

Abstract
Concomitant with the development of polymer nanocomposite (PNC) technologies across numerous industries is an expanding awareness of the uncertainty with which engineered nanoparticles embedded within these materials may be released into the external environment, particularly liquid media. Recently there has been an interest in evaluating potential exposure to nanoscale fillers from PNCs, but existing studies often rely upon uncharacterized, poor quality, or proprietary materials, creating a barrier to making general mechanistic conclusions about release phenomena. In this study we employed semiconductor nanoparticles (quantum dots, QDs) as model nanofillers to quantify potential release into liquid media under specific environmental conditions. QDs of two sizes were incorporated into low-density polyethylene by melt compounding and the mixtures were extruded as free-standing fluorescent films. These films were subjected to tests under conditions intended to accelerate potential release of embedded particles or dissolved residuals into liquid environments. Using inductively-coupled plasma mass spectrometry and laser scanning confocal microscopy, it was found that the acidity of the external medium, exposure time, and small differences in particle size (on the order of a few nm) all play pivotal roles in release kinetics. Particle dissolution was found to play a major if not dominant role in the release process. This paper also presents the first evidence that internally embedded nanoparticles contribute to the mass transfer, an observation made possible via the use of a model system that was deliberately designed to probe the complex relationships between nanoparticle-enabled plastics and the environment.
Introduction
Polymer nanocomposites (PNCs) are hybrid materials in which nanoscale filler elements (nanofillers) are dispersed within an organic polymer. Owing to the unique physics and chemistry that occur at the nanoscale, nanocomposites are being developed for numerous technological applications, including construction and infrastructure development;1-4 food5,6 and electronic7,8 packaging; biomedical devices and materials;9-11 and automotive/aerospace materials.12-14
Despite these potential benefits, there is concern that nanofillers dispersed within PNCs may become released into the environment during routine use or disposal of products manufactured from them. Scrutiny of potential nanofiller release is warranted when the release could occur into external media that have a high likelihood of consumer exposure, such as release into foods from nanotechnology-enabled food packaging. Having a robust understanding of PNC release phenomena is also important because the disposal of PNCs could subject these materials to conditions under which leaching of nanofillers constitutes a transfer route of engineered nanomaterials into the environment.
The extent of passive nanofiller release from PNCs is likely to be highest when the PNC is in direct contact with a fluid for an extended period of time. Several studies have focused on PNCs intended for food contact applications,15-27 but release from nanocomposite textiles,28-31 exterior paints,32,33 biomedical devices,34 and other consumer products31,35,36 has also been evaluated. Release of silver nano-particles or residuals from PNCs has received a high amount of attention15-22,27,31,35,37 due to the widespread interest in the antimicrobial properties of silver. A few studies have also been published on nanofiller release from PNCs incorporating exfoliated nanoclays,23-26,38,39 metal oxide nanoparticles,29,32,40 and nanoparticles composed of metals other than silver.16,34,36,40,41 Most of these studies have reported low-level release of nanofillers or their dissolution products into external liquid media under various conditions, but relationships between experimental conditions and release levels have been difficult to generalize. This is in part due to the fact that many release studies rely on proprietary or poorly characterized test materials.
In this article, we report a bottom-up strategy that utilizes a model PNC designed to study nanoparticle release into liquid media. Specifically, we have fabricated and assessed the release properties of free-standing PNC films based on low density polyethylene (LDPE) as the host material and core–shell, cadmium-selenide/zinc-sulfide (CdSe/ZnS) semiconductor nanocrystals (or quantum dots, QDs) as the nano-filler elements. Although QD/LDPE PNCs are unlikely to find wide use in consumer products, LDPE and QDs provide important advantages that motivate their use as model materials to understand PNC release characteristics. LDPE is a commonly used polyolefin that is also processable at reasonably low temperatures, so morphological or chemical changes to the nanofillers during PNC fabrication are likely to be minimized. QDs are well known for their size-dependent luminescence properties, meaning luminescence microscopy can be used to track them within the host polymer and possibly as they are released into the external medium. High quality QDs with low size polydispersities are commercially available, such that relationships between release properties and particle diameter can begin to be elucidated. Finally, the core–shell architecture of the QDs provides an alternative way to distinguish, to some extent, between whole particle release and release of dissolved ions by tracking the ratio of zinc (found in the shell) to cadmium (found in the core) in the external medium over time. In a scenario in which a portion of embedded QDs are released by dissolution and transport of dissolved ions to the external medium, the zinc to cadmium ratio will exceed that of the whole QDs because dissolution of particles occurs from the outside inward.
Herein we have characterized the physical properties of these novel QD/LDPE PNCs and subjected them to release tests inspired by guidance documents published by the US Food and Drug Administration (FDA) for migration of components from food contact materials.42 We have focused on two QD colors/sizes and two liquid media: distilled water and 3% (v/v) aqueous acetic acid. In addition to monitoring the external medium for the constituent elements of QDs (primarily cadmium and zinc) as a function of exposure time, we used fluorescence microscopy to investigate the embedded QDs during the course of the release experiments, so as to reveal key mechanistic details. While the experimental conditions and liquid matrices have been chosen for their relevance to food contact scenarios, our results contribute to a broader knowledge of nanofiller release from PNCs, which should be of general interest to the health and environmental science communities.
Materials and methods
A full account of the experimental methods for this work is provided in the ESI† section. Here we report only the source of materials used, nanocomposite fabrication methods, and details on the release experiments.
Chemicals and materials
LDPE (density = 0.925 g mL−1 at 25 °C, melt index = 25 g/10 min) was purchased from Sigma-Aldrich, and the molecular weight was analyzed by gel permeation chromatography to be Mn = 10 800, Mw = 79 400 and Mz = 411 800. QDs were purchased from two manufacturers: Ocean Nanotech and Nano-Optical Materials. In both cases, QDs were of the CdSe/ZnS core/shell type, surface modified with aliphatic amine (octadecyl amine and oleylamine, respectively). QDs from Ocean Nanotech were shipped in “powder” form and reconstituted in toluene; those purchased from NanoOptical Materials were shipped in a toluene solution and used as received. Throughout this article, QD546 and QD617 will be used to refer to samples incorporating QDs from Ocean Nanotech and QD539 and QD609 will refer to samples incorporating QDs from NanoOptical Materials. The numerical designation corresponds to the peak luminescence maximum in toluene solution. Toluene (Acros organics, 99.8%, extra dry) used to disperse the QDs and glacial acetic acid used as a release medium (Optima grade) was purchased from Fisher Scientific. All water used was deionized to 18.2 MΩ cm and dispensed from an ELGA Purelab Ultra Mk2 system.
Fabrication of QD/LDPE nanocomposite films
A DSM Xplore micro-compounder with a volume capacity of 15 mL was used to mix the QDs with the polymer melt and subsequently extrude the nanocomposite into freestanding films. The micro-compounder is a scaled down version of a conical co-rotating twin screw extruder that can be fitted with a cast film extrusion die. A valve controls the flow of the polymer melt to either a recirculation channel within the mixing chamber or an exit channel.
Preparation of freestanding QD/LDPE nanocomposite films
Before mixing with the polymer, a stock dispersion of QDs in toluene was prepared. For QD546 and QD617, which were purchased as a solid, 25 mg was dispersed in 2 mL toluene. This mass includes both the inorganic QDs as well as the octadecyl amine surface modifier. The inorganic weight percentage of QD546 and QD617 was determined by ICP-AES (see the ESI† for details) to be 62% and 74%, respectively. The final concentration of inorganic QD mass in the PNC films of 1.08 mg mL−1 and 1.30 mg mL−1 (the standard deviation is about 3%), respectively. Based on 12 g LDPE host matrix and a polymer density of 0.925 g mL−1, these final QD concentrations mean that ~10% of the QD mass is lost during the compounding, probably by boiling out of the mixing barrel during the addition step. For QD539 and QD609, which were purchased already dispersed in toluene, the concentration of QD dispersion added to the polymer was adjusted to give a final inorganic mass fraction (again, based on ICP-AES results to determine the inorganic mass concentration in the as-purchased dispersion) in the polymer of 2.08 mg mL−1 for both QD types.
To prepare QD/LDPE nanocomposite resin, the micro-compounder was attached with extensional flow screws and heated to 150 °C. Once the heating blocks were equilibrated, the screw speed was set to speed control mode at 6.28 rad s−1 (60 RPM) with a maximum force tolerance of 5000 N. 12 g neat LDPE pellets was weighed out and half of this was added to the mixing chamber using a feed hopper. After mixing for 2 min, the screw rotation was stopped and a QD stock solution in toluene was added to the mixing chamber drop-wise with intermittent mixing. This step is important to avoid the boiling out of the QD solution from the hot mixing chamber. Once the entire QD solution was added to the mixing chamber, the remaining half of the LDPE pellets was added. The screw speed was then increased to 12.57 rad s−1 (120 RPM) and the mixture processed for 15 min. The screw speed was then reduced to 10 RPM and the valve was opened to the exit channel. The melt was extruded as a strand and cut into small pellets. The pellets were dried in a vacuum at 60 °C for 24 h to remove residual solvent.
Before the next step, the cast film extrusion die was attached to the exit channel of the micro-compounder. Both the micro-compounder and the die were heated to 150 °C, the screw speed was set to 6.28 rad s−1 (60 RPM), and the dried QD/LPDE pellets were added to the mixing chamber using the feed hopper. The pellets were mixed at 6.28 rad s−1 (60 RPM) for 5 min, after which the screw speed control was set to force control mode with a force setting of 550 N, and a maximum speed setting of 12.57 rad s−1 (120 RPM). The two rollers of the cast film extrusion die accessory (the first RPM controlled and the second torque controlled) were set to values of 400 and 70, respectively. The polymer melt exiting the film die was grabbed with tweezers and guided on to the rollers for spooling. The polymer melt exiting the film die was cooled with an air-knife set to a flow rate of 35 L min−1.
Release experiments
To assess release of QDs from QD/LDPE PNCs, the extruded films were cut into circular discs with a diameter of 42 mm, weighed, and measured for thickness using digital calipers (Mitutoyo model #406-350). The film sections were then rinsed 3 times in water, dried, and placed upright in 50 mL polypropylene tubes (Becton, Dickinson & Co., Franklin Lakes NJ USA) filled with 25 mL of either water or 3% (v/v) acetic acid. The samples were stored at 75 °C in a circulating oven for time periods of (1, 3, 5, 7, 10, or 15) days. Note that aliquots were not removed at each time point from a single set of samples: each time point involved using a separate set of test films, in order to eliminate the potential for introduction of environmental contamination during sampling. After the allotted time for a particular experiment was elapsed, the test films were removed from the tube and the residual liquid was assayed for the presence of QDs or their dissolved residuals (procedure provided in the ESI† section).
Results and discussion
Characterization of quantum dots
We initially purchased two QD samples that have peak emission wavelengths of 546 nm (QD546) and 617 nm (QD617). These QDs consist nominally of cadmium selenide cores surrounded by shells composed of zinc sulfide, although according to the manufacturer, intermediate and alternating layers of cadmium sulfide and zinc sulfide are used to improve the chemical stability and optical properties of these QDs. The absorption and emission spectra of these QDs, as well as representative TEM images, are shown in the ESI,† Fig. S1 and S2 respectively. The small size of QDs leads to quantum mechanical effects whereby the electronic state energies that give rise to optical transitions are discrete and heavily modulated by the physical dimensions of the particles. The size-dependent band gaps are manifested in the luminescence spectra, with smaller QD546 particles exhibiting blue-shifted absorption and emission transitions relative to the larger QD617 particles. According to compositional analysis by ICP-AES, and confirmed by STEM/EDS, the mass ratios of cadmium (core element) to zinc (shell element) differ quite substantially between QD546 and QD617, as shown in Table 1. Table 1 also reveals that neither the Cd/Se nor Zn/S mass ratios of these QDs equal the projected 1 : 1 stoichiometric mass ratios (1.42 and 2.04, respectively), in part due to the aforementioned CdS layers deposited during shell fabrication
Table 1.
Size, luminescence quantum yield, and compositional analysis of as-purchased QDs
| QD typea | Mean diameterb [nm] | Quantum yieldc QY [%] | Elemental mass fractiond
|
Mass ratioe
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| Cd | Se | Zn | S | Zn/S | Zn/Cd | Cd/Se | |||
| QD617 | 8.5 | 58.1f | 0.751 | 0.117 | 0.002 | 0.130 | 0.019 | 0.003 | 6.435 |
| QD546 | 5.6 | 18.5f | 0.734 | 0.040 | 0.053 | 0.174 | 0.304 | 0.072 | 18.297 |
| QD609 | 5.5 | 1.2 | 0.201 | 0.106 | 0.439 | 0.255 | 1.724 | 2.187 | 1.900 |
| QD539 | 4.3 | 6.0 | 0.211 | 0.029 | 0.455 | 0.305 | 1.494 | 2.161 | 7.269 |
The numerical part of the QD designation reflects the emission maximum in nm recorded in dilute toluene solution; the luminescence spectral bandwidths (full-width-at-half-maximum) for QD539, QD546, QD609, and QD617 were determined to be (0.160, 0.144, 0.117 and 0.074) eV, respectively.
The mean particle diameters were determined either by STEM or analysis of the optical properties; see the ESI for details.
The errors of QY values measured in this fashion are typically assumed to be on the order of ±10%.44
This value reflects the mass of each element divided by the summed mass of all four analyzed elements in an analyzed quantity of material, as determined by ICP-AES analysis of QDs after microwave digestion. Compositional analysis data reported here represents the mean values (n = 3); standard deviations are provided in the ESI section.
The ideal (1 : 1) stoichiometric Cd/Se and Zn/S mass ratios are 1.42 and 2.04, respectively.
The QY values of QD617 and QD546 may have been higher at the time they were incorporated into QD/LDPE films, as the QY values for QD617 and QD546 were not measured until after QD609 and QD539 were purchased.
During preliminary release tests, we found that the alternating core/shell architecture of QD546 and QD617 complicated analysis and interpretation of release data from QD/LDPE PNCs manufactured with these QDs. As a result, we purchased a second set of QDs (QD539 and QD609) that were subjected to additional purification of the isolated CdSe cores and utilized TOPO-free syntheses, as acid impurities in TOPO (trioctyl phosphine oxide) have been implicated in surface enrichment of CdSe core particles with cadmium.43 Synthesis of QD539 and QD609 also did not involve any intermediate CdS or other layers to optimize stability and intensity of luminescence properties. Absorption and emission spectra of these modified QDs are provided in the ESI,† Fig. S3A, and basic spectral and geometric information for all the QDs used in this study are provided in Table 1. Compared to QD546 and QD617, QD539 and QD609 have Cd/Se and Zn/S mass ratios much closer to 1 : 1 stoichiometric ideality (Table 1). Unfortunately, QD539 and QD609 also exhibit significantly reduced luminescence quantum yields, possibly because the process used to eliminate surface cadmium enrichment was not optimized to reduce surface defects on the CdSe cores that quench luminescence. As a result, QD/LDPE materials incorporating QD539 and QD609 were used primarily for release experiments, where the simpler internal architecture of these QDs facilitated data analysis and interpretation, but QD/LDPE materials incorporating QD546 and QD617 were used when luminescence properties were important, such as the laser scanning confocal microscopy (LSCM) experiments described later on.
Fabrication and characterization of QD/LDPE nanocomposites
Incorporation of QDs into LDPE via melt-compounding and extrusion of the molten polymer nanocomposite through a 65 mm heated film die affords free-standing QD/LDPE nano-composite films that are on the order of 40 μm thick. Photographs of QD/LDPE films made with highly luminescent QD546 and QD617 are shown in Fig. 1. Analysis of the QD/LDPE films by ICP-AES showed similar elemental compositions compared to the QDs in solution (see ESI,† Table S1). Thermal measurements (ESI,† Fig. S4 and S5, Table S2) revealed that incorporation of QDs into LDPE resulted in very small changes to the melting points, % crystallinity values, or viscoelastic properties compared to those for neat LDPE. Electron microscopy (ESI,† Fig. S2) revealed that QDs were incorporated into LDPE both as clusters and as individually dispersed particles. The composition of the dispersed particles was confirmed to be CdSe/ZnS QDs by energy dispersive X-ray spectroscopy (EDS) mapping (ESI† section, Fig. S6 and S7).
Fig. 1.
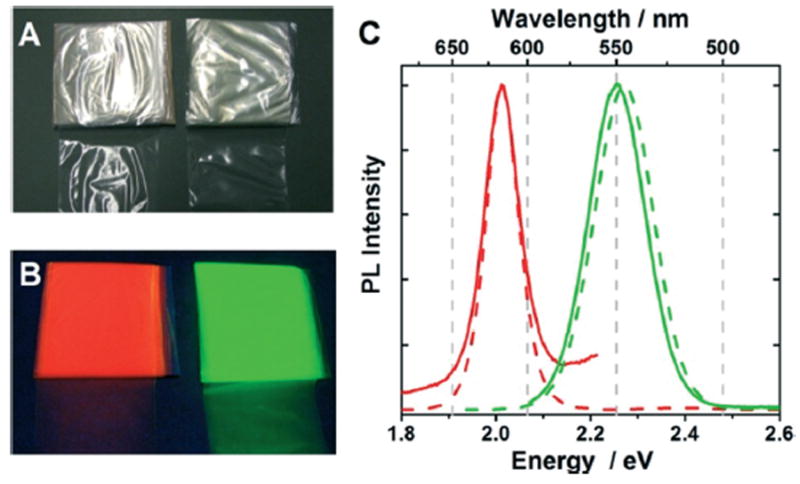
Photographs of QD/LDPE films under ambient room light (A) and during illumination with a hand-held UV-lamp (B). The QDs incorporated within the films on the left have emission maxima at 617 nm (QD617/LDPE, red luminescence) and those incorporated within the films on the right have emission maxima at 546 nm (QD546/LDPE, green luminescence). The inorganic weight fraction in QD617/LDPE and QD546/LDPE was 1.30 mg mL−1 and 1.08 mg mL−1, respectively. Panel (C) shows normalized luminescence spectra of QD/LDPE films. The solid red and green lines correspond to QD617/LDPE and QD546/LDPE films, respectively. For comparison, the spectra of neat QD617 and QD546 in dilute toluene solution are shown as dotted lines. The excitation wavelength for these experiments was 460 nm and 550 nm for QD546 and QD617, respectively.
Under irradiation with a handheld UV lamp, QD/LDPE films manufactured with QD546 and QD617 exhibit bright luminescence, the color of which depends on the incorporated QD size (Fig. 1B). Luminescence spectroscopy (Fig. 1C) reveals that both QD546/LDPE and QD617/LDPE PNCs have nearly the same peak emission maxima and spectral bandwidths that the respective QD samples possess in dilute toluene solution, indicating that the optically active portion of incorporated QDs remain electronically isolated when dispersed within the polymer and that their luminescence properties are not significantly changed by melt-compounding. Efficient light scattering by the semicrystalline LDPE host matrix precluded quantitative determination of luminescence intensity of the compounded materials. Nevertheless, we determined that constant irradiation of the QD/LDPE films with a hand-held UV lamp did not appreciably alter their luminescence intensity or peak emission wavelengths over a period of several days, and the luminescence properties are stable in the dark for months, indicating that the polymer protects against photo-oxidative or other deleterious processes that have been observed to impact the optical properties of QDs dispersed in organic solvents. LSCM experiments also confirmed that QDs are still luminescent after incorporation into LDPE, and are distributed regularly throughout the polymer matrix (ESI,† Fig. S8).
Release of QDs from QD/LDPE PNCs
Release tests were based on FDA recommended procedures to assess release of potential components from food contact substances.42,44 The general idea is to submerge sections of QD/LDPE films in a liquid environment at a specified temperature and monitor the amount of QDs released from the polymer over time. Our primary interest is to evaluate release from a model nanotechnology-enabled food packaging material, but we also desired that our results would be general enough to be applicable to release into other environmental matrices, and so we have selected distilled water and dilute (3% v/v) aqueous acetic acid as our test media. These media are specified to simulate aqueous and acidic foods, respectively. We have performed our experiments at 75 °C, which is high enough to accelerate any QD release, but not so high as to be close to the melting point of these materials (≈110 °C, Fig. S4†). The release data reported below pertain mostly to QD/LDPE materials fabricated with QD539 or QD609, which were designed to have less cadmium surface enrichment in the core region and a more distinct boundary between the core and shell layers. We performed identical release experiments with QD546/LDPE and QD617/LDPE and generally made the same conclusions from them, although interpretation of the data was more complicated due to their alternating shell layers and compositional dissimilarities. Release data for QD546/LDPE and QD617/LDPE are provided in the ESI,† Fig. S17 and S18 and Table S6.
As an initial means of determining the manner in which QDs were released from the QD/LDPE test materials during storage in aqueous solvent at elevated temperature, we used ICP-MS to quantitate cadmium, selenium, zinc, and sulfur in the external liquid medium as a function of immersion time. The results of these analyses are shown in Fig. 2 and are presented as the total amount of mass of each element released into the immersion volume of 25 mL per unit film surface area. Fig. 3 presents the total inorganic mass released in these experiments, which is essentially a sum total of the released quantities of each QD component element. Tabulated data for these experiments, including data calculated on a per-film-mass basis, are provided in the ESI,† Tables S3–S5. We also performed control experiments using LDPE films without QDs and sample tubes with no films. These experiments revealed negligible quantities of zinc, cadmium, and selenium under most environmental conditions. Some sulfur was released (1–15 μg L−1, see ESI†) from these control materials in increasing quantities as a function of time, particularly in acidic media, indicating that a small portion of the released sulfur depicted in Fig. 2 and 3 derived either from the host polymer or sample tubes. The presence of this sulfur background does not change the conclusions derived from the release data, particularly when comparing films loaded with different QD types, as the same low background would be present in all samples included in a given time/media condition.
Fig. 2.
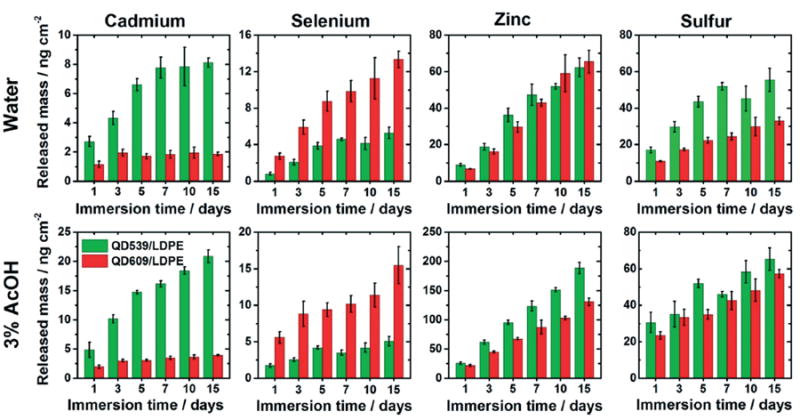
Release of cadmium, selenium, zinc, and sulfur from QD/LDPE nanocomposite films into water (top row) and 3% aqueous acetic acid (bottom row) held at 75 °C as a function of immersion time, expressed as total released mass of the analyte in the 25 mL analyzed solution per unit surface area of exposed film. Green and red bars refer to release experiments using QD539/LDPE and QD609/LDPE, respectively. The inorganic weight fraction for both films was 2.08 mg mL−1. Each reported value is the average of measurements from four independent samples. Error bars represent standard deviations.
Fig. 3.
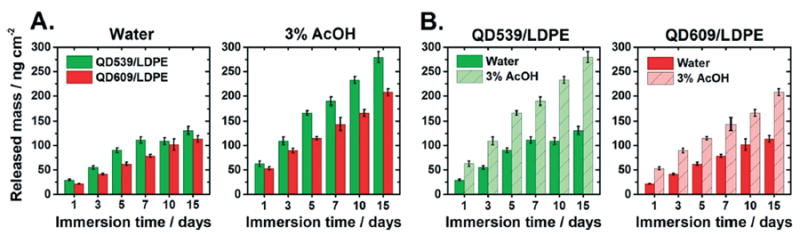
Total released mass from 2.08 mg mL−1 QD/LDPE nanocomposite films into external liquid media held at 75 °C as a function of immersion time, expressed as total released mass of the analyte in the 25 mL analyzed solution per unit surface area of exposed film. (A) and (B) plot the same data in two different ways so as to highlight specific comparisons. The two graphs in (A) compare release of QD539 (green) and QD609 (red) in the two different liquid media. The two graphs in (B) compare the effect of the external medium for the two different particle types. Here, green and red bars refer to the same QD identities as in (A), with lighter colored bars referring to experiments in 3% aqueous acetic acid and darker colored bars representing experiments in water. Each reported value is the sum of the averaged values of cadmium, selenium, zinc, and sulfur measurements from four independent samples. Error bars represent a propagated error expressed as the square root of the sum of the standard deviations for each individual element component.
A few general observations from Fig. 2 and 3 can initially be made. First there is an increase in concentration of each analyte with respect to the immersion time, although the release rate slows as time elapses. This deceleration can be observed by dividing the released elemental masses by the elapsed time, as shown in Fig. S9.† Second, there is generally more released inorganic mass into the acidified matrix compared to release into water, particularly notable in the data plotted in Fig. 3B, suggesting that a portion of the release is driven by dissolution of embedded particles and diffusion of ions (e.g., Cd2+ and Zn2+).This is in agreement with several other studies, which have also observed enhanced dissolution-mediated nanofiller release when PNCs are stored in acidified media.18,20,23,27,36,39 FTIR-ATR spectra of the test materials were unchanged before and after the immersion tests (see the ESI,† Fig. S10B), indicating that acid-mediated oxidative degradation of the polymer likely cannot explain the greater release into acidified media. Third, after immersion in water, the cadmium release appears to level off after a few days, particularly in the case of QD609/LDPE (Fig. 2, top row). This may indicate a process by which QDs initially located at the polymer-environment interface desorb from the film surface. As water is likely not an aggressive enough solvent to fully dissolve through the ZnS QD shell regions and into the cadmium-containing cores of QD embedded within the interior of test films, after this initial desorption process is complete, there is no longer a significant source for transport of cadmium into the external medium; compare this to the analogous data acquired in acetic acid (Fig. 2, bottom row), in which cadmium levels continue to increase throughout the duration of the experiment. This leveling off effect is not clearly observed for shell-containing elements (e.g., zinc), which are perhaps more prone to dissolution in water, a process that would be expected to mask the desorption effect. We note that selenium concentrations also do not appear to level off at long immersion times, even in water. We have some preliminary evidence (not shown) that suggests that dissolved sulfur stimulates selenium release from the core via a substitution process; we are currently running additional experiments to test this hypothesis and will publish this data elsewhere, but these effects underscore the complexity of the particle dissolution and release process.
Additional evidence for particle dissolution prior to release
Although the enhanced release mass observed in acidic media suggests that particle-dissolution contributes significantly to the mass transfer process, we wished to explore this issue further. The most direct way to distinguish between diffusion of whole particles and transport of dissolved ions is to determine whether the QD compositional elements detected by ICP-MS in the liquid medium after prolonged immersion of the test materials derive from whole QDs. Because any material introduced into the high temperature argon plasma will be atomized prior to detection, conventional ICP-MS is not adequate to make this determination. Furthermore, time-resolved or single-particle ICP-MS is likely not sensitive enough to detect nanoparticles as small as the QDs dispersed in our materials. Initial attempts to identify whole QDs in the residual solids obtained after evaporating a droplet of the test media after the immersion experiments using STEM with EDS analysis (see ESI,† Fig. S11–S15) revealed the presence of nano-sized particles, but they did not exhibit sizes, morphological features, or compositions that were consistent with QDs. However, this does not prove that all, or even any, of the released material was in ionic form at the moment it was released.
To obtain more direct evidence that dissolution of embedded particles contributes to the observed release, we turned to the core–shell architecture of QDs. Our hypothesis was that if diffusion of whole QDs drives the mass transfer process, then the mass ratio of QD compositional elements in the liquid medium after release will be close to the mass ratio of QD compositional elements of the QDs inside the test film prior to the immersion experiment. On the contrary, because QDs will dissolve from the outside inward, partial dissolution of QDs will result in a higher amount of shell material (ZnS) than core material (CdSe) in the liquid medium after immersion tests. Therefore, we have used the ratio of Zn to Cd in the external liquid medium as a metric to track (partial) dissolution of QDs prior to release. Note that in the event that whole QDs are released from the test materials, any dissolution or partial dissolution that occurs after release will not impact the ICP-MS analysis, because all QDs in the assayed liquid, no matter the degree of dissolution, will be atomized in the ICP-MS plasma. Therefore if Zn/Cd in the liquid medium after immersion exceeds Zn/Cd of QDs in solution, this is evidence of QD dissolution prior to release.
Fig. 4 plots the mass concentration ratios of Zn to Cd measured in water and 3% (v/v) aqueous acetic acid after immersion of QD/LDPE test films at 75 °C, as a function of immersion time. For comparison, baseline Zn/Cd mass ratios of neat QD539 and QD609 are 2.161 ± 0.041 and 2.187 ± 0.007, respectively (Table 1); these values are depicted in Fig. 4 as a dashed black line. The ICP-MS data show that, at all immersion times, the Zn/Cd mass ratios in the liquid media after immersion are higher than the respective Zn/Cd ratios of the neat QDs and QD-dispersed test materials prior to immersion. (This trend was also observed in materials made from QD546 and QD617, which have a more complex core–shell structure). This is unequivocal evidence that dissolution of embedded QDs plays a major role in the mass transfer process because in such a scenario, ZnS located exclusively in the shell regions of the core–shell QDs would be expected to dissolve before CdSe cores. Note that, especially for the QD609-based samples, the Zn/Cd mass concentration ratio is smallest at the 1 day time point and increases thereafter; this increase in the relative fraction of zinc in the external medium at longer immersion times is consistent with a model of an initial rapid desorption of surface-bound QDs followed by slow dissolution of embedded QDs, ZnS shells first followed by CdSe cores, as the polar liquid media permeates into the interior of the nonpolar host matrix and the dissolution products gradually diffuse back into the low-concentration exterior environment. In other words, permeation of solvent into the interior of the PNCs, dissolution of the embedded QDs, and diffusion of dissolved material back to the liquid medium are all kinetically controlled processes that contribute to the gradual evolution of the Zn/Cd ratios over time.
Fig. 4.
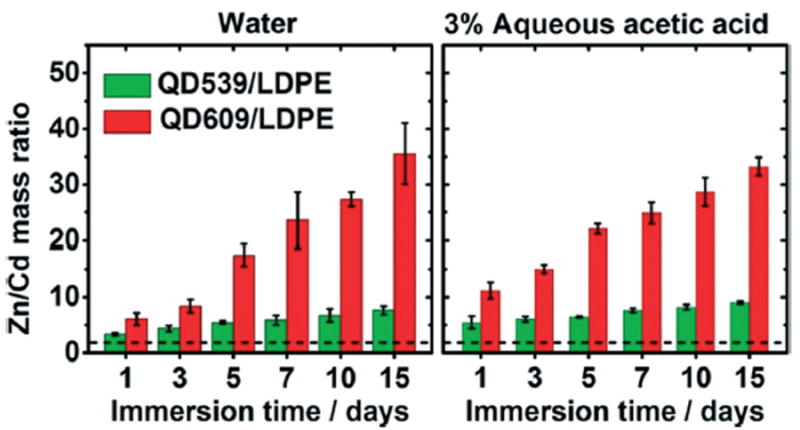
The mass ratio of released zinc to cadmium from QD539/LDPE (green) and QD609/LDPE (red) into water (left panel) and 3% (v/v) aqueous acetic acid (right panel) held at 75 °C, as a function of immersion time. The approximate location of the Zn/Cd mass ratio of neat QDs (2.161 ± 0.041 and 2.187 ± 0.007 for QD539 and QD609, respectively) is shown as a dashed black line. Each reported value is the average of measurements from four independent samples. Error bars represent standard deviations.
We note here that the ratio S/Se could also be used to differentiate dissolution of QD shell and cores regions, but we chose to focus on zinc and cadmium due to the aforementioned sulfur background and the substitution behavior of selenium and sulfur. For example, S/Se ratios were found to generally support the same conclusions that we draw from Fig. 4 – at every time point, the S/Se ratios in the immersion media exceed the S/Se ratios of neat QDs, again implying that elements in the QD shell regions are diffusing into the environmental media faster than those in the QD core regions – but the evolution of the S/Se ratios with immersion time proceeds in a different direction from that of the Zn/Cd ratios depicted in Fig. 4. S/Se ratios are reported in the ESI† for the interested reader. Additional experiments are being conducted to more fully understand the behavior of dissolved selenium and sulfur concentrations; the results of these experiments and their implications for nanoparticle release phenomena will be reported in a separate manuscript.
Contribution of interior and surface-bound particles to release
While a few studies have provided experimental evidence that particle dissolution contributes to the composition of mass transferred from polymer nanocomposites into liquid media,17,18,39 no studies have directly addressed whether the mass transfer of dissolved ions is limited to nanoparticles that are incidentally present at the interface between the polymer and the external environment. If particles buried in the polymer interior are immune to dissolution because of their spatial separation from the dissolving media, then certain strategies (such as coating the PNC material with a barrier polymer layer) could possibly be effective at limiting environmental exposure. On the other hand, such strategies would be less effective if the dissolving medium penetrates sufficiently into the polymer interior to interact with internally-embedded particles. This is a difficult question to answer with conventional nanocomposite test materials and analytical tools.
To some extent, the positive change in Zn/Cd mass ratio as a function of time (vide supra) suggests a release mechanism that includes dissolution of internally bound QDs, as dissolution of QDs incidentally present at the interface between the PNC and the external medium would be expected to be faster than a timescale of days and weeks. However, the fluorescent properties of our model system facilitates a more direct means of probing where the dissolved material observed in the environment after immersion originated. To this end, we used LSCM to observe how the luminescent properties of embedded QDs change during the immersion experiment. We hypothesized that if the external medium (water or dilute acetic acid) is permeating into the polymer interior and dissolving embedded QDs then the QD luminescent properties of those QDs will be attenuated because of the sensitivity of QD luminescence to surface defects. On the other hand, if QDs within the LDPE interior are undisturbed and the mass transfer occurs only from QDs located near or on the polymer surface, then this should manifest as no change in the luminescent properties of QDs located deep within the PNC before and after the immersion tests were performed.
The results of our LSCM analysis of luminescent QD546/LDPE and QD617/LDPE films before and after immersion in water and 3% (v/v) aqueous acetic acid at 75 °C for 17 days are summarized in Fig. 5. The total averaged photo-luminescence (PL) intensities of QDs subjected to the different treatments are quantitated in panels (B) and (C), and panels (D) and (E) break the data down by penetration depth from the upper film surface. Three dimensional LCSM images corresponding to the depth profiling experiments are provided in the ESI,† Fig. S16. The first observation that can be made from these experiments, particularly the quantitative data plotted in Fig. 5B and C, is that the immersion treatment reduces the overall average PL intensity of embedded QDs, and the amount of luminescence quenching is more pronounced when the QD/LDPE films are submerged in 3% (v/v) acetic acid than when they are submerged in water. This finding is consistent with the ICP-MS results presented above, which show a higher level of released mass when the PNCs were exposed to dilute acid compared to water or no treatment at all, and reinforces the conclusion that exposure of the test films to dilute acid accelerates dissolution of embedded QDs.
Fig. 5.
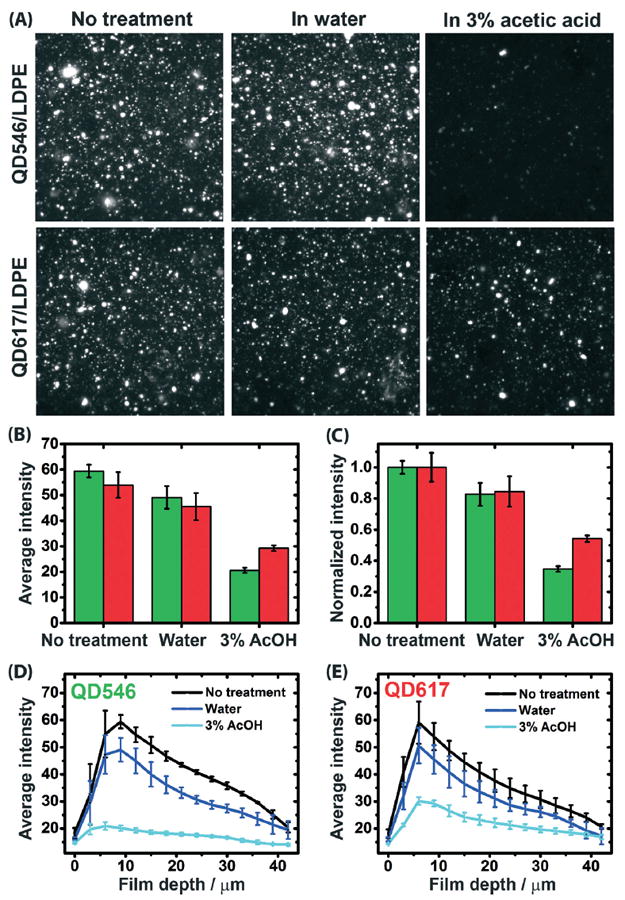
Panel (A) shows comparative LSCM images of QD546/LDPE (top row) and QD617/LDPE (bottom row) after no immersion treatment (left column) and after immersion in water (middle column) or 3% aqueous acetic acid (right column) at 75 °C for 17 days. The images correspond to a focal plane depth of approximately 9 μm below the film surface and have lateral dimensions of 56 μm × 56 μm. Panels (B) and (C), respectively, plot average and normalized (to the “no treatment” condition) photoluminescence intensity for experiments depicted in the LSCM images. Panels (D) and (E) plot the average photoluminescence intensity values for the various treatment types, broken down by focal plane depth, for QD546/LDPE and QD617/LDPE materials, respectively. The neat LDPE control (not shown) exhibits no luminescent intensity either before or after immersion treatments. The error bars represent standard deviations, based on n = 3. Note that the drop off in PL intensity at progressively lower penetration depths is due to the less efficient light capture efficiency of the microscope, so only comparisons between treatment types at specific penetration depths are meaningful. The excitation wavelength was 405 nm in all experiments.
A more notable finding is that the LSCM images shown in Fig. 5A were captured at 9 μm below the film surface and reveal reduced PL intensity from QDs embedded at this depth after immersion in a liquid medium under the specified test conditions. Moreover, Fig. 5D and E show that the PL intensities of QDs embedded at all probed depths (between 0 μm and 42 μm below one film surface) are significantly attenuated by the immersion treatment, particularly when the immersion medium is dilute acetic acid (note that the initial rise and subsequent decline in PL intensity as a function of penetration depth is an experimental artifact arising from the efficiency of light penetration into and out of the test film; therefore only the comparative values for the three test conditions at any given penetration depth have any absolute meaning). This is direct evidence that nanoparticles embedded deep within the PNC contribute to the amount of material released into the liquid medium. Importantly, the difference in luminescence intensity deep within the samples exposed to water and to dilute acid shows that the luminescence quenching is not simply related to the storage of these materials for prolonged periods at high temperature. If this was the case, then the nature of the external medium should make no difference, which is not what was observed.
To observe a measurable difference between the PL quenching of deeply embedded QDs as a function of the immersion medium, the immersion medium must be able to permeate into or otherwise interact with the hydrophobic LDPE interior to mediate QD dissolution. Previous work has shown that the rate of water diffusion through LDPE is very fast, even though the solubility of water in LDPE is low.45 Higher degrees of polymer oxidation have been found to reduce the water diffusivity and increase water solubility due to the water-trapping ability of carbonyl and hydroxide groups that form along the polymer backbones during polymer oxidation.45 FTIR-ATR spectra of our QD/LDPE films show evidence of a very small amount of polymer oxidation (Fig. S10†), at least on the film surface, but even assuming a high 1% oxygen content of the polymer backbone, the diffusion constant of water in LDPE is still estimated to be in the range of 1.4 × 10−6 cm2 s−1,45 which means that the timescale for water to diffuse from the film surface to 40 μm deep (the permeation lag time) when submerged at 75 °C is less than 2 seconds (see the ESI† for a brief description of this calculation). The presence of QDs or other additives in the commercial LDPE resin may attenuate these water diffusivity values to some degree, but nevertheless it is likely that permeation of the external liquid medium throughout the entire PNC film volume is accomplished on much faster timescales than the release of dissolution products observed during the QD/LDPE immersion experiments, which occurs over a period of days. Likewise, lag times for ion diffusion through LDPE have also been reported to be on the order of seconds.46 Therefore, we speculate that the QD release kinetics measured in the ICP-MS experiments are limited by slow nanoparticle dissolution rates caused by low local concentrations of dissolving media (water or dilute acetic acid) present in the hydrophobic LDPE interior.
The influence of particle size
QD539 and QD609 were incorporated within LDPE at close to the same inorganic weight fractions to facilitate direct comparisons between the size of embedded particles and the resulting release characteristics. Such comparisons are made possible in our unique model system because of the fine control over structural properties and elemental composition achievable for commercially available QDs. Fig. 3A directly compares the total mass released (experimental sum total of cadmium, selenium, zinc, and sulfur) from QD539/LDPE and QD609/LDPE after storage at 75 °C in water and 3% aqueous acetic acid. The mean diameters of QD539 and QD609, respectively, are 4.3 nm and 5.5 nm, excluding any contribution from the bound oleylamine surfactants. Fig. 3A data show a clear inverse relationship between the size of the embedded particle and the amount of released mass: QD/LDPE PNCs made with smaller QD539 particles release significantly more mass than those embedded with larger QD609 particles. This effect is particularly evident in acidic media.
As an independent test to confirm that PNCs incorporating smaller particles release more mass to the environment than those incorporating larger particles, we conducted similar release experiments using PNCs based on highly luminescent QD546 and QD617, the QDs used for the LSCM experiments. Release data for these PNCs for select immersion times, normalized for the inorganic mass concentration within the films, are plotted in Fig. S18 of the ESI.† Here we also observed far greater mass transfer into both water and dilute acetic acid from LDPE films incorporating relatively small QD546 particles (diameter = 5.6 nm ± 0.8 nm) than from those incorporating much larger QD617 particles (diameter = 8.5 ± 0.8 nm). Importantly, the physical dimensions of QD546 and QD617 particles were accurately determined by electron microscopy methods, removing any ambiguity about the inverse correlation between particle size and release rate that may be caused by the optical-based method used to determine the sizes of QD539 and QD609. We also note that the difference in magnitude of mass transferred to the environment between QD546 and QD617 (Fig. S18†) is larger than that of the pair QD539 and QD609 (Fig. 3A), consistent with the fact that the difference in mean diameter between QD546 and QD617 is 2.9 nm and the difference in diameter between QD539 and QD609 is only 1.2 nm. For example, at seven days immersion time in dilute acetic acid, the ratio of total released mass (normalized for loading concentration) for QD546 compared to Q617 is 2.11 ± 0.08; for QD539 and QD609, which are closer together in size, this ratio is only 1.31 ± 0.13. It’s also worth pointing out that the relationship between size and mass transfer observed in QD546 and QD617 is consistent with the LSCM experiments (Fig. 5B and C), which show a significantly greater luminescence quenching of polymer-embedded QD546 after exposure of the PNCs to dilute acetic acid compared to that observed in polymer-embedded QD617.
Given that particle dissolution likely plays a dominant role in the mass transfer process, it is reasonable to conclude that the greater extent of mass release from QD539/LDPE versus QD609/LDPE, especially in the relatively aggressive low pH environment, derives from the ≈27.5% greater specific surface area (SSA, ratio of surface area to volume) of the former (1.40 nm−1 versus 1.09 nm−1, based on mean particle radii and assuming spherical shape). This argument also explains the greater mass release from LDPE films containing QD546 (1.15 nm−1) compared to LDPE containing the larger-sized compositional analog QD617 (0.71 nm−1). For most time points, the greater amount of mass release from QD546/LDE versus QD617/LDPE and the greater amount of mass release from QD539/LDPE versus QD609/LDPE exceeds what might be expected from surface area alone (based on comparing the ratio of total released mass to the pristine-particle SSA ratio for each QD pair). We note that the SSA will grow exponentially as the dissolution of embedded particles progresses, and this growth in available relative surface area for subsequent QD dissolution would be more pronounced for initially smaller particles. However, dissolution kinetics and hence total mass released as a function of immersion time may also be complicated by the relative stability of the differing compositional elements in these QDs (e.g., per unit surface area, ZnS shells may dissolve faster than CdSe cores), as well as the growing concentration of dissolved material. Such complications are evident, for example, in Fig. 4, which show considerably higher Zn/Cd ratios in QD609/LDPE than QD539/LDPE, possibly signifying that in the smaller QD539-based materials, the higher SSA results in faster dissolution of ZnS shells and quicker exposure (and partial dissolution) of CdSe core regions. On the other hand, QD539/LDPE cores are more surface-enriched with cadmium (Table 1), which may also complicate direct comparison of the dissolution kinetics for individual elemental components for different QDs.
Although it is clear that particle size plays an important role in the release kinetics, it difficult to generalize the relative importance of particle size compared to other factors that also influence release. Fig. 3 data for QD539/LDPE and QD609/LDPE suggest that the environmental medium impacts the total mass release to a moderately larger degree than the particle size does. However this is contrasted with the release data for QD546 and QD617 (Fig. S18†), in which it is the particle size that seems to play the more important role. This may be due to the larger difference in size between QD546 and QD617, but it is also possible that the different synthetic routes (and hence compositions) of these particles also make a difference. Additionally, a factor not considered in this paper, but which could be potentially more important than either particle size or environmental medium, is the host matrix; materials less permeable than LDPE (such as acrylic glass or other low permeability transparent polymers, which could be used to incorporate QDs for commercial display technologies) may greatly attenuate dissolution and dissolved ion transport kinetics. It may be tempting to conclude that higher barrier polymers should exhibit lower rates of embedded particle dissolution, but as the dissolution process is likely a complex function of the embedded particle composition and size, solubility of the environmental medium in the host matrix, and polymer dielectric properties and permeability, it is not possible to confidently extrapolate our results to other host materials. This certainly deserves further exploration.
In the end, the multi-element nature of QDs and complexity of the release process (including the possibility that some whole QDs desorb or diffuse into the external medium) may preclude a full ability to quantitatively predict the relative proportions of material released from PNCs as a function of embedded particle diameter alone, particularly when embedded nanoparticles possess complex internal architectures. Moreover, the presence of particle aggregates in the polymer matrix and the fact that we only have two particle sizes to directly compare for compositionally similar QDs limits our observation of a relationship between particle size and release rate to a mostly qualitative one at this point. Nevertheless, our data provide some of the first clear evidence that small differences in mean particle size (on the order of only a few nm) can impact the release behavior of nanoparticle-enabled polymer materials, particularly in acidified media. We are currently exploring a wider range of nanoparticle sizes using particles with less complex elemental composition to better quantify these important relationships.
Conclusions
We have successfully fabricated nanocomposites incorporating semiconductor nanocrystals (QDs) into free-standing LDPE materials, which have been developed as models to assess potential exposure to nanoparticles from commercial nanocomposite plastics. This model system affords finer control over the properties of the embedded particles than those employed in many previous exposure studies, which allows us to investigate important structure-impact relationships. Using immersion-type release experiments, our results show that reducing the size of embedded particles increases the amount of material released during long term storage of these materials in aqueous media. Moreover, we have shown that dissolution contributes significantly to the release process, because the compositional ratio of zinc from the QD shells and cadmium from the QD cores is not the same in the external medium as it is in the neat QDs, as it must be if whole QDs were being released. Finally, we have shown for the first time that nanofiller dissolution is not limited to those nanoparticles incidentally bound at the polymer interface. We note that our results are consistent with an earlier effort by Liu et al.36 that utilized QD/acrylate polymer materials as a case study to assess the potential environmental exposure to nanoparticles from nanocomposite-based consumer products; however this prior work provides a different view of release mechanisms that our study offers, because it only investigated a single QD size and also focused predominantly on release of cadmium.
It is important, before closing, to put our results into perspective and to note a few limitations of this study. First, the total amount of released material we have observed is low, especially considering the high temperatures that we have utilized. The concentrations of released mass are typically in the parts-per-billion level. Under the most extreme conditions tested (15 day exposure, acetic acid) the released mass comprises on the order of 10% (depending on QD type) of the initially dispersed QD mass, although a majority of this released mass (approximately 64% to 67%) is zinc, probably zinc ions. Second, the immersion times and temperatures we have used are more extreme than any realistic use scenario for a consumer product, which means that actual release from a nanotechnology-enabled product (e.g., at lower sustained temperatures) is likely to be less than what we report here. The exposure conditions tested were selected to accelerate the release process and allow us to explore release mechanisms. Such information could be critical in the event that consumer products utilizing PNCs find their way to market, as the form of released material may be a key factor in determining the ultimate effects of exposure. Third, while our data show that the released material is likely to be largely in the form of dissolution products of embedded or surface-desorbed nanoparticles, our study cannot rule out the presence of some quantity of whole or partially undissolved nano-particles released either through desorption or diffusion. However, a number of theoretical studies and some experimental work has concluded that diffusion of nanoparticles through organic polymers is too slow to be significant on commercially-relevant timescales, with diffusion constants calculated for 4 nm diameter particles in LDPE to be on the order of 1.6 × 10−20 cm2 s−1 at 40 °C.47,48 Therefore, given the extensive body of knowledge about diffusion physics in polymers, along with our data here, we feel confident in concluding that particle dissolution likely plays the dominant role in the release of QDs from our QD/LDPE model PNCs. Ultrafiltration of the exposure medium may provide additional valuable experimental information on the fraction of released material, if any, that remains particulate at the time of analysis, and we plan to investigate this possibility in future work. We note that in the event that dissolution is indeed the predominant mechanism for release, downstream toxicological assessments of released materials may be able to be made based on conventional or ionic materials rather than nanoparticles.
As a final remark, while our CdSe/ZnS model system provides generally relevant information about nanofiller exposure scenarios, it is possible that PNCs embedded with other nanofillers or manufactured by other means may exhibit different release mechanisms or dissolution/diffusion kinetics. Some particle types (e.g., carbonaceous particles) may not dissolve at all, and for those that do dissolve, even if we restrict the sphere of concern to purely ionic dissolution products, it may be difficult to predict the relative quantity of released products from PNCs incorporating nanoparticles with different compositions. In particular, dissolution and mass transport kinetics of particles with complex (multi-element) compositions may be sensitive to the relative proportion of elemental components present. For example, our data suggests that the relative dissolution kinetics of cadmium and selenide in CdSe/ZnS nanoparticles differ substantially from these elements in bulk CdSe, an aspect that we are exploring further. Also, it is unknown how the particle dispersion state (e.g., degree of aggregation in the polymer) may influence the observe release rate: for instance, nanocomposites that exhibit a smaller degree of particle aggregation may give rise to enhanced rates of release because such individually dispersed particles would have more surface area available for dissolution. In this way, nanocomposite fabrication methods or particle surface chemistry may influence the dispersion characteristics and therefore change the release behavior in unexpected ways. Such issues are endemic to nanoparticles due to their small size and complex chemistry. While we recognize that the model system we have employed here cannot account for all of these factors, we have demonstrated the value of continued investment in bottom-up strategies based on controlled model systems to study these challenging but important issues.
Supplementary Material
Nano impact.
There is growing interest in determining whether consumers can be exposed to nanoparticles during the lifecycles of polymer nanocomposites (PNCs). This study employs a model PNC system based on luminescent semiconductor nanocrystals (quantum dots) dispersed in low density polyethylene, and exposes these materials to liquid media under conditions intended to accelerate the release kinetics. The key findings are that the release of nanofillers is particle-size dependent, and that dissolution of embedded particles and diffusion of dissolution products (ions) into the surrounding environment likely dominates the mass transfer. In addition, mass transfer is not restricted to particles incidentally located at the polymer-environment interface. The pilot study shows that model systems designed to systematically study specific structure–function relationships may be used to gain new insight into the environmental release of nanocomposites.
Acknowledgments
The authors gratefully acknowledge Glenn J. Bastiaans, Ph.D., President of NanoOptical Materials, for helpful discussions related to QD performance and composition. The authors also thank FDA/CFSAN for financial support of this work. This work made use of the EPIC facility (NUANCE Center-Northwestern University), which has received support from the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); and the State of Illinois, through the IIN.
Footnotes
Electronic supplementary information (ESI) available: A full account of experimental methods, information on thermal characteristics of QD/LDPE PNC materials, tabulated release data, tabulated characterization data for all QDs and QD/LDPE materials, and supplemental figures showing: STEM/EDS analysis of post-immersion residuals; absorption, photoluminescence and FTIR spectra of QD/LDPE materials; time-averaged released data; and additional LSCM data. See DOI: 10.1039/c6en00064a
Disclaimer
This article has been reviewed in accordance with the FDA’s and NIST’s peer and administrative review policies and approved for publication. The statements made in this report do not necessarily represent the official position of any of the employers or affiliated organizations of the experts. Certain commercial equipment, instruments, or materials are identified in this article to foster understanding. Such identification does not imply recommendation or endorsement by FDA or NIST, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
Conflict of interest
The authors declare no competing financial interest.
References
- 1.Feldman D. Polymer Nanocomposites in Building, Construction. J Macromol Sci, Part A: Pure Appl Chem. 2014;51:203–209. [Google Scholar]
- 2.Lee J, Mahendra S, Alvarez PJJ. Nanomaterials in the Construction Industry: A Review of Their Applications and Environmental Health and Safety Considerations. ACS Nano. 2010;4:3580–3590. doi: 10.1021/nn100866w. [DOI] [PubMed] [Google Scholar]
- 3.Peyvandi A, Soroushian P, Balachandra AM, Sobolev K. Enhancement of the Durability Characteristics of Concrete Nanocomposite Pipes with Modified Graphite Nanoplatelets. Constr Build Mater. 2013;47:111–117. [Google Scholar]
- 4.Parameshwaran R, Kalaiselvam S. Energy Efficient Hybrid Nanocomposite-Based Cool Thermal Storage Air Conditioning System for Sustainable Buildings. Energy. 2013;59:194–214. [Google Scholar]
- 5.Duncan TV. Applications of Nanotechnology in Food Packaging and Food Safety: Barrier Materials, Antimicrobials and Sensors. J Colloid Interface Sci. 2011;363:1–24. doi: 10.1016/j.jcis.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhim J-W, Park H-M, Ha C-S. Bio-Nanocomposites for Food Packaging Applications. Prog Polym Sci. 2013;38:1629–1652. [Google Scholar]
- 7.Chen C, Tang Y, Ye YS, Xue Z, Xue Y, Xie X, Mai YW. High-Performance Epoxy/Silica Coated Silver Nanowire Composites as Underfill Material for Electronic Packaging. Combust Sci Technol. 2014;105:80–85. [Google Scholar]
- 8.Xu S, Habib AH, Pickel AE, McHenry ME. Magnetic Nanoparticle-Based Solder Composites for Electronic Packaging Applications. Prog Mater Sci. 2015;67:95–160. [Google Scholar]
- 9.Hule RA, Pochan DJ. Polymer Nanocomposites for Biomedical Applications. MRS Bull. 2007;32:354–358. [Google Scholar]
- 10.Armentano I, Dottori M, Fortunati E, Mattioli S, Kenny JM. Biodegradable Polymer Matrix Nanocomposites for Tissue Engineering: A Review. Polym Degrad Stab. 2010;95:2126–2146. [Google Scholar]
- 11.Rizzello L, Cingolani R, Pompa PP. Nanotechnology Tools for Antibacterial Materials. Nanomedicine. 2013;8:807–821. doi: 10.2217/nnm.13.63. [DOI] [PubMed] [Google Scholar]
- 12.Koo JH, Natali M, Tate J, Allcorn E. Polymer Nanocomposites as Ablative Materials - a Comprehensive Review Int. Int J Energ Mater Chem Propul. 2013;12:119–162. [Google Scholar]
- 13.Macke A, Schultz BF, Rohatgi P. Metal Matrix Composites Offer Automotive Industry Opportunity to Reduce Vehicle Weight, Improve Performance. Adv Mater Processes. 2012;170:19–23. [Google Scholar]
- 14.Baur J, Silverman E. Challenges and Opportunities in Multifunctional Nanocomposite Structures for Aerospace Applications. MRS Bull. 2007;32:328–334. [Google Scholar]
- 15.Cushen M, Kerry J, Morris M, Cruz-Romero M, Cummins E. Migration and Exposure Assessment of Silver from a Pvc Nanocomposite. Food Chem. 2013;139:389–397. doi: 10.1016/j.foodchem.2013.01.045. [DOI] [PubMed] [Google Scholar]
- 16.Cushen M, Kerry J, Morris M, Cruz-Romero M, Cummins E. Evaluation and Simulation of Silver and Copper Nanoparticle Migration from Polyethylene Nanocomposites to Food and an Associated Exposure Assessment. J Agric Food Chem. 2014;62:1403–1411. doi: 10.1021/jf404038y. [DOI] [PubMed] [Google Scholar]
- 17.Cushen M, Kerry J, Morris M, Cruz-Romero M, Cummins E. Silver Migration from Nanosilver and a Commercially Available Zeolite Filler Polyethylene Composites to Food Simulants. Food Addit Contam, Part A. 2014;31:1132–1140. doi: 10.1080/19440049.2014.905874. [DOI] [PubMed] [Google Scholar]
- 18.von Goetz N, Fabricius L, Glaus R, Weitbrecht V, Günther D, Hungerbüler K. Migration of Silver from Commercial Plastic Food Containers and Implications for Consumer Exposure Assessment. Food Addit Contam, Part A. 2013;30:612–620. doi: 10.1080/19440049.2012.762693. [DOI] [PubMed] [Google Scholar]
- 19.Echegoyen Y, Nerín C. Nanoparticle Release from Nano-Silver Antimicrobial Food Containers. Food Chem Toxicol. 2013;62:16–22. doi: 10.1016/j.fct.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 20.Song H, Li B, Lin Q-B, Wu H-J, Chen Y. Migration of Silver from Nanosilver-Polyethylene Composite Packaging into Food Simulants. Food Addit Contam, Part A. 2011;28:1758–1762. doi: 10.1080/19440049.2011.603705. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y, Chen S, Bing X, Gao C, Wang T, Yuan B. Nanosilver Migrated into Food-Simulating Solutions from Commercially Available Food Fresh Containers. Packag Technol Sci. 2011;24:291–297. [Google Scholar]
- 22.Artiaga G, Ramos K, Ramos L, Cámara, Gómez-Gómez M. Migration and Characterisation of Nanosilver from Food Containers by AF4-ICP-MS. Food Chem. 2015;166:76–85. doi: 10.1016/j.foodchem.2014.05.139. [DOI] [PubMed] [Google Scholar]
- 23.Mauricio-Iglesias M, Peyron S, Guillard V, Gontard N. Wheat Gluten Nanocomposite Films as Food-Contact Materials: Migration Tests and Impact of a Novel Food Stabilization Technology (High Pressure) J Appl Polym Sci. 2010;116:2526–2535. [Google Scholar]
- 24.Farhoodi M, Mousavi SM, Sotudeh-Gharebagh R, Emam-Djomeh Z, Oromiehie A. Migration of Aluminum and Silicon from PET/Clay Nanocomposite Bottles into Acidic Food Simulant. Packag Technol Sci. 2014;27:161–168. [Google Scholar]
- 25.Schmidt B, Petersen JH, Koch CB, Plackett D, Johansen NR, Katiyar V, Larsen EH. Combining Asymmetrical Flow Field-Flow Fractionation with Light-Scattering and Inductively Coupled Plasma Mass Spectrometric Detection for Characterization of Nanoclay Used in Biopolymer Nanocomposites. Food Addit Contam, Part A. 2009;26:1619–1627. doi: 10.1080/02652030903225740. [DOI] [PubMed] [Google Scholar]
- 26.Xia Y, Rubino M, Auras R. Release of Nanoclay and Surfactant from Polymer-Clay Nanocomposites into a Food Simulant. Environ Sci Technol. 2014;48:13617–13624. doi: 10.1021/es502622c. [DOI] [PubMed] [Google Scholar]
- 27.Ntim SA, Thomas TA, Begley TH, Noonan GO. Characterisation and Potential Migration of Silver Nanoparticles from Commercially Available Polymeric Food Contact Materials. Food Addit Contam, Part A. 2015;32:1003–1011. doi: 10.1080/19440049.2015.1029994. [DOI] [PubMed] [Google Scholar]
- 28.Lorenz C, Windler L, von Goetz N, Lehmann RP, Schuppler M, Hungerbühler K, Heuberger M, Nowack B. Characterization of Silver Release from Commercially Available Functional (Nano)Textiles. Chemosphere. 2012;89:817–824. doi: 10.1016/j.chemosphere.2012.04.063. [DOI] [PubMed] [Google Scholar]
- 29.Windler L, Lorenz C, von Goetz N, Hungerbuhler K, Amberg M, Heuberger M, Nowack B. Release of Titanium Dioxide from Textiles During Washing. Environ Sci Technol. 2012;46:8181–8188. doi: 10.1021/es301633b. [DOI] [PubMed] [Google Scholar]
- 30.Benn TM, Westerhoff P. Nanoparticle Silver Released into Water from Commercially Available Sock Fabrics. Environ Sci Technol. 2008;42:4133–4139. doi: 10.1021/es7032718. [DOI] [PubMed] [Google Scholar]
- 31.Benn T, Cavanagh B, Hristovski K, Posner JD, Westerhoff P. The Release of Nanosilver from Consumer Products Used in the Home. J Environ Qual. 2010;39:1875–1882. doi: 10.2134/jeq2009.0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaegi R, Ulrich A, Sinnet B, Vonbank R, Wichser A, Zuleeg S, Simmler H, Brunner S, Vonmont H, Burkhardt M, Boller M. Synthetic TiO(2) Nanoparticle Emission from Exterior Facades into the Aquatic Environment. Environ Pollut. 2008;156:233–239. doi: 10.1016/j.envpol.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Kaegi R, Sinnet B, Zuleeg S, Hagendorfer H, Mueller E, Vonbank R, Boller M, Burkhardt M. Release of Silver Nanoparticles from Outdoor Facades. Environ Pollut. 2010;158:2900–2905. doi: 10.1016/j.envpol.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Xia X, Tang Y, Xie C, Wang Y, Cai S, Zhu C. An Approach to Give Prospective Life-Span of the Copper/Low-Density-Polyethylene Nanocomposite Intrauterine Device. J Mater Sci: Mater Med. 2011;22:1773–1781. doi: 10.1007/s10856-011-4347-y. [DOI] [PubMed] [Google Scholar]
- 35.Quadros ME, Pierson R, Tulve NS, Willis R, Rogers K, Thomas TA, Marr LC. Release of Silver from Nanotechnology-Based Consumer Products for Children. Environ Sci Technol. 2013;47:8894–8901. doi: 10.1021/es4015844. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Katahara J, Li G, Coe-Sullivan S, Hurt RH. Degradation Products from Consumer Nanocomposites: A Case Study on Quantum Dot Lighting. Environ Sci Technol. 2012;46:3220–3227. doi: 10.1021/es204430f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J, Sonshine DA, Shervani S, Hurt RH. Controlled Release of Biologically Active Silver from Nanosilver Surfaces. ACS Nano. 2010;4:6903–6913. doi: 10.1021/nn102272n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diaz CA, Xia Y, Rubino M, Auras R, Jayaraman K, Hotchkiss J. Fluorescent Labeling and Tracking of Nanoclay. Nanoscale. 2013;5:164–168. doi: 10.1039/c2nr32978f. [DOI] [PubMed] [Google Scholar]
- 39.Echegoyen Y, Rodríguez S, Nerín C. Nanoclay Migration from Food Packaging Materials. Food Addit Contam, Part A. 2016;33:530–539. doi: 10.1080/19440049.2015.1136844. [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, Xie C, Xia X, Cai S. Zn2+ Release Behavior and Surface Characteristics of Zn/LDPE Nanocomposites and ZnO/LDPE Nanocomposites in Simulated Uterine Solution. J Mater Sci: Mater Med. 2008;19:3319–3326. doi: 10.1007/s10856-008-3467-5. [DOI] [PubMed] [Google Scholar]
- 41.Longano D, Ditaranto N, Cioffi N, Di Niso F, Sibillano T, Ancona A, Conte A, Del Nobile MA, Sabbatini L, Torsi L. Analytical Characterization of Laser-Generated Copper Nanoparticles for Antibacterial Composite Food Packaging. Anal Bioanal Chem. 2012;403:1179–1186. doi: 10.1007/s00216-011-5689-5. [DOI] [PubMed] [Google Scholar]
- 42. [04/27/2106];FDA Guidance for Industry: Preparation of Premarket Submissions for Food Contact Substances: Chemistry Recommendations. http://www.fda.gov/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/IngredientsAdditivesGRASPackaging/ucm081818.htm.
- 43.Morris-Cohen AJ, Frederick MT, Lilly GD, McArthur EA, Weiss EA. Organic Surfactant-Controlled Composition of the Surfaces of Cdse Quantum Dots. J Phys Chem Lett. 2010;1:1078–1081. [Google Scholar]
- 44.Duncan TV, Pillai K. Release of Engineered Nanoparticles from Polymer Nanocomposite Materials: Diffusion, Dissolution, and Desorption. ACS Appl Mater Interfaces. 2015;7:2–19. doi: 10.1021/am5062745. [DOI] [PubMed] [Google Scholar]
- 45.McCall DW, Douglass DC, Blyler LL, Johnson GE, Jelinski LW, Bair HE. Solubility and Diffusion of Water in Low-Density Polyethylene. Macromolecules. 1984;17:1644–1649. [Google Scholar]
- 46.Oguzie EE, Onuchukwu AI, Ekpe UJ. Ionic Permeability of Polymeric Membranes: Part 1-Steady State Transport of Binary Electrolytes through Polyethylene Films. J Appl Electrochem. 2007;37:1047–1053. [Google Scholar]
- 47.Bott J, Störmer A, Franz R. A Model Study into the Migration Potential of Nanoparticles from Plastics Nanocomposites for Food Contact. Food Packaging and Shelf Life. 2014;2:73–80. [Google Scholar]
- 48.Šimon P, Chaudhry Q, Bakoš D. Migration of Engineered Nanoparticles from Polymer Packaging to Food -a Physicochemical View. J Food Nutr Res. 2008;47:105–113. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.