

Abstract
Before a pathogen even enters a cell, intrinsic immune defenses are active. This first-line defense is mediated by a variety of constitutively expressed cell proteins collectively termed “restriction factors” (RFs), and they form a vital element of the immune response to virus infections. Over time, however, viruses have evolved in a variety ways so that they are able to overcome these RF defenses via mechanisms that are specific for each virus. This review provides a summary of the universal characteristics of RFs, and goes on to focus on the strategies employed by some of the most important RFs in their attempt to control human cytomegalovirus (HCMV) infection. This is followed by a discussion of the counter-restriction mechanisms evolved by viruses to circumvent the host cell’s intrinsic immune defenses. RFs include nuclear proteins IFN-γ inducible protein 16 (IFI16) (a Pyrin/HIN domain protein), Sp100, promyelocytic leukemia, and hDaxx; the latter three being the keys elements of nuclear domain 10 (ND10). IFI16 inhibits the synthesis of virus DNA by down-regulating UL54 transcription - a gene encoding a CMV DNA polymerase; in response, the virus antagonizes IFI16 via a process involving viral proteins UL97 and pp65 (pUL83), which results in the mislocalizing of IFI16 into the cytoplasm. In contrast, viral regulatory proteins, including pp71 and IE1, seek to modify or disrupt the ND10 proteins and thus block or reverse their inhibitory effects upon virus replication. All in all, detailed knowledge of these HCMV counter-restriction mechanisms will be fundamental for the future development of new strategies for combating HCMV infection and for identifying novel therapeutic agents.
Keywords: Human cytomegalovirus, Intrinsic immunity, Restriction factors, Viral escape mechanisms, DNA sensors
Core tip: Cellular “restriction factors”, active before human cytomegalovirus (HCMV) enters the cells, form a component of the intrinsic resistance to virus infection. Examples of such factors are hDaxx, promyelocytic leukemia, Sp100 - components of ND10 - and IFN-γ inducible protein 16 (IFI16), an Interferon-inducible protein of the Pyrin/HIN domain protein family. Over time, viruses have developed mechanisms to counteract ND10 and IFI16 through viral proteins, such as IE1 and pp71, or UL97 and pp65, respectively. Detailed knowledge of these mechanisms will provide new competencies useful to control HCMV infection and, in turn, contribute to the development of novel therapeutic approaches.
INTRODUCTION
Viral replication in the infected cell is the result of complex interactions between host and viral proteins. Indeed, in the course of evolution, mammalian immune systems have evolved to response via an array of cellular defense mechanisms, which include both innate and adaptive immune responses, designed to protect against and remove invading pathogens[1]. The innate immune system, mediated by specialized cells such as natural killer cells (NK), dendritic cells, and macrophages[2], is the first to respond, but it is not very specific and does not lead to a long-lasting memory of the response to the pathogen. A specific form of innate immunity, termed “intrinsic immunity”, has also been identified of late, thus generating a third branch of the immune system that was until now considered a bipartite system. Intrinsic immunity involved a set of defense mechanisms that operate on the cellular level[3], realized by cellular proteins known as “restriction factors” (RFs), as they can interfere with various steps of the virus replication cycle[4,5]. The word restriction factor was first coined by research groups studying the murine immune response to retroviruses. Work conducted over 40 years ago revealed that “friend virus susceptibility factor-1” was responsible for conferring resistance to infection by retroviruses[6]. Retroviruses consequently became a model system for investigating intrinsic immunity and have been instrumental in deepening our knowledge of the interaction between viruses and their hosts[7,8]. Over time, the notion of “intrinsic immunity” get up from the finding that the cells attacked by primate lentiviruses are able to resist infection, despite the fact that no signaling event appeared to be necessary for this form of defense, and from the finding that these cells constitutively express prototype human antiretroviral RFs, including the APOBEC3 family of cytidine deaminases[9,10], TRIM5a[11], Tetherin[7], SAMHD1[12], and BST-2[13,14]. RFs are thus germline-encoded proteins mediating the intrinsic cellular immune response against viral replication. Type Iinterferons (IFN) have been demonstrated to increase the expression of RFs, however cells targeted by IFN do not rely on its activity for constitutive antiretroviral activity[15].
INTRINSIC IMMUNITY
The sensing of “pathogen-associated molecular patterns” (PAMPs) - typically microbe nucleic acids and proteins (usually absent from healthy hosts and thus hallmarks of infection) - by germline encoded proteins serving as “pattern recognition receptors” (PRRs) constitutes the earliest step in the innate immune response[2,16]. Viral nucleic acid including DNA containing CpG motifs, and RNA species, including both double-stranded and single-stranded RNA, can be detected by Toll-like receptors TLR3, TLR7-8 and TLR9, or PRR in the cytoplasmatic or the nuclear compartment[17,18]. Two different innate immunity signaling cascades are triggered by detecting exogenous nucleic acid. In the first, transcription factors (TFs) are activated, such as NF-κB and IRF3, culminating in the production of chemokines, cytokines, and IFN-type I[19,20]. The second signaling cascade leads to inflammasome complex formation; this activates caspase-1, an enzyme that generates active cytokines set for secretion by proteolytically cutting pro-IL-1β and pro-IL-18[21-23]. Whereas PRRs activate signals that inhibit infection indirectly, RFs provide front-line defense by interfering directly with the activity of genes essential for the virus’s replication. Indeed, this is often computed before the production of antiviral cytokines has even been activated. Thus, the properties of RFs are clearly distinct to those of PRRs. First and foremost, while RFs are basally expressed in many cell types, their expression may be increased by IFN signaling. Second, isolated RFs have been shown to exhibit antiviral activity in cells, maintaining their capacity to inhibit precise steps in the viral life cycle. Third, viral proteins have evolved to antagonize certain RFs. Finally, genetic selection driven by host vs pathogen coevolution has undoubtedly operated on the genes for RFs[3]. Thus, according to the concept of intrinsic immunity, we can define cell as either “restrictive” or “permissive” depending on viruses ability to replicate efficiently within them[5,24]. Retroviruses have presented a model that has played a pivotal role in the development of our understanding of virus-host interactions[4,8,14,15,18]. However, evidence now shows that several other viruses are also counteracted by intrinsic immunity, including herpesviruses[25,26].
Here, we focus on the newest findings about human cytomegalovirus (HCMV), which belongs to the Herpesviridae family[27,28], and provide a summary of the RFs that perturb its replication (Table 1). Interestingly, HCMV appears to have evolved a number of mechanisms to counteract the action of restriction factors, ultimately leading to the successful replication of viruses in cells[29-32].
Table 1.
Overview of host restriction factors for human cytomegalovirus
| Host restriction factors PYHIN family | Regulation | CMV inhibition | HCMV counter measure | Ref. |
| IFI16 | Type I IFN inducible | HCMV-DNA sensing in the nucleus | Sequestration by HCMV pp65 for MIEP activation | [42,43,46,60,72] |
| Interaction with HCMV pp65 to inhibit UL54 promoter | Protection from proteasome degradation by pp65 | |||
| Antiviral cytokine expression | Delocalization upon phosphorylation by HCMV UL97 | |||
| AIM2 | Type I IFN inducible | MCMV-DNA sensing in the cytoplasm | Not known | [66] |
| Inflammasome activation | ||||
| ND10 family | ||||
| PML | Cell cycle dependent | Transcriptionally inactive chromatin state of MIEP induction | Targeting HCMV IE1 for degradation hDaxx binding by pp71 for proteasome degradation | [93-116] |
| hDaxx | ||||
| Sp100 | ||||
| KDMs | Cell cycle dependent | Inhibition of HCMV latency | Prevention of KDM association with the MIEP by HCMV UL138 | [117] |
CMV: Cytomegalovirus; HCMV: Human cytomegalovirus; MCMV: Murine cytomegalovirus; IFI16: IFN-γ inducible protein 16; PYHIN: Pyrin/HIN domain; AIM2: Absent in melanoma 2; KDMs: Lysine-specific demethylases; MIEP: Major immediate early promoter.
THE IFN-γ INDUCIBLE PROTEIN 16 PROTEIN
The IFN inducible IFI16 protein is a member of the Pyrin and HIN domain containing proteins (PYHIN) family; it is coded by an IFN-inducible group of genes residing on chromosome 1q23[33-35]. In humans, this family includes five PYHIN proteins: The recently discovered “Pyrin domain only protein 3”, “Pyrin and HIN domain family member 1” (PYHIN1), “absent in melanoma 2” (AIM2), “myeloid cell nuclear differentiation antigen” (MNDA), and “IFN-γ inducible protein 16” (IFI16)[36,37]. All five of these proteins possess an N-terminal PYRIN domain, and at least four possess a conserved domain of 200-amino acid repeats (HIN-200) within the C-terminal region (in single or tandem copies), thus they are collectively known as PYHIN. The PYD (or PAAD or DAPIN) domain is a member of the death domain family and consists of an α-helical motif that interacts with other PYD-containing proteins[38]. The HIN domain contains consensus motifs encompassing the 200-amino acid repeats, according to which it is classified into 3 subtypes, designated A, B, C[39,40]. PYHIN1, MNDA, and IFI16 all contain nuclear localization sequences located within their N-termini, and as such are primarily expressed in the cell nucleus[35,39,41]. However, in response to environmental stimuli, such as viral infection, they undergo post-translational modifications, i.e., acetylation, and translocate into the cytoplasm[42,43]. Alternative splicing of the IFI16 gene produces three isoforms[39]; each isoform is made up of two domains, designated A and B, each 200-amino acid long. These domains are divided by a spacer region that may vary in its length. The B isoform is the most predominant and has been detected in an array of histologically distinct cell types (i.e., immune, endothelial, and epithelial cells[44]). The IFI16 N-terminal region display a bi-partite “nuclear localization signal”[45], responsible for its nuclear subcellular localization in quiescent cells, such as: Fibroblasts[46], endothelial cells, and keratinocytes (for a review see[47]). It is of interest that IFI16 protein has also been identified within the nucleolus[34]. However, in fibroblasts, macrophages, and keratinocytes, IFI16 is able to relocate from the nucleus to the cytoplasm. In fibroblasts and macrophages, this occurs following infection by herpesvirus[42,43]; while in keratinocytes, exposure to ultraviolet B light is able to trigger this redistribution[48,49]. In herpesvirus infection, IFI16 redistribution is associated with inflammasome, and after UVB exposure, it is associated with apoptosis. IFI16 is able to form homodimers or bind to other proteins to form heterodimers; its partners include: p53[38,50,51], Rb[52], BRCA1[53], ASC[54] and STING[55]. Indeed, protein-protein interactions are now thought to determine the subcellular localization of proteins; however, the molecular mechanisms regulating the redistribution of IFI16 from the nuclear to the cytoplasmic compartment remain unknown. Finally, a role of viral DNA sensor has also been attributed to cytoplasmic IFI16[47,56]. Indeed, the capacity of IFI16 to bind to viral DNA has been confirmed both in vitro and in vivo[40,55-60]. It is now believed that IFI16 may actually tune the innate immune response by stimulating IFN-type I release[47,56]. Thus, in addition to the various types of protein-protein interactions involving IFI16, another factor that may lead or contribute to IFI16 redistribution within the cell is its binding to microbial DNA.
Inhibition of HCMV replication by IFI16 and viral evasion
AIM2 and IFI16 are the two PYHIN members that have been demonstrated to act as PRRs of intracellular DNA of virus origin[51,60-66]. In particular, in cells infected with Kaposi Sarcoma Associated herpesvirus, IFI16 was revealed to form a functional inflammasome by interacting with ASC together with procaspase-1[54]. Moreover, this virus triggered NF-κB and IRF3 expression and activation [TFs routinely observed to be activated after DNA transfection or the infection with herpes simplex virus type 1 (HSV-1)] could be inhibited by reducing IFI16 expression (or its mouse counterpart p204) using siRNA[67-70]. Besides its role as a PRR, IFI16 had previously been recognized to carry out a variety of other functions in the cell, although none in relation to antiviral activity (reviewed in[35]). However, our understanding of the functions of IFI16 in the cell has dramatically changed over modern years; this is largely due to the results gained from the application of two different experimental approaches (reviewed in[42]). The first involves IFI16 knockdown through the use of specific siRNA or IFI16 inactivation achieved by transfecting cells with a lentivirus carrying a dominant negative mutant form of the protein[71]. For example, eliminating functional IFI16 protein in fibroblasts isolated from human embryo lung (HELFs), via either methodology, was shown to significantly increase herpesvirus replication, including HCMV. The second approach involves augmenting the quantity of IFI16. The overexpression of IFI16 in HELFs infected with HCMV was associated with a 2.5 log reduction in viral yield. However, light had yet to be shed on the molecular mechanisms responsible for the antiviral role of IFI16, prompting an investigation into the consequences of overexpression on the distinct phases of virus replication. Exploiting the luciferase reporter gene methodology, transfection experiments were used to study the effects of deleting the viral polymerase (UL54) or UL44 promoters or introducing mutated forms of the two[72]. These studies indicated the IR-1 locus (inverted repeat element 1), located upstream of the polymerase transcription start-site, to be the object of IFI16-induced virus suppression. Chromatin immunoprecipitation and EMSA revealed that Sp1-like factors were effectively blocked by IFI16 and that this in turn led to UL54 suppression. This result was confirmed by deleting the element within the UL44 promoter responsive to Sp1, which accordingly eliminated the suppressive effect of IFI16 on HCMV replication (UL44 protein associates with UL54 during viral DNA replication). Thus, in addition to confirming IFI16’s role as a DNA sensor, for the first time IFI16 had also been demonstrated to act as a restriction factor of herpesvirus replication[72] (Figure 1).
Figure 1.
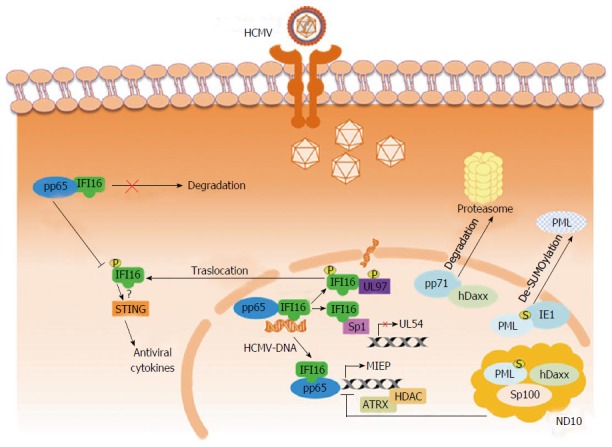
Simplified model of host restrictions against human cytomegalovirus and mechanisms of viral escape. IFI16 recognizes HCMV-DNA in the nucleus. In the early phases of infection, HCMV-pp65 hijacks IFI16 to activate MIEP expression; later on the complex IFI16-pp65 blocks HCMV replication by inhibiting the action of Sp1 on UL54 promoter. IFI16 is not degraded, but it was stabilized by its interaction with pp65. To evade IFI16 antiviral activity, HCMV induces nuclear translocation of IFI16 upon recruitment of HCMV kinase UL97 to IFI16. In the cytoplasm, IFI16 activates STING-mediated antiviral cytokine expression. ND10 components (hDaxx, Sp100, PML) limit viral replication by silencing viral IE genes, inducing a transcriptionally inactive chromatin state of the MIEP via recruitment of ATRX or HDACs to the viral DNA. The viral pp71 binds to hDaxx for proteasome degradation and relieves MIEP repression. Degradation of hDaxx is preceded by the release of ATRX from ND10. IE1 promotes the dispersion of the ND10, by de-SUMOylation, with the consequent inhibition of PML oligomerization. HCMV: Human cytomegalovirus; IFI16: IFN-γ inducible protein 16; PYHIN: Pyrin/HIN domain; AIM2: Absent in melanoma 2; MIEP: Major immediate early promoter; ND10: Nuclear domain 10; ATRX: Alpha thalassemia and mental retardation syndrome X-linked; PML: Promyelocytic leukemia.
HCMV is nevertheless able to replicate in host cells despite the restrictive capacity of IFI16. This suggests that HCMV has developed evasion strategies to respond to the effects of IFI16[42,43]. The first evidence sustaining a plausible HCMV evasion strategy was obtained by infecting fibroblasts with a BAC mutant unable to express UL97 phosphoprotein[42]. Early on during infection, IFI16 binds to virus DNA, but at a later time point during viral DNA synthesis, IFI16 undergoes relocalization from the nucleus to the cytoplasm. It was also revealed that this virus-induced movement of IFI16 out of the nucleus required that UL97 (a viral protein kinase) bound to IFI16. Upon binding to UL97 phosphoprotein, IFI16 undergoes phosphorylation, which in turn promotes its nucleo-cytoplasmic relocalization. IFI16’s ensuing transfer into the virus assembly complex is regulated by the endosomal sorting complex required for transport machinery. Finally, IFI16 becomes integrated into newly assembled virions during the process of virus maturation and budding, effectively expelling IFI16 from the infected host[42]. However, recent studies have revealed that IFI16 phosphorylation by UL97 is not the only mechanism for HCMV escape from IFI16 restriction activity. Using a BAC mutant virus unable to express the tegument protein pp65 (pUL83) Biolatti et al[73] (2016, unpublished results) have demonstrated that IFI16 interacts with pp65 targeting early gene promoters including that of the viral DNA polymerase pUL54. The capability of IFI16 to downregulate virus growth was found to depend on its interaction with pp65 at the UL54 promoter, as shown by the growth properties of the HMCV mutant v65Stop in IFI16 knockdown cells. Interestingly, at later time points of HCMV infection, IFI16 was not degraded, as observed in HSV-1 - infected cells, but it was protected by its interaction with pp65. These data reveal a dual role for pp65. Initially it modulates IFI16 activity at the promoter of immediate-early and early genes, and subsequently, it delocalizes IFI16 from the nucleus, thereby protecting it from proteasomal degradation. Overall, these data identify a novel activity displayed by the pp65/IFI16 interactome in the regulation of UL54 gene expression and IFI16 protein stability during HCMV replication.
In summary, these experiments point toward IFI16 nuclear egression, subsequent to its binding to UL97 and pp65, as the mechanism through which HCMV is successfully able to evade IFI16 restriction activity; this removal of IFI16 from its site of restriction activity is finalized with its incorporation into newly formed virions and expulsion from the cell altogether[42]. This is most likely the event that, to all intents and purposes, underlies the HMCV’s successful evasion of IFI16 antiviral activity (Figure 1, Table 1).
NUCLEAR DOMAIN 10
The nuclear matrix, hypothesized by some to organize and regulate a number of nuclear functions within the nucleus of eukaryotes[74,75], contains discrete bodies designated “nuclear domain 10” (ND10), “promyelocytic leukemia (PML) nuclear bodies”, or PODS. These bodies appear as sphere-like, measuring between 0.1-1 μm in diameter, and in some circumstances they present a granular center. ND10 can be found within the nucleoplasmic domains collectively termed the interchromosomal space, often next to proteinaceous bodies. Sp100, hDaxx, and PML protein are three of the protein constituents of ND10. These proteins recruit additional proteins that are SUMOylated[76,77]. One of these additional proteins is SUMO, a protein related to ubiquitin, and its conjugation to PML is implicated in the further recruitment of yet more binding partners[76,77]. ND10 are devoid of RNA or DNA and they typically gather into clusters of 5 to 15[78]. PML protein forms the outer “casing” of the structure, and its protein partners are usually concentrated inside. Functionally, ND10 play a regulatory role, influencing diverse key processes, such as DNA damage repair[79], oncogenesis[80-82], apoptosis[83-85], senescence[86,87], and gene expression regulation[75,76]. The fact that ND10 are subject to profound biochemical modification during virus infection is of particular interest[81,87-90]. During the infection of quiescent cells, evidence indicates that NB10 accumulates viral DNA within their central core and/or at their periphery. Moreover, considering the fact that IFNs stimulate an increase expression of PML, hDaxx, and Sp100[91-93], it would appear that ND10 play a key role in the innate antiviral response.
ND10 restriction of HCMV replication vs viral evasion tactics
Although aggregates of HCMV IE gene transcripts only form adjacent to ND10 - originally considered the ideal cellular location for HCMV to initiate its program of IE gene expression - it was soon realized that early during infection HCMV actually targets ND10 in order to destroy them[94-97] (Figure 1, Table 1). The viral regulatory protein IE1 is responsible for this activity; moreover ND10 destruction by IE1 correlates with efficient lytic replication[98]. ND10 are now understood to be key cellular restriction factors playing an effective biological role in the inhibition of virus replication. The silencing of PML expression by siRNA and thus depletion of this ND10 constituent, for instance, was shown to increase the susceptibility of “human primary foreskin fibroblasts” (HFF) to HCMV infection[92]. Indeed, the capacity of ND10 bodies to restrict HCMV is achieved by down-regulating IE gene expression[93]. Of note, when PML-null HFF were used, HCMV infection resulted in the formation of Sp100 and hDaxx[99]. It has since emerged that it is actually the virus itself that stimulates the recruitment of ND10 constituents to nucleoprotein complex of HCMV[98]. What is more, studies suggest that this ND10-instigated intrinsic immune response probably entails other ND10 associated proteins, like Sp100 and hDaxx[100-102].
In summary, the results of studies carried out by various groups together depict a scenario where all the three key ND10 proteins - i.e., Sp100, hDaxx, and PML- are involved in restricting viral replication through the silencing of viral IE gene expression[97,102-104]. In addition, infection experiments comparing the effects of single vs double knock-down of ND10 constituents found that HCMV gene expression was actually lower in the single knock-down cells compared with the respective double knock-down conditions[101,102]. This undoubtedly points toward the individual proteins playing independent roles in HCMV restriction. Epigenetic mechanisms able to block viral genome transcription and replication may be involved[105,106]. During early infection, viral DNA exists in a repressive chromatin state, a result of posttranslational modifications of histones[106]. Through its ability to recruit to the viral DNA chromatin modifying enzymes, including the histone deacetylases, or chromatin remodeling protein “alpha thalassemia and mental retardation syndrome X-linked” (ATRX)[105,107], hDaxx has been demonstrated to convert the HCMV immediate-early enhancer/promoter (MIEP) into a transcriptionally inactive chromatin state[108]. Sp100 and PML have similarly been confirmed to interact with enzymes that modify chromatin; once again implicating the contribution of possible epigenetic modifications in the intrinsic immune repression of IE gene expression[109,110].
Until now, the role of ND10 has largely been studied in the context of productive HCMV infection. Interestingly, Saffert and Kalejta[110], using three different cellular settings, including NT2 and THP-1 cells, primary human CD34+ cells, and two myeloblastic cell lines (Kasumi-3 and KG-1), provided evidence that hDaxx is also involved in IE gene silencing in latent HCMV infections. By contrast, the group led by Sinclair provide evidence indicating that hDaxx protein is only marginally involved in MIEP regulation during latent infection, since its knockdown in NT2 cells block IE gene expression[105]. To solve this apparent discrepancy, Stamminger’s group[111] used the THP-1 monocytes recognized as reliable in vitro latency model of HCMV. In non-differentiated THP-1 monocytes, HCMV undergoes latency; while in THP-1 cells induced to undergo differentiation towards a macrophage-like phenotype, achieved using PMA, HCMV enters its lytic cycle[111]. The results obtained showed that the silencing of PML, hDaxx, or Sp100 expression by small hairpin RNA in non-differentiated THP-1 monocytes did not have an effect on IE gene expression. In contrast, the silencing of ND10 in differentiated THP-1 significantly augmented cells positive for IE gene expression. Altogether, these conclusions indicate that hDaxx, PML, and Sp100 serve as restriction factors of IE gene expression, but are only marginally involved in the establishment of HCMV latency[111].
Nevertheless, we know that HCMV is still able to undergo successful lytic replication in spite of the restrictive behavior of the ND10 constituent proteins; this tells us that HCMV has co-evolved to circumvent this aspect of the innate immune response. Indeed, we now know that shortly after virus penetration, pp71 (the viral transactivator tegument protein) moves to ND10 bodies where it interacts with ND10 constituent proteins[112]; in particular, it associates with hDaxx, which, as discussed above, is capable of down-regulating IE gene expression by silencing MIEP[113]. The interaction between pp71 and hDaxx results in the latter being directed down the path of proteasome degradation, thereby relieving MIEP repression[112]. However, investigations by other groups have recently shown that the scenario is actually much more complex. Degradation of hDaxx is preceded by the pp71-stimulated release of ATRX from ND10, and it is the displacement of ATRX that alleviates the repression of IE gene expression[113].
While pp71 is fundamental to counteract the capacity of both ATRX and hDaxx to silence virus genes, it is the action of IE1 protein that seems to eliminate the restrictive effects of PML protein. Indeed, IEI stimulates ND10 body dispersal, with the associated displacement of both PML and Sp100. At low MOI, IE1 synergizes with IE2 and promotes the activation of various viral gene expression[114,115]. However, only IE1 is required for ND10 dispersal. Moreover, the disruption of PML by IE1 is not followed by degradation of PML via proteosome; instead it becomes de-SUMOylated; which effectively inhibits PML oligomerization and thus its ability to re-associate within ND10 bodies[116] (Figure 1, Table 1).
LYSINE-SPECIFIC DEMETHYLASES
Lysine-specific demethylases (KDMs) inhibit the establishment of HCMV latency by getting rid of epigenetic “tags” present on histones associated with the repression of MIEP. Interestingly, the viral UL138 protein counteracts this defense by interfering with the association of KDMs with the MIEP[117] (Table 1).
Thus, the presence of viral factors neutralizing cognate host restriction factors indicate that HCMV has developed multiple escape strategies over lifelong colonization at the cellular level.
CONCLUSION
In conclusion, frontline cell defense against HCMV replication is now known to be accomplished by different proteins through different pathways. Moreover, the viral countermeasures to overcome these restriction factors are now clearly understood to involve a number of viral proteins, including pp71, IE1, UL97, and pp65. Ongoing research is presently being focused at compiling a more in-depth picture of the molecular mechanisms involving ND10 that underlie the host cell’s restrictive response the viral evasion strategies.
Footnotes
Conflict-of-interest statement: The authors declare that they have no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Manuscript source: Invited manuscript
Specialty type: Virology
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: March 21, 2016
First decision: April 19, 2016
Article in press: July 13, 2016
P- Reviewer: Chan CH, Roohvand F S- Editor: Qiu S L- Editor: A E- Editor: Li D
References
- 1.McCormick AL, Mocarski ES. Viral modulation of the host response to infection. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press. [accessed 2016 Apr 19] In: Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, et al., editors. Available from: http://www.ncbi.nlm.nih.gov/books/NBK47417. [Google Scholar]
- 2.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Bieniasz PD. Intrinsic immunity: a front-line defense against viral attack. Nat Immunol. 2004;5:1109–1115. doi: 10.1038/ni1125. [DOI] [PubMed] [Google Scholar]
- 4.Bieniasz PD. Restriction factors: a defense against retroviral infection. Trends Microbiol. 2003;11:286–291. doi: 10.1016/s0966-842x(03)00123-9. [DOI] [PubMed] [Google Scholar]
- 5.Johnson WE. Rapid adversarial co-evolution of viruses and cellular restriction factors. Curr Top Microbiol Immunol. 2013;371:123–151. doi: 10.1007/978-3-642-37765-5_5. [DOI] [PubMed] [Google Scholar]
- 6.Pincus T, Rowe WP, Lilly F. A major genetic locus affecting resistance to infection with murine leukemia viruses. II. Apparent identity to a major locus described for resistance to friend murine leukemia virus. J Exp Med. 1971;133:1234–1241. doi: 10.1084/jem.133.6.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 8.Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol. 2015;16:546–553. doi: 10.1038/ni.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Compton AA, Hirsch VM, Emerman M. The host restriction factor APOBEC3G and retroviral Vif protein coevolve due to ongoing genetic conflict. Cell Host Microbe. 2012;11:91–98. doi: 10.1016/j.chom.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos Trans R Soc Lond B Biol Sci. 2009;364:675–687. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grütter MG, Luban J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr Opin Virol. 2012;2:142–150. doi: 10.1016/j.coviro.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan J, Kaur S, DeLucia M, Hao C, Mehrens J, Wang C, Golczak M, Palczewski K, Gronenborn AM, Ahn J, et al. Tetramerization of SAMHD1 is required for biological activity and inhibition of HIV infection. J Biol Chem. 2013;288:10406–10417. doi: 10.1074/jbc.M112.443796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hammonds J, Wang JJ, Spearman P. Restriction of Retroviral Replication by Tetherin/BST-2. Mol Biol Int. 2012;2012:424768. doi: 10.1155/2012/424768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jakobsen MR, Olagnier D, Hiscott J. Innate immune sensing of HIV-1 infection. Curr Opin HIV AIDS. 2015;10:96–102. doi: 10.1097/COH.0000000000000129. [DOI] [PubMed] [Google Scholar]
- 15.Neil S, Bieniasz P. Human immunodeficiency virus, restriction factors, and interferon. J Interferon Cytokine Res. 2009;29:569–580. doi: 10.1089/jir.2009.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240–73, Table of Contents. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thaiss CA, Levy M, Itav S, Elinav E. Integration of Innate Immune Signaling. Trends Immunol. 2016;37:84–101. doi: 10.1016/j.it.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. 2011;3:920–940. doi: 10.3390/v3060920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. 2013;38:870–880. doi: 10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Unterholzner L. The interferon response to intracellular DNA: why so many receptors? Immunobiology. 2013;218:1312–1321. doi: 10.1016/j.imbio.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao TS. The nucleic acid-sensing inflammasomes. Immunol Rev. 2015;265:103–111. doi: 10.1111/imr.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dempsey A, Bowie AG. Innate immune recognition of DNA: A recent history. Virology. 2015;479-480:146–152. doi: 10.1016/j.virol.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diner BA, Lum KK, Cristea IM. The emerging role of nuclear viral DNA sensors. J Biol Chem. 2015;290:26412–26421. doi: 10.1074/jbc.R115.652289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol. 2011;11:143–154. doi: 10.1038/nri2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boeckh M, Geballe AP. Cytomegalovirus: pathogen, paradigm, and puzzle. J Clin Invest. 2011;121:1673–1680. doi: 10.1172/JCI45449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffiths P, Baraniak I, Reeves M. The pathogenesis of human cytomegalovirus. J Pathol. 2015;235:288–297. doi: 10.1002/path.4437. [DOI] [PubMed] [Google Scholar]
- 29.Beck K, Meyer-König U, Weidmann M, Nern C, Hufert FT. Human cytomegalovirus impairs dendritic cell function: a novel mechanism of human cytomegalovirus immune escape. Eur J Immunol. 2003;33:1528–1538. doi: 10.1002/eji.200323612. [DOI] [PubMed] [Google Scholar]
- 30.Browne EP, Shenk T. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci USA. 2003;100:11439–11444. doi: 10.1073/pnas.1534570100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Child SJ, Hakki M, De Niro KL, Geballe AP. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol. 2004;78:197–205. doi: 10.1128/JVI.78.1.197-205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall EE, Geballe AP. Multifaceted evasion of the interferon response by cytomegalovirus. J Interferon Cytokine Res. 2009;29:609–619. doi: 10.1089/jir.2009.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trapani JA, Dawson M, Apostolidis VA, Browne KA. Genomic organization of IFI16, an interferon-inducible gene whose expression is associated with human myeloid cell differentiation: correlation of predicted protein domains with exon organization. Immunogenetics. 1994;40:415–424. doi: 10.1007/BF00177824. [DOI] [PubMed] [Google Scholar]
- 34.Dawson MJ, Trapani JA. The interferon-inducible autoantigen, IFI 16: localization to the nucleolus and identification of a DNA-binding domain. Biochem Biophys Res Commun. 1995;214:152–162. doi: 10.1006/bbrc.1995.2269. [DOI] [PubMed] [Google Scholar]
- 35.Gariglio M, Mondini M, De Andrea M, Landolfo S. The multifaceted interferon-inducible p200 family proteins: from cell biology to human pathology. J Interferon Cytokine Res. 2011;31:159–172. doi: 10.1089/jir.2010.0106. [DOI] [PubMed] [Google Scholar]
- 36.Connolly DJ, Bowie AG. The emerging role of human PYHIN proteins in innate immunity: implications for health and disease. Biochem Pharmacol. 2014;92:405–414. doi: 10.1016/j.bcp.2014.08.031. [DOI] [PubMed] [Google Scholar]
- 37.Jakobsen MR, Paludan SR. IFI16: At the interphase between innate DNA sensing and genome regulation. Cytokine Growth Factor Rev. 2014;25:649–655. doi: 10.1016/j.cytogfr.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Liao JC, Lam R, Brazda V, Duan S, Ravichandran M, Ma J, Xiao T, Tempel W, Zuo X, Wang YX, et al. Interferon-inducible protein 16: insight into the interaction with tumor suppressor p53. Structure. 2011;19:418–429. doi: 10.1016/j.str.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnstone RW, Kershaw MH, Trapani JA. Isotypic variants of the interferon-inducible transcriptional repressor IFI 16 arise through differential mRNA splicing. Biochemistry. 1998;37:11924–11931. doi: 10.1021/bi981069a. [DOI] [PubMed] [Google Scholar]
- 40.Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, Jiang Z, Horvath G, Rathinam VA, Johnstone RW, et al. Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36:561–571. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gariglio M, Azzimonti B, Pagano M, Palestro G, De Andrea M, Valente G, Voglino G, Navino L, Landolfo S. Immunohistochemical expression analysis of the human interferon-inducible gene IFI16, a member of the HIN200 family, not restricted to hematopoietic cells. J Interferon Cytokine Res. 2002;22:815–821. doi: 10.1089/107999002320271413. [DOI] [PubMed] [Google Scholar]
- 42.Dell’Oste V, Gatti D, Gugliesi F, De Andrea M, Bawadekar M, Lo Cigno I, Biolatti M, Vallino M, Marschall M, Gariglio M, et al. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J Virol. 2014;88:6970–6982. doi: 10.1128/JVI.00384-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li T, Diner BA, Chen J, Cristea IM. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci USA. 2012;109:10558–10563. doi: 10.1073/pnas.1203447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei W, Clarke CJ, Somers GR, Cresswell KS, Loveland KA, Trapani JA, Johnstone RW. Expression of IFI 16 in epithelial cells and lymphoid tissues. Histochem Cell Biol. 2003;119:45–54. doi: 10.1007/s00418-002-0485-0. [DOI] [PubMed] [Google Scholar]
- 45.Briggs LJ, Johnstone RW, Elliot RM, Xiao CY, Dawson M, Trapani JA, Jans DA. Novel properties of the protein kinase CK2-site-regulated nuclear- localization sequence of the interferon-induced nuclear factor IFI 16. Biochem J. 2001;353:69–77. [PMC free article] [PubMed] [Google Scholar]
- 46.Cristea IM, Moorman NJ, Terhune SS, Cuevas CD, O’Keefe ES, Rout MP, Chait BT, Shenk T. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J Virol. 2010;84:7803–7814. doi: 10.1128/JVI.00139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veeranki S, Choubey D. Interferon-inducible p200-family protein IFI16, an innate immune sensor for cytosolic and nuclear double-stranded DNA: regulation of subcellular localization. Mol Immunol. 2012;49:567–571. doi: 10.1016/j.molimm.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bawadekar M, De Andrea M, Gariglio M, Landolfo S. Mislocalization of the interferon inducible protein IFI16 by environmental insults: implications in autoimmunity. Cytokine Growth Factor Rev. 2015;26:213–219. doi: 10.1016/j.cytogfr.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Costa S, Borgogna C, Mondini M, De Andrea M, Meroni PL, Berti E, Gariglio M, Landolfo S. Redistribution of the nuclear protein IFI16 into the cytoplasm of ultraviolet B-exposed keratinocytes as a mechanism of autoantigen processing. Br J Dermatol. 2011;164:282–290. doi: 10.1111/j.1365-2133.2010.10097.x. [DOI] [PubMed] [Google Scholar]
- 50.Gugliesi F, Mondini M, Ravera R, Robotti A, de Andrea M, Gribaudo G, Gariglio M, Landolfo S. Up-regulation of the interferon-inducible IFI16 gene by oxidative stress triggers p53 transcriptional activity in endothelial cells. J Leukoc Biol. 2005;77:820–829. doi: 10.1189/jlb.0904507. [DOI] [PubMed] [Google Scholar]
- 51.Johnstone RW, Wei W, Greenway A, Trapani JA. Functional interaction between p53 and the interferon-inducible nucleoprotein IFI 16. Oncogene. 2000;19:6033–6042. doi: 10.1038/sj.onc.1204005. [DOI] [PubMed] [Google Scholar]
- 52.Xin H, Curry J, Johnstone RW, Nickoloff BJ, Choubey D. Role of IFI 16, a member of the interferon-inducible p200-protein family, in prostate epithelial cellular senescence. Oncogene. 2003;22:4831–4840. doi: 10.1038/sj.onc.1206754. [DOI] [PubMed] [Google Scholar]
- 53.Aglipay JA, Lee SW, Okada S, Fujiuchi N, Ohtsuka T, Kwak JC, Wang Y, Johnstone RW, Deng C, Qin J, et al. A member of the Pyrin family, IFI16, is a novel BRCA1-associated protein involved in the p53-mediated apoptosis pathway. Oncogene. 2003;22:8931–8938. doi: 10.1038/sj.onc.1207057. [DOI] [PubMed] [Google Scholar]
- 54.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. 2011;9:363–375. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brázda V, Coufal J, Liao JC, Arrowsmith CH. Preferential binding of IFI16 protein to cruciform structure and superhelical DNA. Biochem Biophys Res Commun. 2012;422:716–720. doi: 10.1016/j.bbrc.2012.05.065. [DOI] [PubMed] [Google Scholar]
- 57.Ni X, Ru H, Ma F, Zhao L, Shaw N, Feng Y, Ding W, Gong W, Wang Q, Ouyang S, et al. New insights into the structural basis of DNA recognition by HINa and HINb domains of IFI16. J Mol Cell Biol. 2016;8:51–61. doi: 10.1093/jmcb/mjv053. [DOI] [PubMed] [Google Scholar]
- 58.Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, Chan J, Bartholomeu DC, Lauw F, Hall JP, et al. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity. 2011;35:194–207. doi: 10.1016/j.immuni.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stratmann SA, Morrone SR, van Oijen AM, Sohn J. The innate immune sensor IFI16 recognizes foreign DNA in the nucleus by scanning along the duplex. Elife. 2015;4:e11721. doi: 10.7554/eLife.11721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horan KA, Hansen K, Jakobsen MR, Holm CK, Søby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol. 2013;190:2311–2319. doi: 10.4049/jimmunol.1202749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ansari MA, Singh VV, Dutta S, Veettil MV, Dutta D, Chikoti L, Lu J, Everly D, Chandran B. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J Virol. 2013;87:8606–8623. doi: 10.1128/JVI.00805-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson KE, Chikoti L, Chandran B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol. 2013;87:5005–5018. doi: 10.1128/JVI.00082-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Tengchuan J, Laustsen A, Hansen K, Ostergaard L, Fitzgerald KA, et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci USA. 2013;110:E4571–80. doi: 10.1073/pnas.1311669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lo Cigno I, De Andrea M, Borgogna C, Albertini S, Landini MM, Peretti A, Johnson KE, Chandran B, Landolfo S, Gariglio M. The Nuclear DNA Sensor IFI16 Acts as a Restriction Factor for Human Papillomavirus Replication through Epigenetic Modifications of the Viral Promoters. J Virol. 2015;89:7506–7520. doi: 10.1128/JVI.00013-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol. 2016;46:269–280. doi: 10.1002/eji.201545839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJ. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol. 2012;5:173–183. doi: 10.1038/mi.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orzalli MH, DeLuca NA, Knipe DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci USA. 2012;109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, Knipe DM. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci USA. 2013;110:E4492–E4501. doi: 10.1073/pnas.1316194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Søby S, Laursen RR, Ostergaard L, Melchjorsen J. HSV-1-induced chemokine expression via IFI16-dependent and IFI16-independent pathways in human monocyte-derived macrophages. Herpesviridae. 2012;3:6. doi: 10.1186/2042-4280-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gariano GR, Dell’Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, Gribaudo G, Gariglio M, Landolfo S. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 2012;8:e1002498. doi: 10.1371/journal.ppat.1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rivera-Molina YA, Martínez FP, Tang Q. Nuclear domain 10 of the viral aspect. World J Virol. 2013;2:110–122. doi: 10.5501/wjv.v2.i3.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Biolatti M, Dell'Oste V, Pautasso S, von Einem J, Marschall M, Plachter B, Gariglio M, De Andrea M, Landolfo S. Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability. J Virol. 2016 doi: 10.1128/JVI.00923-16. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stuurman N, Meijne AM, van der Pol AJ, de Jong L, van Driel R, van Renswoude J. The nuclear matrix from cells of different origin. Evidence for a common set of matrix proteins. J Biol Chem. 1990;265:5460–5465. [PubMed] [Google Scholar]
- 75.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 76.Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. The mechanisms of PML-nuclear body formation. Mol Cell. 2006;24:331–339. doi: 10.1016/j.molcel.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boisvert FM, Hendzel MJ, Bazett-Jones DP. Promyelocytic leukemia (PML) nuclear bodies are protein structures that do not accumulate RNA. J Cell Biol. 2000;148:283–292. doi: 10.1083/jcb.148.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ, Verbel DA, Cordon-Cardo C, Pandolfi PP. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst. 2004;96:269–279. doi: 10.1093/jnci/djh043. [DOI] [PubMed] [Google Scholar]
- 79.Koken MH, Linares-Cruz G, Quignon F, Viron A, Chelbi-Alix MK, Sobczak-Thépot J, Juhlin L, Degos L, Calvo F, de Thé H. The PML growth-suppressor has an altered expression in human oncogenesis. Oncogene. 1995;10:1315–1324. [PubMed] [Google Scholar]
- 80.Terris B, Baldin V, Dubois S, Degott C, Flejou JF, Hénin D, Dejean A. PML nuclear bodies are general targets for inflammation and cell proliferation. Cancer Res. 1995;55:1590–1597. [PubMed] [Google Scholar]
- 81.Everett RD. Interactions between DNA viruses, ND10 and the DNA damage response. Cell Microbiol. 2006;8:365–374. doi: 10.1111/j.1462-5822.2005.00677.x. [DOI] [PubMed] [Google Scholar]
- 82.Bernardi R, Pandolfi PP. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene. 2003;22:9048–9057. doi: 10.1038/sj.onc.1207106. [DOI] [PubMed] [Google Scholar]
- 83.Bernardi R, Papa A, Pandolfi PP. Regulation of apoptosis by PML and the PML-NBs. Oncogene. 2008;27:6299–6312. doi: 10.1038/onc.2008.305. [DOI] [PubMed] [Google Scholar]
- 84.Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi PP. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000;2:730–736. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- 85.Bischof O, Kirsh O, Pearson M, Itahana K, Pelicci PG, Dejean A. Deconstructing PML-induced premature senescence. EMBO J. 2002;21:3358–3369. doi: 10.1093/emboj/cdf341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108:165–170. doi: 10.1016/s0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 87.Carvalho T, Seeler JS, Ohman K, Jordan P, Pettersson U, Akusjärvi G, Carmo-Fonseca M, Dejean A. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J Cell Biol. 1995;131:45–56. doi: 10.1083/jcb.131.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Everett RD. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene. 2001;20:7266–7273. doi: 10.1038/sj.onc.1204759. [DOI] [PubMed] [Google Scholar]
- 89.Maul GG, Guldner HH, Spivack JG. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0) J Gen Virol. 1993;74(Pt 12):2679–2690. doi: 10.1099/0022-1317-74-12-2679. [DOI] [PubMed] [Google Scholar]
- 90.Szekely L, Pokrovskaja K, Jiang WQ, de The H, Ringertz N, Klein G. The Epstein-Barr virus-encoded nuclear antigen EBNA-5 accumulates in PML-containing bodies. J Virol. 1996;70:2562–2568. doi: 10.1128/jvi.70.4.2562-2568.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Negorev DG, Vladimirova OV, Maul GG. Differential functions of interferon-upregulated Sp100 isoforms: herpes simplex virus type 1 promoter-based immediate-early gene suppression and PML protection from ICP0-mediated degradation. J Virol. 2009;83:5168–5180. doi: 10.1128/JVI.02083-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Regad T, Chelbi-Alix MK. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene. 2001;20:7274–7286. doi: 10.1038/sj.onc.1204854. [DOI] [PubMed] [Google Scholar]
- 93.Ahn JH, Hayward GS. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J Virol. 1997;71:4599–4613. doi: 10.1128/jvi.71.6.4599-4613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kelly C, Van Driel R, Wilkinson GW. Disruption of PML-associated nuclear bodies during human cytomegalovirus infection. J Gen Virol. 1995;76(Pt 11):2887–2893. doi: 10.1099/0022-1317-76-11-2887. [DOI] [PubMed] [Google Scholar]
- 95.Kim YE, Lee JH, Kim ET, Shin HJ, Gu SY, Seol HS, Ling PD, Lee CH, Ahn JH. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J Virol. 2011;85:11928–11937. doi: 10.1128/JVI.00758-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tavalai N, Adler M, Scherer M, Riedl Y, Stamminger T. Evidence for a dual antiviral role of the major nuclear domain 10 component Sp100 during the immediate-early and late phases of the human cytomegalovirus replication cycle. J Virol. 2011;85:9447–9458. doi: 10.1128/JVI.00870-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Korioth F, Maul GG, Plachter B, Stamminger T, Frey J. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp Cell Res. 1996;229:155–158. doi: 10.1006/excr.1996.0353. [DOI] [PubMed] [Google Scholar]
- 98.Tavalai N, Papior P, Rechter S, Leis M, Stamminger T. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J Virol. 2006;80:8006–8018. doi: 10.1128/JVI.00743-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Glass M, Everett RD. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J Virol. 2013;87:2174–2185. doi: 10.1128/JVI.02950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tavalai N, Stamminger T. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim Biophys Acta. 2008;1783:2207–2221. doi: 10.1016/j.bbamcr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 101.Tavalai N, Papior P, Rechter S, Stamminger T. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J Virol. 2008;82:126–137. doi: 10.1128/JVI.01685-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Adler M, Tavalai N, Müller R, Stamminger T. Human cytomegalovirus immediate-early gene expression is restricted by the nuclear domain 10 component Sp100. J Gen Virol. 2011;92:1532–1538. doi: 10.1099/vir.0.030981-0. [DOI] [PubMed] [Google Scholar]
- 103.Everett RD, Chelbi-Alix MK. PML and PML nuclear bodies: implications in antiviral defence. Biochimie. 2007;89:819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 104.Lukashchuk V, McFarlane S, Everett RD, Preston CM. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J Virol. 2008;82:12543–12554. doi: 10.1128/JVI.01215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J Biol Chem. 2006;281:37652–37660. doi: 10.1074/jbc.M604273200. [DOI] [PubMed] [Google Scholar]
- 106.Preston CM, Nicholl MJ. Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J Gen Virol. 2006;87:1113–1121. doi: 10.1099/vir.0.81566-0. [DOI] [PubMed] [Google Scholar]
- 107.Reeves M, Woodhall D, Compton T, Sinclair J. Human cytomegalovirus IE72 protein interacts with the transcriptional repressor hDaxx to regulate LUNA gene expression during lytic infection. J Virol. 2010;84:7185–7194. doi: 10.1128/JVI.02231-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim EJ, Park JI, Nelkin BD. IFI16 is an essential mediator of growth inhibition, but not differentiation, induced by the leukemia inhibitory factor/JAK/STAT pathway in medullary thyroid carcinoma cells. J Biol Chem. 2005;280:4913–4920. doi: 10.1074/jbc.M410542200. [DOI] [PubMed] [Google Scholar]
- 109.Shin HJ, Kim YE, Kim ET, Ahn JH. The chromatin-tethering domain of human cytomegalovirus immediate-early (IE) 1 mediates associations of IE1, PML and STAT2 with mitotic chromosomes, but is not essential for viral replication. J Gen Virol. 2012;93:716–721. doi: 10.1099/vir.0.037986-0. [DOI] [PubMed] [Google Scholar]
- 110.Saffert RT, Kalejta RF. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol. 2006;80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wagenknecht N, Reuter N, Scherer M, Reichel A, Müller R, Stamminger T. Contribution of the Major ND10 Proteins PML, hDaxx and Sp100 to the Regulation of Human Cytomegalovirus Latency and Lytic Replication in the Monocytic Cell Line THP-1. Viruses. 2015;7:2884–2907. doi: 10.3390/v7062751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hofmann H, Sindre H, Stamminger T. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J Virol. 2002;76:5769–5783. doi: 10.1128/JVI.76.11.5769-5783.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cantrell SR, Bresnahan WA. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J Virol. 2005;79:7792–7802. doi: 10.1128/JVI.79.12.7792-7802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Greaves RF, Mocarski ES. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus ie1 mutant. J Virol. 1998;72:366–379. doi: 10.1128/jvi.72.1.366-379.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gawn JM, Greaves RF. Absence of IE1 p72 protein function during low-multiplicity infection by human cytomegalovirus results in a broad block to viral delayed-early gene expression. J Virol. 2002;76:4441–4455. doi: 10.1128/JVI.76.9.4441-4455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu Y, Ahn JH, Cheng M, apRhys CM, Chiou CJ, Zong J, Matunis MJ, Hayward GS. Proteasome-independent disruption of PML oncogenic domains (PODs), but not covalent modification by SUMO-1, is required for human cytomegalovirus immediate-early protein IE1 to inhibit PML-mediated transcriptional repression. J Virol. 2001;75:10683–10695. doi: 10.1128/JVI.75.22.10683-10695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee SH, Albright ER, Lee JH, Jacobs D, Kalejta RF. Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci Adv. 2015;1:e1501164. doi: 10.1126/sciadv.1501164. [DOI] [PMC free article] [PubMed] [Google Scholar]