

ABSTRACT
Staphylococcus aureus is a ubiquitous opportunistic human pathogen and a major health concern worldwide, causing a wide variety of diseases from mild skin infections to systemic disease. S. aureus is a major source of severe secondary bacterial pneumonia after influenza A virus infection, which causes widespread morbidity and mortality. While the phenomenon of secondary bacterial pneumonia is well established, the mechanisms behind the transition from asymptomatic colonization to invasive staphylococcal disease following viral infection remains unknown. In this report, we have shown that S. aureus biofilms, grown on an upper respiratory epithelial substratum, disperse in response to host physiologic changes related to viral infection, such as febrile range temperatures, exogenous ATP, norepinephrine, and increased glucose. Mice that were colonized with S. aureus and subsequently exposed to these physiologic stimuli or influenza A virus coinfection developed pronounced pneumonia. This study provides novel insight into the transition from colonization to invasive disease, providing a better understanding of the events involved in the pathogenesis of secondary staphylococcal pneumonia.
IMPORTANCE
In this study, we have determined that host physiologic changes related to influenza A virus infection causes S. aureus to disperse from a biofilm state. Additionally, we report that these same host physiologic changes promote S. aureus dissemination from the nasal tissue to the lungs in an animal model. Furthermore, this study identifies important aspects involved in the transition of S. aureus from asymptomatic colonization to pneumonia.
INTRODUCTION
Staphylococcus aureus, a ubiquitous Gram-positive bacterium and opportunistic pathogen, causes a myriad of diseases ranging from minor to severe skin infections, toxic shock syndrome, osteomyelitis, implant-associated infections, and sepsis (1, 2). S. aureus is a substantial burden in the health care setting, as it is one of the leading causes of health care-associated infections and has become much more difficult to manage and treat due to the expansion of antibiotic resistance (3–5).
S. aureus is also one of the most common causes of secondary bacterial pneumonia in cases of seasonal influenza and especially during influenza pandemics (6–15). Secondary bacterial pneumonia is a serious condition often associated with respiratory virus infections, particularly influenza A virus (16–19). Secondary staphylococcal pneumonia is often very severe, resulting in extensive damage to the respiratory tract, including necrosis of lung tissue, and is closely linked to increased rates of mortality in young children with or without underlying health issues (14, 20–23). Epidemiologic reports show that nasal colonization occurs in approximately 30 to 80% of the overall population and is prevalent among children. This colonization state serves as a reservoir for invasive staphylococcal disease (8, 19, 24–35). In most instances, colonizing bacteria form biofilms on nasal tissue, allowing S. aureus to persist by evading host defenses and antibiotic treatment (36).
Several elegant studies have investigated the link between influenza virus infection and staphylococcal pneumonia, and these studies have uncovered many facets of the interplay between the virus, host, and S. aureus. Using a mouse model of coinfection, Iverson et al. and Lee et al. have shown that viral infection primes mice for staphylococcal pneumonia and that these mice displayed higher levels of tissue damage, increased inflammation, and immune cell infiltrates, as well as significantly increased rates of mortality over single-agent infections (37, 38). Additionally, Lijek and Weiser have shown that influenza virus infection prior to staphylococcal infection modulates both innate and adaptive immune responses (39). Viral infection appears to modulate type 1 interferon responses, lower interleukin 1β production with implications for interleukin 27, 10, and 17, reducing the phagocytic clearance ability by inhibiting NADPH oxidase, and reducing the production of antimicrobial peptides (40–45). In addition to immune modulation, influenza virus infection has been shown to compromise the epithelial cells of the respiratory tract, leading to dissemination of staphylococci, thereby contributing to systemic infection (46).
While these studies have provided important insight on secondary staphylococcal pneumonia at the site of infection, little is known about the causes of transition from asymptomatic colonization to the invasive disease state. As such, we have investigated several physiologic changes in the host that are directly related to influenza A virus infection. Our results have identified a number of host factors that cause S. aureus to disperse from an asymptomatic colonization state in the nasal tissue and disseminate to the lungs, resulting in pneumonia.
RESULTS
S. aureus forms robust biofilms in vitro on fixed H292 cell substrata.
Although previous studies have demonstrated that S. aureus UAMS-1 forms biofilms on various abiotic surfaces, S. aureus most commonly colonizes the human nasal mucosa. Therefore, we performed studies to determine whether UAMS-1 formed biofilms using an in vitro model system consisting of prefixed human respiratory epithelial cell substrata. Figure 1 indicates that S. aureus UAMS-1 developed biofilms using this system, with greater than 1010 CFU per well after 48 h with comparatively less bacteria, approximately 108 CFU in the surrounding supernatant. Biofilms grown for only 24 h had approximately 10-fold-less bacteria in the supernatant compared to biofilm, suggesting that biofilms are not fully formed at this point (Fig. 1). These quantitative data were subsequently supported by scanning electron microscopy (SEM) analysis. Figure 2 clearly shows that strain UAMS-1 formed robust and mature biofilms displaying hallmark characteristics, such as tower formations, water channels, and a well-developed matrix under these more physiologically relevant conditions.
FIG 1 .

S. aureus UAMS-1 forms biofilms in vitro on respiratory epithelium substrata. (Left) Biofilm-associated (closed circles) and planktonic (open circles) bacteria were enumerated from individual wells after 24 h (n = 4) and 48 h (n = 6). Each symbol represents the value for an individual well. The line shows the mean for the wells. The dotted line indicates the limit of CFU that could be detected. (Right) Bacteria were predominately biofilm associated at 48 h. Ratios of supernatant CFU/biofilm CFU were calculated for individual wells. Data presented are representative of three independent replicate experiments. Values that are significantly different are indicated by asterisks as follows: *, P = 0.013; **, P = 0.0095.
FIG 2 .

SEM micrographs of S. aureus UAMS-1 biofilms grown on H292 cells for 48 h. Arrows indicate water channels (A), tower formations (B), and extracellular matrix (C and D).
S. aureus responds to host physiologic changes related to virus infection in vitro.
Influenza A virus infection leads to a wide variety of physiologic changes in the host due to virus-induced inflammatory responses, including increased body temperature and release of nutrients and cellular “danger signals” associated with extensive tissue damage (47, 48). We investigated whether stimuli related to influenza infection induced S. aureus UAMS-1 dispersal from established biofilms. S. aureus UAMS-1 responded to increased physiologically relevant temperature by exhibiting approximately fourfold-higher dispersal from the biofilm compared to the control (Fig. 3). Biofilm dispersal due to febrile-range temperatures occurred in clinical isolates NRS8, NRS20, NRS70, NRS100, and NRS123, indicating that this is a species-wide phenomenon and not strain specific (data not shown). Interestingly, exogenous ATP, norepinephrine, and glucose failed to elicit appreciable dispersal on their own, in contrast to data reported for other Gram-positive pathogens (48). Paired combinations of these stimuli also failed to elicit increased dispersal (data not shown). However, when all four of these stimuli were used in combination, S. aureus dispersed from the biofilm, suggesting that there was a synergistic effect in vitro (Fig. 3). The combination of these conditions is more consistent with a viral infection, as these “danger signals” would be induced simultaneously in the host.
FIG 3 .

In vitro dispersal of 48-h biofilms following 4 h of heat treatment (38.5°C) or exposure to ATP (1 mM), glucose (25 mM), norepinephrine (100 nM), or all four treatments. Dispersal values are presented as the fold change of ratios of supernatant CFU to biofilm CFU values compared to the untreated sample (control). Data presented are cumulative data from four independent replicate experiments. Values that are significantly different are indicated by bars and asterisks as follows: *, P = 0.018; **, P = 0.007.
S. aureus strain UAMS-1 colonizes the murine nasal tissue.
Previous coinfection studies with S. aureus and influenza A virus involve initial aspiration of the virus followed by aspiration of a large inoculating dose of bacteria 3 to 7 days later (37, 38). While these studies have provided important insight into the pathogenesis of secondary pneumonia, this method circumvents the initial colonizing state that has been shown to be important in disease. Therefore, we established an in vivo S. aureus nasal tissue colonization model to study the transition from colonization to invasive disease in the upper respiratory tract. Figure 4 demonstrates that strain UAMS-1 successfully colonizes the murine nasopharynx after intranasal inoculation. The nasal tissues of the challenged mice were stably colonized with approximately 103 CFU per mouse at 48 h with no bacteria detected in the lung tissue (Fig. 4). After 7 days, three of five mice remained colonized with strain UAMS-1 in the nasal tissues, with no UAMS-1 detected in the lungs.
FIG 4 .
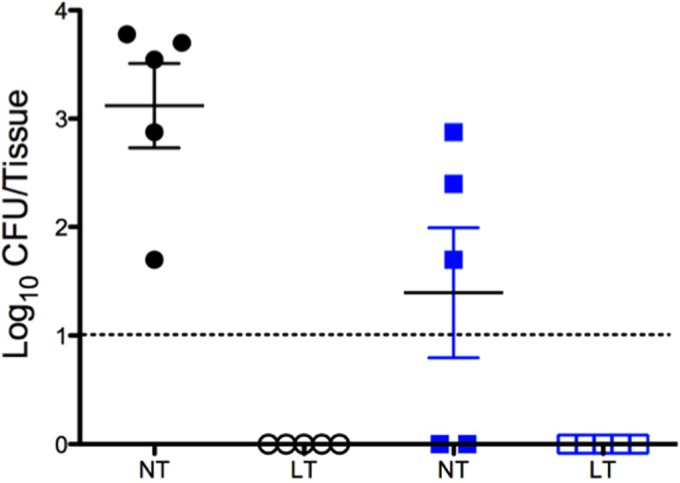
S. aureus UAMS-1 colonization of the murine nasal mucosa. The CFU recovered from nasal tissue (NT) and lung tissue (LT) harvested 48 h (black circles) and 7 days (blue squares) after intranasal inoculation. Each symbol represents the CFU recovered from an individual mouse. The dashed line indicates the limit of CFU detection. Data presented are representative of four independent replicate experiments.
These quantitative data were supported by SEM analysis of nasal tissue harvested from mice colonized for 48 h. Figure 5 shows that strain UAMS-1 colonizes the murine nasal tissue in the form of a biofilm with hallmark structures, such as water channels, tower formation, and an extensive extracellular matrix. Taken together, these data indicate that UAMS-1 stably colonizes the murine nasal mucosa for 48 h in biofilms, and in some cases for 1 week, without detectable dissemination into the lungs or development of invasive disease.
FIG 5 .

SEM micrographs of nasal-tissue-associated S. aureus UAMS-1 biofilms after 48 h of colonization in vivo. Arrows indicate water channels (A), tower formations (B), and extracellular matrix (C and D).
S. aureus responds to host physiologic changes induced by virus infection in vivo.
Our in vitro studies showed that S. aureus UAMS-1 dispersed from the biofilm when exposed to host physiologic changes associated with influenza A virus infection. Using our colonization model, we analyzed the effects of virus-induced host factors on strain UAMS-1 in vivo. Figure 6 shows that mice exposed to febrile-range temperatures had high levels of UAMS-1 in the lungs. This result supports the conclusion that UAMS-1 disseminates from the nasal tissue and is consistent with our in vitro data that UAMS-1 disperses from the biofilm after exposure to febrile-range temperature. However, in contrast to our in vitro results, the other host factors also induced dissemination of strain UAMS-1 to the lungs, suggesting that these individual stimuli promote staphylococcal dispersal from nasal biofilms to the lung. Although a strong overall dispersal-response trend was observed in vivo, these results were not statistically significant. Taken together, these data suggest that host factors associated with viral infection could stimulate UAMS-1 to leave the stably colonized nasal tissues and invade the lungs after only 4 h of exposure to exogenous stimuli that recapitulates virus-induced host physiologic changes.
FIG 6 .
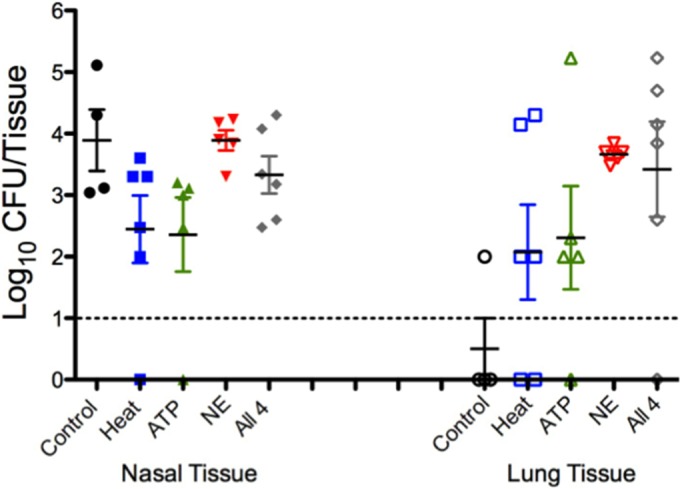
S. aureus UAMS-1 dissemination in colonized mice treated with dispersal stimuli. Mice were treated as described in Materials and Methods with heat (blue squares), ATP (green triangles), norepinephrine (NE) (red triangles), or a combination of the stimuli (gray diamonds) or were not challenged or treated (control) (black circles). Each symbol represents the log10 CFU/tissue recovered from a tissue sample harvested from an individual mouse (closed symbols, nasal tissue; open symbols, lungs). The dashed line indicates the limit of CFU detection. Data presented are representative of two independent replicate experiments.
Concomitant influenza A virus infection in asymptomatically colonized mice leads to staphylococcal pneumonia.
Our results indicated that host factors released in response to viral infection induced bacterial dissemination in vivo, thus we performed subsequent studies to determine whether influenza A virus infection elicited secondary staphylococcal pneumonia using our mouse colonization model. Figure 7 shows that after 48 h of colonization, the control group that received S. aureus UAMS-1 alone had approximately 103 CFU in the nasal tissue. In this experiment, bacteria were detected in the lungs of 4 of the 28 colonized animals (~14%); however, none of these mice exhibited any symptoms of disease, such as piloerection, lethargy, or pyrexia. Mice colonized with strain UAMS-1 for 48 h and subsequently coinfected with influenza A virus show comparable levels of bacteria in nasal tissue; however, these mice also developed a significant secondary bacterial pneumonia with lung bacterial burdens averaging 102 CFU. The coinfected mice showed overt signs of morbidity, including overall lethargy, piloerection, and increased body temperature consistent with the development of invasive disease in murine models compared to control cohorts. UAMS-1 was not recovered from blood samples from either control mice or mice coinfected with influenza A virus. These results indicate that influenza virus infection in mice asymptomatically colonized with UAMS-1 in their nasal tissues induces a transition from colonization to invasive disease and causes pronounced secondary staphylococcal pneumonia.
FIG 7 .

S. aureus UAMS-1 dissemination following influenza A virus coinfection. Each symbol represents the CFU recovered from a tissue sample from an individual mouse. The closed points represent nasal tissue (NT), and the open symbols represent lung tissue (LT). The black circles show the results for control mice that received only intranasal bacteria, and the blue diamonds show the results for mice that were colonized and coinfected with influenza virus 48 h later. The dashed line indicates the limit of CFU detection. Data presented are cumulative data from four independent replicate experiments. Values that are significantly different are indicated by bars and asterisks as follows: *, P = 0.0325; ***, P = 0.0002.
DISCUSSION
Staphylococcal pneumonia is often quite severe, resulting in gross tissue damage and necrosis in the lungs, as well as dissemination into the bloodstream, resulting in sepsis. While influenza A virus itself causes tissue damage and disease, the development of secondary bacterial pneumonia is the most significant source of both morbidity and mortality in cases of seasonal influenza and especially during influenza pandemics (6–15). Moreover, epidemiologic studies of the 2009 H1N1 pandemic indicated that secondary bacterial pneumonia with S. aureus in particular was a strong indicator of mortality in previously healthy children and patients in intensive care units (13, 14). While there are other etiologic agents of secondary bacterial pneumonia, recent studies have shown concerning increases in the incidence of staphylococcal pneumonia (22, 49–51).
Given the severe nature and increasing prevalence of secondary staphylococcal pneumonia, recent research has focused on discerning the steps involved in pathogenesis. Multiple studies designed to investigate the process of pathogenesis have shown that influenza A virus infection primes mice for staphylococcal pneumonia, often resulting in severe tissue damage and increased mortality, and that the virus modulates the immune systems of infected mice, allowing S. aureus to persist in the lungs (40–45, 52). While these studies show aspects of the intricate interplay between these two very different pathogens, directly inoculating bacteria and virus into the lung bypasses the natural route of infection that occurs when colonizing bacteria in the nasal tissues disseminate into the lower respiratory tract. Several studies have shown that colonization precedes invasive disease, implicating this stage as a very important aspect of pathogenesis (35).
We sought to address this gap in knowledge by investigating the transition of S. aureus from nasal tissue colonization to invasive disease. While the above-mentioned studies have described the effect of influenza virus infection on modulating the host immune response to allow for S. aureus persistence, the effect of “danger signals” produced by viral infection on the bacteria had not been investigated. We established an in vitro model using a human respiratory epithelial cell substratum and demonstrated that S. aureus forms robust biofilms after a 48-h incubation period. Inclusion of the epithelial substratum and incubation at 34°C more closely mimic the ecological niche of the upper respiratory tract, a common site of S. aureus colonization. Biofilms grown in this in vitro model displayed characteristic hallmarks of mature staphylococcal biofilms developed in vivo on nasal mucosa with organized and defined tower-like structures complete with channels for nutrient exchange and a well-developed biofilm matrix.
Utilizing a physiologically relevant biofilm model system allowed us to investigate the effects of host “danger signals” produced in response to viral infection on S. aureus biofilms. Our results showed that S. aureus UAMS-1 responds to exogenous stimuli by dispersing from the biofilm. Increased temperature mimicking fever produced a nearly fourfold increase in dispersal, while a combination of four viral-infection-related stimuli (increased temperature, ATP, glucose, and norepinephrine) showed an additive effect, leading to a sixfold increase in dispersal. These results indicate that the physiologic changes that occur in a virally infected host could lead to dispersal from a sessile biofilm community.
These results were further confirmed in vivo with our mouse model of nasal colonization. By using intranasal inoculation, we demonstrated that strain UAMS-1 could stably colonize the nasal tissue of mice for 48 h and in some animals up to 7 days without detectable migration to the lungs and subsequent development of invasive disease. However, exposure to the above-mentioned “danger signals” stimulated S. aureus to disseminate into the lungs from the nasal tissue and resulted in the development of staphylococcal pneumonia. These results indicate that virus-related physiologic changes serve not only as signaling within the host itself but also across kingdoms to the colonizing bacteria.
While previous studies have demonstrated that viral infections are linked to secondary staphylococcal pneumonia, very little is known about the factors that induce S. aureus to transition from colonization to invasive disease. Our novel method of coinfection of the nasal mucosa, the native site of bacterial and viral entry into the host respiratory tract, has provided new insights into possible mechanisms of dissemination. To our knowledge, these are the first studies demonstrating that S. aureus can colonize the nasal tissue of mice and subsequently disperse and migrate to the lungs following viral infection leading to secondary staphylococcal pneumonia. These physiologically relevant studies have provided insight into intricate interplay between the host, S. aureus, and influenza A virus. While dissemination from the lungs into the bloodstream leading to sepsis is common in the pathogenesis of secondary staphylococcal pneumonia, we were unable to detect strain UAMS-1 in the bloodstream of mice 48 h after virus infection. It may be that in mice, sepsis does not develop until later during infection. Taken together, these data underscore the importance of transition from asymptomatic colonization to invasive disease and highlight the complex nature of cross-kingdom signaling in the development of pneumonia.
Future studies will investigate transcriptional changes that occur in S. aureus in response to virally induced host “danger signals” and how these changes are involved in the pathogenesis of secondary staphylococcal pneumonia. Additionally, future studies will investigate whether mice develop sepsis and whether mice succumb to, or clear, these infections.
MATERIALS AND METHODS
Ethics statement.
These studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (53), of the National Institutes of Health and the protocols were approved by the Institutional Animal Care and Use Committee at the University at Buffalo, Buffalo, NY. Experiments were performed under conditions to ensure minimal suffering of the animals involved.
Reagents.
Cell culture reagents were from Invitrogen, Carlsbad, CA. Bacterial and cell culture materials were from VWR Inc., Radnor, PA. Chemically defined streptococcal growth medium (CDM) was prepared as previously described (54). Sheep blood was purchased from Biolink, Liverpool, NY.
Cells and bacterial and viral strains.
NCI-H292 bronchial epithelial cells were grown as described previously (55). Staphylococcus aureus strain UAMS-1, an osteomyelitis clinical isolate (ATCC 49230) (56), was graciously provided by Steve Gill (University of Rochester, Rochester, NY) for use in these studies. Staphylococcus aureus strains NRS8, NRS20, NRS70, NRS100, and NRS123 have been previously described (57) and were used in biofilm formation and dispersal experiments. S. aureus was cultured in CDM at 37°C unless otherwise noted. Influenza A virus strain A/PR8/34 (H3N2, ATCC VR-777) was used for the viral coinfection studies.
Static biofilms on fixed epithelial substratum.
Static staphylococcal biofilms were grown as previously described with slight modifications (58, 59). In brief, 1 × 107 CFU were seeded onto a fixed H292 cell substratum and incubated at 34°C, nasopharyngeal temperature, for 48 h to allow for biofilm development with changes of culture medium at 12-h intervals. A minimum of three independent assays with six replicates each were performed.
In vitro biofilm dispersal.
Biofilm dispersal was performed using previously described virus-induced host response signals that cause Streptococcus pneumoniae biofilm dispersal and bacterial release (48). Briefly, static biofilms of S. aureus were exposed for 4 h to the following exogenous stimuli: ATP (1 mM), glucose (25 mM), norepinephrine (100 nM), a shift to febrile-range temperature (38.5°C), or all four stimuli together. Supernatants from each biofilm were removed, vortexed, and plated onto blood agar plates for bacterial quantification. The remaining biofilm bacteria were sonicated in phosphate-buffered saline (PBS), vortexed, and plated onto blood agar plates for bacterial quantification. Reported values are the calculated ratio of supernatant to biofilm CFU as a measure of bacterial dispersal.
Mouse nasal tissue colonization and infection model.
Nasal tissue colonization experiments were performed as previously described with limited modifications (60). Briefly, nonanesthetized 5-week-old BALB/cByJ mice were intranasally inoculated with ~2 × 108 CFU of S. aureus UAMS-1 in a 20-µl volume by pipetting the inoculum into the nares. For dispersal studies, mice were colonized for 48 h and exposed to the above-mentioned dispersal stimuli for 4 h exactly as described previously (48). Nasal tissue and lungs were collected, homogenized, and dilution plated onto blood agar plates to assess bacterial burden (48, 61, 62). For influenza virus coinfection experiments, mice colonized with strain UAMS-1 for 48 h as indicated above were anesthetized with isofluorane, and 80 PFU of influenza virus A/PR8/34 in 20 µl was pipetted into the nares. Nasal tissue and lungs were collected 48 h later, homogenized, and plated onto blood agar plates to assess bacterial burden. Assays were performed using cohorts of at least three mice per condition on at least two separate occasions.
Statistical analysis.
The data were analyzed for statistical significance by Mann-Whitney test using Prism 5 software (La Jolla, CA) (38, 46, 63, 64). A P value of <0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Lisa Hansen and Shauna Sauberan for their technical assistance with processing mouse samples for the in vivo experiments
This study was funded in part by NIH R01DC013554 awarded to Anthony A. Campagnari and VR 2015-03704 awarded to Anders P. Hakansson.
Footnotes
Citation Reddinger RM, Luke-Marshall NR, Hakansson AP, Campagnari AA. 2016. Host physiologic changes induced by influenza A virus lead to Staphylococcus aureus biofilm dispersion and transition from asymptomatic colonization to invasive disease. mBio 7(4):e01235-16. doi:10.1128/mBio.01235-16.
REFERENCES
- 1.Becker RE, Bubeck Wardenburg J. 2015. Staphylococcus aureus and the skin: a longstanding and complex interaction. Skinmed 13:111–120. [PubMed] [Google Scholar]
- 2.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr.. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spagnolo AM, Orlando P, Panatto D, Amicizia D, Perdelli F, Cristina ML. 2014. Staphylococcus aureus with reduced susceptibility to vancomycin in healthcare settings. J Prev Med Hyg 55:137–144. [PMC free article] [PubMed] [Google Scholar]
- 4.Dryden M, Andrasevic AT, Bassetti M, Bouza E, Chastre J, Baguneid M, Esposito S, Giamarellou H, Gyssens I, Nathwani D, Unal S, Voss A, Wilcox M. 2015. Managing skin and soft-tissue infection and nosocomial pneumonia caused by MRSA: a 2014 follow-up survey. Int J Antimicrob Agents 45(Suppl 1):S1–S14. doi: 10.1016/S0924-8579(15)30002-9. [DOI] [PubMed] [Google Scholar]
- 5.López-Alcalde J, Mateos-Mazón M, Guevara M, Conterno LO, Solà I, Cabir Nunes S, Bonfill Cosp X. 2015. Gloves, gowns and masks for reducing the transmission of meticillin-resistant Staphylococcus aureus (MRSA) in the hospital setting. Cochrane Database Syst Rev 7:CD007087. doi: 10.1002/14651858.CD007087.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shieh WJ, Blau DM, Denison AM, Deleon-Carnes M, Adem P, Bhatnagar J, Sumner J, Liu L, Patel M, Batten B, Greer P, Jones T, Smith C, Bartlett J, Montague J, White E, Rollin D, Gao R, Seales C, Jost H, Metcalfe M, Goldsmith CS, Humphrey C, Schmitz A, Drew C, Paddock C, Uyeki TM, Zaki SR. 2010. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am J Pathol 177:166–175. doi: 10.2353/ajpath.2010.100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Punpanich W, Chotpitayasunondh T. 2012. A review on the clinical spectrum and natural history of human influenza. Int J Infect Dis 16:e714–e723. doi: 10.1016/j.ijid.2012.05.1025. [DOI] [PubMed] [Google Scholar]
- 8.van den Bergh MR, Biesbroek G, Rossen JW, de Steenhuijsen Piters WA, Bosch AA, van Gils EJ, Wang X, Boonacker CW, Veenhoven RH, Bruin JP, Bogaert D, Sanders EA. 2012. Associations between pathogens in the upper respiratory tract of young children: interplay between viruses and bacteria. PLoS One 7:e47711. doi: 10.1371/journal.pone.0047711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chertow DS, Memoli MJ. 2013. Bacterial coinfection in influenza: a grand rounds review. JAMA 309:275–282. doi: 10.1001/jama.2012.194139. [DOI] [PubMed] [Google Scholar]
- 10.Murray RJ, Robinson JO, White JN, Hughes F, Coombs GW, Pearson JC, Tan HL, Chidlow G, Williams S, Christiansen KJ, Smith DW. 2010. Community-acquired pneumonia due to pandemic A(H1N1)2009 influenzavirus and methicillin resistant Staphylococcus aureus co-infection. PLoS One 5:e8705. doi: 10.1371/journal.pone.0008705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawood FS, Fiore A, Kamimoto L, Nowell M, Reingold A, Gershman K, Meek J, Hadler J, Arnold KE, Ryan P, Lynfield R, Morin C, Baumbach J, Zansky S, Bennett NM, Thomas A, Schaffner W, Kirschke D, Finelli L, Emerging Infections Program (EIP) Network. 2010. Influenza-associated pneumonia in children hospitalized with laboratory-confirmed influenza, 2003–2008. Pediatr Infect Dis J 29:585–590. doi: 10.1097/INF.0b013e3181d411c5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dawood FS, Chaves SS, Pérez A, Reingold A, Meek J, Farley MM, Ryan P, Lynfield R, Morin C, Baumbach J, Bennett NM, Zansky S, Thomas A, Lindegren ML, Schaffner W, Finelli L, Emerging Infections Program Network. 2014. Complications and associated bacterial coinfections among children hospitalized with seasonal or pandemic influenza, United States, 2003–2010. J Infect Dis 209:686–694. doi: 10.1093/infdis/jit473. [DOI] [PubMed] [Google Scholar]
- 13.Rice TW, Rubinson L, Uyeki TM, Vaughn FL, John BB, Miller RR III, Higgs E, Randolph AG, Smoot BE, Thompson BT, NHLBI ARDS Network. 2012. Critical illness from 2009 pandemic influenza A virus and bacterial coinfection in the United States. Crit Care Med 40:1487–1498. doi: 10.1097/CCM.0b013e3182416f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randolph AG, Vaughn F, Sullivan R, Rubinson L, Thompson BT, Yoon G, Smoot E, Rice TW, Loftis LL, Helfaer M, Doctor A, Paden M, Flori H, Babbitt C, Graciano AL, Gedeit R, Sanders RC, Giuliano JS, Zimmerman J, Uyeki TM, Pediatric Acute Lung Injury and Sepsis Investigator’s Network and the National Heart, Lung, and Blood Institute ARDS Clinical Trials Network. 2011. Critically ill children during the 2009–2010 influenza pandemic in the United States. Pediatrics 128:e1450–e1458. doi: 10.1542/peds.2011-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papanicolaou GA. 2013. Severe influenza and S. aureus pneumonia: for whom the bell tolls? Virulence 4:666–668. doi: 10.4161/viru.26957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuiken T, Riteau B, Fouchier RA, Rimmelzwaan GF. 2012. Pathogenesis of influenza virus infections: the good, the bad and the ugly. Curr Opin Virol 2:276–286. doi: 10.1016/j.coviro.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Morens DM, Taubenberger JK, Fauci AS. 2008. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blyth CC, Webb SA, Kok J, Dwyer DE, van Hal SJ, Foo H, Ginn AN, Kesson AM, Seppelt I, Iredell JR, ANZIC Influenza Investigators, COSI Microbiological Investigators. 2013. The impact of bacterial and viral co-infection in severe influenza. Influenza Other Respir Viruses 7:168–176. doi: 10.1111/j.1750-2659.2012.00360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bosch AA, Biesbroek G, Trzcinski K, Sanders EA, Bogaert D. 2013. Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog 9:e1003057. doi: 10.1371/journal.ppat.1003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park SS, Kim SH, Kim M, Kim JW, Ko YM, Kim SK, Kim SH, Kim CH. 2015. A case of severe pseudomembranous tracheobronchitis complicated by co-infection of influenza A (H1N1) and Staphylococcus aureus in an immunocompetent patient. Tuberc Respir Dis (Seoul) 78:366–370. doi: 10.4046/trd.2015.78.4.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed C, Kallen AJ, Patton M, Arnold KE, Farley MM, Hageman J, Finelli L. 2009. Infection with community-onset Staphylococcus aureus and influenza virus in hospitalized children. Pediatr Infect Dis J 28:572–576. doi: 10.1097/INF.0b013e31819d8b71. [DOI] [PubMed] [Google Scholar]
- 22.Bhat N, Wright JG, Broder KR, Murray EL, Greenberg ME, Glover MJ, Likos AM, Posey DL, Klimov A, Lindstrom SE, Balish A, Medina MJ, Wallis TR, Guarner J, Paddock CD, Shieh WJ, Zaki SR, Sejvar JJ, Shay DK, Harper SA, Cox NJ, Fukuda K, Uyeki TM. 2005. Influenza-associated deaths among children in the United States, 2003–2004. N Engl J Med 353:2559–2567. doi: 10.1056/NEJMoa051721. [DOI] [PubMed] [Google Scholar]
- 23.Denison AM, Deleon-Carnes M, Blau DM, Shattuck EC, McDougal LK, Rasheed JK, Limbago BM, Zaki SR, Paddock CD. 2013. Molecular characterization of Staphylococcus aureus and influenza virus coinfections in patients with fatal pneumonia. J Clin Microbiol 51:4223–4225. doi: 10.1128/JCM.02503-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wertheim HF, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JA, van Keulen PH, Vandenbroucke-Grauls CM, Meester MH, Verbrugh HA. 2004. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet 364:703–705. doi: 10.1016/S0140-6736(04)16897-9. [DOI] [PubMed] [Google Scholar]
- 25.Fukutani KF, Nascimento-Carvalho CM, Van der Gucht W, Wollants E, Khouri R, Dierckx T, Van Ranst M, Houspie L, Bouzas ML, Oliveira JR, Barral A, Van Weyenbergh J, de Oliveira CI. 2015. Pathogen transcriptional profile in nasopharyngeal aspirates of children with acute respiratory tract infection. J Clin Virol 69:190–196. doi: 10.1016/j.jcv.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bogaert D, van Belkum A, Sluijter M, Luijendijk A, de Groot R, Rümke HC, Verbrugh HA, Hermans PW. 2004. Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children. Lancet 363:1871–1872. doi: 10.1016/S0140-6736(04)16357-5. [DOI] [PubMed] [Google Scholar]
- 27.Spijkerman J, Prevaes SM, van Gils EJ, Veenhoven RH, Bruin JP, Bogaert D, Wijmenga-Monsuur AJ, van den Dobbelsteen GP, Sanders EA. 2012. Long-term effects of pneumococcal conjugate vaccine on nasopharyngeal carriage of S. pneumoniae, S. aureus, H. influenzae and M. catarrhalis. PLoS One 7:e39730. doi: 10.1371/journal.pone.0039730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biesbroek G, Wang X, Keijser BJ, Eijkemans RM, Trzciński K, Rots NY, Veenhoven RH, Sanders EA, Bogaert D. 2014. Seven-valent pneumococcal conjugate vaccine and nasopharyngeal microbiota in healthy children. Emerg Infect Dis 20:201–210. doi: 10.3201/eid2002.131220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.García-Rodríguez JA, Fresnadillo Martínez MJ. 2002. Dynamics of nasopharyngeal colonization by potential respiratory pathogens. J Antimicrob Chemother 50(Suppl S2):59–73. doi: 10.1093/jac/dkf506. [DOI] [PubMed] [Google Scholar]
- 30.Masuda K, Masuda R, Nishi J, Tokuda K, Yoshinaga M, Miyata K. 2002. Incidences of nasopharyngeal colonization of respiratory bacterial pathogens in Japanese children attending day-care centers. Pediatr Int 44:376–380. doi: 10.1046/j.1442-200X.2002.01587.x. [DOI] [PubMed] [Google Scholar]
- 31.Muthukrishnan G, Lamers RP, Ellis A, Paramanandam V, Persaud AB, Tafur S, Parkinson CL, Cole AM. 2013. Longitudinal genetic analyses of Staphylococcus aureus nasal carriage dynamics in a diverse population. BMC Infect Dis 13:221. doi: 10.1186/1471-2334-13-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harputluoglu U, Egeli E, Sahin I, Oghan F, Ozturk O. 2005. Nasopharyngeal aerobic bacterial flora and Staphylococcus aureus nasal carriage in deaf children. Int J Pediatr Otorhinolaryngol 69:69–74. doi: 10.1016/j.ijporl.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Esposito S, Terranova L, Zampiero A, Ierardi V, Rios WP, Pelucchi C, Principi N. 2014. Oropharyngeal and nasal Staphylococcus aureus carriage by healthy children. BMC Infect Dis 14:723. doi: 10.1186/s12879-014-0723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graham PL III, Lin SX, Larson EL. 2006. A U.S. population-based survey of Staphylococcus aureus colonization. Ann Intern Med 144:318–325. doi: 10.7326/0003-4819-144-5-200603070-00006. [DOI] [PubMed] [Google Scholar]
- 35.von Eiff C, Becker K, Machka K, Stammer H, Peters G. 2001. Nasal carriage as a source of Staphylococcus aureus bacteremia. N Engl J Med 344:11–16. [DOI] [PubMed] [Google Scholar]
- 36.Scherr TD, Heim CE, Morrison JM, Kielian T. 2014. Hiding in plain sight: interplay between staphylococcal biofilms and host immunity. Front Immunol 5:37. doi: 10.3389/fimmu.2014.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iverson AR, Boyd KL, McAuley JL, Plano LR, Hart ME, McCullers JA. 2011. Influenza virus primes mice for pneumonia from Staphylococcus aureus. J Infect Dis 203:880–888. doi: 10.1093/infdis/jiq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee MH, Arrecubieta C, Martin FJ, Prince A, Borczuk AC, Lowy FD. 2010. A postinfluenza model of Staphylococcus aureus pneumonia. J Infect Dis 201:508–515. doi: 10.1086/650204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lijek RS, Weiser JN. 2012. Co-infection subverts mucosal immunity in the upper respiratory tract. Curr Opin Immunol 24:417–423. doi: 10.1016/j.coi.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, Di YP, Clay ME, Enelow RI, Dubin PJ, Alcorn JF. 2015. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol 309:L158–L167. doi: 10.1152/ajplung.00338.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Warnking K, Klemm C, Loffler B, Niemann S, van Kruchten A, Peters G, Ludwig S, Ehrhardt C. 2015. Super-infection with Staphylococcus aureus inhibits influenza virus-induced type I IFN signalling through impaired STAT1-STAT2 dimerization. Cell Microbiol 17:303–317. doi: 10.1111/cmi.12375. [DOI] [PubMed] [Google Scholar]
- 42.Robinson KM, Choi SM, McHugh KJ, Mandalapu S, Enelow RI, Kolls JK, Alcorn JF. 2013. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J Immunol 191:5153–5159. doi: 10.4049/jimmunol.1301237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson KM, Lee B, Scheller EV, Mandalapu S, Enelow RI, Kolls JK, Alcorn JF. 2015. The role of IL-27 in susceptibility to post-influenza Staphylococcus aureus pneumonia. Respir Res 16:10. doi: 10.1186/s12931-015-0168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun K, Metzger DW. 2014. Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and enhances susceptibility to secondary methicillin-resistant Staphylococcus aureus infection. J Immunol 192:3301–3307. doi: 10.4049/jimmunol.1303049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson KM, McHugh KJ, Mandalapu S, Clay ME, Lee B, Scheller EV, Enelow RI, Chan YR, Kolls JK, Alcorn JF. 2014. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J Infect Dis 209:865–875. doi: 10.1093/infdis/jit527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang C, Armstrong SM, Sugiyama MG, Tabuchi A, Krauszman A, Kuebler WM, Mullen B, Advani S, Advani A, Lee WL. 2015. Influenza-induced priming and leak of human lung microvascular endothelium upon exposure to Staphylococcus aureus. Am J Respir Cell Mol Biol 53:459–470. doi: 10.1165/rcmb.2014-0373OC. [DOI] [PubMed] [Google Scholar]
- 47.Grebe KM, Takeda K, Hickman HD, Bailey AL, Bailey AM, Embry AC, Bennink JR, Yewdell JW. 2010. Sympathetic nervous system increases proinflammatory cytokines and exacerbates influenza A virus pathogenesis. J Immunol 184:540–544. doi: 10.4049/jimmunol.0903395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marks LR, Davidson BA, Knight PR, Hakansson AP. 2013. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 4:e00438-13. doi: 10.1128/mBio.00438-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crum NF, Lee RU, Thornton SA, Stine OC, Wallace MR, Barrozo C, Keefer-Norris A, Judd S, Russell KL. 2006. Fifteen-year study of the changing epidemiology of methicillin-resistant Staphylococcus aureus. Am J Med 119:943–951. doi: 10.1016/j.amjmed.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 50.Fritz SA, Garbutt J, Elward A, Shannon W, Storch GA. 2008. Prevalence of and risk factors for community-acquired methicillin-resistant and methicillin-sensitive Staphylococcus aureus colonization in children seen in a practice-based research network. Pediatrics 121:1090–1098. doi: 10.1542/peds.2007-2104. [DOI] [PubMed] [Google Scholar]
- 51.Kallen AJ, Reed C, Patton M, Arnold KE, Finelli L, Hageman J. 2010. Staphylococcus aureus community-onset pneumonia in patients admitted to children’s hospitals during autumn and winter of 2006-2007. Epidemiol Infect 138:666–672. doi: 10.1017/S095026880999135X. [DOI] [PubMed] [Google Scholar]
- 52.Rynda-Apple A, Robinson KM, Alcorn JF. 2015. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect Immun 83:3764–3770. doi: 10.1128/IAI.00298-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.National Research Council 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 54.van de Rijn I, Kessler RE. 1980. Growth characteristics of group A streptococci in a new chemically defined medium. Infect Immun 27:444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Schilfgaarde M, van Alphen L, Eijk P, Everts V, Dankert J. 1995. Paracytosis of Haemophilus influenzae through cell layers of NCI-H292 lung epithelial cells. Infect Immun 63:4729–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 63:3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marks LR, Clementi EA, Hakansson AP. 2013. Sensitization of Staphylococcus aureus to methicillin and other antibiotics in vitro and in vivo in the presence of HAMLET. PLoS One 8:e63158. doi: 10.1371/journal.pone.0063158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marks LR, Parameswaran GI, Hakansson AP. 2012. Pneumococcal interactions with epithelial cells are crucial for optimal biofilm formation and colonization in vitro and in vivo. Infect Immun 80:2744–2760. doi: 10.1128/IAI.00488-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marks LR, Reddinger RM, Hakansson AP. 2012. High levels of genetic recombination during nasopharyngeal carriage and biofilm formation in Streptococcus pneumoniae. mBio 3:e00200-12. doi: 10.1128/mBio.00200-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tyx RE, Roche-Hakansson H, Hakansson AP. 2011. Role of dihydrolipoamide dehydrogenase in regulation of raffinose transport in Streptococcus pneumoniae. J Bacteriol 193:3512–3524. doi: 10.1128/JB.01410-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pritchard MT, Ostberg JR, Evans SS, Burd R, Kraybill W, Bull JM, Repasky EA. 2004. Protocols for simulating the thermal component of fever: preclinical and clinical experience. Methods 32:54–62. doi: 10.1016/S1046-2023(03)00187-7. [DOI] [PubMed] [Google Scholar]
- 62.Ostberg JR, Taylor SL, Baumann H, Repasky EA. 2000. Regulatory effects of fever-range whole-body hyperthermia on the LPS-induced acute inflammatory response. J Leukoc Biol 68:815–820. [PubMed] [Google Scholar]
- 63.Haynes L, Szaba FM, Eaton SM, Kummer LW, Lanthier PA, Petell AH, Duso DK, Luo D, Lin JS, Lefebvre JS, Randall TD, Johnson LL, Kohlmeier JE, Woodland DL, Smiley ST. 2012. Immunity to the conserved influenza nucleoprotein reduces susceptibility to secondary bacterial infections. J Immunol 189:4921–4929. doi: 10.4049/jimmunol.1201916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keller LE, Jones CV, Thornton JA, Sanders ME, Swiatlo E, Nahm MH, Park IH, McDaniel LS. 2013. PspK of Streptococcus pneumoniae increases adherence to epithelial cells and enhances nasopharyngeal colonization. Infect Immun 81:173–181. doi: 10.1128/IAI.00755-12. [DOI] [PMC free article] [PubMed] [Google Scholar]