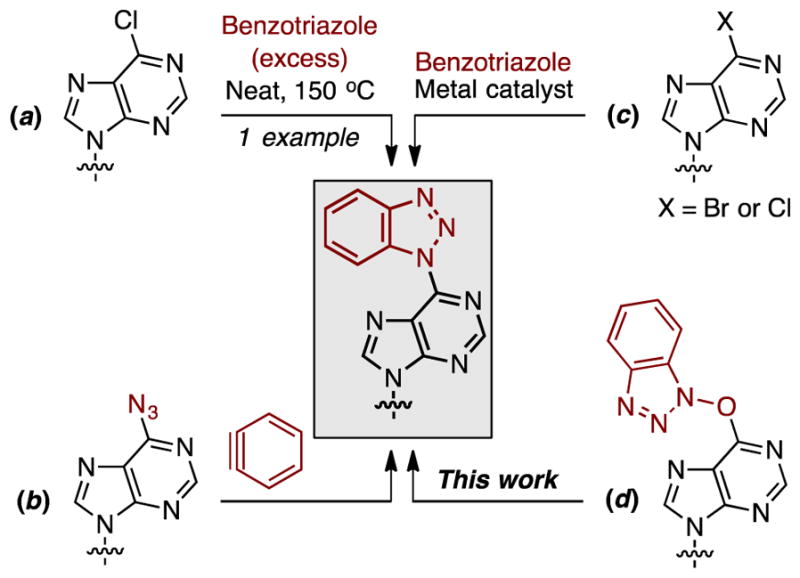
Four possible approaches to 6-(benzotriazol-1-yl)purine nucleosides.
Because we have previously developed the synthesis of 6-azidopurine nucleosides from O6-(benzotriazol-1-yl) nucleoside derivatives,24 we decided to evaluate the aryne-azide cycloaddition first. 6-Azido-9-(2-deoxy-3,5-di-O-acetyl-β-D-ribofuranosyl)purine was synthesized as described,24 and this compound was exposed to benzyne generated from o-(trimethylsilylphenyl) trifluoromethanesulfonate and CsF in MeCN. In contrast to the facile reaction of various organic azides with benzyne, this nucleoside azide did not yield any product either in the presence or absence of 18-Cr-6. One plausible reason could be that nucleoside azides can exist in the alternate tetrazolyl forms.24
The failure of the aryne-azide cycloaddition only left the diboron-mediated deoxygenation of O6-(benzotriazol-1-yl) nucleosides to be investigated as the alternate metal-free approach. In our previous report, under unoptimized conditions, compound 4 was observed to undergo conversion to product 5 with (pinB)2/Cs2CO3 in PhMe at 100 °C.12 This formed the basis for our present investigations and optimizations (Table 1). Precursors 1 and 4 are readily available via our published procedures.11
Table 1.
Evaluation of conditions for the deoxygenation of O6-(benzotriazol-1-yl)inosine and 2′-deoxyinosine derivativesa
| ||||
|---|---|---|---|---|
| Entry | Substrate | Conditions | Result | Total yield |
| 1 | 1 | Cs2CO3, (pinB)2, PhMe, 100 °C | 2: 98%, 3: 0% | 98% |
| 2 | 1 | K3PO4, (pinB)2, PhMe, 100 °C | 2: 82%, 3: 8% | 90%b,c |
| 3 | 1 | K2CO3, (pinB)2, PhMe, 100 °C | 2: 74%, 3: 0% | 74%d |
| 4 | 1 | no base, (pinB)2, PhMe, 100 °C | 2: 0%, 3: 0% | 0%e |
| 5 | 1 | Cs2CO3, (pinB)2, MeCN, 75 °C | 2: 38%, 3: 0% | 38% |
| 6 | 1 | K3PO4, (pinB)2, MeCN, 75 °C | 2: 94%, 3: 4% | 98% |
| 7 | 1 | K2CO3, (pinB)2, MeCN, 75 °C | 2: 81%, 3: 11% | 92%c |
| 8 | 4 | Cs2CO3, (pinB)2, PhMe, 100 °C | 5: 90%, 6: 0% | 90% |
| 9 | 4 | K3PO4, (pinB)2, MeCN, 75 °C | 5: 63%, 6: 6% | 69%b,c |
Reactions were conducted on 76.5 μmol of the nucleoside at 0.1 M concentration in the reaction solvent, with 2 equiv. of base, and 1.1 equiv. of (pinB)2. Yields reported are after chromatographic isolation of products or product mixtures.
Reaction did not reach completion.
The N1 and N2 isomers were collected as a combined fraction and the ratio was assessed by the integration of their purinyl resonances.
The reaction was incomplete, and the starting material and the N1 isomer were collected as a single fraction. The % of the N1 isomer was estimated from the integration of the H-1′ resonances.
No reaction was observed and compound 1 was still present.
The results in Table 1 point to several important facts. First, base is essential for the reaction (entries 1–3), in the absence of which no reaction occurred (entry 4). Cs2CO3 is best, giving only the N1 isomer (entry 1), and although K3PO4 gave a good overall yield, formation of the N2 isomer was observed (entry 2). It is easy to ascertain structures of the N1 and N2 isomers on the basis of the symmetry or lack thereof in the benzotriazolyl unit. Use of MeCN as solvent for the Cs2CO3/(pinB)2 combination proved to be inferior (entry 5). In contrast to this, with both K3PO4 and K2CO3, reactions in MeCN gave good yields, but the N2 isomer was formed in both (entries 6 and 7). The Cs2CO3/(pinB)2 combination in PhMe at 100 °C again provided an excellent yield of the N1 isomer 5 from precursor 4 (entry 8). But the K3PO4/(pinB)2 combination in MeCN, which was quite good in the case of the ribose substrate 1, proved to be inferior (entry 9). Use of B2(OH)2 in place of (pinB)2, under otherwise identical conditions, gave an incomplete reaction and multiple products. Thus, this avenue was not pursued.
For further exploration, several precursors (Figure 1) were prepared by reactions of commercially available nucleosides, protected as TBDMS ethers, with BOP, PyAOP, and PyClock. Syntheses of compounds 1, 4, 7, and 8 have been published previously,11,15 and compounds 9–14 were synthesized via analogous procedures.
Fig. 1.

Structures of commercially available peptide coupling agents and precursors prepared with them. PG = TBDMS.
In addition to these ten precursors, one more unnatural nucleoside-based ribosyl precursor was synthesized as shown in Scheme 3. Conversion of O6-(benzotriazol-1-yl)guanosine derivative 7 to the O6-allyl compound 15 was accomplished as described.15 Diazotization-bromination then gave the 2-bromo-O6-allyl guanosine derivative 16.25 Notably, when this reaction was conducted in CH2Cl2 a minor byproduct (17) was observed that was consistent with the allyl group dibromination, on the basis of 1H NMR and HRMS analysis. In CH2Br2, not only was the yield of bromide 16 lower (36%) but the amount of the tribromo byproduct 17 also increased (13%). The C2 chlorination product formed in CH2Cl2 would not be a problem as this would be reactive in the subsequent step. Via a minor modification of our previous results,26 C–C cross coupling and deprotection of the O6-allyl group was performed in a single step to yield 2-phenylinosine derivative 18. Finally, reaction of the amide group with BOP, using DBU as base, in MeCN gave the desired precursor 19.
Scheme 3.

Synthesis of an O6-(benzotriazol-1-yl)-2-phenylinosine derivative. PG = TBDMS.
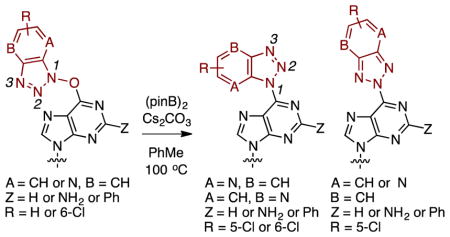
With substrates 1, 4, 7–14, and 19 the next step was an evaluation of the ability of (pinB)2/Cs2CO3 to deoxygenate these in PhMe at 100 °C. We were aware that with substrates 9–14, we had to pay particular attention to the benzotriazolyl substitution in the product, i.e., we had to query whether the locations of the pyridyl N atom and the Cl atom were identical to those in the precursors or whether some rearrangement had occurred. With all these considerations, each substrate was subjected to the deoxygenation conditions and the results from these experiments are shown in Table 2.
Table 2.
Deoxygenation of various O6-(benzotriazol-1-yl)purine nucleoside derivatives to 6-(benzotriazolyl)purine nucleosidesa
| ||||
|---|---|---|---|---|
| Entry | Substrate | N1 isomer | N2 isomer | Other |
| 1b | 1: A = B = CH, Z = H, R = H (ribo) | 2: 90% | – | – |
| 2b | 4: A = B = CH, Z = H, R = H (deoxyribo) | 5: 87% | – | – |
| 3b | 7: A = B = CH, Z = NH2, R = H (ribo) | 20: 82% | 21: Trace | – |
| 4b | 8: A = B = CH, Z = NH2, R = H (deoxyribo) | 22: 91% | 23: Trace | – |
| 5b | 9: A = N, B = CH, Z = H, R = H (ribo) | – | – | 25: 25%c |
| 6b | 10: A = N, B = CH, Z = H, R = H (deoxyribo) | 24: 11% | – | 26: 29%c |
| 7b | 11: A = B = CH, Z = H, R = 6-Cl (ribo) | 27a+b: 64% | – | – |
| 8b | 12: A = B = CH, Z = H, R = 6-Cl (deoxyribo) | 28a+b: 84%d,e | – | – |
| 9b | 13: A = B = CH, Z = NH2, R = 6-Cl (ribo) | 29a+b: 84%d,e | – | – |
| 10b | 14: A = B = CH, Z = NH2, R = 6-Cl (deoxyribo) | 30a+b: 79% | – | – |
| 11f | 19: A = B = CH, Z = Ph, R = H (ribo) | 31: 89% | – | – |
Yields of the N1 and N2 products are of isolated, purified products.
Reactions were conducted at 0.1 M nucleoside (0.167 mmol) in PhMe, with 2 equiv. of Cs2CO3, and 1.1 equiv. of (pinB)2.
Formation of dimers (25 and 26) was observed in these cases (see text).
Two N1 isomers were observed (see text).
Small amounts of each isomer were separated for characterization.
Reaction was conducted with 0.05 mmol of compound 19 at a 0.1 M concentration in PhMe.
As can be seen from Table 2, successful product formation was observed in nine of the eleven cases (see Figure 2 for product structures). Reactions of precursors 1 and 4 proceeded uneventfully on the larger scales yielding products 2 and 5, respectively. No N2 products were observed. Comparably, reactions of the O6-(benzotriazol-1-yl)guanine nucleosides 7 and 8 also proceeded well to give the C6 benzotriazolyl products 20 and 22. In these cases, trace amounts of what could be N2 isomers 21 and 23 were observed by TLC. Also, the appearance of symmetrical benzotriazolyl resonances in one chromatographically obtained fraction from the reaction of precursor 7 supports this.
Fig. 2.

Structures of products formed from the deoxygenation reactions. PG = TBDMS.
In contrast to these, in reactions of the 7-azabenzotriazolyl substrates 9 and 10, only a small amount of deoxygenated product 24 was isolated from the deoxyribose precursor 10. In both cases, the major products isolated were dimers 25 and 26 (see Figure 2). We have previously independently prepared 2611 and identified its formation in a failed C–C cross-coupling reaction.12 Here, comparable product 25 was identified from substrate 9. The exact structure of product 24 arising from compound 10 has not been unequivocally assigned, but the N1 structure is proposed because N1 products predominate in all other cases. We also cannot unequivocally comment on the location of the pyridyl N atom, i.e., whether A = N and B = CH or whether A = CH and B = N. In a second reaction of compound 10, use of 2 equiv. of (pinB)2 only led to an increased amount of dimer 26 (32%). We have previously shown that deoxygenations of 1-hydroxy-1H-4-aza and 1-hydroxy-1H-7-azabenzotriazoles with B2(OH)4 occur slowly.27 Therefore, dimer formation, rather than slow deoxygenation may be a competing reaction.
Monochloro precursors 11–14 can potentially yield three products, two different N1 isomers and one N2 isomer. Deoxygenation of 11 gave two products in 1:1.6 ratio and the reaction of 12 also gave two products in 1:1.4 ratio. Small amounts of products from precursor 12 were separated for characterization and, first, their NOESY spectra were analyzed. The chromatographically early-eluting product showed a weak but definite correlation between a benzotriazolyl doublet (δ = 8.72 ppm, J = 8.8 Hz) and a purinyl singlet (δ 8.99 ppm). Hence, we propose that this compound is the 5-chloro isomer 28a. No NOESY correlation was observed in the case of the slower-eluting product, which we assign as 28b. That 28b is also likely a N1 isomer comes from its hydrogenation with 10% Pd-C and Et3N, in EtOH, at room temperature. From this reduction, a 51% yield of compound 5 was obtained, indicating that 28b is a N1 benzotriazolyl compound (a purple byproduct, which was sensitive to CDCl3, was also obtained in the reduction and it could not be characterized). Comparable reduction of 28a also gave compound 5 in 66% yield, and along with the NOESY data above, this supports the assigned structure. Reduction of product 27b gave a 54% yield of compound 2, again indicating that this was an N1 isomeric product (formation of a purple byproduct occurred and due to its sensitivity to CDCl3 it could not be characterized). Structures of regioisomeric ribose derivatives 27a and 27b were assigned by comparison of the benzotriazolyl and purinyl proton resonances to those of the deoxy analogues 28a and 28b (see Table 3).
Table 3.
Chemical shifts of the benzotriazolyl and purinyl proton resonances in chloro benzotriazolyl products 27–30a
| Compound | Benzotriazolyl proton resonances | Purinyl proton resonances |
|---|---|---|
| 27a | δ = 8.74, 8.21, 7.64 ppm | δ = 8.99, 8.63 ppm |
| 28a | δ = 8.72, 8.21, 7.64 ppm | δ = 8.99, 8.59 ppm |
| 29a | δ = 8.54, 8.17, 7.57 ppm | δ = 8.28 ppm |
| 30a | δ = 8.48, 8.14, 7.52 ppm | δ = 8.23 ppm |
| 27b | δ = 8.85, 8.15, 7.50 ppm | δ = 9.01, 8.63 ppm |
| 28b | δ = 8.81, 8.12, 7.48 ppm | δ = 8.99, 8.58 ppm |
| 29b | δ = 8.64, 8.11, 7.46 ppm | δ = 8.29 ppm |
| 30b | δ = 8.59, 8.08, 7.42 ppm | δ = 8.22 ppm |
Proton NMR spectra were obtained in CDCl3.
Deoxygenation of precursors 13 and 14 each gave two products, in a 1:4 and 1:1 ratio, respectively. The two products obtained from 14 were inseparable and were characterized as a mixture. However, the products arising from 13 were separable and NOESY spectra of the individual products were analyzed. Similar to data obtained with 28a and 28b, the chromatographically early-eluting product showed a weak but definite correlation between a benzotriazolyl doublet (δ = 8.54 ppm, J = 9.3 Hz) and the NH2 resonance (δ 5.20 ppm). Hence, this product was assigned the 5-chloro structure 29a. By analogy, we surmise 29b to be the 6-chloro isomer. Structures of the inseparable deoxy derivatives 30a and 30b were made by comparison of chemical shifts of the benzotriazolyl and purinyl proton resonances to those of 29a and 29b. Table 3 shows the chemical shifts of the benzotriazolyl and purinyl proton resonances in compounds 27–30. Finally, the deoxygenation of O6-(benzotriazol-1-yl)-2-phenylinosine 19 derivative proceeded efficiently to yield product 31 in excellent yield.
All preceding reactions were conducted with substrates prepared from commercially available peptide-coupling agents. A legitimate question, therefore, would be the applicability of this chemistry to access products where peptide-coupling agents are not available. To investigate this there are three potential approaches, all requiring access to aryl-ring substituted 1-hydroxy-1H-benzotriazoles. (a) The peptide-coupling reagent is independently synthesized from the hydroxybenzotriazole and then reacted with the nucleoside, (b) the nucleoside is converted to a phosphonium salt, which is then reacted with the hydroxybenzotriazole, and (c) a peptide-coupling agent is prepared in situ from the hydroxybenzotriazole and then reacted with the nucleoside. Although synthesis of BOP and PyAOP are well known and can be applied to the synthesis of similar compounds,28,29 we excluded option (a) as being more labor intensive. We have previously investigated approach (b) and initially attempted this.12,13
Synthesis of 1-hydroxy-1H-benzotriazoles are well documented in the literature (Caution!21,30).27,31 On this basis, we chose to prepare 5,6-dichloro-1-hydroxy-1H-benzotriazole (34 in Scheme 4) and test this for additional reactions. Although this hydroxybenzotriazole is listed as commercially available, it is not readily obtained. Thus, it was prepared from the relatively cheap and commercially available 1,2,4-trichlorobenzene (32) via nitration (96%)32 and reaction with hydrazine33 (56%, Scheme 4). In addition to the routine spectroscopic analyses, compound 34 was also assessed by its reduction to 5,6-dichloro-1H-benzotriazole (35) under conditions we have recently reported.27
Scheme 4.
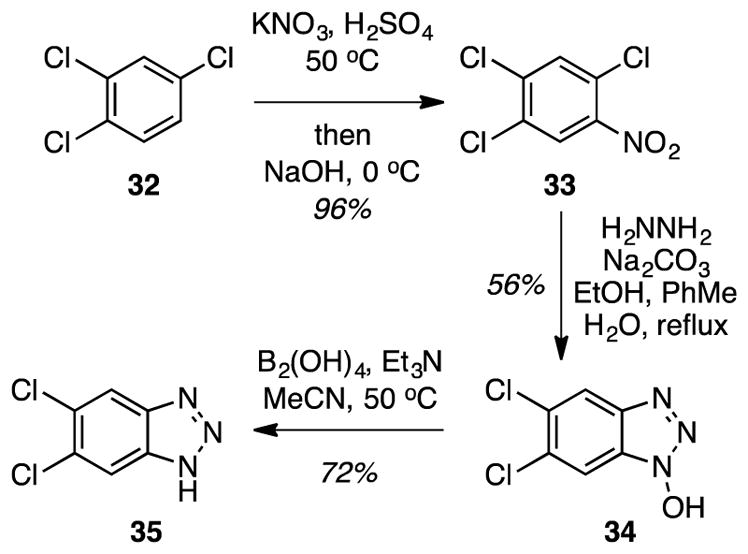
Synthesis of 5,6-dichloro-1-hydroxy-1H-benzotriazole.
Next, a series of attempts were directed towards conversion of TBDMS-protected inosine (I) and 2′-deoxyinosine (dI) to the O6-(5,6-dichloro-1H-benzotriazol-1-yl)inosine derivatives via the nucleoside phosphonium salt (Scheme 5). (a) Reaction of hydroxy benzotriazole 34 with HMPT, I2, di-O-silyl dI, DIPEA in CH2Cl2 at 50 °C resulted in the phosphordiamidite 36 (Scheme 5) in 42% yield (see the ESI for data). (b) The same reaction was then conducted stepwise. HMPT was first reacted with I2, followed by the sequential addition of DIPEA and di-O-silyl dI, followed by stirring at room temperature for 15 minutes. Finally, hydroxybenzotriazole 34 was added, and the reaction conducted at 50 °C for 20 h. In this event, among several products that formed, two that could be characterized were dichlorobenzotriazole 35 and silylated N6,N6-dimethyl-2′-deoxyadenosine 37. Formation of compound 37 is not entirely unanticipated, as we have previously observed its formation in reactions of HMPT and I2.34 (c) In variations of the procedure described in (b), PPh3 was used in place of HMPT, and reactions were conducted at room temperature and at 45 °C. In both reactions dichlorobenzotriazole 35 was formed and significant amounts of the nucleoside precursor were left unreacted.
Scheme 5.

Products formed in the various attempted conversions of TBDMS-protected I and dI to the O6-(5,6-dichlorobenzotriazol-1-yl)inosine derivatives.
Because of these unsuccessful attempts, we next focused on the use of commercially available bromotripyrrolidinophosphonium hexafluorophophate (PyBroP) for activating the purinyl amide and to form the peptide-coupling agent in situ. Both reactions can lead to the desired O6-(5,6-dichlorobenzotriazol-1-yl)inosine derivatives as shown in Scheme 6.
Scheme 6.
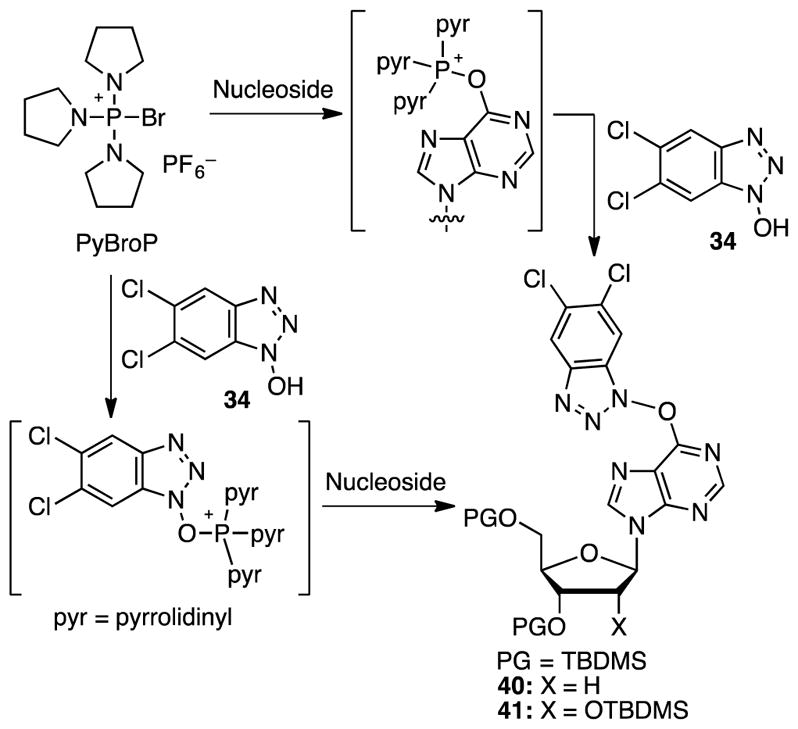
Use of PyBroP for conversion of TBDMS-protected dI and I to the corresponding O6-(5,6-dichlorobenzotriazol-1-yl) derivatives.
Initial attempts were the conversion of TBMDS-protected dI to the desired product 40 with 3.8 equiv. of PyBroP and 4.5 equiv. of hydroxybenzotriazole 34. In these reactions, expected product 40 predominated. However, in addition, various other products were also observed, such as the C6 pyrrolidino nucleoside 38, the C6 benzotriazolyl derivative 39, and the reduced benzotriazole 35 (all shown in Scheme 5). The best yield of the desired 40 was 48%. Similarly, reaction of TBDMS-protected I gave a 49% yield of product 41. Further experiments revealed that by lowering the amount of PyBroP to 1.9 equiv., and with 4.5 equiv. of hydroxybenzotriazole 34, substantial yield improvements and cleaner reactions could be attained (70% each of 40 and 41). Reactions of 40 and 41 with (pinB)2 and Cs2CO3 under the optimized conditions proceeded smoothly to provide products 42 and 43 in 61 and 56% yield, respectively (Scheme 7).
Scheme 7.
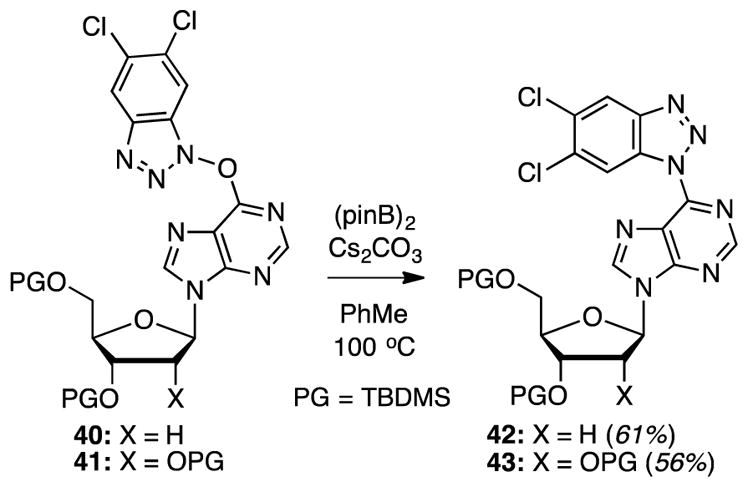
Deoxygenation of O6-(5,6-dichloro-1H-benzotriazol-1-yl)inosine
At this point we had shown the generality of this deoxygenative approach to C6 benzotriazolyl purine nucleoside derivatives with substrates obtained from commercially available benzotriazole-based peptide-coupling agents, analyzed the isomeric products formed in some cases, and demonstrated the applicability of the approach when peptide-coupling agents are unavailable. The next focus was to gain some understanding of a plausible mechanistic pathway and there are two plausible mechanisms. One is an intramolecular process where the benzotriazolyloxy unit does not become completely detached from the nucleoside but transfers the oxygen atom to the diboron. In a second pathway, the benzotriazolyloxy moiety can become detached from the purine, undergo deoxygenation as hydroxybenzotriazoles do,27 and then become reattached to the purine.
We reasoned that a crossover experiment involving two precursors with differently substituted benzotriazoles could shed light on the underlying mechanism. If an intramolecular process were operational, then each substrate would yield the corresponding deoxygenation product, and only two products would be obtained. However, should the mechanism involve detachment of the benzotriazolyloxy group from the purine, then four products could be expected. After TLC analysis of prospective starting materials and potential products, we found that O6-(benzotriazol-1-yl)-2′-deoxyinosine derivative 4 and O6-(5,6-dichlorobenzotriazol-1-yl)inosine derivative 41 would be optimal. An equimolar mixture of compounds 4 and 41 was subjected to the deoxygenation conditions. Four separable products 2, 5, 42, and 43 were obtained (Scheme 8) and the mole fractions of these were 0.30:0.38:0.17:0.14, respectively. This outcome indicated that the benzotriazolyloxy and 5,6-dichlorobenzotriazolyloxy units likely become detached from the purine and then undergo deoxygenation. Subsequently, the ensuing reduced benzotriazolyl and 5,6-dichlorobenzotriazolyl anions can function as nucleophiles, reacting via the SNAr mechanism with either substrate, again producing the two benzotriazolyloxy anions. The N–O reduction/SNAr process can then continue until each substrate is consumed. Such a mechanism would readily account for the observed crossover results. The product distribution shows, as would be expected, that the dichlorobenzotriazole species is less reactive than the unhalogenated analogue. The ribose appears less reactive than the 2′-deoxy derivative, plausibly for electronic and steric reasons.
Scheme 8.
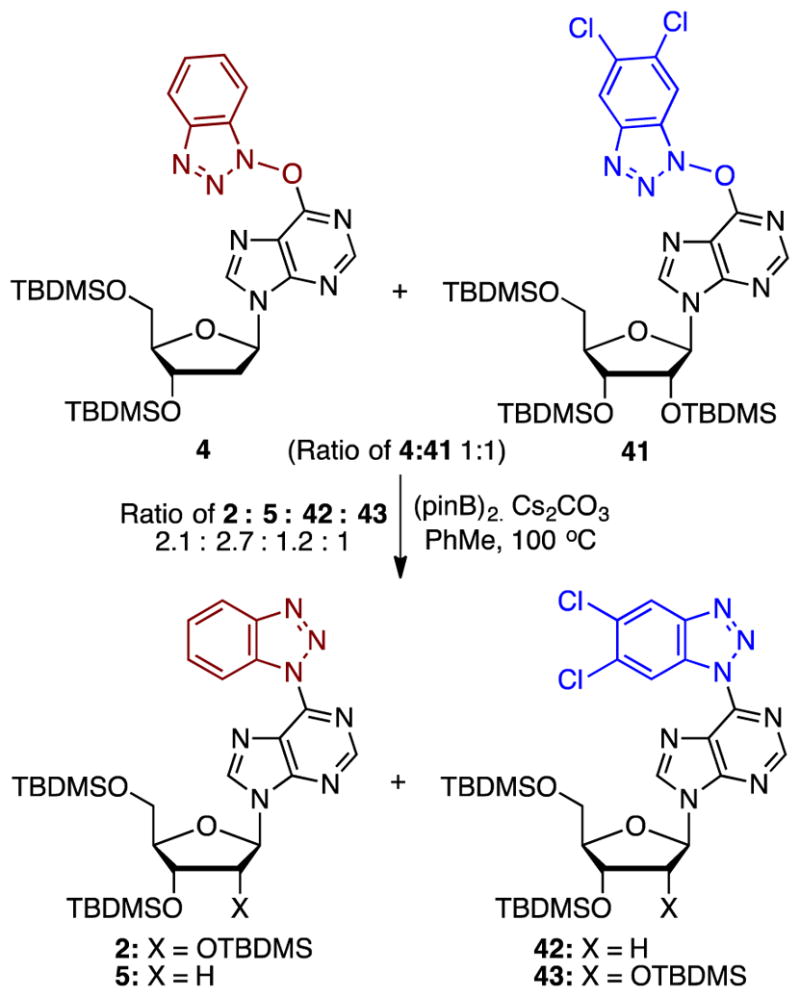
The crossover experiment and the products obtained.
Two final tests were conducted (Scheme 9). In the first, a reaction of substrate 1 was conducted in the absence of (pinB)2. A 29% yield of the dimer 25 was obtained, indicating that there is the possibility of a background reaction leading to the release of the benzotriazolyloxy group. This can then potentially be a mechanism that initiates the N–O reduction/SNAr cycle. For this background reaction we then considered the role of trace amounts of moisture that could hydrolyze the O6-(benzotriazol-1-yl) substrates, releasing the benzotriazolyloxy group. Such hydrolysis could also readily account for the dimer formation. Thus, a final experiment was conducted wherein substrate 1 was exposed to (pinB)2 (1.1 equiv.) and Cs2CO3 (2 equiv.) in 9:1 PhMe-H2O at 100 °C. In a 4-hour reaction time, product 2 was isolated in 81% yield, a result that is slightly lower than the 90% yield obtained in the absence of water (entry 1 in Table 2). Contrary to expectation though, no significant hydrolysis of the substrate to silyl-protected I was observed. Therefore, at the present time we are not entirely certain of the mechanism by which the benzotriazolyloxy group becomes detached from the purine (perhaps a very minor hydrolysis reaction), but the crossover experiment does indicate that such a process could be occurring.
Scheme 9.
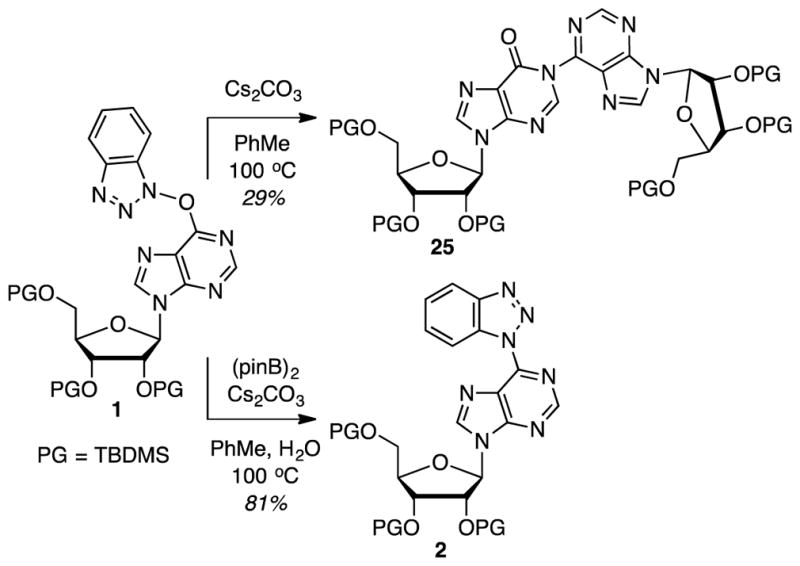
Two additional experiments, in the absence of (pinB)2, and in the presence of water.
In attempting to elucidate underlying mechanisms, two plausible pathways can be postulated (a and b in Scheme 10). It is currently not possible to comment on pathway a involving a one-step deoxygenation with or without intermediacy of a benzotriazolyl N-oxide intermediate that can be formed via rearrangement (shown in the box in Scheme 10).
Scheme 10.

Plausible reactions pathways that can result in the deoxygenation.
On the other hand, even if a low concentration of 1H-benzotriazol-1-olate (BtO−) is initially accessible (pathway b), this can undergo reduction with (pinB)2. The resulting benzotriazolyl anion (Bt−) can then yield additional BtO− by SNAr displacement, and the reaction can continue along the sequential reduction/SNAr reactions. The N3 atom of the benzotriazolyl unit in one substrate can function as a nucleophile in displacing BtO− from another, and this can be one way for the formation of BtO−. Notably, if a N-oxide intermediate (shown in the box in Scheme 10) is formed in any pathway, this will also undergo reduction by (pinB)2. Pathway b would be consistent with the crossover results and, in support of this, we have previously shown that benzotriazole is capable of displacing BtO− from compound 4.12 The role of Cs2CO3 in these reactions is not clear at this time.
Conclusions
Herein we have demonstrated a new approach to the preparation of C6 benzotriazolyl purine nucleoside derivatives. The chemistry hinges on an unusual conversion of a C–O–N linkage to a C–N via the use of (pinB)2 and Cs2CO3. Thus, this is effectively a metal-free C–N bond-forming reaction. The requisite precursors were readily prepared by reactions of amide linkages in purine nucleosides with commercially available, benzotriazole-based peptide coupling agents, or with in situ-formed reagents when peptide-coupling agents are not available commercially. Although isomeric N1 benzotriazolyl products are formed from unsymmetrical benzotriazolyl peptide-coupling agents, and traces of N2 products are seen in rare cases, regioisomeric product formation also occurs in metal-catalyzed C–N bond-forming reactions of benzotriazoles and in aryne-azide cycloadditions leading to N-substituted benzotriazoles.22,23 The use of diboron reagents for deoxygenating N–O bonds can be traced back to the reductions of 4-methylmorpholine, pyridine, and 4-phenylpyridine N-oxides, by (pinB)2 and bis(catecholato)diboron [(catB)2].35 In these cases, the free amines remained complexed to the electrophilic boron center in the B–O–B product. Silica gel chromatography was adequate to release the uncomplexed pyridines, but the 4-methylmorpholine adduct of (pinB)2O was isolated in good yield.35 In 2011, we comprehensively demonstrated broadly-applicable synthetic use of (pinB)2 and (catB)2 for the reduction of alkyl amino, pyridyl, and anilino N-oxides.36 The use of B2(OH)4 for reduction of pyridyl N-oxides was demonstrated in 2013,37 and in 2015 we successfully demonstrated a general synthesis of 1H-benzotriazoles from 1-hydroxy-1H-benzotriazoles with this reagent.27 In the present work, B2(OH)4 was ineffective in comparison to (pinB)2. From a mechanism standpoint, a crossover experiment indicated that the benzotriazolyloxy group likely becomes detached from the purine, undergoes reduction with (pinB)2, and the formed benzotriazole can then function as a nucleophile for SNAr displacement. The observation of two isomeric N1-aryl benzotriazoles from unsymmetrically substituted precursors, and the formation of N2 products in some cases is consistent with this mechanistic manifold.
The results described here contribute to an emerging area on the synthetic applicability of diboron reagents in reductive processes. Importantly, as shown here and as we clearly demonstrated previously,36 the reductions are generally facile and insensitive to water. This feature, when combined with the benign nature of the boron-based byproducts, makes such reactions broadly applicable in a variety of contexts. For instance, (pinB)2 has been used for the reduction of convergine to hippodamine, both ladybeetle alkaloids, allowing for an otherwise difficult quantification of these alkaloids due to the thermal lability of convergine.38 Also, (pinB)2, and plausibly B2(OH)4 formed from the hydrolysis of (pinB)2, have shown biocompatibility in aqueous reactions.39 It should be noted that despite the recently claimed novelty of amine N-oxide reduction by diboron-based reagents,39 that report is really an application of prior discoveries by others and by us.12,35–37 The present results further amplify the utility of diboron reducing agents for effecting a range of N–O deoxygenations under a variety of conditions. This type of diboron-reagent-mediated deoxygenation appears to be quite broadly applicable, and additional results of this chemistry are forthcoming from our laboratories.
Experimental Section
General information
Thin layer chromatography was performed on 200 μm aluminum-foil-backed silica gel plates, and column chromatography was performed using 230–400 mesh silica gel. PhMe was distilled from sodium, MeCN and DME were distilled from CaH2. Et3N and DIPEA were distilled from CaH2 and stored over KOH. 6-Azido-9-(2-deoxy-3,5-di-O-acetyl-β-D-ribofuranosyl)purine was synthesized as described.24 O6-(Benzotriazol-1-yl) derivatives of inosine, 2′-deoxyinosine, guanosine, and 2′-deoxyguanosine (1, 4, 7, and 8) were synthesized by reported procedures.11,15 The remaining analogues shown in Figure 1 were synthesized by application of these known routes (see the ESI). O6-Allyl-2′,3′,5′-tri-O-(t-butyldimethylsilyl)guanosine (15) was prepared from guanosine15 and converted to the O6-allyl-2-bromo nucleoside (16) on the basis of our previously published route.26 All other reagents were used as received from commercial suppliers. 1H NMR spectra were obtained at 500 MHz in CDCl3 and are referenced to the residual protonated solvent resonance. 13C NMR spectra were obtained at 125 MHz in CDCl3 and are referenced to the solvent resonance. Chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are in hertz (Hz). Standard abbreviations are used to designate resonance multiplicities.
O6-Allyl-2′,3′,5′-tri-O-(t-butyldimethylsilyl)-2-bromoinosine (16)
A stirred mixture of nucleoside derivative 15 (300.0 mg, 0.45 mmol) and SbBr3 (227.7 mg, 0.63 mmol) in dry CH2Cl2 (5 mL) was cooled to −10 °C. To the cooled mixture t-BuONO (374.0 μL, 3.15 mmol) was added dropwise and the stirring was continued at −10 °C for 3 h. The mixture was then poured into cold saturated aq. NaHCO3, filtered, and the residual solid was washed with CH2Cl2. The filtrate was washed with water and brine. The aqueous layer was back extracted with CH2Cl2. The organic layer was dried with anhydrous Na2SO4, filtered, and evaporated under reduced pressure. Purification of the crude material by flash chromatography (SiO2, eluted with 5% EtOAc in hexanes) gave 123.0 mg (55% yield) of product 16 (and the C2 chloro analogue) as a white, fluffy solid. In this reaction compound 17 (26.8 mg, 7% yield) was formed as a minor separable product. Rf (20% EtOAc in hexanes) = 0.64. 1H NMR of pure 16 (500 MHz, CDCl3): δ 8.27 (s, 1H, Ar-H), 6.17–6.12 (m, 1H, =CH), 5.98 (d, J = 4.9 Hz, 1H, H-1′), 5.48 (d, J = 17.1 Hz, 1H, =CHtrans), 5.32 (dd, J = 0.9, 10.0 Hz, 1H, =CHcis), 5.10 (d, J = 5.8 Hz, 2H, OCH2), 4.61 (t, J = 4.4 Hz, 1H, H-2′), 4.31 (t, J = 3.9 Hz, 1H, H-3′), 4.16–4.13 (m, 1H, H-4′), 4.05 (dd, J = 4.4, 11.3 Hz, 1H, H-5′), 3.80 (dd, J = 2.0, 11.3 Hz, 1H, H-5′), 0.95, 0.93, and 0.83 (3s, 27H, t-Bu), 0.15, 0.14, 0.10, 0.09, −0.01, and −0.17 (6s, 18H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 160.6, 152.8, 143.2, 141.7, 131.8, 121.4, 119.4, 89.0, 85.4, 75.8, 71.6, 68.6, 62.3, 26.1, 25.8, 25.7, 18.5, 18.1, 17.9, −4.3, −4.7, −4.8, −5.0, −5.4. HRMS (ESI/TOF) calcd for C31H57BrN4O5Si3Na [M + Na]+ 751.2712, found 751.2714.
2′,3′,5′-Tri-O-(t-butyldimethylsilyl)-2-phenylinosine (18).26
In a clean, dry screw-cap vial containing a stirring bar were placed the bromo nucleoside 16 (140.0 mg, 0.192 mmol), PhB(OH)2 (70.1 mg, 0.575 mmol), and anhydrous K3PO4 (122.0 mg, 0.575 mmol). Anhydrous 1,4-dioxane (4 mL) was added, the vial was flushed with nitrogen gas for 5 min followed by the addition of (dcpf)PdCl2 (43.5 mg, 0.0575 mmol). The vial was capped and the mixture was stirred at 100 °C for 7 h. Then the mixture was cooled to room temperature, filtered through Celite, and the filtrate was evaporated to dryness. Purification of the crude material by flash chromatography (SiO2, eluted with 3% CH3OH in CH2Cl2) gave 124.5 mg (95% yield) of product 18 as a pale-brown, fluffy solid. Rf (5% CH3OH in CH2Cl2) = 0.37. 1H NMR (500 MHz, C6D6): δ 10.30 (br s, 1H, NH), 8.23 (s, 1H, Ar-H), 8.06–8.02 (m, 2H, Ar-H), 7.59–7.55 (m, 3H, Ar-H), 6.10 (d, J = 4.4 Hz, 1H, H-1′), 4.54 (t, J = 4.1 Hz, 1H, H-2′), 4.36–4.34 (m, 1H, H-3′), 4.16–4.13 (m, 1H, H-4′), 4.00 (dd, J = 3.6, 11.3 Hz, 1H, H-5′), 3.82 (dd, J = 1.9, 11.0 Hz, 1H, H-5′), 0.96, 0.94, and 0.82 (3s, 27H, t-Bu), 0.14, 0.13, 0.12, 0.10, −0.05, −0.14 (6s, 18H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 158.0, 153.1, 149.2, 139.3, 134.0, 131.8, 129.2, 127.2, 88.3, 85.2, 76.4, 71.7, 62.4, 26.1, 25.8, 25.7, 20.2, 18.5, 18.1, 17.9, −4.3, −4.6, −4.9, −5.3.
O6-(Benzotriazol-1yl)-2′,3′,5′-tri-O-(t-butyldimethylsilyl)-2-phenylinosine (19)
In a clean, dry, screw-cap vial a mixture of 2-phenylinosine derivative 18 (120.0 mg, 0.175 mmol), BOP (154.5 mg, 0.349 mmol), and DBU (52.0 μL, 0.349 mmol) was stirred in anhydrous MeCN (1.5 mL) for 24 h, under a nitrogen atmosphere. The mixture was diluted with EtOAc and washed with water containing a small amount of NaCl. The aqueous layer was back extracted with EtOAc, the combined organic layer was dried with anhydrous Na2SO4, filtered, and evaporated under reduced pressure. Purification of the crude material by flash chromatography (SiO2, eluted with 10% EtOAc in hexanes) gave 49.0 mg (40% yield) of pure product 19 as a white, fluffy solid. (An equal amount of product containing trace amounts of impurities was also obtained. The more polar starting material was isolated by elution with 90% EtOAc in acetone.) Rf (10% EtOAc in hexanes) = 0.27. 1H NMR (500 MHz, C6D6): δ 8.52 (s, 1H, Ar-H), 8.15 (dd, J = 1.5, 8.0 Hz, 2H, Ar-H), 7.92–7.90 (m, 1H, Ar-H), 7.05–7.01 (m, 3H, Ar-H), 6.94 (dd, J = 2.6, 6.5 Hz, 1H, Ar-H), 6.89–6.86 (m, 2H, Ar-H), 6.22 (d, J = 3.6 Hz, 1H, H-1′), 4.74 (t, J = 3.9 Hz, 1H, H-2′), 4.55 (t, J = 4.8 Hz, 1H, H-3′), 4.25–4.28 (m, 1H, H-4′), 3.99 (dd, J = 4.0, 11.3 Hz, 1H, H-5′), 3.74 (dd, J = 2.2, 11.4 Hz, 1H, H-5′), 0.98 and 0.91 (2s, 27H, t-Bu), 0.11, 0.10, 0.08, 0.01, −0.016, −0.023 (6s, 18H, SiCH3). 13C NMR (125 MHz, C6D6): δ 159.2, 158.5, 154.9, 143.9, 136.6, 130.6, 128.4, 128.3, 128.0, 127.7, 127.6, 124.1, 120.5, 119.2, 110.0, 108.6, 89.2, 84.5, 76.2, 71.2, 61.9, 26.0, 25.8, 25.6, 18.4, 18.0, 17.9, −4.4, −4.7, −4.8, −4.9, −5.5, −5.6. HRMS (ESI/TOF) calcd for C40H61N7O5Si3Na [M + Na]+ 826.3934, found 826.3925.
Representative procedure: deoxygenation of O6-(benzotriazol-1-yl)inosine derivative (1)
In a clean, dry, screw-cap vial a mixture of compound 1 (121.9 mg, 0.167 mmol), (pinB)2 (46.1 mg, 0.182 mmol), and Cs2CO3 (109.0 mg, 0.335 mmol) was stirred in anhydrous PhMe (1.5 mL) at 100 °C over 4 h, under a nitrogen atmosphere. The mixture was cooled, diluted with EtOAc, filtered through Celite, and evaporated under reduced pressure. The crude material was purified by flash chromatography (SiO2, eluted with 15% EtOAc in hexanes) to give 107.4 mg (90% yield) of product 212 as a white, fluffy solid. Rf (20% EtOAc in hexanes) = 0.46. 1H NMR (500 MHz, CDCl3): δ 8.99 (s, 1H, Ar-H), 8.77 (d, J = 8.3 Hz, 1H, Ar-H), 8.61 (s, 1H, Ar-H), 8.23 (d, J = 8.2 Hz, 1H, Ar-H), 7.68 (t, J = 7.8 Hz, 1H, Ar-H), 7.52 (t, J = 7.8 Hz, 1H, Ar-H), 6.26 (d, J = 5.4 Hz, 1H, H-1′), 4.71 (t, J = 4.6 Hz, 1H, H-2′), 4.35 (br s, 1H, H-3′), 4.19 (br s, 1H, H-4′), 4.03 (dd, J = 3.0, 11.2 Hz, 1H, H-5′), 3.83 (dd, J = 2.0, 11.2 Hz, 1H, H-5′), 0.97, 0.95, and 0.79 (3s, 27H, t-Bu), 0.16, 0.15, 0.12, −0.02, and −0.25 (5s, 18H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 154.3, 151.9, 147.8, 146.4, 144.7, 132.3, 129.5, 125.7, 123.9, 120.5, 115.3, 88.3, 86.2, 76.3, 72.4, 62.8, 26.3, 26.0, 25.8, 18.7, 18.2, 18.0, −4.3, −4.4, −4.5, −4.8, −5.1, −5.2.
Deoxygenation of O6-(benzotriazol-1-yl)-2′-deoxyinosine derivative (4)
As described for the deoxygenation of compound 1, this reaction was carried out with compound 4 (110.0 mg, 0.167 mmol), (pinB)2 (46.1 mg, 0.182 mmol), and Cs2CO3 (109.0 mg, 0.335 mmol) in PhMe (1.5 mL) at 100 °C over 4 h. Purification by flash chromatography (SiO2, eluted with 20% and 25% EtOAc in hexanes) gave 84.4 mg (87% yield) of product 512 as a white, fluffy solid. Rf (20% EtOAc in hexanes) = 0.16. 1H NMR (500 MHz, CDCl3): δ 9.00 (s, 1H, Ar-H), 8.76 (d, J = 8.3 Hz, 1H, Ar-H), 8.57 (s, 1H, Ar-H), 8.23 (d, J = 7.8 Hz, 1H, Ar-H), 7.68 (t, J = 7.5 Hz, 1H, Ar-H), 7.53 (t, J = 7.5 Hz, 1H, Ar-H), 6.64 (app t, J = 6.3 Hz, 1H, H-1′), 4.68–4.65 (m, 1H, H-3′), 4.10–4.08 (m, 1H, H-4′), 3.90 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.82 (dd, J = 2.9, 11.2 Hz, 1H, H-5′), 2.70 (app quint, Japp ~ 6.4 Hz, 1H, H-2′), 2.54 (ddd, J = 3.3, 6.1, 13.1 Hz, 1H, H-2′), 0.94 and 0.92 (2s, 18H, t-Bu), 0.13, 0.10, and 0.09 (3s, 12H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 153.9, 151.8, 147.7, 146.4, 144.3, 132.2, 129.5, 125.7, 124.1, 120.5, 115.2, 88.4, 84.8, 72.3, 63.0, 41.6, 26.1, 25.9, 18.6, 18.2, −4.5, −4.6, −5.2, −5.3.
Deoxygenation of O6-(benzotriazol-1-yl)guanosine derivative (7)
As described for the deoxygenation of compound 1, this reaction was carried out with compound 7 (124.4 mg, 0.167 mmol), (pinB)2 (46.1 mg, 0.182 mmol), and Cs2CO3 (109.0 mg, 0.335 mmol) in PhMe (1.5 mL) at 100 °C over 4 h. Purification by flash chromatography (SiO2, eluted with 20% EtOAc in hexanes) gave 100.0 mg (82% yield) of product 20 as a pale-brown, fluffy solid. A trace amount of N2 isomer 21 was observed in this reaction. Rf (30% EtOAc in hexanes) = 0.40. 1H NMR (500 MHz, CDCl3): δ 8.59 (d, J = 8.3 Hz, 1H, Ar-H), 8.30 (s, 1H, Ar-H), 8.20 (d, J = 8.3 Hz, 1H, Ar-H), 7.63 (t, J = 7.4 Hz, 1H, Ar-H), 7.49 (t, J = 7.4 Hz, 1H, Ar-H), 6.08 (d, J = 5.4 Hz, 1H, H-1′), 5.28 (br s, 2H, NH2), 4.59 (t, J = 4.9 Hz, 1H, H-2′), 4.31 (t, J = 4 Hz, 1H, H-3′), 4.15–4.13 (m, 1H, H-4′), 3.98 (dd, J = 3.4, 11.3 Hz, 1H, H-5′), 3.81 (dd, J = 2.7, 11.5 Hz, 1H, H-5′), 0.95, 0.94, and 0.82 (3s, 27H, t-Bu), 0.15, 0.14, 0.12, 0.00, −0.03, and −0.16 (6s, 18H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 158.9, 156.2, 148.1, 146.2, 141.4, 132.0, 128.9, 125.3, 120.3, 118.1, 115.0, 87.5, 85.6, 76.2, 72.2, 62.8, 26.1, 25.9, 25.7, 18.6, 18.1, 17.9, −4.3, −4.5, −4.6, −4.9, −5.2, −5.3. HRMS (ESI/TOF) calcd for C34H58N8O4Si3Na [M + Na]+ 749.3787, found 749.3781.
Deoxygenation of O6-(benzotriazol-1-yl)-2′-deoxyguanosine derivative (8)
As described for the deoxygenation of compound 7, this reaction was carried out with compound 8 (102.6 mg, 0.167 mmol), (pinB)2 (46.1 mg, 0.182 mmol), and Cs2CO3 (109.0 mg, 0.335 mmol) in PhMe (1.5 mL) at 100 °C over 4 h. Purification by flash chromatography (SiO2, eluted with 30% EtOAc in hexanes) gave 90.9 mg (91% yield) of product 22 as an off-white, fluffy solid. A trace amount of N2 isomer 23 was observed in this reaction. Rf (40% EtOAc in hexanes) = 0.37. 1H NMR (500 MHz, CDCl3): δ 8.58 (d, J = 8.3 Hz, 1H, Ar-H), 8.28 (s, 1H, Ar-H), 8.21 (d, J = 8.3 Hz, 1H, Ar-H), 7.64 (t, J = 7.6 Hz, 1H, Ar-H), 7.50 (t, J = 7.7 Hz, 1H, Ar-H), 6.47 (t, J = 6.6 Hz, 1H, H-1′), 5.24 (br s, 2H, NH2), 4.63–4.60 (m, 1H, H-3′), 4.06–4.04 (m, 1H, H-4′), 3.86–3.78 (m, 2H, 2H-5′), 2.60 (app quint, Japp ~ 6.7 Hz, 1H, H-2′), 2.46 (ddd, J = 3.4, 5,8, 13.2 Hz, 1H, H-2′), 0.94 and 0.92 (2s, 18H, t-Bu), 0.13, 0.12, 0.10, and 0.09 (4s, 12H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 158.9, 155.9, 146.2, 141.2, 132.0, 129.0, 125.3, 120.2, 118.1, 114.9, 88.0, 83.8, 113.8, 72.2, 62.9, 41.0, 26.0, 25.8, 24.9, 18.4, 18.0, −4.6, −4.7, −5.3, −5.4. HRMS (ESI/TOF) calcd for C28H44N8O3Si2Na [M + Na]+ 619.2973, found 619.2979.
Attempted deoxygenation of O6-(7-azabenzotriazol-1-yl)inosine derivative (9)
As described for the deoxygenation of compound 1, this reaction was attempted with compound 9 (58.8 mg, 81.0 μmol), (pinB)2 (22.24 mg, 87.6 μmol), and Cs2CO3 (52.6 mg, 0.161 mmol) in PhMe (0.75 mL) at 100 °C over 4 h. Evaporation of the reaction mixture and purification by flash chromatography (SiO2, eluted with 40% EtOAc in hexanes) gave 24.6 mg (25% yield) of compound 25 as an oily product. Rf (40% EtOAc in hexanes) = 0.84. 1H NMR (500 MHz, CDCl3): δ 8.99 (s, 1H, Ar-H), 8.60 (s, 1H, Ar-H), 8.26 (s, 1H, Ar-H), 8.25 (s, 1H, Ar-H), 6.17 (d, J = 3.4 Hz, 1H, H-1′), 6.02 (d, J = 3.9 Hz, 1H, H-1′), 4.57 (t, J = 3.6 Hz, 1H, H-2′), 4.50 (t, J = 4.2 Hz, 1H, H-2′), 4.37 (t, J = 4.4 Hz, 1H, H-3′), 4.33 (t, J = 4.4 Hz, 1H, H-3′), 4.18–4.14 (m, 2H, H-4′), 4.06–4.01 (m, 2H, H-5′), 3.83–3.79 (m, 2H, H-5′), 0.96, 0.93, 0.86, and 0.85 (4s, 54H, t-Bu), 0.15, 0.14, 0.12, 0.11, 0.10, 0.03, −0.04, −0.07 (8s, 36H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 155.1, 153.5, 152.2, 147.8, 147.1, 145.9, 144.9, 138.9, 129.9, 124.9, 89.0, 88.7, 85.0, 84.9, 83.5, 76.4, 71.2, 71.0, 62.1, 62.0, 26.1, 25.9, 25.74, 25.7, 25.0, 18.6, 18.1, 17.9, −4.2, −4.3, −4.6, −4.8, −5.28, −5.33. HRMS (ESI/TOF) calcd for C56H106N8O9Si6Na [M + Na]+ 1225.6596, found 1225.6604.
Attempted deoxygenation of O6-(7-azabenzotriazol-1-yl)-2′-deoxyinosine derivative (10)
As described for the deoxygenation of compound 1, this reaction was attempted with compound 10 (48.3 mg, 80.7 μmol), (pinB)2 (22.2 mg, 87.6 μmol), and Cs2CO3 (52.6 mg, 0.161 mmol) in PhMe (0.75 mL) at 100 °C over 4 h. Evaporation of the reaction mixture and purification by preparative TLC (1 mm, 20 × 20 cm SiO2 plate eluted with 60% EtOAc in hexanes) gave 22.0 mg (29% yield) of compound 26 as an oily product, and 5.0 mg (10% yield) of compound 24 as an off-white, fluffy solid. Rf of 26 (50% EtOAc in hexanes) = 0.17. 1H NMR of 26 (500 MHz, CDCl3): δ 8.98 (s, 1H, Ar-H), 8.50 (s, 1H, Ar-H), 8.25 (s, 1H, Ar-H), 8.14 (s, 1H, Ar-H), 6.56 (t, J = 6.4 Hz, 1H, H-1′), 6.42 (t, J = 6.4 Hz, 1H, H-1′), 4.64–4.59 (m, 2H, H-3′), 4.06–4.02 (m, 2H, H-4′), 3.89–3.84 (m, 2H, H-5′), 3.80–3.77 (dd, J = 2.6, 11.4 Hz, 2H, H-5′), 2.67 (app quint, Japp ~ 6.5 Hz, 1H, H-2′), 2.56 (app quint, Japp ~ 6.4 Hz, 1H, H-2′), 2.50 (ddd, J = 3.9, 6.4, 13.0 Hz, 1H, H-2′), 2.45 (ddd, J = 3.9, 6.2, 13.0 Hz, 1H, H-2′), 0.92, 0.917, 0.914, and 0.89 (4s, 36H, t-Bu), 0.12, 0.11, 0.10, 0.09, 0.08, and 0.07 (6s, 36H, SiCH3). HRMS of 26 (ESI/TOF) calcd for C44H78N8O7Si4Na [M + Na]+ 965.4968, found 965.4963. Compound 26 is known in the literature.11 Rf of 24 (50% EtOAc in hexanes) = 0.07. 1H NMR of 24 (500 MHz, CDCl3): δ 9.09 (d, J = 8.3 Hz, 1H, Ar-H), 8.99 (s, 1H, Ar-H), 8.88 (d, J = 4.4 Hz, 1H, Ar-H), 8.62 (s, 1H, Ar-H), 7.62 (dd, J = 4.4, 8.3 Hz, 1H, Ar-H), 6.64 (t, J = 6.6 Hz, 1H, H-1′), 4.67–4.65 (m, 1H, H-3′), 4.11–4.08 (m, 1H, H-4′), 3.90 (dd, J = 3.9, 11.2 Hz, 1H, H-5′), 3.81 (dd, J = 2.5, 11.2 Hz, 1H, H-5′), 2.68 (app quint, Japp ~ 6.5 Hz, 1H, H-2′), 2.54 (ddd, J = 3.4, 5.9, 13.2 Hz, 1H, H-2′), 0.94 and 0.91 (2s, 18H, t-Bu), 0.13 and 0.10 (2s, 12H, SiCH3). HRMS of 24 (ESI/TOF) calcd for C27H42N8O3Si2Na [M + Na]+ 605.2816, found 605.2814. A 13C NMR spectrum could not be obtained because of low quantity of the material isolated.
Deoxygenation of O6-(6-chlorobenzotriazol-1-yl)inosine derivative (11)
As described for the deoxygenation of compound 1, this reaction was carried out with compound 11 (127.6 mg, 0.167 mmol), (pinB)2 (46.1 mg, 0.182 mmol), and Cs2CO3 (109.0 mg, 0.335 mmol) in PhMe (1.5 mL) at 100 °C for 4 h. Purification by flash chromatography (SiO2, eluted with 15% EtOAc in hexanes) gave isomer 27a (31.1 mg) and 27b (48.3 mg), each as an off-white, fluffy solid, for an overall yield of 64%. Rf of 27a (20% EtOAc in hexanes) = 0.55. 1H NMR of 27a (500 MHz, CDCl3): δ 8.99 (s, 1H, Ar-H), 8.74 (d, J = 9.3 Hz, 1H, Ar-H), 8.63 (s, 1H, Ar-H), 8.21 (s, 1H, Ar-H), 7.64 (d, J = 8.7 Hz, 1H, Ar-H), 6.27 (d, J = 5.5 Hz, 1H, H-1′), 4.70 (app t, J = 4.9 Hz, 1H, H-2′), 4.34 (t, J = 3.2 Hz, 1H, H-3′), 4.20–4.18 (m, 1H, H-4′), 4.03 (dd, J = 3.5, 11.2 Hz, 1H, H-5′), 3.83 (d, J = 11.5 Hz, 1H, H-5′), 0.97, 0.96, and 0.79 (3s, 27H, t-Bu), 0.17, 0.16, 0.13, −0.02, and −0.25 (5s, 18H, SiCH3). 13C NMR of 27a (125 MHz, CDCl3): δ 154.4, 151.8, 147.3, 147.0, 144.9, 131.4, 130.9, 130.2, 123.8, 119.9, 116.2, 88.3, 86.2, 76.4, 72.3, 62.8, 26.3, 25.9, 25.7, 18.7, 17.9, −4.3, −4.4, −4.5, −4.9, −5.1, −5.2. HRMS of 27a (ESI/TOF) calcd for C34H56O4ClN7Si3Na [M + Na]+ 768.3282, found 768.3282. Rf of 27b (20% EtOAc in hexanes) = 0.42. 1H NMR of 27b (500 MHz, CDCl3): δ 9.01 (s, 1H, Ar-H), 8.85 (s, 1H, Ar-H), 8.63 (s, 1H, Ar-H), 8.15 (d, J = 8.8 Hz, 1H, Ar-H), 7.50 (d, J = 7.2 Hz, 1H, Ar-H), 6.27 (d, J =5.5 Hz, 1H, H-1′), 4.71 (t, J = 4.9 Hz, 1H, H-2′), 4.35 (t, J = 3.4 Hz, 1H, H-3′), 4.20–4.18 (m, 1H, H-4′), 4.04 (dd, J = 3.3, 11.0 Hz, 1H, H-5′), 3.82 (d, J = 11.5 Hz, 1H, H-5′), 0.97, 0.96, and 0.79 (3s, 27H, t-Bu), 0.17, 0.16, 0.13, −0.02, and −0.25 (5s, 18H, SiCH3). 13C NMR of 27b (125 MHz, CDCl3): δ 154.2, 151.6, 147.1, 144.7, 144.6, 135.8, 132.5, 126.6, 123.6, 121.0, 115.1, 88.1, 86.0, 76.1, 72.2, 62.6, 26.1, 25.8, 25.5, 18.5, 18.0, 17.7, −4.5, −4.6, −4.7, −5.1, −5.3, −5.4. HRMS of 27b (ESI/TOF) calcd for C34H56O4ClN7Si3Na [M + Na]+ 768.3282, found 768.3282.
Deoxygenation of O6-(6-chlorobenzotriazol-1-yl)-2′-deoxyinosine derivative (12)
As described for the deoxygenation of compound 1, this reaction was carried out with compound 12 (105.8 mg, 0.167 mmol), (pinB)2 (46.1 mg, 0.182 mmol), and Cs2CO3 (109.0 mg, 0.335 mmol) in PhME (1.5 mL) at 100 °C for 4 h. Purification by flash chromatography (SiO2, eluted with 20% EtOAc in hexanes) gave isomers 28a (27.2 mg), 28b (14 mg), and a mixture of 28a + 28b (45.6 mg), each as an off-white, amorphous solid, for an overall yield of 64%. Rf of 28a (50% EtOAc in hexanes) = 0.79. 1H NMR of 28a (500 MHz, CDCl3): δ 8.99 (s, 1H, Ar-H), 8.72 (d, J = 8.8 Hz, 1H, Ar-H), 8.59 (s, 1H, Ar-H), 8.21 (s, 1H, Ar-H), 7.64 (dd, J = 2.0, 8.8 Hz, 1H, Ar-H), 6.64 (t, J = 6.4 Hz, 1H, H-1′), 4.66–4.64 (m, 1H, H-3′), 4.09–4.07 (m, 1H, H-4′), 3.89 (dd, J = 3.9, 11.3 Hz, 1H, H-5′), 3.81 (dd, J = 3.0, 11.2 Hz, 1H, H-5′), 2.68 (app quint, Japp ~ 6.5 Hz, 1H, H-2′), 2.54 (ddd, J = 3.5, 5.8, 12.9 Hz, 1H, H-2′), 0.94 and 0.91 (2s, 18H, t-Bu), 0.13, 0.10, and 0.09 (3s, 12H, SiCH3). 13C NMR of 28a (125 MHz, CDCl3): δ 153.8, 151.7, 147.1, 146.8, 144.4, 131.3, 130.8, 130.1, 123.8, 119.7, 116.1, 88.3, 84.8, 72.1, 62.8, 41.5, 26.0, 25.8, 18.4, 18.0, −4.6, −4.7, −5.3, −5.4. HRMS of 28a (ESI/TOF) calcd for C28H43O3ClN7Si2 [M + H]+ 616.2654, found 616.2643. Rf of 28b (50% EtOAc in hexanes) = 0.69. 1H NMR of 28b (500 MHz, CDCl3): δ 8.99 (s, 1H, Ar-H), 8.81 (d, J = 1.0 Hz, 1H, Ar-H), 8.58 (s, 1H, Ar-H), 8.12 (d, J = 8.8 Hz, 1H, Ar-H), 7.48 (dd, J = 1.4, 8.8 Hz, 1H, Ar-H), 6.63 (t, J = 6.6 Hz, 1H, H-1′), 4.66–4.64 (m, 1H, H-3′), 4.09–4.07 (m, 1H, H-4′), 3.89 (dd, J = 4.2, 11.0 Hz, 1H, H-5′), 3.80 (dd, J = 2.9, 11.2 Hz, 1H, H-5′), 2.69 (app quint, Japp ~ 6.4 Hz 1H, H-2′), 2.53 (ddd, J = 3.4, 5.9, 13.1 Hz, 1H, H-2′), 0.93 and 0.90 (2s, 18H, t-Bu), 0.12, 0.09, and 0.08 (3s, 12H, SiCH3). 13C NMR of 28b (125 MHz, CDCl3): δ 153.7, 151.6, 147.1, 144.7, 144.3, 135.8, 132.5, 126.6, 123.6, 121.0, 115.1, 88.2, 84.7, 72.1, 62.8, 41.4, 25.9, 25.7, 18.4, 17.9, −4.7, −4.8, −5.4, −5.5. HRMS of 28b (ESI/TOF) calcd for C28H42O3ClN7Si2Na [M + Na]+ 638.2468, found 638.2467.
Deoxygenation of O6-(6-chlorobenzotriazol-1-yl)guanosine derivative (13)
As described for the deoxygenation of compound 1, this reaction was carried out with compound 13 (130.2 mg, 0.167 mmol), (pinB)2 (46.1 mg, 0.182 mmol), and Cs2CO3 (109.0 mg, 0.335 mmol) in PhMe (1.5 mL) at 100 °C for 4 h. Purification by flash chromatography (SiO2, eluted with 15% EtOAc in hexanes) gave isomers 29a (19.1 mg), 29b (46.4 mg) and a mixture of 29a + 29b (46.4 mg), each as an off-white to white fluffy solid, for an overall yield of 84%. Rf of 29a (20% EtOAc in hexanes) = 0.42. 1H NMR of 29a (500 MHz, CDCl3): δ 8.54 (d, J = 9.3 Hz, 1H, Ar-H), 8.28 (s, 1H, Ar-H), 8.17 (d, J = 1.7 Hz, 1H, Ar-H), 7.57 (dd, J = 1.6, 8.8 Hz, 1H, Ar-H), 6.07 (d, J = 5.4 Hz, 1H, H-1′), 5.20 (s, 2H, NH2), 4.56 (t, J = 4.7 Hz, 1H, H-2′), 4.30 (t, J = 3.8 Hz, 1H, H-3′), 4.13–4.15 (m, 1H, H-4′), 3.98 (dd, J = 3.3, 11.5 Hz, 1H, H-5′), 3.81 (dd, J = 2.2, 11.5 Hz, 1H, H-5′), 0.96, 0.95, and 0.82 (3s, 27H, t-Bu), 0.15, 0.14, 0.12, 0.00, and −0.16 (5s, 18H, SiCH3). 13C NMR of 29a (125 MHz, CDCl3): δ 158.9, 156.4, 147.6, 146.8, 141.6, 131.0, 130.7, 129.6, 119.6, 117.9, 115.9, 87.4, 85.8, 76.3, 72.3, 62.8, 26.1, 25.9, 25.7, 18.6, 18.1, 17.9, −4.3, −4.5, −4.6, −4.9, −5.2, −5.3. HRMS of 29a (ESI/TOF) calcd for C34H58O4ClN8Si3 [M + H]+ 761.3572, found 761.3567. Rf of 29b (20% EtOAc in hexanes) = 0.30. 1H NMR of 29b (500 MHz, CDCl3): δ 8.64 (d, J = 1.7 Hz, 1H, Ar-H), 8.29 (s, 1H, Ar-H), 8.11 (d, J = 8.2 Hz, 1H, Ar-H), 7.46 (dd, J = 1.6, 8.8 Hz, 1H, Ar-H), 6.07 (d, J = 5.4 Hz, 1H, H-1′), 5.22 (s, 2H, NH2), 4.57 (t, J = 4.4 Hz, 1H, H-2′), 4.31–4.30 (m, 1H, H-3′), 4.15–4.13 (m, 1H, H-4′), 3.98 (dd, J = 3.3, 11.5 Hz, 1H, H-5′), 3.81 (dd, J = 2.2, 11.5 Hz, 1H, H-5′), 0.96, 0.95, and 0.83 (3s, 27H, t-Bu), 0.16, 0.15, 0.12, 0.00, and −0.16 (5s, 18H, SiCH3). 13C NMR of 29b (125 MHz, CDCl3): δ 158.8, 156.4, 147.7, 144.7, 141.7, 135.4, 132.5, 126.4, 121.0, 118.0, 115.0, 87.5, 85.7, 76.2, 72.2, 62.7, 26.1, 25.9, 25.7, 18.6, 18.1, 17.9, −4.3, −4.5, −4.6, −4.9, −5.2, −5.3. HRMS of 29b (ESI/TOF) calcd for C34H58O4ClN8Si3 [M + H]+ 761.3572, found 761.3577.
Deoxygenation of O6-(6-chlorobenzotriazol-1-yl)-2′-deoxyguanosine derivative (14)
As described for the deoxygenation of compound 1, this reaction was carried out with compound 14 (108.4 mg, 0.167 mmol), (pinB)2 (46.1 mg, 0.182 mmol), and Cs2CO3 (109.0 mg, 0.335 mmol) in PhMe (1.5 mL) at 100 °C for 4 h. Purification by flash chromatography (SiO2, eluted with 25% EtOAc in hexanes) gave 83 mg (79% yield) a 1:1 mixture of 30a + 30b as an off-white, fluffy solid. Rf (30% EtOAc in hexanes) = 0.37, 0.30. 1H NMR (500 MHz, CDCl3): δ 8.59 (d, J = 1.1 Hz, 1H, Ar–H), 8.48 (d, J = 8.7 Hz, 1H, Ar–H), 8.23 (s, 1H, Ar–H), 8.22 (s, 1H, Ar–H), 8.14 (s, 1H, Ar–H), 8.08 (d, J = 8.3 Hz, 1H, Ar–H), 7.52 (dd, J = 1.1, 8.8 Hz, 1H, Ar–H), 7.42 (dd, J = 1.6, 8.8 Hz, 1H, Ar–H), 6.43 (t, J = 6.0 Hz, 2H, H-1′), 5.32 and 5.27 (2s, 4H, NH2), 4.62–4.60 (m, 2H, H-3′), 4.04–4.02 (m, 2H, H-4′), 3.84–3.73 (m, 4H, H-5′), 2.60 (app quint, Japp ~ 4.4 Hz, 2H, H-2′), 2.45–2.40 (m, 2H, H-2′), 0.92 and 0.90 (2s, 36H, t-Bu), 0.12, 0.08, and 0.07 (3s, 24H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 158.9, 158.8, 156.0, 147.6, 147.5, 146.7, 144.7, 141.5, 141.4, 135.5, 132.5, 131.0, 130.6, 129.7, 126.4, 121.0, 119.6, 118.0, 117.9, 115.8, 114.9, 88.0, 83.8, 62.9, 41.1, 26.0, 25.8, 18.4, 18.0, −4.6, −4.7, −5.3, −5.5. HRMS (ESI/TOF) calcd for C28H44O3ClN8Si2 [M + H]+ 631.2758, found 631.2745.
Deoxygenation of O6-(benzotriazol-1yl)-2′,3′,5′-tri-O-(t-butyldimethylsilyl)-2-phenylinosine (19)
As described for the deoxygenation of compound 1, this reaction was carried out with compound 19 (40.0 mg, 0.05 mmol), (pinB)2 (13.7 mg, 0.054 mmol), and Cs2CO3 (32.4 mg, 0.1 mmol) in PhMe (1 mL) at 100 °C for 4 h. Purification by flash chromatography (SiO2, eluted with 20% and 25% EtOAc in hexanes) gave 35.1 mg (89% yield) of product 31 as a pale pink, fluffy solid. Rf (20% EtOAc in hexanes) = 0.47. 1H NMR (500 MHz, C6D6): δ 8.78 (d, J = 7.4 Hz, 2H, Ar-H), 8.71 (d, J = 8.3 Hz, 1H, Ar-H), 8.63 (s, 1H, Ar-H), 7.97 (d, J = 8.3 Hz, 1H, Ar-H), 7.42 (t, J = 7.8 Hz, 2H, Ar-H), 7.30 (t, J = 7.4 Hz, 1H, Ar-H), 7.23 (t, J = 7.8 Hz, 1H, Ar-H), 7.01 (t, J = 7.6 Hz, 1H, Ar-H), 6.42 (d, J = 4.4 Hz, 1H, H-1′), 4.91 (t, J = 4.2 Hz, 1H, H-2′), 4.60 (t, J = 4.2 Hz, 1H, H-3′), 4.33–4.31 (m, 1H, H-4′), 4.03 (dd, J = 3.5, 11.2 Hz, 1H, H-5′), 3.78 (dd, J = 1.5, 11.3 Hz, 1H, H-5′), 1.02, 0.98, and 0.93 (3s, 27H, t-Bu), 0.16, 0.15, 0.11, 0.05, 0.00, and −0.01 (6s, 12H, SiCH3). 13C NMR (125 MHz, C6D6): δ 158.5, 155.0, 147.9, 146.6, 144.4, 137.9, 132.2, 130.4, 128.5, 128.4, 128.0, 127.8, 127.6, 124.8, 123.6, 120.1, 114.5, 88.7, 84.9, 76.2, 71.7, 62.2, 25.9, 25.7, 25.6, 18.3, 18.0, 17.8, 1.0, −4.4, −4.8, −4.9, −5.6, −5.62. HRMS (ESI/TOF) calcd for C40H62O4N7Si3 [M + H]+ 788.4166, found 788.4151.
Synthesis of 5,6-dichloro-1-hydroxy-1H-benzotriazole (34)
Step 1: preparation of 1,2,4-trichloro-5-nitrobenzene (33).32
In a clean, dry, round-bottomed flask a mixture of KNO3 (8.34 g, 82.5 mmol) in conc. H2SO4 (110 mL) was cooled at 0 °C with stirring. To this stirring mixture was added 1,2,4-trichlorobenzene (6.9 mL, 55.5 mmol) dropwise. The mixture was then warmed to 50 °C for 3 h. The mixture was cooled to 0 °C and neutralized with 2M aq. NaOH. The crystals formed were filtered, washed with water, and crystallized from petroleum ether to yield compound 33 as fine, pale-yellow crystals. To remove impurity traces, further purification was done by flash chromatography (SiO2, eluted with hexanes) to give 12.0 g (96% yield) of compound 33. Rf (20% EtOAc/hexanes) = 0.71. 1H NMR (500 MHz, DMSO-d6): δ 8.49 (s, 1H, Ar-H), 8.25 (s, 1H, Ar-H). 13C NMR (125 MHz, DMSO-d6): δ 146.4, 136.5, 132.8, 131.2, 127.1, 124.9.
Step 2: conversion of 33 to 34
In a clean, dry, round-bottomed flask equipped with a stirring bar were placed compound 33 (3.3 g, 14.6 mmol), EtOH (33 mL), PhMe (16.5 mL), Na2CO3 (2.01 g, 18.9 mmol), and water (1.8 mL) in that order. To the gently stirring mixture was added hydrazine (4.57 g, 145.7 mmol) and the mixture was heated to reflux at 100 °C for 20 h. The mixture was cooled to room temperature and the flask was transferred to an ice-cold water bath. Upon acidification to pH 2 with 1M HCl a precipitate formed, which was filtered, and washed with cold water. The crude material was recrystallized from 1:1 H2O/EtOH to give 1.678 g (56% yield) of product 34 as a pale-brown powder. Rf (10% CH3OH in CH2Cl2) = 0.60. 1H NMR (500 MHz, DMSO-d6): δ 14.05 (br s, 1H, OH), 8.40 (s, 1H, Ar-H), 8.09 (s, 1H, Ar-H). 13C NMR (125 MHz, DMSO-d6): δ 141.8, 130.8, 127.7, 127.02, 120.7, 111.3, 124.9. HRMS (ESI/TOF) calcd for C6H3OCl2N3Na [M + Na]+ 225.9545, found 225.9546.
Reduction of 5,6-dichloro-1-hydroxy-1H-benzotriazole (34) to 5,6-dichloro-1H-benzotriazole (35)
To a mixture of 5,6-dichloro-1-hydroxy-1H-benzotriazole (34, 50.0 mg, 0.245 mmol) in CH3CN (1 mL) was added Et3N (41.0 μL, 0.294 mmol), and the mixture was stirred at room temperature for 30 min. To this mixture was added B2(OH)4 (26.3 mg, 0.294 mmol) and the mixture was stirred at 50 °C for 30 min. The mixture was cooled and evaporated under reduced pressure. Purification of the crude material by flash chromatography (SiO2, eluted with 40% EtOAc in hexanes) to gave 33.3 mg (72% yield) of 5,6-chloro-1-hydroxy-1H-benzotriazole (35) as a pale-yellowish-brown powder. Rf (10% CH3OH in CH2Cl2) = 0.63. 1H NMR (500 MHz, DMSO-d6): δ 16.05 (s, 1H, NH), 8.34 (s, 2H, Ar-H). HRMS (ESI/TOF) calcd for C6H4Cl2N3 [M + H]+ 187.9777, found 187.9779.
O6-(5,6-Dichlorobenzotriazol-1yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyinosine (40)
To a stirred solution of 5,6-dichloro-1-hydroxy-1H-benzotriazole (34, 286.0 mg, 1.404 mmol) in anhydrous DME (3 mL) were added Et3N (195.8 μL, 1.404 mmol) and MeCN (1.5 mL), and the mixture was stirred at room temperature for 15 min. PyBroP (276.3 mg, 0.59 mmol) was added and the stirring was continued at room temperature for 15 min. 3′,5′-Di-O-TBDMS-protected 2′-deoxyinosine (150.0 mg, 0.312 mmol) was added, followed by the dropwise addition of DIPEA (198.0 μL, 1.135 mmol). The mixture was stirred at room temperature for 20 h and then evaporated under reduced pressure. Purification by flash chromatography (SiO2, eluted with 15% EtOAc in hexanes) gave 146.0 mg (70% yield) of product 40 as a yellow, fluffy solid. Rf (20% EtOAc in hexanes) = 0.43. 1H NMR (500 MHz, CDCl3): δ 8.55 (s, 1H, Ar-H), 8.41 (s, 1H, Ar-H), 8.26 (s, 1H, Ar-H), 7.64 (s, 1H, Ar-H), 6.55 (t, J = 6.3 Hz, 1H, H-1′), 4.65–4.63 (m, 1H, H-3′), 4.07–4.05 (m, 1H, H-4′), 3.92 (dd, J = 3.9, 11.0 Hz, 1H, H-5′), 3.80 (dd, J = 3.1, 11.3 Hz, 1H, H-5′), 2.64 (app quint, Japp ~ 6.3 Hz, 1H, H-2′), 2.52 (ddd, J = 4.4, 6.0, 13.2 Hz, 1H, H-2′), 0.92 (1s, 18H, tert-Bu), 0.11 and 0.109 (2s, 12H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 158.6, 153.7, 151.2, 143.9, 142.2, 134.2, 130.0, 128.1, 121.5, 119.8, 110.1, 88.2, 85.1, 71.6, 62.6, 41.7, 26.0, 25.7, 18.4, 18.0, −4.6, −4.8, −5.4, −5.5. HRMS (ESI/TOF) calcd for C28H41Cl2N7O4Si2Na [M + Na] 688.2028, found 688.2029.
O6-(5,6-Dichlorobenzotriazol-1yl)-2′,3′,5′-tri-O-(t-butyldimethylsilyl)inosine (41)
As described for the preparation of compound 40, this synthesis was performed with 5,6-dichloro-1-hydroxy-1H-benzotriazole (34, 286.4 mg, 1.404 mmol), anhydrous DME (3 mL), Et3N (195.0 μL, 1.404 mmol), CH3CN (1.5 mL), PyBroP (276.3 mg, 0.59 mmol), 2′,3′,5′-tri-O-TBDMS-protected inosine (190.6 mg, 0.312 mmol), and DIPEA (198.0 μL, 1.815 mmol). After addition of the last reagent the mixture was stirred at room temperature for 7 h and evaporated under reduced pressure. Purification by flash chromatography (SiO2, eluted with 5% and 10% EtOAc in hexanes) gave 174.0 mg (70% yield) of product 41 as an off-white, fluffy solid. (Trace amounts of 38 and 39 were observed and removed chromatographically). Rf (15% EtOAc in hexanes) = 0.64. 1H NMR (500 MHz, CDCl3): δ 8.64 (s, 1H, Ar-H), 8.41 (s, 1H, Ar-H), 8.26 (s, 1H, Ar-H), 7.63 (s, 1H, Ar-H), 6.15 (d, J = 3.9 Hz, 1H, H-1′), 4.57 (t, J = 3.9 Hz, 1H, H-2′), 4.34 (t, J = 4.4 Hz, 1H, H-3′), 4.19–4.17 (m, 1H, H-4′), 4.05 (dd, J = 3.4, 11.7 Hz, 1H, H-5′), 3.82 (d, J = 11.7 Hz, 1H, H-5′), 0.97, 0.93, and 0.82, (3s, 27H, t-Bu), 0.17, 0.16, 0.11, 0.10, 0.004, and −0.15 (6s, 18H, SiCH3). 13C NMR (125 MHz, CDCl3): δ 158.7, 154.1, 151.5, 144.3, 142.4, 134.4, 130.1, 128.2, 121.6, 110.2, 89.1, 85.6, 76.7, 71.6, 62.3, 26.3, 26.0, 25.8, 18.7, 18.2, 18.0, −4.2, −4.5, −4.6, −4.8, −5.2, −5.3, −11.7. HRMS (ESI/TOF) calcd for C34H55Cl2N7O5Si3Na [M + Na]+ 818.2842, found 818.2862.
Deoxygenation of O6-(5,6-dichlorobenzotriazol-1yl)-3′,5′-di-O-(t-butyldimethylsilyl)-2′-deoxyinosine (40)
As described for the deoxygenation of compound 1, this reaction was carried out with compound 40 (100.0 mg, 0.15 mmol), (pinB)2 (41.3 mg, 0.163 mmol), and Cs2CO3 (97.7 mg, 0.29 mmol) in PhMe (1.5 mL) at 100 °C for 4 h. Purification by flash chromatography (SiO2, eluted with 20% and 30% EtOAc in hexanes) gave 60.0 mg (61% yield) of product 42 as a yellow, fluffy solid. Rf (20% EtOAc in hexanes) = 0.25. 1H NMR (500 MHz, CDCl3): δ 9.01 (s, 1H, Ar-H), 8.94 (s, 1H, Ar-H), 8.60 (s, 1H, Ar-H), 8.33 (s, 1H, Ar-H), 6.64 (t, J = 6.6 Hz, 1H, H-1′), 4.67–4.65 (m, 1H, H-3′), 4.10–4.08 (m, 1H, H-4′), 3.90 (dd, J = 3.6, 11.2 Hz, 1H, H-5′), 3.82 (d, J =11 Hz, 1H, H-5′), 2.68 (app quint, Japp ~ 3.3 Hz, 1H, H-2′), 2.54 (ddd, J = 3.4, 6.7, 12.9 Hz, 1H, H-2′), 0.94 and 0.91 (2s, 18H, t-Bu), 0.13, 0.10, and 0.10 (3s, 12H, SiCH3). HRMS (ESI/TOF) calcd for C28H42Cl2N7O3Si2 [M + H]+ 650.2259, found 650.2260.
Deoxygenation of O6-(5,6-dichlorobenzotriazol-1yl)-2′,3′,5′-tri-O-(t-butyldimethylsilyl)inosine (41)
As described for the deoxygenation of compound 1, this reaction was carried out with compound 41 (119.5 mg, 0.15 mmol), (pinB)2 (41.3 mg, 0.163 mmol), and Cs2CO3 (97.7 mg, 0.29 mmol) in PhMe (1.5 mL) at 100 °C for 4 h. Purification by flash chromatography (SiO2, eluted with 10% EtOAc in hexanes) gave 61.1 mg (56% yield) of product 43 as a pale-yellow powder. Rf (15% EtOAc in hexanes) = 0.46. 1H NMR (500 MHz, CDCl3): δ 9.00 (s, 1H, Ar-H), 8.99 (s, 1H, Ar-H), 8.64 (s, 1H, Ar-H), 8.32 (s, 1H, Ar-H), 6.26 (d, J = 5.4 Hz, 1H, H-1′), 4.69 (t, J = 4.6 Hz, 1H, H-2′), 4.34–4.33 (m, 1H, H-3′), 4.20–4.18 (m, 1H, H-4′), 4.03 (dd, J = 3.6, 11.4 Hz, 1H, H-5′), 3.83 (d, J = 11.7 Hz, 1H, H-5′), 0.97, 0.95, and 0.78 (3s, 27H, t-Bu), 0.17, 0.16, 0.12, −0.03, and −0.26 (5s, 18H, SiCH3). HRMS (ESI/TOF) calcd for C34H56Cl2N7O4Si3 [M + H]+ 780.3073, found 780.3073.
Crossover experiment involving compounds 4 and 41
As described for the deoxygenation of compound 1, a mixture of compounds 4 (50.0 mg, 83.7 μmol), 41 (66.7 mg, 83.7 μmol), (pinB)2 (46.1 mg, 0.182 mmol), and Cs2CO3 (109 mg, 0.335 mmol) in PhMe (1.5 mL) was stirred at 100 °C for 4 h. The mixture was cooled and evaporated under reduced pressure. Chromatographic separation by preparatory TLC (1 mm, 20 × 20 cm SiO2 plate eluted with 30% EtOAc in hexanes) gave compounds 43 (15.4 mg), 2 (28.8 mg), 42 (15.1 mg), and 5 (30.2 mg). Rf values (30% EtOAc in hexanes) of 43: 0.82, 2: 0.70, 42: 0.53, and 5: 0.34.
Supplementary Material
Acknowledgments
Support of this work by NSF Grant CHE-1265687 (MKL) and a PSC CUNY award is gratefully acknowledged. Infrastructural support at CCNY was provided by NIH grant G12MD007603 from the National Institute on Minority Health and Health Disparities. We thank AllyChem for generous samples of (pinB)2, (catB)2, and B2(OH)2.
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, and copies of 1H and 13C NMR spectra.
References
- 1.Herdewijn P, editor. Modified Nucleosides in Biochemistry, Biotechnology and Medicine. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 2.Blackburn GM, Gait MJ, Loakes D, Williams DM, editors. Nucleic Acids in Chemistry and Biology. 3. RSC Publishing; Cambridge, UK: 2006. [Google Scholar]
- 3.Simons C. Nucleoside Mimetics: Their Chemistry and Biological Properties. Gordon and Breach; Amsterdam: 2001. [Google Scholar]
- 4.Kisakürek MV, Rosemeyer H, editors. Perspectives in Nucleoside and Nucleic Acid Chemistry. Verlag Helvetica Chimica Acta, Zurich and Wiley-VCH; Weinheim: 2000. [Google Scholar]
- 5.Suhadolnik RJ. Nucleosides as Biological Probes. Wiley; New York: 1979. [Google Scholar]
- 6.Chu CK, editor. Antiviral Nucleosides: Chiral Synthesis and Chemotherapy. Elsevier; Amsterdam: 2003. [Google Scholar]
- 7.Venkatesan N, Seo YJ, Bang EK, Park SM, Lee YS, Kim BH. Bull Korean Chem Soc. 2006;27:613–630. [Google Scholar]
- 8.Wiebe LI. Braz Arch Biol Technol. 2007;50:445–459. [Google Scholar]
- 9.Bergstrom DE. Curr Protoc Nucleic Acids Chem. 2009;37:1.4:1.4.1–1.4.32. doi: 10.1002/0471142700.nc0104s37. [DOI] [PubMed] [Google Scholar]
- 10.Srivatsan SG, Sawant AA. Pure Appl Chem. 2011;83:213–232. [Google Scholar]
- 11.Bae S, Lakshman MK. J Am Chem Soc. 2007;129:782–789. doi: 10.1021/ja064682n. [DOI] [PubMed] [Google Scholar]
- 12.Bae S, Lakshman MK. J Org Chem. 2008;73:1311–1319. doi: 10.1021/jo7021795. [DOI] [PubMed] [Google Scholar]
- 13.Bae S, Lakshman MK. J Org Chem. 2008;73:3707–3713. doi: 10.1021/jo702558n. [DOI] [PubMed] [Google Scholar]
- 14.Bae S, Lakshman MK. Org Lett. 2008;10:2203–2206. doi: 10.1021/ol8006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lakshman MK, Frank J. Org Biomol Chem. 2009;7:2933–2940. doi: 10.1039/b905298d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satishkumar S, Vuram PK, Relangi SS, Gurram V, Zhou H, Kreitman RJ, Martínez Montemayor MM, Yang L, Kaliyaperumal M, Sharma S, Pottabathini N, Lakshman MK. Molecules. 2015;20:18437–18463. doi: 10.3390/molecules201018437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wan ZK, Binnun E, Wilson DP, Lee J. Org Lett. 2005;7:5877–5880. doi: 10.1021/ol052424+. [DOI] [PubMed] [Google Scholar]
- 18.Wan ZK, Wacharasindhu S, Levins CG, Lin M, Tabei K, Mansour TS. J Org Chem. 2007;72:10194–10210. doi: 10.1021/jo7020373. [DOI] [PubMed] [Google Scholar]
- 19.Ashton TD, Scammells PJ. Aust J Chem. 2008;61:49–58. [Google Scholar]
- 20.Qu GR, Zhang HL, Niu HY, Xue ZK, Lv XX, Guo HM. Green Chem. 2012;14:1877–1879. [Google Scholar]
- 21.Malow M, Wehrstedt KD, Neuenfeld S. Tetrahedron Lett. 2007;48:1233–1235. [Google Scholar]
- 22.See for leading examples: Shi F, Waldo JP, Chen Y, Larock RC. Org Lett. 2008;10:2409–2412. doi: 10.1021/ol800675u.Campbell-Verduyn L, Elsinga PH, Mirfeizi L, Dierckx RA, Feringa BL. Org Biomol Chem. 2008;6:3461–3463. doi: 10.1039/b812403e.
- 23.(a) Antilla JC, Baskin JM, Barder TE, Buchwald SL. J Org Chem. 2004;69:5578–5587. doi: 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]; (b) Ueda S, Su M, Buchwald SL. Angew Chem Int Ed. 2011;50:8944–8947. doi: 10.1002/anie.201103882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lakshman MK, Singh MK, Parrish D, Balachandran R, Day BW. J Org Chem. 2010;75:2461–2473. doi: 10.1021/jo902342z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Harwood EA, Sigurdsson ST, Edfeldt NBF, Reid BR, Hopkins PB. J Am Chem Soc. 1999;121:5081–5082. [Google Scholar]; (b) Qian M, Glaser R. J Am Chem Soc. 2005;127:880–887. doi: 10.1021/ja045108j. [DOI] [PubMed] [Google Scholar]
- 26.Gurram V, Pottbathini N, Garlapati R, Chaudhary AB, Patro B, Lakshman MK. Chem Asian J. 2012;7:1853–1861. doi: 10.1002/asia.201200093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurram V, Akula HK, Garlapati R, Pottabathini N, Lakshman MK. Adv Synth Catal. 2015;357:451–462. doi: 10.1002/adsc.201400889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Dormoy J-R, Castro B. Tetrahedron Lett. 1979;20:3321–3322. [Google Scholar]; (b) Coste J, Le-Nguyen D, Castro B. Tetrahedron Lett. 1990;31:205–208. [Google Scholar]
- 29.Rivero IA, Somanathan R, Hellberg LH. Synth Commun. 1995;25:2185–2188. [Google Scholar]
- 30.Wehrstedt KD, Wandrey PA, Heitkamp D. J Hazard Mater. 2005;126:1–7. doi: 10.1016/j.jhazmat.2005.05.044. [DOI] [PubMed] [Google Scholar]
- 31.Leonard NJ, Golankiewicz K. J Org Chem. 1969;34:359–365. [Google Scholar]
- 32.Legrand YM, Gray M, Cooke G, Rotello VM. J Am Chem Soc. 2003;125:15789–15795. doi: 10.1021/ja036940b. [DOI] [PubMed] [Google Scholar]
- 33.(a) Augustynowicz-Kopec E, Zwolska Z, Orzeszko A. Acta Pol Pharm Drug Res. 2008;65:435–439. [PubMed] [Google Scholar]; (b) Dabhade PS. Asian J Biomed Pharm Sci. 2013;3:29–34. [Google Scholar]
- 34.Lakshman MK, Choudhury A, Bae S, Rochttis E, Pradhan P, Kumar A. Eur J Org Chem. 2009:152–159. doi: 10.1002/ejoc.200800752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carter CAG, John KD, Mann G, Martin RL, Cameron TM, Baker RT, Bishop KL, Broene RD, Westcott SA. Bifunctional Lewis acid reactivity of diol-derived diboron reagents. In: Shapiro PJ, Atwood DA, editors. Group 13 Chemistry/From Fundamentals to Applications. American Chemical Society; Washington, DC: 2002. pp. 70–87. ACS Symposium Series 822. [Google Scholar]
- 36.Kokatla HP, Thomson PF, Bae S, Lakshman MK. J Org Chem. 2011;76:7842–7848. doi: 10.1021/jo201192c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Londregan AT, Piotrowski DW, Xiao J. Synlett. 2013;24:2695–2700. [Google Scholar]
- 38.Wheeler CA, Millar JG, Cardé RT. Chemoecology. 2015;25:123–133. [Google Scholar]
- 39.Kim J, Bertozzi CR. Angew Chem Int Ed. 2016;54:15777–15781. doi: 10.1002/anie.201508861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.