

Abstract
Background
Recombinant protein production in the methylotrophic yeast Pichia pastoris largely relies on integrative vectors. Although the stability of integrated expression cassettes is well appreciated for most applications, the availability of reliable episomal vectors for this host would represent a useful tool to expedite cloning and high-throughput screening, ameliorating also the relatively high clonal variability reported in transformants from integrative vectors caused by off-target integration in the P. pastoris genome. Recently, heterologous and endogenous autonomously replicating sequences (ARS) were identified in P. pastoris by genome mining, opening the possibility of expanding the available toolbox to include efficient episomal plasmids. The aim of this technical report is to validate a 452-bp sequence (“panARS”) in context of P. pastoris expression vectors, and to compare their performance to classical integrative plasmids. Moreover, we aimed to test if such episomal vectors would be suitable to sustain in vivo recombination, using fragments for transformation, directly in P. pastoris cells.
Results
A panARS-based episomal vector was evaluated using blue fluorescent protein (BFP) as a reporter gene. Normalized fluorescence from colonies carrying panARS-BFP outperformed the level of signal obtained from integrative controls by several-fold, whereas endogenous sequences, identified from the P. pastoris genome, were not as efficient in terms of protein production. At the single cell level, panARS-BFP clones showed lower interclonal variability but higher intraclonal variation compared to their integrative counterparts, supporting the idea that heterologous protein production could benefit from episomal plasmids. Finally, efficiency of 2-fragment and 3-fragment in vivo recombination was tested using varying lengths of overlapping regions and molar ratios between fragments. Upon optimization, minimal background was obtained for in vivo assembled vectors, suggesting this could be a quick and efficient method to generate of episomal plasmids of interest.
Conclusions
An expression vector based on the panARS sequence was shown to outperform its integrative counterparts in terms of protein productivity and interclonal variability, facilitating recombinant protein expression and screening. Using optimized fragment lengths and ratios, it was possible to perform reliable in vivo recombination of fragments in P. pastoris. Taken together, these results support the applicability of panARS episomal vectors for synthetic biology approaches.
Keywords: P. pastoris, panARS, BFP, Episomal plasmid, Interclonal variability, Digital droplet PCR, In vivo recombination, Synthetic biology
Background
The methylotrophic yeast Pichia pastoris is widely considered an industrial workhorse for recombinant protein production (RPP, [1–3]); insights into P. pastoris genomic arrangement [4, 5] and metabolism [6, 7] are starting to accumulate in the literature, as a testament to the interest in this host. Despite an increasing repertoire of molecular tools to enable efficient protein production, including newly identified natural promoters [8], engineered sequences [9, 10], and secretion signals [11], the vast majority of vectors available for RPP are based on genomic integration of expression cassettes in the P. pastoris genome. Although in general stable clones derived from genomic integration are preferred for RPP, disadvantages of their use are related to relatively low efficiency of transformation (recently at least mitigated by technical developments [12]), to a certain degree of heterogeneity in protein production due to non-specific integration and to genetic instability of multi-copy integration in presence of stress conditions [13]. The classical solution to this problem—episomal expression vectors—is deemed to alleviate only the chromosomal instability of multi-copy integrative clones, since other reasons of heterogeneity, recently addressed analysing micro engraved P. pastoris secretive clones, are hypothesized to be related to stochastic post-translational events, especially relevant in secreted protein expression [14, 15]. Nonetheless, episomal expression systems present advantages such as simpler protocols and higher efficiencies of transformation; however, such tools are unavailable in most non-canonical protein production hosts, often due to lack of efficient replication origins that promote in vivo plasmid replication and maintenance. Recent high-throughput work has identified novel autonomously replicating sequences (ARSs) in different organisms, to bring to light novel regions conferring self-replicating properties and understand their features [16–18]. These elements may help expedite RPP efforts through addition of stable expression plasmids to the available molecular toolkit; moreover, another intriguing possibility is represented by the possibility of self-assembly recombinant DNA fragments in its nucleus, eliminating the cloning process for plasmid assembly [19]; such strategy is theoretically facilitated by self-replication of the assembled fragment in the recipient cells, and has been successfully applied to Saccharomyces cerevisiae [20, 21]. In vivo recombination in P. pastoris was first observed when a library of Rhizopus chinensis lipase mutants was assembled directly by the host and generated a linear expression cassette integrated at the targeted genomic locus. Overlapping ends as short as 50 nucleotides were reported to be sufficient to promote assembly at a relatively high efficiency [22].
In this technical report, we aimed to functionally characterize the use of panARS, a 452-nt element originally isolated from Kluyveromyces lactis and synthetically optimized, as well as two endogenous sequences derived from P. pastoris, using blue fluorescent protein (BFP) as a reporter protein. Moreover, we evaluated in vivo recombination of a panARS-based vector, to establish its technical feasibility for efficient gene assembly in P. pastoris.
Results and discussion
Evaluation of ARSs in Pichia pastoris GS115
Recently, endogenous autonomously replicating sequences from P. pastoris have been identified and described [23]. In order to evaluate the general performance of two sequences in comparison with a wide-range ARS (panARS, [16]), sequences A76 and C937, previously described to possess respectively strong and moderately weak self-replicating activity, were tested in the same genetic context as panARS. To do so, blue fluorescent protein (BFP)-expressing plasmids, containing AOX1 promoter, zeocin resistance cassette and each of the three different ARS sequences, were constructed starting from the commercial vector pSEC-SUMO (see ‘‘Methods’’ section). BFP was selected as reporter gene, due to its fast maturation, high photostability and pH-stability [24–26].
Transforming P. pastoris GS115 with a circular plasmid carrying either A76, C937 or panARS sequences, a substantial number of colonies was obtained. At least 16 independent colonies carrying A76-, C937- or panARS-based BFP-expressing plasmids were tested for fluorescence emission at 452 nm after 48 h of cultivation using sorbitol and methanol as carbon sources (Fig. 1a). Co-feeding with sorbitol and methanol allowed shortened cultivation time, as sorbitol is in general more easily assimilated than methanol, without repressing the AOX1 promoter [27]. Fluorescence emission was then normalized on OD values as an estimation of biomass. Interestingly, the BFP expression level for A76 or C937-carrying strains was significantly lower than either panARS-based or integrative vector strains (Fig. 1a, b). As shown in Fig. 1a, three independent colonies of P. pastoris GS115, carrying an integrative version of the same plasmid (pSEC) and selected from the middle of the expression landscape (data not shown), were used as positive controls and as benchmarks for episomal vectors evaluation (Fig. 1a). Such average clones, typically carrying a single copy of expression cassette genomically integrated, were outperformed by panARS-based clones, while showing higher and more consistent BFP expression level than either A76- or C937-based vectors (Fig. 1a). In an attempt to determine the reason of such performance, the average copy number of the different constructs was determined using digital droplet PCR. Two primer sets were designed either on the zeocin—pEM72 promoter fragment or within the BFP coding sequence. Three representative transformants per construct (integrative, panARS-, A76- and C937-based vectors) were tested (Table 1). Clones D4 and E2 (which normalized BFP fluorescence showed a nearly identical level) approximately integrated one copy per genome, while clone G5, despite a very similar fluorescence level, presented approximately two copies of BFP-expressing vector per genome. A possible explanation for this observation is that the extra integrated copy might have been integrated in a poorly transcriptionally active genome region: although attempts to identify such transcriptionally inactive region were carried out, it was not possible to clearly identify the location of the second integrated copy, leaving the possibility of low transcription rate for the second copy a speculation at the moment. PanARS-based vector transformants showed significant variation in copy number between different clones, despite a very similar normalized fluorescence level, suggesting the involvement of post-translational (or epigenetic) factors in protein expression for P. pastoris, consistently with the recent literature [14], as previously mentioned; further investigations will be required to fully clarify the bottlenecks involved. Finally, clones carrying either A76- or C937-based vectors showed copy number lower than one, strongly indicating an asymmetric distribution of plasmids within the population; for this reason, and since the average expression level of plasmids carrying A76 or C937 was significantly lower than panARS-based vectors, only the latter was selected for further evaluation and development.
Fig. 1.
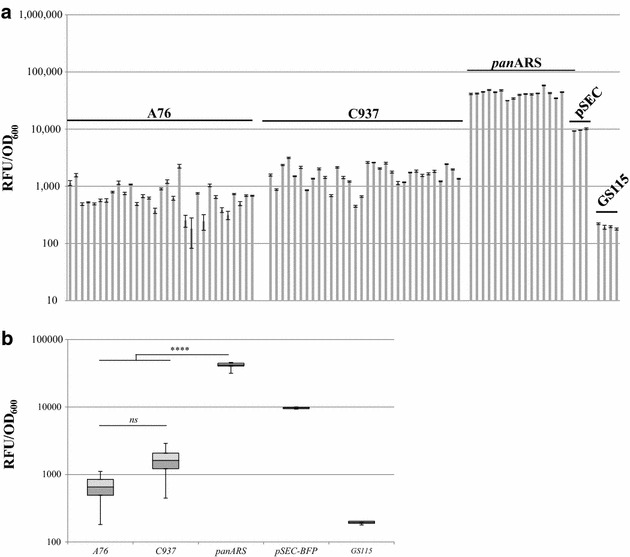
Evaluation of normalized fluorescence of panARS-, A76- or C937-based plasmids. a Histogram and b representation of normalized fluorescence level (RFU/OD600) of individual clones transformed with A76-, C937-, panARS-based episomal vectors, compared to integrative clones (pSEC) and parental strain (GS115) (ns Sidak’s multiple comparison test on one-way ANOVA, α 0.05; *one-way ANOVA, p < 0.05)
Table 1.
Copy number determination for integrated or episomal BFP-expressing vectors
| Construct/clone | Copy number Zeo Set 1 | Copy number BFP Set 1 |
|---|---|---|
| Integrative/D4 | 0.87 | 1.06 |
| Integrative/G5 | 1.91 | 2.20 |
| Integrative/E2 | 0.98 | 1.20 |
| panARS/B8 | 18.26 | 19.08 |
| panARS/E8 | 13.00 | 14.14 |
| panARS/C10 | 6.11 | 6.48 |
| A76/D1 | 0.42 | 0.39 |
| A76/F2 | 0.66 | 0.66 |
| A76/F3 | 0.45 | 0.41 |
| C937/D3 | 0.89 | 0.88 |
| C937/C1 | 0.67 | 0.59 |
| C937/A6 | 0.74 | 0.78 |
Evaluation of potential positional effect of panARS
Autonomously replicating sequences can influence RNA transcription machinery in addition to their role in DNA replication [28, 29]: we therefore evaluated the positional effect of panARS on protein expression. PanARS was placed either upstream of the methanol-inducible AOX1 promoter, downstream of the AOX1 terminator (pAOX-BFP-ARS) or downstream of the cytochrome c terminator, on the opposite side of the BFP coding sequence (pARS-oppBFP) (Fig. 2a). When panARS was placed opposite of BFP or upstream of the AOX1 promoter, the same BFP expression level was detected (data not shown); however, a slight beneficial positional effect was observed when origins of replication were placed downstream terminator sequences in episomal plasmids (Fig. 2b), (unpaired t test, p < 0.05). As the substantial equivalence of expression levels detected with pARS-AOX-BFP and pARS-oppBFP suggests, further analysis will be required to properly clarify whether the vicinity of panARS to the AOX1 terminator is responsible for the observed slight performance improvement, possibly reinforcing transcription termination of mRNA.
Fig. 2.
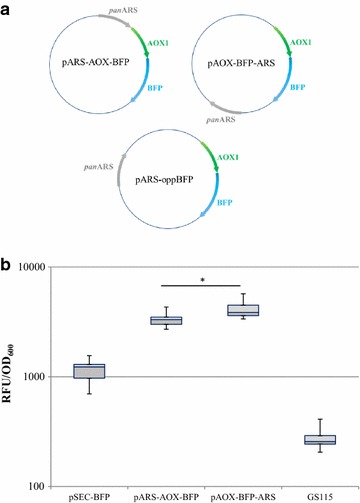
Evaluation of positional effects for panARS. a Schematic representation of constructs generated to evaluate putative positional effects for the panARS sequence. b Box-plot representation of normalized fluorescence emission (RFU/OD600) of P. pastoris independent colonies carrying pARS-AOX-BFP or pAOX-BFP-ARS (*, unpaired t-test, p < 0.05)
Specific fluorescence evaluation at the single cell level
To evaluate the intra population variation in the BFP expression from either episomal on integrative plasmids, samples from deep-well plate cultivation of integrative or episomal BFP producers were analysed by flow cytometry. 40 individual colonies, transformed with either integrative or episomal BFP expression plasmids, were cultivated in 96-well format and induced for BFP expression. After 48 h of cultivation, colonies were tested for specific fluorescence emission (normalized on OD600 as an estimation of biomass) and analysed via flow cytometry. The fluorescence channel corresponding to BFP emission at 452 nm was gated applying an equidistant threshold, providing the percentage of cells populating 10 different gates (named from G1 to G10), from zero (G1) to highest (G10) BFP emission. The percentage of fluorescent cells populating the different gates is depicted in a heat map (Fig. 3a). When hierarchial clustering was applied, 97.5 % of episomal clones were grouped in the same cluster (cluster 12, Fig. 3b), whilst integrative clones were more heterogeneous, strongly suggesting increased variation across the population of several independent clones (approximately 45 % of integrative clones were clustered in cluster 14, Fig. 3b). The increase in BFP production at the macroscopic level could have resulted from two different events at the microscopic level: (a) every single cell in the population could present a moderate increase in BFP emission, resulting in an increased median value for emission, or (b) a sub-population of high producers, driven by asymmetric plasmid distribution within the growing population. Analysing flow cytometry profiles, it appears that episomal expression of BFP in the whole population is higher, when compared to integrative expression, due to a polarization of the cell population, resulting in a higher density of both low- and high-producers. Such an observation was confirmed by analysing single clones, cultivated in deep-well plates or shake flasks, where the percentage of fluorescent events corresponding to high BFP emission was significantly higher for clones carrying episomal vectors (data not shown). Among the reasons of such instability there is plasmid stability, even in presence of selective pressure. Preliminary results on a subset of episomal clones showed that plasmid stability was 97.3 % ± 3.80 and 39.04 ± 5.54 (expressed as percentage of cells retaining the plasmid) when growth was performed for 10.8 generations in YPD media supplemented or not with the selective marker zeocin, respectively. Since cultivation before BFP assessment took place in presence of zeocin after propagation of <10 generations, this suggests that such propagation rates for panARS vectors appear to exclude plasmid instability as the reason for the relatively large number of non-fluorescent single cells measured, since cultivation took place in presence of zeocin and for lower generation numbers. Further study will be required to analyse such effects over longer generation times, and how the selection marker might influence this outcome, since it must be noted that copy number variation was noted taken into account when plasmid stability was determined.
Fig. 3.
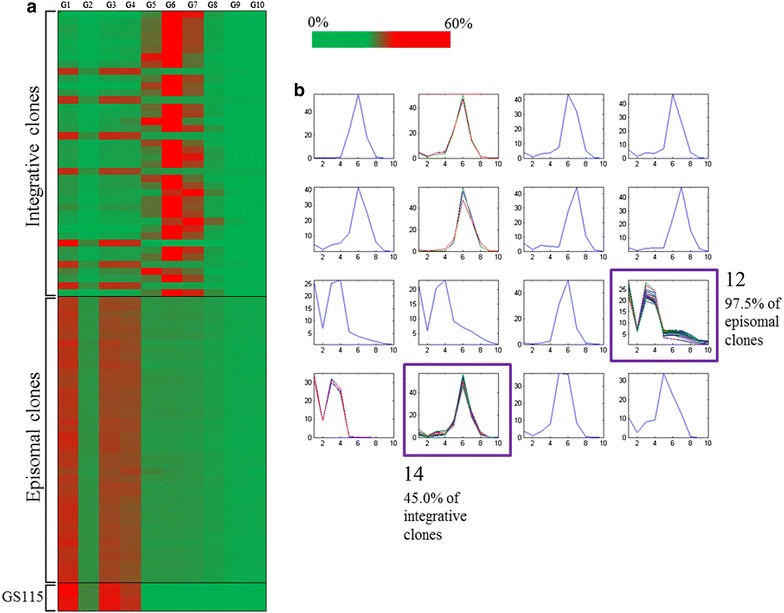
Flow cytometry analysis and hierarchial clustering of individual clones carrying episomal or integrative BFP-expressing vectors. a The overall BFP fluorescence signal was distributed between 10 equally spaced gates as percentage of fluorescence (being the total fluorescence for every clone equal to 100 %). Color-coded gating for frequencies of BFP fluorescence events in cultures derived from 40 individual clones expressing either integrative or episomal vectors (Green 0 %, Red 60 %). b Hierarchial clustering of fluorescence distribution; cluster 12 (containing 97.5 % of episomal clones, and 14, containing 45 % of integrative clones, are highlighted—see text and ‘‘Methods’’ section for details)
In vivo recombination: proof-of-principle
One of the goals of developing an efficient episomal vector for P. pastoris was the possible application of in vivo recombination (self-assembly of an expression cassette or plasmid directly by the cell machinery) in P. pastoris, as such techniques would allow faster, cheaper, and higher throughput screening of bioparts from this organism for synthetic biology, as in S. cerevisiae [30, 31]. In order to test whether panARS-based expression would be a suitable tool for in vivo recombination in Pichia pastoris, the following strategy was conceived (Fig. 4a). As a proof-of-principle, 4 fragments were PCR-amplified using panARS-BFP as a template and gel-purified. To minimize the integration of the fragment carrying the zeocin marker, which would have resulted in a background of non-fluorescent colonies (given the tendency of P. pastoris to integrate linear DNA fragments in its genome), the coding sequence for the selection marker was split between fragment 1 and either fragments 2, 3 or 4. These fragments were designed to overlap each other by 20, 50, or 100 nucleotides over a backbone fragment (fragment 1; Fig. 4a). Theoretically, only an in vivo reconstituted plasmid could provide an intact selection marker, (zeocin resistant, non-fluorescent colonies), whilst the individual fragments (1 plus either 2, 3 or 4), even if integrating, would not be able to do so. Transformation efficiency, which in this case implies that DNA must be not only incorporated, but also assembled by the cell machinery to yield a growing colony, was measured. When single fragments were used to transform P. pastoris GS115, no colonies were obtained (data not shown), whereas when two fragments were co-transformed a significant efficiency of transformation was detected (Fig. 4b). Such efficiency dropped when the overlapping region was trimmed to 20 nucleotides: the total number of colonies obtained in this case was an average of 152.4 ± 41.7, compared with approximately a number ten times higher when the overlapping regions were of 100 and 50 nucleotides in length. Although the efficiency of transformation was lower than in other cases, the 20-nt overlap strategy provides a transformation/ligation efficiency still compatible with recombinant protein production or colony screening, with the advantage that 20-nucleotide overlaps can more cheaply and easily be introduced into PCR primers than 100- or 50-nucleotide overlaps. In addition, as shown in Fig. 3b, the macroscopic interclonal variability of transformants with the panARS vector is limited, minimizing the required screening throughput for productive clones. Eight independent colonies, obtained from various transformation events with different fragments, were tested for BFP production (Fig. 4c), confirming that the resulting clones were expressing BFP at a statistically identical level in respect to the control strains transformed using the full vector pARS-BFP (one-way ANOVA test, p < 0.05, and Sidak’s multiple comparison test on one-way ANOVA). Genomic DNA was extracted from three independent clones from the different sets and used to transform E. coli. Sequencing of plasmid DNA extracted from bacterial colonies confirmed the identity of the reconstituted plasmid. No E. coli colonies were obtained when genomic DNA from integrated pSEC-BFP clones was used.
Fig. 4.
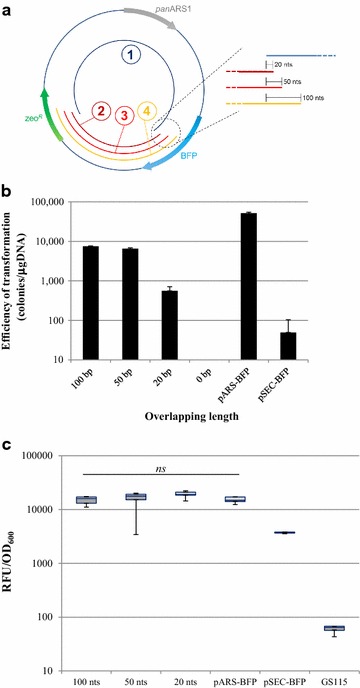
Proof-of-principle of in vivo recombination (two-fragment assembly). a Schematic diagram of fragments used for testing in vivo recombination using pARS-BFP as template. b Frequencies of transformation obtained using overlaps of 100, 50, or 20 nucleotides between fragment 1 and fragments 2, 3, or 4. c BFP fluorescence signal obtained from 8 independent colonies randomly selected from the different pools (ns non-significant, according to Sidak’s multiple comparison test on one-way ANOVA, α 0.05)
In vivo recombination: cloning simulation
The successful proof-of-principle of two-fragment in vivo recombination in P. pastoris (Fig. 4) was based on a pre-assembled expression vector. In order to verify whether cloning could be performed adopting such a strategy, a second in vivo recombination experiment was conceived (Fig. 5). A three-fragment strategy was implemented, where two fragments (1 and 2) were designed to reconstitute the backbone vector, and a third one (number 3) encoded the gene of interest (in this instance, BFP). The junctions between the three fragments, marked with a star symbol in Fig. 5, consisted of either 50-nt or 20-nt overlapping regions between the various fragments. Since genome integration of a reconstituted selection marker cassette resulting from recombined fragments 1 and 2 could potentially provide significant background, different molar ratios (from 1:1:1 to 1:1:3 to 1:1:6, respectively for fragments 1, 2 and 3) were tested, for both 20 and 50 nucleotide overlapping regions. Clones were considered positive if their BFP fluorescence was at least higher than 80 % of the difference between negative and positive control (see ‘‘Methods’’ section). Increasing the molar ratio between the backbone fragments and the “gene-of-interest” (BFP) fragment from 1:1:1 to 1:1:3 increased the relative amount of positive clones to >85 % over 72 independent clones tested (Table 2). When the molar ratio was further increased to 1:1:6, ~90 % of the colonies were positive for BFP expression. Taken together, these results suggest that although some background is observed, this method allows fast cloning-independent synthetic biology approaches in P. pastoris. Interestingly, when comparing 50-nts overlaps versus 20-nts overlaps, no apparent improvement in performance was observed. To further validate the method, an analysis on the relative abundance of the different fragments was undertaken. Total DNA extracted from randomly-picked clones carrying BFP-expressing vector as integrative, episomal or in vivo recombined constructs was tested for an “assembly ratio” (expressed as the ratio of Ct numbers for Zeo and BFP amplicons, normalized by the relative efficiency of the respective primers). In almost all cases, the assembly ratio obtained was close to 1 (Fig. 6); a single clone, characterized by low fluorescent emission failed to yield any plasmid despite repeated efforts, suggesting a relation between plasmid presence and fluorescence level. The fluctuation obtained around a value of assembly ratio of ~1.0) was not statistically different when a fragment ratio of 1:1:3 ratio was compared against 1:1:6; interestingly, a 1:1:1 fragment ratio yielded a statistically significant increase over 1.0 in the assembly ratio (One way ANOVA test, α: 0.05), suggesting a tendency of the zeocin fragment to be present in slightly higher numbers compared to BFP fragment, in accordance with the fact that the theoretical relative abundance of zeocin fragments versus BFP fragment is in fact higher in a 1:1:1 ratio than in the other cases. In vivo recombined vectors were amplified in E. coli and sequenced, confirming the successful assembly in all cases: in all, these data suggest that exogenously provided fragments could be assembled in vivo and that the fragment relative abundance plays a marginal role in limiting genomic off-target integration.
Fig. 5.
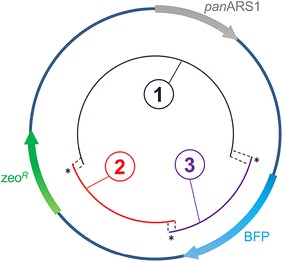
Proof-of-principle of in vivo recombination (three-fragment assembly). Schematic diagram of fragments used for testing in vivo recombination using pARS-BFP as template
Table 2.
Summary of results for in vivo recombination (3-fragments assembly)
| Fragments | Molar ratio | Overlapping ends (nts) | Fluorescent coloniesa | Coefficient of variation | Fluorescent colonies (%)b |
|---|---|---|---|---|---|
| 1 + 2 + 3 | 1:1:1 | 50 | 47 | 2.13 | 65.3 |
| 1 + 2 + 3 | 1:1:1 | 20 | 42.5 | 3.53 | 59 |
| 1 + 2 + 3 | 1:1:3 | 50 | 62.5 | 4 | 86.8 |
| 1 + 2 + 3 | 1:1:3 | 20 | 63 | 4.76 | 87.5 |
| 1 + 2 + 3 | 1:1:6 | 50 | 65.5 | 2.29 | 91 |
| 1 + 2 + 3 | 1:1:6 | 20 | 65 | 3.07 | 90.3 |
aAverage of colony numbers obtained from 3 independent transformations
bClones are defined as positive if their normalized fluorescent resulted >80 % of the difference between positive and negative controls
Fig. 6.
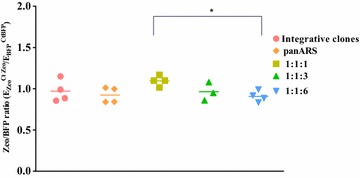
Fragment ratio determination for integrative, episomal and in vivo recombined plasmids (three fragments assembly). Cluster plot for Zeo/BFP fragment ratio for 4 independent clones carrying integrative (red dot), episomal (orange diamond) or three-fragments in vivo recombined (1:1:1 lime square; 1:1:3 green triangle—3 independent clones; 1:1:6 blue inverted triangle). All distributions were evaluated using one-factor ANOVA analysis (α 0.05); the average distribution between sample 1:1:1 and 1:1:6 (bracketed and indicated by a star) resulted statistically significant by ANOVA test
Conclusions
In this technical report, a 452-nt sequence (panARS) was tested for its capacity to confer stable replicative maintenance to a commercial plasmid used for recombinant protein production in P. pastoris. Plasmids carrying the panARS sequence complement the existing toolbox for RPP in P. pastoris. It is hereby showed that episomal panARS plasmids outperformed the corresponding integrative plasmids in terms of protein production, efficiency of transformation, and macroscopic clonal homogeneity. Endogenous P. pastoris sequences were not nearly as efficient in BFP expression compared to panARS-based constructs. At the single cell level, discrete subpopulations of high- and low-producers were consistently detected in clones carrying episomal panARS vectors. In addition, panARS episomal plasmids could be successfully used for quick in vivo self-ligation cloning directly using fragments with overlapping DNA sequences. This method was validated for gene assembly with panARS-based vectors of varying overlapping lengths and molar ratios between fragments: the resulting minimal amount of background, and reported high reproducibility of clone behaviour will allow the application of panARS plasmids to in vivo recombination for synthetic biology applications, where complex, multigene cassettes might be more easily assembled in P. pastoris. Taken together, this report characterizes for the first time the use of panARS episomal plasmids to improve the P. pastoris molecular toolbox for synthetic biology.
Methods
Strains, plasmids, and materials
Pichia pastoris strain GS115 (Life Technologies, USA) and E. coli Top10F (Life Technologies, USA) were used in this study. Plasmid pSEC-SUMO (Lifesensors, USA) was used as a backbone vector to generate pARS vector for P. pastoris. Vector Gateway® TagBFP-AS-C (Evrogen, Russia) was used as the source of a Blue Fluorescent Protein (BFP) gene codon-optimized for S. cerevisiae. All primers and gBlocks, available as supplementary information, were synthesized by IDT (Singapore). All restriction enzymes, as well as Gibson cloning kits, were purchased from NEB (Singapore). DNA amplification was performed using Q5 DNA polymerase from NEB (Singapore), while colony PCR for screening was performed with Dream Taq polymerase (Promega, USA) following standard molecular biology protocols [32]. DNA sequencing was performed by Axil Scientific Support (Singapore). All chemicals were purchased from Sigma (USA).
Cloning and transformation
The BFP gene was amplified from the Gateway® TagBFP-AS-C vector using primers AA_B1_BstB1_BFP_fw and AA_B3_EcoR1_BFP_rev (Table 3). Plasmid pSEC-SUMO was amplified using Q5 polymerase and primers AB_A8_AOX1end_Rv and AB_A9_pARS1_GOIend_Fw, removing the α-mating factor and SUMO coding sequences, and BFP was joined to the backbone vector via Gibson cloning to generate the vector pSEC-BFP. A 452-nt fragment, corresponding to the panARS sequence [16], was synthesized as a gBlock (IDT, Singapore), and cloned in various positions on the pSEC-BFP vector via Gibson cloning, generating vectors pARS-AOX1-BFP, pAOX1-BFP-ARS, and pARS-oppBFP. All vectors were fully sequenced for confirmation. Approximately 50 ng of the various vectors were used to transform P. pastoris GS115, using the condensed electroporation protocol [12]. For transformation with integrative vectors, plasmids were digested at 30 °C for 2 h with SwaI, and column purified with Wizard SV Gel and PCR Clean-up System (Promega, USA) prior to transformation. All transformations for in vivo recombination evaluation were performed in duplicate.
Table 3.
Primers used in this study
| Primer name | Primer sequence | Notes |
|---|---|---|
| AA_B1_BstB1_BFP_fw | AAGTTCGAAACGATGTCTGAATTGATTAAAGAGAATATG | |
| AA_B3_EcoR1_BFP_rev | AGGGAATTCTCATTAGTTCAATTTGTGTCCTAACTTA | |
| AB_A8_AOX1end_Rv | CGTTTCGAATAATTAGTTGTTTTTTGATCTTCT | |
| AB_A9_pARS1_GOIend_Fw | GAATTCCAACCTGCGATTGATCT | |
| qPCR-PpACT1-Fw (ACT1, Set 1) | CCATCCATTGTGCACCTCAAG | [35] |
| qPCR-PpACT1-Rv (ACT1, Set 1) | CGTCTAGAAGCTGAACGACAAG | [35] |
| qPCR-PpACT1_2_Fw (ACT1, Set 2) | CGAATCTGGACCATCCATTGTG | |
| qPCR-PpACT1_2_Rv (ACT1, Set 2) | ACGACAAGTAGACACCACCAATC | |
| qPCR_pEM7_1_Fv (Zeo, Set 1) | ACGACAAGGTGAGGAACTAAACC | |
| qPCR_ZEO_1_Rv (Zeo, Set 1) | AAGTCGTCCTCCACGAAGTC | |
| qPCR_pEM7_2_Fv (Zeo, Set 2) | CGACAAGGTGAGGAACTAAACC | |
| qPCR_ZEO_2_Rv (Zeo, Set 2) | GAAGTCGTCCTCCACGAAGTC | |
| qPCR_BFP_1_Fw (BFP, Set 1) | AGACGGTGGAGTTTTGACTGC | |
| qPCR_BFP_1_Rv (BFP, Set 1) | AATGTCTCAGTGAATGCCTCCC | |
| qPCR_BFP_2_Fw (BFP, Set 2) | ACGGTGGAGTTTTGACTGCTAC | |
| qPCR_BFP_2_Rv (BFP, Set 2) | TGAATGCCTCCCATCCCAATG |
Deep well plate cultivation and fluorescence measurement
Pichia pastoris cultures were grown in 96 Well Masterblock, 2 mL, V-bottom plates (Greiner, Germany), sealed with BREATHseal™ gas-permeable sealer (Greiner, Germany) in a variant of BMD1 media [33], where 1 % d-glucose was substituted with 1 % sorbitol (BMS media), and using 100 mg/L of zeocin as selection marker. A final concentration of 1 % methanol was added to the media to induce protein production. All cultivations were performed at least in duplicate. After 48 h cell suspensions were diluted 1:10 in PBS and evaluated for OD600 and fluorescence emission in a Tecan Infinite® 200 PRO series (Tecan, Austria), using Microplate PS, 96 Well, F-Bottom clear plates (Greiner, Germany) or Microplate PS, 96 Well, F-Bottom Black plates (Greiner, Germany), for OD600 or fluorescence respectively; the latter was determined using 402 nm and 452 nm as excitation and emission wavelength, respectively, and normalizing all values against OD600. Every reading of both OD600 and fluorescence was performed in triplicate (technical replicates).
Genomic DNA extraction
20 OD of yeast cultures were pelleted and washed in 500 µL distilled H2O. Pellet was re-suspended in breaking buffer (2 % (v/v) Triton X-100, 1 % (v/v) SDS, 100 mM NaCl, 10 mM Tris–Cl pH 8.0, 1 mM EDTA pH 8.0). 200 µL of glass beads and 200 µL of phenol/chloroform/isoamylalcohol were added. Samples were vortexed at highest speed for 5 min, supplemented with 200 µL of TE Buffer (10 mM Tris–Cl, 0.1 mM EDTA pH 8.0), vortexed briefly, and centrifuged at 13,000g for 5 min at room temperature. Samples were extracted two more times with phenol/chloroform/isoamylalcohol and one final time with chloroform. 1 mL of 100 % freeze-cold ethanol was added to the aqueous phase, mixing by inversion, and samples were incubated at −20 °C for 1 h. Samples were mixed several times by inversion, centrifuged at 13,000g for 3 min at 4 °C and supernatant aspirated. Pellet was air-dried; 400 µL of TE were then added to pellets and incubated at 65 °C for 10 min, followed by addition of 30 µL of RNase A and incubation for 1 h at 37 °C. 10 µL of 4 M ammonium acetate was added, samples were mixed by inversion, and quickly spun. 1 mL of 100 % ethanol (−20 °C) was added and mixed by inversion, followed by 13,000g centrifugation (3 min) at 4 °C. Pellet was air-dried, resuspended in 50 µL of TE buffer and incubated in 65 °C water bath for 10 min.
Picogreen DNA quantification
Lambda DNA (Life Technologies, USA) was serially diluted to produce DNA standards at the following concentrations (ng/µL): DNA Standards Set #1—50, 33.33, 16.66, 8.3, 4.16, 2.08, 1.042, 0.5208, 0; DNA Standards Set #2—75, 50, 25, 12.5, 6.25, 3.125, 1.5262, 0. Quant-iT™ PicoGreen® dsDNA reagent (Life Technologies, USA), diluted 200× in 1x Tris–EDTA (10 mM Tris–HCl, 0.1 mM EDTA, pH 8.0) was transferred to wells of a white 96-well plate (Greiner Bio-One, Germany), 195 μL per well (“DNA sample plate”).
Based on previous spectrophotometer readings (NanoDrop 2000c), genomic DNA samples were diluted 5x, 10x, 20x, or 40x, to fall within the range of the DNA standards made. 2 µL of each lambda DNA standard and diluted genomic DNA sample was transferred to wells of the DNA sample plate. Sample plate was scanned on a SpectraMax M5 microplate reader (Molecular Devices, USA) using the preconfigured Quant-It Picogreen protocol in the accompanying SoftMax Pro software, measuring fluorescence signal at 525 nm (excitation wavelength: 490 nm). Sample concentrations were calculated from RFU readings using the linear regression equation derived from the DNA standards. All samples were evaluated in triplicate.
Digital droplet PCR (ddPCR) copy number determination
Purified genomic DNA was digested with KpnI (NEB, USA) for 1 h at 37 °C. No heat inactivation was performed, following ddPCR manufacturer indications. An Evagreen ddPCR mastermix was assembled, mixing 10 µL of Eva Green 2x master solution (Biorad, Singapore), 2 µL of primer set (100 nM each, Table 3), 1 µL of genomic DNA sample (0.5 ng/µl, as reported in [34]) and mQ H2O up to 20 µL. Primers were designed following published indications [35], although different sets of primers were tested for optimal signal. Following droplet generation with a Droplet generator (Biorad, Singapore), samples were transferred to a 96-well plate and sealed. PCR was performed adjusting the ramp rate on a C1000 Touch Deep Well PCR system (Biorad, Singapore) to 2 °C/s, applying the following cycle: 95 °C, 5 min; 40x (95 °C, 30 s; annealing/extension 57–60 °C); 4 °C, 5 min; 90 °C, 5 min; 4 °C, infinite hold). Droplet detection was carried out in a QX200 Droplet Digital PCR system (Biorad, Singapore) and analysed using the software QuantaSoft v. 1.7.4.0917 (Biorad), following an absolute quantification protocol.
Copy number determination of in vivo recombined fragments
qPCR was conducted to accurately evaluate the copy number of individual fragments upon in vivo recombination. All qPCR reactions were performed in triplicates for each transformed clone and each diluted standard, using FastStart Essential DNA Green Master (Roche, USA) and LightCycler® 96 Instrument (Roche, USA). A 5 ng DNA sample of each transformed clone was used as template for qPCR reaction. Two primer sets, namely Zeo, Set 1 and BFP, Set 2, were designed to determine the copy number of the two transformed fragments. To determine PCR efficiency of the two primer sets, a 5-log dilution series of PARS-BFP plasmid, started from 10 ng, was used. Each diluted standard was mixed with 5 ng of untransformed parental genomic DNA, to compensate for any non-specific amplification arising from genomic DNA contamination in the plasmid preparation. During the qPCR reaction, the enzyme was activated at 95 °C for 10 min, followed by 45 cycles of 95 °C for 10 s, 57 °C for 10 s and 72 °C for 10 s.
Standard curve of each primer set amplified the 5-log diluted plasmid was plotted, and primer amplification efficiency was determined. The adjusted copy number ratio of the two transformed fragments in each transformed clone was calculated using formula: ECt ZEOZeo/ECt BFPBFP, where EZeo and EBFP are primer efficiencies of Zeo and BFP primer set, respectively, while CtZEO and CtBFP are Ct values of a transformed clone amplified using ZEO and BPF primer set, respectively.
FACS analysis and hierarchial clustering
Samples were normalized to OD600 = 0.1 using a Tecan EVO 150 liquid-handling robot. After washing with PBS twice, samples were analysed in 96-well batches on a MACSQuant VYB instrument (Miltenyi), acquiring ~80,000 cells per clone. The Blue Florescence Protein signal profile was gated into 10 equally spaced areas (P1–P10) using FlowJo_vX.0.7 software. The blue fluorescence intensity for each gate was visualized in heat maps built using customized R scripts. Hierarchial clustering was generated based on the relative frequencies of fluorescence events in every gate, using MATLAB R2014a (version 8.3.0.532, Mathworks, Natick, MA). An agglomerative ‘bottom-up’ clustering algorithm [36] was used where the fluorescence of each clone initiated as its own cluster, which then merged with clones possessing similar profiles.
Statistical analysis
Data analysis was performed with GraphPad Prism 6 (GraphPad Software, USA).
Authors’ contributions
AC conceived the outline of the work, and performed all cloning, transformation, and cultivation tasks in collaboration with AG. AG and SWN performed copy number determination with ddPCR and qPCR, under the supervision of AHMT. IL and MJD provided support for sequence selection; GR, AT and GL performed DNA extraction, quantification and flow cytometry experiments (including analysis and data processing). LYY processed and evaluated the clustering data. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
IL and MJD declare a possible competing interest (co-inventors in “Pan-yeast autonomously replicating sequence”, WO 2014131056 A1). AC, AG, LYY, AHMT, SWN, AT, GL and GR declare that they no competing interests.
Availability of data and materials
The datasets during and/or analysed during the current study are available from the corresponding author on reasonable request.
Consent for publication
This manuscript does not contain data from any individual person.
Ethics approval and consent to participate
This manuscript does not report on or involve the use of any animal or human data or tissue.
Funding
This study was supported by the Biomedical Research Council of the Singapore Agency for Science, Technology and Research. GR lab is supported by A-STAR Investigatorship award 1437a00119.
Contributor Information
Andrea Camattari, Email: andrea_camattari@bti.a-star.edu.sg.
Amelia Goh, Email: amelia_goh@bti.a-star.edu.sg.
Lian Yee Yip, Email: yip_lian_yee@bti.a-star.edu.sg.
Andy Hee Meng Tan, Email: andy_tan@bti.a-star.edu.sg.
Sze Wai Ng, Email: ng_sze_wai@bti.a-star.edu.sg.
Anthony Tran, Email: anthony.tran@imb.a-star.edu.sg.
Gaowen Liu, Email: gaowen1987@gmail.com.
Ivan Liachko, Email: il34@uw.edu.
Maitreya J. Dunham, Email: maitreya@uw.edu
Giulia Rancati, Email: Giulia.rancati@imb.a-star.edu.sg.
References
- 1.Cregg JM, Tolstorukov I, Kusari A, Sunga J, Madden K, Chappell T. Expression in the yeast Pichia pastoris. Methods Enzymol. 2009;463:169–189. doi: 10.1016/S0076-6879(09)63013-5. [DOI] [PubMed] [Google Scholar]
- 2.Cregg JM, Cereghino JL, Shi J, Higgins DR. Recombinant protein expression in Pichia pastoris. Mol Biotechnol. 2000;16:23–52. doi: 10.1385/MB:16:1:23. [DOI] [PubMed] [Google Scholar]
- 3.Cereghino JL, Cregg JM. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev. 2000;24:45–66. doi: 10.1111/j.1574-6976.2000.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 4.Chung BK-S, Lakshmanan M, Klement M, Ching CB, Lee D-Y. Metabolic reconstruction and flux analysis of industrial Pichia yeasts. Appl Microbiol Biotechnol. 2013;97:1865–1873. doi: 10.1007/s00253-013-4702-7. [DOI] [PubMed] [Google Scholar]
- 5.De Schutter K, Lin Y-C, Tiels P, Van Hecke A, Glinka S, Weber-Lehmann J, Rouzé P, Van de Peer Y, Callewaert N. Genome sequence of the recombinant protein production host Pichia pastoris. Nat Biotechnol. 2009;27:561–566. doi: 10.1038/nbt.1544. [DOI] [PubMed] [Google Scholar]
- 6.Mattanovich D, Graf A, Stadlmann J, Dragosits M, Redl A, Maurer M, Kleinheinz M, Sauer M, Altmann F, Gasser B. Genome, secretome and glucose transport highlight unique features of the protein production host Pichia pastoris. Microb Cell Fact. 2009;8:29. doi: 10.1186/1475-2859-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klug L, Tarazona P, Gruber C, Grillitsch K, Gasser B, Trötzmüller M, Köfeler H, Leitner E, Feussner I, Mattanovich D, Altmann F, Daum G. The lipidome and proteome of microsomes from the methylotrophic yeast Pichia pastoris. Biochim Biophys Acta. 2014;1841:215–226. doi: 10.1016/j.bbalip.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Liang S, Zou C, Lin Y, Zhang X, Ye Y. Identification and characterization of P GCW14: a novel, strong constitutive promoter of Pichia pastoris. Biotechnol Lett. 2013;35:1865–1871. doi: 10.1007/s10529-013-1265-8. [DOI] [PubMed] [Google Scholar]
- 9.Hartner FS, Ruth C, Langenegger D, Johnson SN, Hyka P, Lin-Cereghino GP, Lin-Cereghino J, Kovar K, Cregg JM, Glieder A. Promoter library designed for fine-tuned gene expression in Pichia pastoris. Nucleic Acids Res. 2008;36:e76. doi: 10.1093/nar/gkn369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin X, Qian J, Yao G, Zhuang Y, Zhang S, Chu J. GAP promoter library for fine-tuning of gene expression in Pichia pastoris. Appl Environ Microbiol. 2011;77:3600–3608. doi: 10.1128/AEM.02843-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kottmeier K, Ostermann K, Bley T, Rödel G. Hydrophobin signal sequence mediates efficient secretion of recombinant proteins in Pichia pastoris. Appl Microbiol Biotechnol. 2011;91:133–141. doi: 10.1007/s00253-011-3246-y. [DOI] [PubMed] [Google Scholar]
- 12.Lin-Cereghino J, Wong WW, Xiong S, Giang W, Luong LT, Vu J, Johnson SD, Lin-Cereghino GP. Condensed protocol for competent cell preparation and transformation of the methylotrophic yeast Pichia pastoris. Biotechniques. 2005;38:44–46, 48. doi: 10.2144/05381BM04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu T, Guo M, Sun C, Qian J, Zhuang Y, Chu J, Zhang S. A systematical investigation on the genetic stability of multi-copy Pichia pastoris strains. Biotechnol Lett. 2009;31:679–684. doi: 10.1007/s10529-009-9917-4. [DOI] [PubMed] [Google Scholar]
- 14.Love KR, Panagiotou V, Jiang B, Stadheim TA, Love JC. Integrated single-cell analysis shows Pichia pastoris secretes protein stochastically. Biotechnol Bioeng. 2010;106:319–325. doi: 10.1002/bit.22688. [DOI] [PubMed] [Google Scholar]
- 15.Love KR, Politano TJ, Panagiotou V, Jiang B, Stadheim TA, Love JC. Systematic single-cell analysis of Pichia pastoris reveals secretory capacity limits productivity. PLoS ONE. 2012;7:e37915. doi: 10.1371/journal.pone.0037915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liachko I, Dunham MJ. An autonomously replicating sequence for use in a wide range of budding yeasts. FEMS Yeast Res. 2014;14:364–367. doi: 10.1111/1567-1364.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liachko I, Bhaskar A, Lee C, Chung SCC, Tye B-K, Keich U. A comprehensive genome-wide map of autonomously replicating sequences in a naive genome. PLoS Genet. 2010;6:e1000946. doi: 10.1371/journal.pgen.1000946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liachko I, Youngblood RA, Keich U, Dunham MJ. High-resolution mapping, characterization, and optimization of autonomously replicating sequences in yeast. Genome Res. 2013;23:698–704. doi: 10.1101/gr.144659.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oldenburg KR, Vo KT, Michaelis S, Paddon C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997;25:451–452. doi: 10.1093/nar/25.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pereira F, de Azevedo F, Parachin NS, Hahn-Haegerdal B, Gorwa-Grauslund MF, Johansson B. The Yeast Pathway Kit: a method for metabolic pathway assembly with automatically simulated executable documentation. ACS Synth Biol. 2016;5(5):386–394. doi: 10.1021/acssynbio.5b00250. [DOI] [PubMed] [Google Scholar]
- 21.Juhas M, Ajioka JW: High molecular weight DNA assembly in vivo for synthetic biology applications. Crit Rev Biotechnol 2016:1–10. [DOI] [PubMed]
- 22.Yu X-W, Wang R, Zhang M, Xu Y, Xiao R. Enhanced thermostability of a Rhizopus chinensis lipase by in vivo recombination in Pichia pastoris. Microb Cell Fact. 2012;11:102. doi: 10.1186/1475-2859-11-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liachko I, Youngblood RA, Tsui K, Bubb KL, Queitsch C, Raghuraman MK, Nislow C, Brewer BJ, Dunham MJ. GC-rich DNA elements enable replication origin activity in the methylotrophic yeast Pichia pastoris. PLoS Genet. 2014;10:e1004169. doi: 10.1371/journal.pgen.1004169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ai H, Shaner NC, Cheng Z, Tsien RY, Campbell RE. Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry. 2007;46:5904–5910. doi: 10.1021/bi700199g. [DOI] [PubMed] [Google Scholar]
- 25.Subach OM, Gundorov IS, Yoshimura M, Subach FV, Zhang J, Grüenwald D, Souslova EA, Chudakov DM, Verkhusha VV. Conversion of red fluorescent protein into a bright blue probe. Chem Biol. 2008;15:1116–1124. doi: 10.1016/j.chembiol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saeed IA, Ashraf SS. Denaturation studies reveal significant differences between GFP and blue fluorescent protein. Int J Biol Macromol. 2009;45:236–241. doi: 10.1016/j.ijbiomac.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 27.Niu H, Jost L, Pirlot N, Sassi H, Daukandt M, Rodriguez C, Fickers P. A quantitative study of methanol/sorbitol co-feeding process of a Pichia pastoris Mut+/pAOX1-lacZ strain. Microb Cell Fact. 2013;12:33. doi: 10.1186/1475-2859-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivessa AS, Zakian VA. To fire or not to fire: origin activation in Saccharomyces cerevisiae ribosomal DNA. Genes Dev. 2002;16:2459–2464. doi: 10.1101/gad.1033702. [DOI] [PubMed] [Google Scholar]
- 29.Kohzaki H, Ito Y, Murakami Y. Context-dependent modulation of replication activity of Saccharomyces cerevisiae autonomously replicating sequences by transcription factors. Mol Cell Biol. 1999;19:7428–7435. doi: 10.1128/MCB.19.11.7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao Z, Zhao H. Construction and engineering of large biochemical pathways via DNA assembler. Methods Mol Biol. 2013;1073:85–106. doi: 10.1007/978-1-62703-625-2_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin Q, Jia B, Mitchell LA, Luo J, Yang K, Zeller KI, Zhang W, Xu Z, Stracquadanio G, Bader JS, Boeke JD, Yuan YJ. RADOM, an efficient in vivo method for assembling designed DNA fragments up to 10 kb long in Saccharomyces cerevisiae. ACS Synth Biol. 2015;4:213–220. doi: 10.1021/sb500241e. [DOI] [PubMed] [Google Scholar]
- 32.Wood E. Molecular cloning. a laboratory manual. Biochem Educ. 1983;11:82. doi: 10.1016/0307-4412(83)90068-7. [DOI] [Google Scholar]
- 33.Weis R, Luiten R, Skranc W, Schwab H, Wubbolts M, Glieder A. Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res. 2004;5:179–189. doi: 10.1016/j.femsyr.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 34.Cámara E, Albiol J, Ferrer P. Droplet digital PCR-aided screening and characterization of Pichia pastoris multiple gene copy strains. Biotechnol Bioeng. 2015;113(7):1542–1551. doi: 10.1002/bit.25916. [DOI] [PubMed] [Google Scholar]
- 35.Yang Y, Fan F, Zhuo R, Ma F, Gong Y, Wan X, Jiang M, Zhang X. Expression of the laccase gene from a white rot fungus in Pichia pastoris can enhance the resistance of this yeast to H2O2-mediated oxidative stress by stimulating the glutathione-based antioxidative system. Appl Environ Microbiol. 2012;78:5845–5854. doi: 10.1128/AEM.00218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets during and/or analysed during the current study are available from the corresponding author on reasonable request.