

Abstract
The vacuolar ATPases (V-ATPases) are a family of proton pumps that couple ATP hydrolysis to proton transport into intracellular compartments and across the plasma membrane. They function in a wide array of normal cellular processes, including membrane traffic, protein processing and degradation, and the coupled transport of small molecules, as well as such physiological processes as urinary acidification and bone resorption. The V-ATPases have also been implicated in a number of disease processes, including viral infection, renal disease, and bone resorption defects. This review is focused on the growing evidence for the important role of V-ATPases in cancer. This includes functions in cellular signaling (particularly Wnt, Notch, and mTOR signaling), cancer cell survival in the highly acidic environment of tumors, aiding the development of drug resistance, as well as crucial roles in tumor cell invasion, migration, and metastasis. Of greatest excitement is evidence that at least some tumors express isoforms of V-ATPase subunits whose disruption is not lethal, leading to the possibility of developing anti-cancer therapeutics that selectively target V-ATPases that function in cancer cells.
I. INTRODUCTION
V-ATPases are ATP-driven proton pumps that function in a wide array of normal physiological processes, many of which are altered in cancer (17, 25, 46, 77, 115, 192). They couple the energy released from ATP hydrolysis to the transport of protons out of the cytosol into either the lumen of intracellular compartments or, for V-ATPases present in the plasma membrane, into the extracellular space. This review is focused on the role of V-ATPases in tumor cell growth, survival, signaling, and metastasis and concludes with a discussion of V-ATPases as a potential target in the development of anti-cancer therapeutics. To understand the function of V-ATPases in cancer and to explore the possibility of exploiting this role to inhibit the growth and metastasis of tumor cells, we will first briefly review the role of V-ATPases in normal processes and some aspects of their structure and regulation.
A. Function of V-ATPases
V-ATPases are present in both intracellular membranes such as lysosomes, endosomes, and secretory vesicles and, for specialized cells, the plasma membrane. V-ATPases within lysosomes create the luminal acidic environment required for the degradation of proteins by acid-dependent proteases called cathepsins (225). The pH gradient across lysosomal membranes is also utilized to drive the coupled transport of many small molecules and ions, including amino acids (which are primarily exported into the cytosol following protein degradation) and Ca2+ (151). The proton gradient across the membranes of secretory vesicles is also used to drive the coupled transport of small molecules, particularly neurotransmitters such as norepinephrine (165). V-ATPases are electrogenic proton pumps (i.e., they create a luminal positive transmembrane potential), and this membrane potential drives the uptake into synaptic vesicles of glutamate, a particularly important neurotransmitter in the brain (130). Proteolytic processing of prohormones in secretory vesicles, such as cleavage of proinsulin, also depends on the acidic pH created by the V-ATPases (157).
V-ATPases within endosomes function in membrane trafficking processes, including receptor-mediated endocytosis and intracellular trafficking of lysosomal enzymes. V-ATPase-dependent acidification of early endosomes provides the low pH signal that causes endocytosed ligands, such as low-density lipoprotein (LDL), to dissociate from their receptors (49). This dissociation is in turn required for recycling of the receptors to the plasma membrane and targeting of the released ligands to the lysosome for degradation. Endosomal acidification is also involved in the budding of endosomal carrier vesicles that transport cargo between early and late endosomes (57) as well as in the trafficking of newly synthesized lysosomal enzymes from the Golgi to the lysosome utilizing the mannose-6-phosphate receptor, which interacts with lysosomal enzymes bearing a mannose-6-phosphate recognition marker in a pH-dependent manner (87). The V-ATPase has also recently been shown to function at the earliest stages of clathrin-coated vesicle formation (88). It should be noted that a number of pathogens, including envelope viruses such as influenza virus and Ebola virus, and toxins, such as diphtheria toxin and anthrax toxin, gain access to the cytoplasm of infected cells via acid-dependent fusion or pore-forming events that occur within endocytic compartments (56). There is also evidence from studies in Saccharomyces cerevisiae, Drosophila melanogaster, Caenorhabditis elegans, and Mus musculus that the integral V0 domain of the V-ATPase (see below) may play a role in membrane fusion independent of acidification (34, 61, 100, 148, 149, 191).
Plasma membrane V-ATPases are primarily present in specialized cells. In osteoclasts, V-ATPases are targeted to the ruffled border in contact with bone and provide the acidic extracellular environment that is essential for bone resorption (102). Defects in the plasma membrane V-ATPase in osteoclasts lead to loss of bone resorption and development of the disease osteopetrosis, which is characterized by highly brittle bone and skeletal defects during embryonic development (102). In renal alpha intercalated cells of the late distal tubule and collecting duct, the V-ATPase is targeted to the apical membrane and is involved in acid secretion into the urine (17). The density of V-ATPases in the apical membrane of intercalated cells is tightly controlled in response to plasma pH through exocytic insertion and endocytic retrieval of pumps. A decrease in plasma pH results in an increase in the number of V-ATPases at the apical surface which, in turn, increases acid secretion into the urine. V-ATPases in the kidney are thus important in pH homeostasis. In the male reproductive tract, V-ATPases are present in the apical membrane of epididymal clear cells and function to maintain an acidic pH in the epididymal lumen that is essential for normal sperm development and storage (17). V-ATPases have also been implicated in left-right patterning during vertebrate development, with the pump's ability to generate a membrane potential apparently key to its function (1, 2).
B. Structure and Mechanism of the V-ATPases
The V-ATPases are large, multisubunit complexes organized into a peripheral V1 domain that carries out ATP hydrolysis and an integral V0 domain responsible for proton transport (Figure 1; see Ref. 232 for a recent model based on cryoelectron microscopy). V1 is composed of eight different subunits (A-H) present in a stoichiometry of A3B3CDE3FG3H, whereas the V0 is composed of five subunits (a,c,c”,d,e) in a stoichiometry of ac9c”de. The yeast enzyme contains an additional proteolipid subunit c' (152), and some mammalian enzymes also contain additional accessory proteins that may not be part of all V-ATPase complexes, including Ac45 (193) and the prorenin receptor (PRR; see below). ATP hydrolysis occurs at three catalytic sites located primarily in subunit A but at the interface of the A and B subunits (110). V-ATPases operate by a rotary mechanism in which ATP hydrolysis in the A3B3 catalytic head causes rotation of a central stalk composed of subunits D and F of V1 attached to subunit d of V0 (65). Subunit d in turn is attached at the center of a 10-membered ring of proteolipid subunits (c9c”) (232). Each proteolipid subunit contains an essential buried glutamic acid residue that undergoes reversible protonation during proton transport (64). ATP-driven rotation of the central stalk causes rotation of the proteolipid ring relative to subunit a of the V0 domain. Subunit a is a 100 kDa transmembrane protein containing an NH2-terminal cytoplasmic domain and a COOH-terminal hydrophobic domain containing eight transmembrane helices (212). The NH2-terminal domain of subunit a together with subunits C and H provide a platform for attachment of the three peripheral stalks (each composed of an EG heterodimer) that hold the A3B3 catalytic head in place during ATP-driven rotation of the central stalk (231). The COOH-terminal domain of subunit a contains two proton-conducting hemi-channels that lead from the cytoplasmic side of the membrane to the membrane interior and from the membrane interior to the luminal (or extracellular) side of the membrane (197). Protons enter from the cytoplasmic compartment through the first hemichannel in subunit a and protonate an essential glutamate residue on one of the proteolipid subunits. ATP-driven rotation of the proteolipid ring brings sequential proteolipid subunits into contact with this cytoplasmic hemichannel where they can undergo protonation. At the same time, rotation brings these protonated glutamate residues into contact with the luminal hemi-channel. Here, as a result of interaction with a positively charged arginine residue in subunit a (80), a proton from each glutamate is released into the luminal channel, which allows this proton to complete translocation to the luminal compartment. Thus ATP-driven rotational movement within the V-ATPase complex is converted into unidirectional proton transport across the membrane. It should be noted that the V-ATPase complex associates with a large variety of proteins, including heme-responsive gene 1 (HRG1) (137), tumor metastasis suppressor gene 1 (TMSG1) (124), the PRR (105), synaptobrevin (128), ARNO/Arf6 (72), the Ragulator/Rag GTPase complex (233), transmembrane 9 superfamily protein member 4 (TM9SF4) (103), the HPV E5 oncoprotein (187), and the glycolytic enzymes aldolase (106) and phosphofructokinase (190a). The functional significance of a number of these interactions is discussed in detail in later sections.
FIGURE 1.
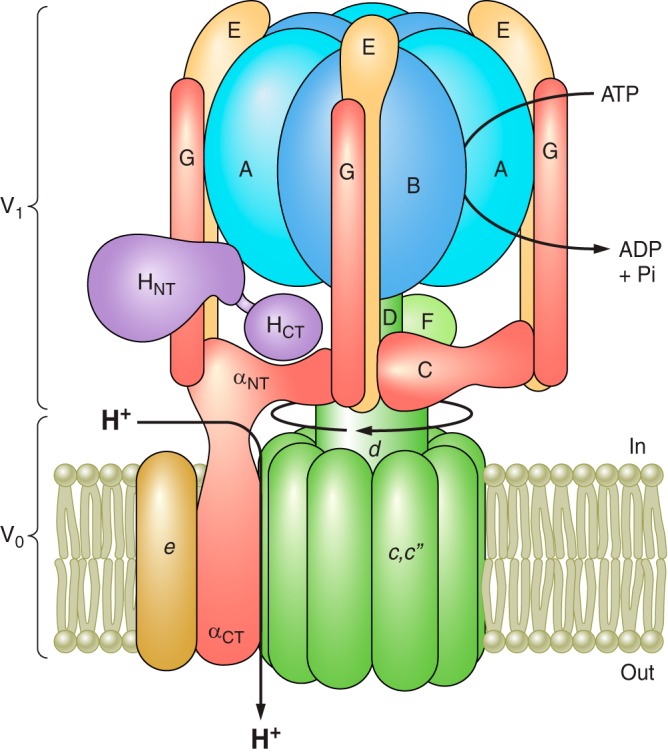
Structure and mechanism of the V-ATPase. The V-ATPase is composed of a peripheral V1 domain that hydrolyzes ATP and an integral V0 domain that translocates protons. ATP hydrolysis occurs at nucleotide binding sites located at the interface of the A and B subunits and drives rotation of a central rotary complex composed of subunits D and F of V1 and subunit d and the ring of proteolipid subunits (c and c”) of V0 relative to the remainder of the complex. Rotation of the proteolipid ring relative to subunit a drives unidirectional proton transport from the cytoplasm to the lumen (see text for details). The A3B3 catalytic head is held fixed relative to subunit a by peripheral stalks composed of three EG heterodimers that connect to subunits C and H and the NH2-terminal cytoplasmic domain of subunit a. Model is adapted from Couoh-Cardel et al. (26). See Reference 232 for a recent model based on cryo-EM of the yeast V-ATPase.
C. Regulation of V-ATPase Activity
Among the most important means of regulating V-ATPase activity in cells are reversible dissociation/reassembly and regulated trafficking (Figure 2). In mammalian cells, an increase in assembly of the V1 and V0 domains is observed in response to a number of stimuli, including elevated glucose concentrations (169), amino acid starvation (190), exposure to growth factors (219), dendritic cell maturation (99, 202), and during viral infection (113). Increased assembly (and activity) at high glucose concentrations may allow the cell to more effectively rid itself of the additional metabolic acid generated by increased rates of glycolysis while decreased assembly at low glucose may conserve cellular stores of ATP. Increased assembly and activity in response to amino acid starvation may increase the rate of lysosomal protein degradation, raising the intracellular pool of free amino acids for essential uses (190). The increased assembly observed in the presence of epidermal growth factor (EGF) has been hypothesized to be necessary to generate adequate levels of amino acids from acid-dependent protein breakdown in lysosomes to stimulate the mechanistic target of rapamycin complex 1 (mTORC1) (219), as will be discussed in further detail below. In dendritic cells, the increased assembly that occurs during maturation facilitates antigen processing by acid-dependent proteases within lysosomes (99, 202). For envelope viruses like influenza, increased assembly in early endosomes enhances acid-dependent fusion of the viral coat with the endosomal membrane, a step necessary for effective transfer of the viral RNA to the host cytoplasm (113). In mammalian cells, PI-3 kinase (PI3K) has been shown to be involved in increased assembly in response to elevated glucose, dendritic cell maturation, and upon viral infection (99, 113, 169), although amino acid-dependent modulation does not involve PI3K (190). Interactions with the glycolytic enzymes aldolase and phosphofructokinase have also been proposed to regulate assembly of the pump (106, 195). In addition, several proteins of the rabconnectin family have been shown to promote V-ATPase assembly and acidification (37, 172). These proteins are homologous to proteins that make up the RAVE complex, which is required for V-ATPase assembly in yeast (177, 184).
FIGURE 2.

Regulation of V-ATPase activity. A: V-ATPase activity is regulated in vivo by reversible dissociation of the complex into its component V1 and V0 domains, which results in inactivation of the complex. In mammalian cells, regulated assembly occurs in response to a number of cues, including changes in glucose concentration, starvation of amino acids, exposure to growth factors, upon maturation of dendritic cells, and during infection of cells by influenza virus. PI3K is involved in controlling assembly in response to changes in glucose concentration, during dendritic cell maturation and during viral infection, but not in response to changes in amino acid levels. B: V-ATPase activity in epithelial cells in the kidney and epididymis is controlled by regulated trafficking of complexes to the apical membrane of these cells. In both systems, trafficking is controlled by PKA downstream of a bicarbonate-sensitive adenylate cyclase.
A second mechanism of controlling V-ATPase activity is regulated trafficking, which occurs primarily in epithelial cells (Figure 2). Renal intercalated cells respond to decreased plasma pH by increasing the fusion of V-ATPase-rich vesicles with the apical membrane (17). Similarly, epididymal clear cells control the pH of the epididymal lumen by controlling the density of V-ATPases in the apical membrane through vesicle-mediated insertion and retrieval of pump-laden vesicles. In both of these systems, vesicle fusion and endocytosis of V-ATPase-containing vesicles is controlled through PKA-mediated phosphorylation, which is in turn modulated via a bicarbonate-sensitive adenylate cyclase (4, 145). Knowledge of the structure and modes of regulation of V-ATPase activity will likely prove crucial in the development of drugs to control their activity in cancer cells.
II. V-ATPASES AND CELL SIGNALING
A. Role of the V-ATPase in Cell Signaling
The ability of cells to sense and respond to extrinsic and intrinsic cues relies on a complex network of cellular signaling pathways. When these pathways are altered, homeostasis cannot be maintained. This loss of normal homeostatic control underlies tumorigenesis and cancer progression. The V-ATPase plays both direct and indirect roles in the control of cellular signaling. Critical pathways of growth, survival, and differentiation that are frequently altered in cancer rely on the V-ATPase, highlighting the appeal of V-ATPase modulation as a therapeutic strategy. Control of vesicular pH by the V-ATPase is essential for proper signaling by many plasma membrane receptors that traffic through the endomembrane network, including Notch, Wnt, G protein-coupled receptors (GPCRs), and receptor tyrosine kinases (RTKs). We will first consider these cases, and then discuss the direct role of the pump in nutrient sensing and mTORC1 activation.
B. V-ATPase Functions in Plasma Membrane Receptor Signaling and Recycling
Plasma membrane receptors, such as RTKs and GPCRs, stimulate intracellular pathways controlling growth, survival, and migration, and their misregulation is highly selected for in cancer development (156, 196). Decreasing pH in the endovesicle system plays an important role in receptor signaling and recycling. EGF receptor and insulin receptor signaling rely on low endosomal pH for ligand dissociation and receptor trafficking (3). Treatment with V-ATPase inhibitors inhibits endosomal acidification and blocks proper receptor trafficking, and can prolong signaling of activated receptors that accumulate in endosomal compartments (3, 219) or lead to apoptosis (166). GPCR signaling can also be regulated by endosomal pH changes. Parathyroid hormone (PTH) signaling is negatively regulated by decreasing pH. After internalization, as the activated receptor moves through the endocytic pathway, lower endosomal pH causes a shift in which proteins bind to the receptor. During active signaling, arrestin is bound, but at low pH, retromer binding is enhanced and PTH receptor signaling is turned off. V-ATPase activity promotes this shift, positioning it as a negative regulator of GPCR signaling (50).
C. V-ATPase and Wnt/β-catenin Signaling
Wnt signaling controls many aspects of cellular function, influencing gene expression, polarity, movement, intracellular calcium levels, and proliferation during normal development as well as in cancer (6). Wnt signaling requires V-ATPase function for proper trafficking and activation (Figure 3A). There are three classes of Wnt signaling. Each is initiated by binding of a Wnt ligand to its receptor Frizzled (Fz) in complex with a specific co-receptor (6). The canonical Wnt signaling pathway involves the co-receptor LRP6 (6). Binding activates the Fz-LRP6 receptor complex, which stimulates recruitment of the Disheveled protein and Axin from the β-catenin destruction complex. This results in stabilization of β-catenin (6). β-Catenin then accumulates, translocates to the nucleus, and promotes the transcription of Wnt target genes. Genetic or chemical inhibition of the V-ATPase blocks activation of the Wnt pathway by blocking the activating phosphorylation of the receptor following ligand binding (13, 27, 48, 182). In addition, V-ATPase function is necessary for internalization of the receptor after ligand binding, which is required for complete receptor activation (14, 179, 221). Furthermore, Rabconnectin 3, a positive regulator of the V-ATPase, is also necessary for Wnt signaling (205). Rabconnectin knockout leads to an initial decrease in Wnt signaling due to impaired receptor activation. However, prolonged inhibition causes receptor accumulation and spatial disorganization within the plasma membrane due to defects in endocytosis, leading to upregulated signaling, highlighting the role of the V-ATPase in both receptor activation and trafficking (205).
FIGURE 3.

The V-ATPase is required for cellular signaling. A: the role of the V-ATPase in Wnt signaling. Wnt ligand binds to the plasma membrane receptor Frizzled in complex with a coreceptor, here LRP6. LRP6 and Frizzled are bound by the prorenin receptor (PRR), which acts as an adapter between the receptor complex and the V-ATPase. Upon ligand binding, the complex must be internalized into a signaling endosome for full activation via LRP6 phosphorylation, which requires V-ATPase activity. This activation inhibits activity of the β-catenin destruction complex, allowing β-catenin to accumulate, translocate to the nucleus, and alter gene transcription. B: the role of the V-ATPase in Notch signaling. The Notch receptor is activated by binding of Notch ligand expressed on the surface of an adjacent cell. Ligand binding stimulates a series of proteolytic cleavages which release the Notch intracellular domain (NICD) from the membrane, allowing it to be imported into the nucleus and drive transcription of Notch target genes. The cleavage of the Notch receptor is enhanced by V-ATPase-dependent acidification of endosomes after receptor internalization. C: the role of the V-ATPase in mTORC1 signaling. The V-ATPase is part of the amino acid sensing machinery associated with the surface of the lysosome. In the absence of amino acids (left panel), the V-ATPase is tightly complexed with the Ragulator, and the RagGTPase heterodimer is inactive. As a result, mTORC1 is inactive in the cytosol, thus repressing anabolic processes and cellular growth. When amino acids are sufficient in the cell (right panel), the V-ATPase-Ragulator complex undergoes a conformational change, stimulating Ragulator to act as a GEF, activating the RagGTPases. The active Rags recruit mTORC1 from the cytosol to the lysosomal surface where it is activated by Rheb. Active mTORC1 then promotes cellular growth.
Additional evidence for the importance of the V-ATPase in Wnt signaling comes from its association with the PRR (also know as ATP6AP2). LPR6 and Fz interact strongly with PRR (13, 19, 27, 60), which was initially discovered by its association with the V-ATPase (105). Subsequent work demonstrated that PRR, which may serve as an accessory subunit of the V-ATPase complex, functions as an adaptor between LRP6, Fz, and the V-ATPase (27) and is necessary for Wnt receptor localization and activation (13, 19, 27, 60). PRR is essential for survival, even in organisms that do not express renin (5, 85, 97, 158), indicating that its essential functions are not those associated with its role in the renin-angiotensin system. Instead, it appears to be crucial for coordinating the V-ATPase-Wnt interaction (85). Although the V-ATPase and PRR most often appear to be positive regulators of Wnt signaling, PRR can act as a repressor in some contexts (13).
Because V-ATPase activity is required for receptor activation, it is also important for noncanonical Wnt signaling, which controls planar cell polarity through regulation of the cytoskeleton. Knockdown or overexpression of PRR leads to altered cell polarity and patterning defects in Drosophila (60). Similarly, knockdown of individual V-ATPase subunits causes morphological defects (60).
D. V-ATPase and Notch Signaling
Notch signaling is important in controlling cell fate and proliferation throughout development and adult life. This pathway is integrally linked to cancer stemness and metastatic potential (29, 42), and tumor expression correlates with poor survival (35). Like Wnt, Notch signaling relies on the V-ATPase for proper trafficking and activation (Figure 3B). The Notch receptor is a single-pass transmembrane protein expressed at the surface of signal-receiving cells. Receptor activation occurs after interaction of the receptor with a transmembrane ligand expressed on the plasma membrane of a juxtaposed cell (228). Ligand binding to the extracellular domain stimulates processing of the receptor via successive proteolytic cleavage events that release the intracellular domain (NICD). The NICD translocates to the nucleus where it alters gene transcription to alter cell identity and growth (228). Correct processing and trafficking of the Notch receptor requires V-ATPase function (40, 86, 155, 178, 206, 207, 223). Notch receptor expression is also impaired by prolonged V-ATPase inhibition (198). The first evidence for the involvement of the V-ATPase in Notch signaling came from studies in Drosophila, which demonstrated that mutations in V-ATPase genes or in Rabconnectin-3A and 3B, both regulators of V-ATPase function, diminished Notch signaling and altered receptor trafficking (206, 223). Expression of the NICD rescued these effects, indicating that receptor processing was inhibited in mutant cells. In particular, S3 cleavage by γ-secretase in early endosomes that releases the NICD was shown to be V-ATPase dependent (206, 223). This cleavage defect upon V-ATPase inhibition could be due to the dependence of γ-secretase on an acidic environment to cleave the Notch receptor, or it could be that the acidic pH generated by the V-ATPase serves to activate cofactors that stimulate cleavage. Similar effects were also observed in normal and transformed human cell lines, as well as during mouse development (86, 92, 178, 198). In Notch-addicted breast tumor cell lines, V-ATPase inhibition reduced cell proliferation (86). However, while treatment of T-ALL cells with the V-ATPase inhibitor bafilomycin did reduce proliferation, there was no effect on Notch signaling, and the growth effects were attributed to alterations in Akt signaling (86). A recent study in endocrine-resistant breast cancer further links the upregulation of DMXL2, the human homolog of Rabconnectin-3, and subsequent activation of the V-ATPase, to Notch-driven epithelial to mesenchymal transition (EMT) (40). DMXL2 is upregulated in many ERα positive tumors that progress during treatment with endocrine therapy as well as in metastatic lesions (40). However, contrary to these previous studies, it was recently reported that inhibition of the V-ATPase increased Notch signaling in triple negative breast cancers. In this case, a similar accumulation of Notch receptors due to loss of lysosomal degradation is observed, but the consequence of this accumulation is persistent Notch signaling (143). Further studies will clarify the factors underlying this difference.
E. mTORC1 Activation and the Role of the V-ATPase in Nutrient Sensing
V-ATPase function is necessary for activation of mechanistic target of rapamycin complex 1 (mTORC1) (11, 30, 51, 74, 112, 190, 213, 219, 229, 233). The mTOR pathway is one of the most frequently dysregulated pathways in cancer (93). mTOR, which is a protein kinase, nucleates two complexes, mTOR complex 1 and complex 2 (mTORC1 and mTORC2). mTORC1 is a central node of cellular signaling, receiving inputs from growth factor receptors, nutrient availability, and energy status. mTORC2 is involved in survival signaling, and its activity is independent of V-ATPase function, unlike mTORC1. The role of the V-ATPase in mTORC1 signaling is unique in that the pump directly contributes to signaling of the pathway, although the precise mechanism of its involvement is unknown. It was first reported that inhibition of the V-ATPase induced autophagy and caused accumulation of autophagosomes, a process repressed by mTORC1. Indeed, these authors observed that mTORC1 signaling was repressed by V-ATPase inhibition (112). Later work demonstrated that the V-ATPase functions as a critical component of the nutrient sensing machinery that signals amino acid sufficiency to mTORC1 (Figure 3C) (11, 36, 233). During amino acid starvation, mTORC1 is primarily localized in the cytoplasm. When amino acids are present, mTORC1 is recruited to the surface of the lysosome by the RagGTPases (83, 168). Rag localization is controlled by the pentameric Ragulator complex, which is bound to the lysosomal membrane. The V-ATPase directly interacts with the Ragulator (30, 36, 233). Associations of the Ragulator with V1 are strengthened during amino acid starvation, while association with V0 is constant regardless of amino acid availability (233). In the amino acid starved state, the Rags, Ragulator, and V-ATPase are tightly associated, and mTORC1 is excluded from the lysosome. After amino acid addition, the activating signal is somehow transmitted via the V-ATPase to the Ragulator, which alters its interaction with the RagGTPases to allow mTORC1 to be recruited to the lysosomal surface, where it is activated (11).
It was recently demonstrated that V-ATPase assembly and activity in lysosomes are increased in response to amino acid starvation (190). This effect appears to be unique from the amino acid-dependent change in the V-ATPase-Ragulator complex involved in mTORC1 activation for several reasons. First, amino acid starvation increases V-ATPase activity under conditions where mTORC1 activity is reduced (190). Second, changes in V-ATPase activity in response to starvation of individual amino acids is variable and does not correlate with changes in mTORC1 activity under the same single amino acid starvation conditions (190). Finally, the effects of chloroquine on these two processes are not consistent with a causal relationship between V-ATPase assembly and mTORC1 activity. Thus, while complete inhibition of V-ATPase activity prevents mTORC1 activation, the amino acid-dependent change in activity and assembly are not required for amino acids to stimulate mTORC1 activity (190). Instead, it is possible that elevated assembly and activity of the V-ATPase in lysosomes leads to lower lysosomal pH, increased breakdown of proteins within lysosomes by acid-dependent proteases, and increased amino acid availability as a result of this increased lysosomal proteolysis (190).
Amino acid availability is necessary but not sufficient for complete activation of mTORC1, which also receives inputs from many indicators of cell and organismal state, such as growth factors. Interestingly, the V-ATPase has been found to be involved in mTORC1 activation in response to a number of stimuli, including growth factors and GPCR activation (213, 219). EGF was shown to increase V-ATPase activity by stimulating increased assembly (39). This increased V-ATPase activity led to increased lysosomal acidification and increased levels of essential amino acids in the cell, which is suggested to be important in EGF-induced stimulation of mTORC1. Cycloheximide, which inhibits translation and thus raises intracellular amino acid levels, appears to rescue the effect of bafilomycin on mTORC1 activation (219). This is in contrast to a previous report that salicylamide A does inhibit cycloheximide-induced mTORC1 activation (233). Stimulation of the GPCRs OX1R and OX2R by orexin also leads to activation of mTORC1 that is V-ATPase dependent (213).
The V-ATPase-Ragulator complex is also implicated in activation of AMPK, a critical sensor of cellular energy which is activated during cellular stress and energy depletion (41). This suggests that the V-ATPase may be a crucial part of a metabolic switch (229). The V-ATPase-Ragulator complex appears to serve as a docking site for activators of AMPK on the lysosome during glucose starvation. When the V-ATPase is knocked down, AMPK is not activated at low glucose concentrations. However, inhibition of the V-ATPase by concanamycin A treatment is suggested to induce the Ragulator-V-ATPase conformation found during starvation, leading to increased association of AMPK activators with the lysosomal components.
Lysosomes undergo a variety of changes during carcinogenesis. Cancer cells often display enhanced lysosomal biogenesis, lysosomal protease activity, and lysosomal trafficking towards the leading edge (53, 76, 164). Enhanced V-ATPase expression in cancer cells allows lysosomes to participate in processes critical to carcinogenesis. Interestingly, V-ATPase expression is also regulated by mTORC1 via the MiT/TFE family of transcription factors, MITF, TFE3, and TFEB (116). These highly related transcription factors are master regulators of lysosomal biogenesis, and their expression is largely tissue specific. TFEB is the best studied as a regulator of V-ATPase gene expression (146, 180). Phosphorylation of TFEB by mTORC1 sequesters it in the cytoplasm. However, when mTORC1 activity is lost, TFEB translocates into the nucleus and stimulates transcription of genes necessary for autophagy and lysosome biogenesis, including V-ATPase genes (51, 84, 146, 180). Similar results have been found for melanocyte-specific family member MITF in primary melanocytes and melanoma cells (230). Recent studies in Drosophila demonstrate that regulation of V-ATPase gene expression by MiT/TFE transcription factors is conserved (16, 198, 230), and that control of lysosomal function by these factors plays an important role in cellular fate determination, particularly by influencing Notch signaling (198).
Together, these studies emphasize the importance of the V-ATPase in the signaling network that maintains cellular homeostasis and that is often dysregulated during tumor formation.
III. V-ATPASES AND CELL DEATH
A. V-ATPases and Apoptosis of Cancer Cells
Induction of apoptosis by V-ATPase inhibition has been reported in both human and murine models encompassing many tumor types (31–33, 55, 67, 68, 112, 123, 129, 135, 139, 170, 171, 174). The V-ATPase plays essential roles in fundamental cellular processes, as detailed above. It is therefore not surprising that prolonged inhibition of V-ATPase activity is lethal to cells. However, there is evidence that cancer cells are particularly reliant on the V-ATPase for survival, exhibiting greater sensitivity to V-ATPase inhibition than nontransformed cells (12, 129, 135, 174).
Poor vascular density of tumors, localized hypoxia, and increased glycolytic flux produce a high acid load in tumor tissue (28). V-ATPase activity is important for excretion of excess acid into the extracellular environment, thus maintaining a neutral cytosolic pH (24, 117, 134). To aid this, V-ATPase expression on the whole may be upregulated (12), or the pump may be localized to the plasma membrane through the expression of specific V-ATPase subunit a isoforms known to direct the pump to the cell surface (21, 63). This activity contributes to reversal of the normal cellular pH gradient across the plasma membrane, specifically alkalinization of the cytosol and acidification of the extracellular space in tumor cells (181, 214). This reversal allows cancer cells to evade apoptosis, and increases multidrug resistance, cell proliferation, and cell migration and invasion, which are discussed in detail below. Treatment with V-ATPase inhibitors causes loss of the pH gradient across the plasma membrane, increasing the external pH and decreasing the internal pH, leading to slowed growth and increased cell death (31). The V-ATPase is particularly important in hypoxic environments typical of solid tumors and early, poorly vascularized neoplasias, as evidenced by the finding that treatment with V-ATPase inhibitors increased hypoxia-induced cell death in breast cancer xenografts (55).
In addition to its role in cytosolic pH homeostasis, the V-ATPase is important for maintaining the pH of intracellular compartments. Dysregulation of protein synthesis and nutrient scarcity make cancer cells particularly prone to endoplasmic reticulum (ER) stress. During ER stress, Bax inhibitor-1 (BI-1) activates lysosomal activity to increase protein turnover as part of the unfolded protein response. Inhibition of the V-ATPase prevents this compensation, and cells undergo apoptosis (95). RTK signaling may also confer susceptibility to V-ATPase inhibition. In cells overexpressing the epidermal growth factor receptor (EGFR), addition of a V-ATPase inhibitor during stimulation of the receptor by EGF caused cell death (226). This is likely due to an accumulation of activated EGFR-EGF complexes in endosomes that results in apoptosis (166). In addition, indirect alteration of V-ATPase activity causes apoptosis. Interestingly, downregulation in gene expression, as with diphyllin treatment (182), or increases in V-ATPase activity by disruption of microtubules (12) both cause apoptotic cell death, highlighting the necessity for precise V-ATPase regulation in maintaining cell viability.
Mechanistically, V-ATPase inhibition first stimulates an increase in reactive oxygen species (ROS) (32, 67, 112, 123, 170, 210). In RAW 264.7 murine leukemia cells, bafilomycin or concanamycin A treatment induced nitric oxide (NO) production via upregulation of NO synthase expression (67). Blocking the increase in ROS prevents mitochondrial depolarization and subsequent apoptosis (32, 67). While ROS production is a critical stimulus for apoptosis, it also serves as a positive signal to cytoprotective mechanisms, such as autophagy (112, 174) and activation of survival pathways, including induction of HIF1α (174), stimulation of Akt (170), and proteasomal degradation of proapoptotic factors (170). V-ATPase inhibition can also induce protective MAPK inhibition (55). Blockage of such protective processes increases V-ATPase inhibition-induced apoptosis. In addition to ROS induction, V-ATPase inhibition induces an increase in lysosome pH, resulting in a compromise of lysosomal membrane integrity and a decrease in cytosolic pH (32, 112). Finally, mitochondrial membrane potential is lost and cytochrome c release activates intrinsic apoptosis pathways (32, 67, 112, 123, 174).
While the broad induction of apoptosis by V-ATPase inhibition makes it an attractive target for cancer therapy, it is important to note that V-ATPases can play a proapoptotic role. Endosomal acidification is important for TNF-related apoptosis-inducing ligand (TRAIL) signaling. Cotreatment of cells with TRAIL and V-ATPase inhibitors attenuates the apoptotic response (69), highlighting that rational application of V-ATPase inhibition will be necessary to best exploit it as a target in cancer treatment.
B. V-ATPases and Autophagy
Autophagy is induced during periods of starvation and stress. Autophagy involves the formation of double membrane vesicles (autophagosomes) that sequester cytoplasmic contents and deliver them to lysosomes, allowing recycling of their contents by degradative lysosomal enzymes. This process reduces cellular stress by degrading long-lived or damaged cellular components, including proteins and organelles. Additionally, it can provide cellular energy and building blocks during periods of nutrient deprivation and hypoxia (203, 222). However, prolonged or overactive autophagy is detrimental and induces cell death (203, 174).
Autophagy requires V-ATPase activity to support the activity of pH-dependent proteases and possibly lysosome/autophagosome fusion. Thus inhibition of the V-ATPase blocks autophagic flux (75, 79, 121, 122, 126, 133, 138, 220). Furthermore, induction of autophagy involves upregulation of V-ATPase gene expression via the mTORC1-mediated activity of the TFE family of transcription factors (discussed above). This enables increased lysosomal biogenesis and function to support autophagy (16, 222).
The role of autophagy in cancer is complex, as it has been implicated as both a tumor suppressive and tumor promoting mechanism. For example, reduction of autophagy is correlated with the accumulation of cellular stressors such as misfolded proteins, protein aggregates, and defective mitochondria. This can result in the generation of ROS, which damage DNA and thus may promote tumorigenesis (120, 203, 222). Prolonged autophagy can also induce cell death (203, 174). However, cancer cells can also utilize autophagy as a survival mechanism to cope with starvation, high metabolic demand, hypoxia, acidosis, and drug treatment. Autophagy promotes recycling of essential biomolecules that supports the energy demands of the tumor cell. The removal of damaged organelles and proteins also helps to prevent apoptosis by maintaining mitochondrial function and limiting the levels of ROS (222). Autophagy is also induced upon treatment with anti-cancer agents, such as radiation and chemotherapy, as an attempt to avoid cell death (203). In this sense, the V-ATPase may enhance tumor cell survival through its participation in autophagy. Interestingly, treatment with low concentrations of V-ATPase inhibitors induces autophagy in tumor cells, primarily as a means to prolong survival. However, prolonged treatment with low concentrations of V-ATPase inhibitors eventually results in apoptosis. Cell death occurs more rapidly when tumor cells are treated with a combination of V-ATPase and autophagy inhibitors (109, 174).
IV. V-ATPASES AND DRUG RESISTANCE
Multidrug resistance (MDR) presents a huge challenge in the successful treatment of cancer. Most cancer deaths are caused by recurrence or metastasis of tumors that have escaped initial treatment and have become drug resistant. MDR occurs through a number of mechanisms. Transporters, such as P-glycoprotein/MDR1, can be upregulated which allows cells to directly remove drugs from the cytoplasm. MDR can also be induced by changes in the normal pH gradient across the plasma membrane (183), which is significantly influenced by the activity of V-ATPases in the plasma membrane. Early work to understand physiological factors that could determine chemotherapeutic efficacy found that acidic extracellular pH reduced the ability of many drugs, particularly weak bases such as anthracyclines, to effectively kill cancer cells (15, 44, 62, 211). Mechanistically, at low pH, these drugs are protonated and lose the ability to enter cells or become trapped inside acidic vesicles within the cell, thus protecting the DNA of the tumor cell from the action of the drug (44, 62). Furthermore, cytosolic pH is highly correlated with response to chemotherapy (81). Indeed, an alkaline cytosolic pH and acidic extracellular pH is common across tumors and has been proposed to be a new hallmark of cancer (181, 214).
Mechanistic characterization of how cells develop and maintain an alkaline cytosolic pH despite an abnormally acidic extracellular pH has highlighted the important role of the V-ATPase in this process. It was first observed that treatment of resistant cells with a V-ATPase inhibitor allowed vincristine or doxorubicin to accumulate within the cell and prevented drug efflux out of the cell from occurring (114). While resistant cells exhibited robust V-ATPase activity at the plasma membrane, this was in part due to endomembrane exocytosis, facilitating the release of vesicle contents, including sequestered chemotherapy drugs, into the extracellular environment (118, 153). Additionally, inhibition of the V-ATPase decreases cytosolic pH, which hinders drug sequestration and enhances cell killing (98).
The V-ATPase is commonly activated in drug-resistant settings. Lung and esophageal tumors expressing higher levels of V-ATPase were more likely to be drug resistant (71, 107). Similarly, high expression in ovarian tumors was associated with worse survival outcomes (96). Some cancer stem cells are highly drug resistant and are reported to express high levels of V-ATPase genes (167). V-ATPase expression increases in response to drug treatment, which is accompanied by an increase in cellular pH and lysosomal drug accumulation (70, 131, 199). In vitro binding studies found that cisplatin binds DNA with much higher affinity at lower pH (131). In the case of the gene encoding the small c subunit, upregulation in response to drug treatment can be caused by an increase in transcription or enhanced mRNA stability (199). However, it is important to note that expression of V-ATPase genes does not always correlate with drug resistance, even when cotreatment with V-ATPase inhibitors and chemotherapies overcomes resistance (8). Expansion of the lysosomal compartment has also been observed to correlate with drug resistance due to the increased capacity to sequester drugs. In cells with this phenotype, V-ATPase inhibition can restore drug sensitivity (111, 142).
Cancer cells upregulate V-ATPase activity directly and also modify V-ATPase regulatory effectors. TMSG1 (also known as LASS2) interacts directly with the V-ATPase and acts as a negative regulator (39, 124, 217, 218). Its expression is downregulated in drug-resistant breast cancer cells, and overexpression increases chemosensitivity (38). In patients, low TMSG1 expression is correlated with poor prognosis (38). As mentioned above, DMXL2 is a positive regulator of V-ATPase activity (178). In ERα+ breast cancer patients, a molecular subtype which makes up ∼70% of all breast cancers, DMXL2 is upregulated in the 40% of these patients who continue to progress on therapy (40). Targeting the transmembrane protein TM9SF4, another positive enhancer of V-ATPase activity, increases drug sensitivity (103).
Combining V-ATPase inhibition and cytotoxic drugs overcomes the protective change in pH and causes synergistic killing (70, 96, 131, 167, 199, 227). The combination is effective in resistant lines, but does not enhance drug efficacy in sensitive lines (96). Combination of V-ATPase inhibition with targeted therapy can also overcome resistance, inducing apoptosis and inhibiting reactivation of the targeted pathway (173). Pretreatment of cells exhibiting MDR in vitro as well as cancer cells in mouse xenograft models with acid-activated proton pump inhibitors before administration of chemotherapeutic drugs caused a large and significant increase in cell killing, increased extracellular and lysosomal pH, and improved retention of drugs within the cell (104). Importantly, cotreatment did not have the same effect, suggesting that timing of V-ATPase inhibition and drug treatment may be important in employing this strategy (32, 104).
Manipulation of tumor pH may present a significant therapeutic opportunity in overcoming drug resistance. Inhibiting the V-ATPase interferes with an important mechanism by which cells can escape drug toxicity, namely, the sequestration of drugs in the extracellular environment or in acidic intracellular vesicles.
V. V-ATPASES AND TUMOR METASTASIS
A. Expression and Localization of V-ATPases in Tumor Cells
V-ATPases were first identified as participating in the control of both cytoplasmic and lysosomal pH in a variety of human tumor cell lines from functional studies using fluorescent pH indicators that localized to specific cellular compartments (117). Subsequent work demonstrated that the V-ATPase is present on the plasma membrane of invasive breast cancer cells, but not noninvasive breast cancer cells, and participates in control of cytoplasmic pH (175). The V-ATPase has since been found to be expressed on the plasma membranes of numerous invasive cancer cell types, including melanoma, Ewing sarcoma, and breast, liver, lung, ovarian, esophageal, prostate, and pancreatic cancers (9, 21, 23, 24, 63, 71, 90, 107, 125, 136, 176, 216). Furthermore, overexpression of particular V-ATPase subunits has been observed in both human cancer cell lines and human tumor samples. For example, subunit V1C is overexpressed in oral squamous cell carcinoma samples (47, 141, 147), and subunit V0c is overexpressed in human hepatocellular carcinoma samples (216) and pancreatic cancer samples (139). Expression of subunit V1E and subunit V1A is increased in pancreatic cancer and gastric cancer, respectively, and expression correlates with tumor grade (23, 101). V-ATPase subunit overexpression has also been observed in breast, lung, and esophageal cancer tissues (59, 71, 107). The 100 kDa V0 subunit a, which is present in four isoforms in mammalian cells (a1–a4) and is responsible for localizing the V-ATPase to specific membranes within the cell, has also been found to be overexpressed in a number of cancer cell types, and will be discussed further below.
As these studies did not report the expression levels of multiple subunits of the V-ATPase, it is possible that several (or all) of the V-ATPase subunits are overexpressed in these contexts. Alternatively, overexpression of a single subunit may have important implications in tumor cells. For example, alteration in the expression of the subunit a isoforms could alter V-ATPase localization. Interestingly, a recent study has found that overexpression of the V1G1 subunit in glioblastoma samples correlates with poor survival. The remaining V-ATPase subunits were not overexpressed in these samples (33). It is important to note that it is unknown whether overexpression of a single subunit results in increased V-ATPase activity. This would suggest that the overexpressed subunit is limiting for activity and that the other subunits are present in excess.
Future work is required to assess the expression patterns of multiple subunits in cancer tissues as well as whether activity can be modulated through variations in expression of a single subunit. While it is known that TFEB and Mitf regulate V-ATPase subunit gene expression (discussed above), additional factors may be involved, and these need to be identified and characterized. Furthermore, genetic alterations may also lead to subunit overexpression via amplification of V-ATPase genes. This may occur due to selection for increased subunit expression, or possibly through expansion driven by selection for amplification of nearby cancer related genes. For example, the gene encoding the a3 isoform of subunit a is on the long arm of chromosome 11 near the gene encoding cyclin D1, which is frequently amplified across tumor types (132).
B. V-ATPases Are Important for Tumor Cell Invasion and Migration
The presence of the V-ATPase on the plasma membrane of invasive cancer cells and the overexpression of V-ATPase subunits in human cancer samples has suggested a possible role for the V-ATPase in cancer cell migration and invasion. Several studies have demonstrated that both in vitro invasion and migration of human breast cancer cells is dependent on V-ATPase activity. Treatment of highly invasive MB231 and MCF10CA1a human breast cancer cells with the specific V-ATPase inhibitors concanamycin A and bafilomycin results in a significant decrease in in vitro invasion as measured by transwell assays (21, 24, 63, 175). Furthermore, the in vitro migratory capabilities of MB231, SKBR3, and 4T1 breast cancer cells are reduced upon treatment with V-ATPase inhibitors (24, 43, 175, 215). Similar results have been obtained with other cancer cell types. The invasion and migration of pancreatic cancer cells are reduced upon treatment with concanamycin A (23), and V-ATPase inhibition decreases in vitro invasion of prostate cancer cells and migration of ovarian cancer cells (90, 125). Finally, knockdown of subunit c of V0 and subunit A of V1 using siRNA treatment, which results in complete ablation of V-ATPase activity, reduces the invasiveness of liver and gastric cancer cells, respectively (101, 108, 216). Interestingly, overexpression of subunit C in Drosophila wing epithelia results in a tumorlike transformation of the wing disc, marked by epithelial cell overgrowth and enhanced invasiveness, further supporting a role for the V-ATPase in tumor cell invasion (150).
C. Function of a Subunit Isoforms and Plasma Membrane V-ATPases in Tumor Cell Invasion and Migration
Because the available V-ATPase inhibitors, such as concanamycin A, bafilomycin, and archazolid, are all membrane permeable, treatment of cells with these inhibitors reduces the activity of both intracellular and plasma membrane V-ATPases. A similar limitation applies to studies in which expression of subunits common to all V-ATPases, such as subunit c, is reduced by treatment of cells with siRNA. These studies are therefore unable to determine whether intracellular or plasma membrane pumps, or both, are critical for cancer cell invasion and migration in vitro. Evidence for a role of plasma membrane V-ATPases in breast cancer cell invasion was first provided through studies examining the expression and function of isoforms of subunit a of the V0 domain. The cytoplasmic NH2-terminal domain of subunit a is responsible for localizing the pump to specific cellular membranes (80). It has been shown that the subunit a isoforms a3 and a4, which localize the pump to the plasma membranes of osteoclasts and renal intercalated cells, respectively, are upregulated at the mRNA level in invasive MB231 breast cancer cells relative to noninvasive MCF7 cells (63, 140, 185, 186, 200, 201). Significantly, MB231 cells show reduced invasiveness when a3 and a4 are knocked down by isoform-specific siRNAs, and loss of a4 reduces localization of the V-ATPase at the plasma membrane (63). A more recent study has found that subunit a3 is overexpressed in invasive MCF10CA1a breast cancer cells relative to MCF10a breast epithelial cells and that siRNA-mediated knockdown of a3 again reduces in vitro invasiveness (21). Importantly, overexpression of a3 in the noninvasive MCF10a cell line increases both invasiveness and localization of the V-ATPase to the plasma membrane (21). These results suggest that overexpression of specific a subunit isoform a3 by invasive breast cancer cells increases trafficking of V-ATPases to the plasma membrane where they enhance tumor cell invasion (Figure 4). Moreover, these findings are not limited to breast cancer cells. For example, a3 is expressed on the plasma membrane of invasive pancreatic cells and has been shown to be involved in the invasion and migration of melanoma cells (23, 136). Furthermore, a recent study that examined the role of a2 in ovarian cancer also demonstrated significant overexpression of a3 in ovarian cancer tissues relative to normal samples (90). Similarly, a4 is overexpressed in human glioma samples and is required for glioma cell migration and invasion (52). While studies thus far suggest that overexpression of subunit a isoforms can alter V-ATPase localization and enhance invasiveness in cancer cells, the mechanism by which these isoforms are upregulated is poorly understood. Additionally, the localization patterns of subunit a isoforms in cancer cells remain uncertain due to the scarcity of reliable, isoform-specific antibodies.
FIGURE 4.

The promotion of metastasis via plasma membrane V-ATPase activity. Upregulation of V-ATPase subunits such as a3 or a4 localize V-ATPases to the plasma membrane, where they participate in a number of processes that enhance cancer cell invasion and migration. Plasma membrane V-ATPases are believed to activate pH-dependent proteases, promote trafficking of pro-invasive factors, and enhance extracellular protease activity. They are also thought to interact with and alter actin dynamics, enhance trafficking of pro-migratory factors, and alter ion channel conductance to promote migration. Enhanced invasion and migration of cancer cells eventually leads to the escape of tumor cells from the primary tumor site and the development of metastases.
These studies do not distinguish between the direct participation of particular a subunit isoforms in targeting of V-ATPases to the plasma membrane and a more indirect role in which V-ATPases within intracellular compartments participate in trafficking events that are necessary for correct targeting of V-ATPases to the cell surface. To distinguish between these possibilities, a recent study tested the effect of specific ablation of plasma membrane V-ATPase activity using either an inhibitory antibody or a membrane-impermeant small molecule inhibitor (24). The results show that selective inhibition of plasma membrane V-ATPase activity inhibits the invasion and migration of MB231 cells (24), thus providing strong support for an important functional role of cell surface V-ATPases in these processes. Nevertheless, it remains possible that intracellular pumps may also function in invasion or migration of cancer cells. For example, general V-ATPase inhibition reduced invasiveness of poorly metastatic prostate cancer cells with low plasma membrane V-ATPase expression (114).
D. Mechanism of V-ATPase Promotion of Tumor Cell Invasion and Migration
The mechanisms by which V-ATPases contribute to cancer cell migration and invasion have yet to be fully elucidated, although several hypotheses exist. One possibility is that V-ATPases promote the activity of acid-dependent proteases that are involved in invasion (24, 46) (Figures 4 and 5). Such proteases may either directly cleave the extracellular matrix necessary to escape a primary tumor or to invade a new site or activate other proteases that participate in these processes. Two important classes of proteases that have been implicated in tumor metastasis are cathepsins and matrix metalloproteases (MMPs). Cathepsins are proteases that normally reside in lysosomes and that require a low pH for both their activity and their proteolytic activation (115, 180, 190), while MMPs, although not directly pH dependent, can nevertheless be activated at low pH, in particular by cathepsin-dependent cleavage (83, 166, 190). Both cathepsins and MMPs are secreted by tumor cells and have been shown to participate in invasion (8, 54, 110, 115, 143, 185). Moreover, both intracellular and plasma membrane V-ATPases may contribute to the promotion of protease activity. Intracellular V-ATPases appear to promote the activation of invasion-promoting proteases such as MMP-2 and cathepsin B within secretory compartments of breast cancer cells (59, 89). In contrast, plasma membrane V-ATPases may create a local acidic microenvironment outside of the cell that promotes the activation and/or activity of proteases involved in invasion (24) (Figure 5). Thus plasma membrane V-ATPases participate in the creation of the acidic extracellular environment of tumor cells, and this environment supports a more invasive phenotype. For example, neutralization of the acidic extracellular environment reduces both metastasis of breast cancer cells in mice and extracellular MMP activity in breast tumors (139, 140). Similarly, inhibition of V-ATPase activity reduces extracellular MMP-2 or MMP-9 activity in a variety of invasive cancer cell lines (21, 99, 121).
FIGURE 5.

Mechanisms by which the V-ATPase enhances tumor cell invasiveness and survival. The primary mechanism by which the V-ATPase is thought to promote tumor cell invasion is through the promotion of extracellular pH-dependent protease activity. Intracellularly, V-ATPase-mediated acidification of intracellular compartments may allow for activation of proteases that are then trafficked to the cell surface. V-ATPases localized to the plasma membrane of cancer cells upon upregulation of the subunit a isoforms a3 or a4 may create an acidic microenvironment that allows for activation of pH-dependent proteases and/or enhances pH-dependent protease activity outside of the cell. Enhanced plasma membrane V-ATPase activity also increases acid secretion out of the cytosol. This is important for tumor cell survival as tumor cells generate large amounts of metabolic acid due to their reliance on glycolytic metabolism and are able to survive in highly acidic microenvironments.
A possible mechanism by which V-ATPases may promote cancer cell migration is through interaction with the actin cytoskeleton. V-ATPase subunits have been shown to interact or colocalize with actin in a variety of cell types, including invasive breast cancer cells (18, 20, 62, 85, 186, 209). Furthermore, loss of subunit C of V1 disrupts proper actin arrangement in breast cancer cells (20). It remains unclear whether pharmacological inhibition of the V-ATPase alters pump-actin interactions and actin arrangement. In microvascular endothelial cells, plasma membrane V-ATPases have been speculated to contribute to migration by localizing to the leading edge, where they could promote actin polymerization by creating a local alkaline cytosolic pH (142). It is possible that plasma membrane V-ATPases may contribute to cancer cell migration in a similar manner. Alternatively, V-ATPases may facilitate the proper localization of factors involved in cell migration. For example, treatment of SKBR3 breast cancer cells with specific V-ATPase inhibitors alters the proper localization of Rac1 and EGFR, both of which are involved in cell migration (191). Finally, the V-ATPase has been implicated in regulating membrane potential, which in turn could alter ion channel conductance. V-ATPase activity at the plasma membrane may alter calcium influx into the cell, which has been linked to enhanced migratory capability (Figure 4) (42, 152).
Interactions with other cellular targets may also be important in the function of V-ATPases in tumor cell invasion and migration. It has been suggested that TMSG1 interacts with and inhibits V-ATPase activity (39, 112, 193, 194). In breast cancer cells, overexpression of TMSG1 decreases V-ATPase activity, reduces cell growth, and inhibits in vitro invasion and migration (39, 112). In poorly metastatic prostate cancer cells, silencing of TMSG1 increases V-ATPase activity, in vitro invasion and migration, and in vivo lymph node metastasis (193, 194). The mechanisms by which TMSG1 regulates V-ATPase activity are not yet understood. In contrast to TMSG1, the HRG-1 protein has been reported to interact with and promote V-ATPase activity. HRG-1 colocalizes with the V-ATPase at the plasma membrane of invasive, but not noninvasive, cancer cells. Loss of HRG-1 reduces V-ATPase expression at the plasma membrane and reduces the in vitro invasion, extracellular acidification, and extracellular protease activity of MB231 breast cancer cells. Conversely, HRG-1 overexpression enhances the invasiveness of noninvasive MCF7 breast cancer cells (45). Finally, the transmembrane protein TM9SF4 has been shown to interact with the V-ATPase and modulate activity through regulation of pump assembly. Silencing of TM9SF4 in colon cancer cells reduces in vitro invasiveness (103). Thus the roles of V-ATPases in promoting tumor cell invasion and migration may be complex, and additional studies will be required to fully elucidate these roles.
VI. V-ATPASE AS A DRUG TARGET
The observations that V-ATPase inhibition reduces in vitro invasion and migration of a variety of cancer cell types and that the pump is involved in signaling, growth, survival, and drug resistance of cancer cells suggest that the V-ATPase represents an important and novel anti-cancer target (Figure 6). A limited number of studies have examined the effects of V-ATPase inhibition on in vivo cancer growth and metastasis. In a mouse model of liver cancer, knockdown of subunit c of V0 resulted in reduced primary tumor growth and decreased intrahepatic and pulmonary metastasis (99, 192). Treatment of mice with the V-ATPase inhibitor archazolid dramatically reduced the ability of intravenously injected 4T1 breast cancer cells to colonize the lung (191). Similarly, 4T1 cells pretreated with archazolid have a reduced ability to disseminate to the lung after tail vein injection in mice (150). Knockdown of subunit C of V1 in 4T1 cells implanted into the mammary fat pad of mice reduced tumor formation; decreased metastasis to the lung, liver, and bone; and improved mouse survival after xenograft (43). Finally, knockdown of subunit a3 in a mouse model of melanoma significantly reduces metastasis to both the lung and bone, highlighting the importance of the subunit a isoforms in cancer cell invasion and migration (121).
FIGURE 6.
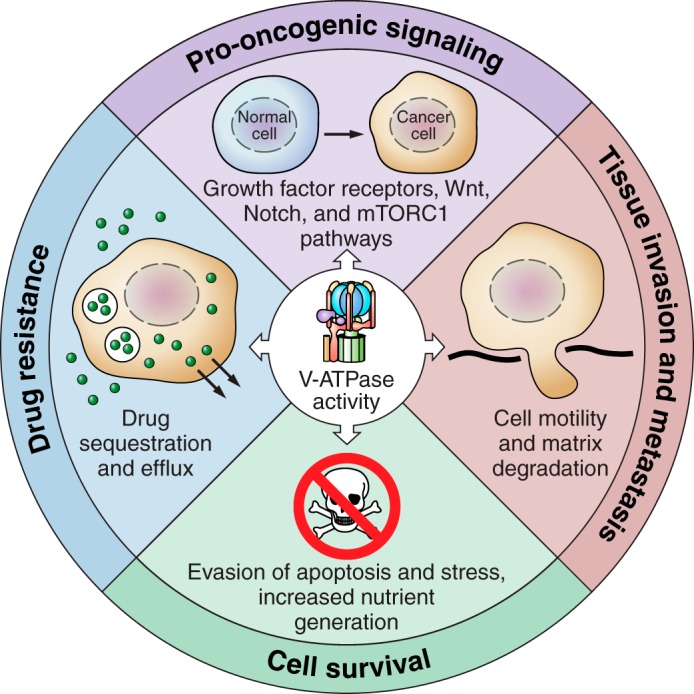
The role of V-ATPases in cancer. V-ATPase activity supports pro-oncogenic signaling pathways, including growth factor receptors, Wnt, Notch, and mTORC1 (top panel). V-ATPase activity allows tumor cells to evade apoptosis and various cellular stressors (right panel). V-ATPase activity promotes the ability of tumor cells to invade surrounding tissue and metastasize to secondary sites within the body by promoting cell motility and extracellular matrix degradation (bottom panel). V-ATPase activity contributes to drug resistance by promoting drug trapping in acidic cellular spaces and promoting drug efflux out of the cell (left panel).
The mechanisms by which inhibition of V-ATPase activity or loss of subunit a3 reduces in vivo tumor growth and metastasis are not fully understood, but likely involve participation of the pump in protease activation as well as tumor cell migration, survival, and proliferation. Treatment with the V-ATPase inhibitor archazolid reduces cathepsin B abundance and activity in tumors in a mouse model of breast cancer, supporting the hypothesis that the V-ATPase contributes to metastasis through the promotion of protease activity (89). Reduced metastasis of breast cancer cells after V-ATPase inhibition may also be a result of the induction of anoikis or increased susceptibility to apoptosis under hypoxic conditions (51, 150).
Interestingly, V-ATPase inhibition can also activate protective cellular responses (51, 103, 150, 154). For example, treatment of MB231 cells with bafilomycin causes upregulation of prosurvival MAP kinases (55). Combination treatment of bafilomycin with the kinase inhibitor sorafenib inhibits the activity of these MAP kinases and reduces in vivo breast tumor growth and metastasis to a greater degree than bafilomycin or sorafenib alone. This suggests that V-ATPase inhibitors may have their optimal anti-tumor effects in combination with other anti-cancer agents.
While the V-ATPases have clearly been implicated in cancer cell signaling, survival, drug resistance, and metastasis, the question remains whether V-ATPases can serve as a safe target for anticancer therapeutic agents, particularly in light of the importance of V-ATPases in a wide range of normal physiological processes. Several lines of evidence suggest this is the case. Prolonged exposure to specific V-ATPase inhibitors is lethal to all mammalian cell types (54, 73, 78). However, cancer cell survival has been reported to be more sensitive to V-ATPase inhibition than that of normal cells (10, 116, 120, 154), likely due to their heavy reliance on V-ATPases to remove the large amount of metabolic acid generated by the extensive glycolytic metabolism that occurs in cancer cells. Furthermore, patients with the genetic disease X-linked myopathy with excessive autophagy (XMEA), which is caused by a mutation in a V-ATPase chaperone protein required for assembly of V0, exhibit a global reduction in V-ATPase activity of 70–90%. Despite this, patients typically only have defects in skeletal muscle function. This suggests that most normal cells may be able to survive and function with reduced V-ATPase activity during cancer treatment (134). In this sense, treatment with low doses of available V-ATPase inhibitors may represent a safe and effective anti-cancer treatment. This study also highlights the possibility of targeting dedicated V-ATPase chaperones required for assembly of the complex in the development of drugs that reduce V-ATPase activity.
It may also be feasible to reduce the tumorigenic effects of the V-ATPase by repurposing already available drugs. For example, proton pump inhibitors such as omeprazole are commonly used in the clinic and have been shown to reduce V-ATPase activity (104). Additionally, chloroquine, a weak base that neutralizes acidic compartments, is often used as a treatment for malaria. It is feasible that chloroquine treatment could dissipate the effects of enhanced intracellular V-ATPase activity in cancer cells, allowing for inhibition of autophagy, induction of apoptosis, and reduced sequestration of weakly basic chemotherapeutic agents (144).
It is noteworthy that V-ATPases containing particular isoforms of subunit a (especially a3 and a4, which target V-ATPases to the plasma membrane) are critical for the invasiveness of certain cancer cells (19, 22, 59, 121). Plasma membrane V-ATPases are found primarily in specialized cell types, including osteoclasts, renal intercalated cells, and epididymal clear cells (16, 176). Membrane-impermeable V-ATPase inhibitors designed to specifically inhibit plasma membrane V-ATPases would thus be expected to have much more restricted and manageable side effects in cancer patients than membrane-permeable inhibitors, which would have access to all V-ATPases in the cell. With respect to isoforms of subunit a, the phenotypic consequences of complete disruption of a3 and a4 are known from genetic studies; loss of a3 leads to osteopetrosis (5, 122, 172), whereas loss of a4 leads to renal tubule acidosis (167, 184). Renal tubule acidosis is treatable by maintenance of adequate levels of plasma bicarbonate (141). Calcium homeostasis is a major concern of osteopetrosis. Osteopetrosis patients are thus typically treated with calicitrol to help manage proper calcium levels (82, 102). Impairment of osteoclast function through inhibition of a3-containing V-ATPases may be also beneficial for the treatment of cancers such as breast cancer that commonly undergo osteolytic metastasis to the bone, a process that relies on osteoclast function (199). Thus a3 and a4 containing V-ATPases represent particularly attractive and viable drug targets for the development of anti-cancer therapeutics. As noted above, to counteract the induction of prosurvival pathways by aggressive cancer cells after V-ATPase inhibition, combination treatment may be needed to optimize the anti-metastatic effect of such drugs in vivo (55). Further studies are required to determine how the V-ATPase can be effectively and safely targeted to prevent cancer cell growth, drug resistance, and metastasis.
GRANTS
This work was supported by National Institutes of Health Grant GM34478 (to M. Forgac), predoctoral Ruth L. Kirschstein National Research Service Award CA189321 (to L. Stransky), and predoctoral Ruth L. Kirschstein National Research Service Award CA192500 (to K. Cotter).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
acknowledgments
We thank Christina McGuire for many helpful discussions.
L. Stransky and K. Cotter contributed equally.
Address for reprint requests and other correspondence: M. Forgac, Dept. of Developmental, Molecular and Chemical Biology, Tufts University School of Medicine, 136 Harrison Ave., Boston, MA 02111 (e-mail: michael.forgac@tufts.edu).
REFERENCES
- 1.Adams DS, Masi A, Levin M. H+ pump-dependent changes in membrane voltage are an early mechanism necessary and sufficient to induce Xenopus tail regeneration. Development 134: 1323–1335, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Adams DS, Robinson KR, Fukumoto T, Yuan S, Albertson RC, Yelick P, Kuo L, McSweeney M, Levin M. Early, H+-V-ATPase-dependent proton flux is necessary for consistent left-right patterning of non-mammalian vertebrates. Development 133: 1657–1671, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alwan HAJ, van Zoelen EJJ, van Leeuwen JEM. Ligand-induced lysosomal epidermal growth factor receptor (EGFR) degradation is preceded by proteasome-dependent EGFR de-ubiquitination. J Biol Chem 278: 35781–35790, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Alzamora R, Thali RF, Gong F, Smolak C, Li H, Baty CJ, Bertrand CA, Auchli Y, Brunisholz RA, Neumann D, Hallows KR, Pastor-Soler NM. PKA regulates vacuolar H+-ATPase localization and activity via direct phosphorylation of the a subunit in kidney cells. J Biol Chem 285: 24676–24685, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci USA 101: 12792–12797, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13: 11–26, 2012. [DOI] [PubMed] [Google Scholar]
- 7.Anderson SL, Jalas C, Fedick A, Reid KF, Carpenter TO, Chirnomas D, Treff NR, Ekstein J, Rubin BY. A founder mutation in the TCIRG1 gene causes osteopetrosis in the Ashkenazi Jewish population. Clin Genet 88: 74–79, 2015. [DOI] [PubMed] [Google Scholar]
- 8.Asakura T, Imai A, Ohkubo-Uraoka N, Kuroda M, Iidaka Y, Uchida K, Shibasaki T, Ohkawa K. Relationship between expression of drug-resistance factors and drug sensitivity in normal human renal proximal tubular epithelial cells in comparison with renal cell carcinoma. Oncol Rep 14: 601–607, 2005. [PubMed] [Google Scholar]
- 9.Avnet S, Di Pompo G, Lemma S, Salerno M, Perut F, Bonuccelli G, Granchi D, Zini N, Baldini N. V-ATPase is a candidate therapeutic target for Ewing sarcoma. Biochim Biophys Acta 1832: 1105–1116, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Balduyck M, Zerimech F, Gouyer V, Lemaire R, Hemon B, Grard G, Thiebaut C, Lemaire V, Dacquembronne E, Duhem T, Lebrun A, Dejonghe MJ, Huet G. Specific expression of matrix metalloproteinases 1, 3, 9 and 13 associated with invasiveness of breast cancer cells in vitro. Clin Exp Metastasis 18: 171–178, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150: 1196–1208, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernard D, Gebbia M, Prabha S, Gronda M, MacLean N, Wang X, Hurren R, Sukhai MA, Cho EE, Manolson MF, Datti A, Wrana J, Minden MD, Al-Awar R, Aman A, Nislow C, Giaever G, Schimmer AD. Select microtubule inhibitors increase lysosome acidity and promote lysosomal disruption in acute myeloid leukemia (AML) cells. Apoptosis 20: 948–959, 2015. [DOI] [PubMed] [Google Scholar]
- 13.Bernhard SM, Seidel K, Schmitz J, Klare S, Kirsch S, Schrezenmeier E, Zaade D, Meyborg H, Goldin-Lang P, Stawowy P, Zollmann FS, Unger T, Funke-Kaiser H. The (pro)renin receptor [(P)RR] can act as a repressor of Wnt signalling. Biochem Pharmacol 84: 1643–1650, 2012. [DOI] [PubMed] [Google Scholar]
- 14.Blitzer JT, Nusse R. A critical role for endocytosis in Wnt signaling. BMC Cell Biol 7: 28, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Born R, Eichholtz-Wirth H. Effect of different physiological conditions on the action of adriamycin on Chinese hamster cells in vitro. Br J Cancer 44: 241–246, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouché V, Espinosa AP, Leone L, Sardiello M, Ballabio A, Botas J. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy 12: 484–498, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breton S, Brown D. Regulation of luminal acidification by the V-ATPase. Physiology 28: 318–329, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown D, Breton S. H(+)V-ATPase-dependent luminal acidification in the kidney collecting duct and the epididymis/vas deferens: vesicle recycling and transcytotic pathways. J Exp Biol 203: 137–145, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Buechling T, Bartscherer K, Ohkawara B, Chaudhary V, Spirohn K, Niehrs C, Boutros M. Wnt/Frizzled signaling requires dPRR, the Drosophila homolog of the prorenin receptor. Curr Biol 20: 1263–1268, 2010. [DOI] [PubMed] [Google Scholar]
- 20.Cai M, Liu P, Wei L, Wang J, Qi J, Feng S, Deng L. Atp6v1c1 may regulate filament actin arrangement in breast cancer cells. PloS One 9: e84833, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Capecci J, Forgac M. The function of vacuolar ATPase (V-ATPase) a subunit isoforms in invasiveness of MCF10a and MCF10CA1a human breast cancer cells. J Biol Chem 288: 32731–32741, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen SH, Bubb MR, Yarmola EG, Zuo J, Jiang J, Lee BS, Lu M, Gluck SL, Hurst IR, Holliday LS. Vacuolar H+-ATPase binding to microfilaments: regulation in response to phosphatidylinositol 3-kinase activity and detailed characterization of the actin-binding site in subunit B. J Biol Chem 279: 7988–7998, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Chung C, Mader CC, Schmitz JC, Atladottir J, Fitchev P, Cornwell ML, Koleske AJ, Crawford SE, Gorelick F. The vacuolar-ATPase modulates matrix metalloproteinase isoforms in human pancreatic cancer. Lab Invest J Tech Methods Pathol 91: 732–743, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cotter K, Capecci J, Sennoune S, Huss M, Maier M, Martinez-Zaguilan R, Forgac M. Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J Biol Chem 290: 3680–3692, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cotter K, Stransky L, McGuire C, Forgac M. Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem Sci 40: 611–622, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Couoh-Cardel S, Milgrom E, Wilkens S. Affinity purification and structural features of the yeast vacuolar ATPase Vo membrane sector. J Biol Chem 290: 27959–27971, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, Boutros M, Niehrs C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 327: 459–463, 2010. [DOI] [PubMed] [Google Scholar]
- 28.Damaghi M, Wojtkowiak JW, Gillies RJ. pH sensing and regulation in cancer. Front Physiol 4: 370, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D'Angelo RC, Ouzounova M, Davis A, Choi D, Tchuenkam SM, Kim G, Luther T, Quraishi AA, Senbabaoglu Y, Conley SJ, Clouthier SG, Hassan KA, Wicha MS, Korkaya H. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol Cancer Ther 14: 779–787, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dechant R, Saad S, Ibáñez AJ, Peter M. Cytosolic pH regulates cell growth through distinct GTPases, Arf1 and Gtr1, to promote Ras/PKA and TORC1 activity. Mol Cell 55: 409–421, 2014. [DOI] [PubMed] [Google Scholar]
- 31.De Milito A, Canese R, Marino ML, Borghi M, Iero M, Villa A, Venturi G, Lozupone F, Iessi E, Logozzi M, Mina PD, Santinami M, Rodolfo M, Podo F, Rivoltini L, Fais S. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int J Cancer 127: 207–219, 2010. [DOI] [PubMed] [Google Scholar]
- 32.De Milito A, Iessi E, Logozzi M, Lozupone F, Spada M, Marino ML, Federici C, Perdicchio M, Matarrese P, Lugini L, Nilsson A, Fais S. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res 67: 5408–5417, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Di Cristofori A, Ferrero S, Bertolini I, Gaudioso G, Russo MV, Berno V, Vanini M, Locatelli M, Zavanone M, Rampini P, Vaccari T, Caroli M, Vaira V. The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma. Oncotarget 6: 17514–17531, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Giovanni J, Boudkkazi S, Mochida S, Bialowas A, Samari N, Lévêque C, Youssouf F, Brechet A, Iborra C, Maulet Y, Moutot N, Debanne D, Seagar M, El Far O. V-ATPase membrane sector associates with synaptobrevin to modulate neurotransmitter release. Neuron 67: 268–279, 2010. [DOI] [PubMed] [Google Scholar]
- 35.Donnem T, Andersen S, Al-Shibli K, Al-Saad S, Busund LT, Bremnes RM. Prognostic impact of Notch ligands and receptors in nonsmall cell lung cancer: coexpression of Notch-1 and vascular endothelial growth factor-A predicts poor survival. Cancer 116: 5676–5685, 2010. [DOI] [PubMed] [Google Scholar]
- 36.Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493: 679–683, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Einhorn Z, Trapani JG, Liu Q, Nicolson T. Rabconnectin3α promotes stable activity of the H+ pump on synaptic vesicles in hair cells. J Neurosci 32: 11144–11156, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan S, Niu Y, Tan N, Wu Z, Wang Y, You H, Ke R, Song J, Shen Q, Wang W, Yao G, Shu H, Lin H, Yao M, Zhang Z, Gu J, Qin W. LASS2 enhances chemosensitivity of breast cancer by counteracting acidic tumor microenvironment through inhibiting activity of V-ATPase proton pump. Oncogene 32: 1682–1690, 2013. [DOI] [PubMed] [Google Scholar]
- 39.Fan S, Wang Y, Wu Z, Zhang Z, Lu J, Li M, Shan Q, Wu D, Sun C, Hu B, Zheng Y. AGPAT9 suppresses cell growth, invasion and metastasis by counteracting acidic tumor microenvironment through KLF4/LASS2/V-ATPase signaling pathway in breast cancer. Oncotarget 6: 18406–18417, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faronato M, Nguyen VTM, Patten DK, Lombardo Y, Steel JH, Patel N, Woodley L, Shousha S, Pruneri G, Coombes RC, Magnani L. DMXL2 drives epithelial to mesenchymal transition in hormonal therapy resistant breast cancer through Notch hyper-activation. Oncotarget 6: 22467–22479, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett 356: 165–170, 2015. [DOI] [PubMed] [Google Scholar]
- 42.Fender AW, Nutter JM, Fitzgerald TL, Bertrand FE, Sigounas G. Notch-1 promotes stemness and epithelial to mesenchymal transition in colorectal cancer. J Cell Biochem 116: 2517–2527, 2015. [DOI] [PubMed] [Google Scholar]
- 43.Feng S, Zhu G, McConnell M, Deng L, Zhao Q, Wu M, Zhou Q, Wang J, Qi J, Li YP, Chen W. Silencing of atp6v1c1 prevents breast cancer growth and bone metastasis. Int J Biol Sci 9: 853–862, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferguson PJ, Phillips JR, Selner M, Cass CE. Differential activity of vincristine and vinblastine against cultured cells. Cancer Res 44: 3307–3312, 1984. [PubMed] [Google Scholar]
- 45.Fogarty FM, O'Keeffe J, Zhadanov A, Papkovsky D, Ayllon V, O'Connor R. HRG-1 enhances cancer cell invasive potential and couples glucose metabolism to cytosolic/extracellular pH gradient regulation by the vacuolar-H+ ATPase. Oncogene 33: 4653–4663, 2014. [DOI] [PubMed] [Google Scholar]
- 46.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8: 917–929, 2007. [DOI] [PubMed] [Google Scholar]
- 47.García-García A, Pérez-Sayáns García M, Rodríguez MJ, Antúnez-López J, Barros-Angueira F, Somoza-Martín M, Gándara-Rey JM, Aguirre-Urízar JM. Immunohistochemical localization of C1 subunit of V-ATPase (ATPase C1) in oral squamous cell cancer and normal oral mucosa. Biotech Histochem 87: 133–139, 2012. [DOI] [PubMed] [Google Scholar]
- 48.George A, Leahy H, Zhou J, Morin PJ. The vacuolar-ATPase inhibitor bafilomycin and mutant VPS35 inhibit canonical Wnt signaling. Neurobiol Dis 26: 125–133, 2007. [DOI] [PubMed] [Google Scholar]
- 49.Geuze HJ, Slot JW, Strous GJ, Lodish HF, Schwartz AL. Intracellular site of asialoglycoprotein receptor-ligand uncoupling: double-label immunoelectron microscopy during receptor-mediated endocytosis. Cell 32: 277–287, 1983. [DOI] [PubMed] [Google Scholar]
- 50.Gidon A, Al-Bataineh MM, Jean-Alphonse FG, Stevenson HP, Watanabe T, Louet C, Khatri A, Calero G, Pastor-Soler NM, Gardella TJ, Vilardaga JP. Endosomal GPCR signaling turned off by negative feedback actions of PKA and v-ATPase. Nat Chem Biol 10: 707–709, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gleixner EM, Canaud G, Hermle T, Guida MC, Kretz O, Helmstädter M, Huber TB, Eimer S, Terzi F, Simons M. V-ATPase/mTOR signaling regulates megalin-mediated apical endocytosis. Cell Rep 8: 10–19, 2014. [DOI] [PubMed] [Google Scholar]
- 52.Gleize V, Boisselier B, Marie Y, Poëa-Guyon S, Sanson M, Morel N. The renal v-ATPase a4 subunit is expressed in specific subtypes of human gliomas. Glia 60: 1004–1012, 2012. [DOI] [PubMed] [Google Scholar]
- 53.Glunde K, Guggino SE, Solaiyappan M, Pathak AP, Ichikawa Y, Bhujwalla ZM. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia 5: 533–545, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gottlieb RA, Giesing HA, Zhu JY, Engler RL, Babior BM. Cell acidification in apoptosis: granulocyte colony-stimulating factor delays programmed cell death in neutrophils by up-regulating the vacuolar H+-ATPase. Proc Natl Acad Sci USA 92: 5965–5968, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Graham RM, Thompson JW, Webster KA. Inhibition of the vacuolar ATPase induces Bnip3-dependent death of cancer cells and a reduction in tumor burden and metastasis. Oncotarget 5: 1162–1173, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gruenberg J, van der Goot FG. Mechanisms of pathogen entry through the endosomal compartments. Nat Rev Mol Cell Biol 7: 495–504, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Gu F, Gruenberg J. ARF1 regulates pH-dependent COP functions in the early endocytic pathway. J Biol Chem 275: 8154–8160, 2000. [DOI] [PubMed] [Google Scholar]
- 58.Hegedüs L, Cho H, Xie X, Eliceiri GL. Additional MDA-MB-231 breast cancer cell matrix metalloproteinases promote invasiveness. J Cell Physiol 216: 480–485, 2008. [DOI] [PubMed] [Google Scholar]
- 59.Hendrix A, Sormunen R, Westbroek W, Lambein K, Denys H, Sys G, Braems G, Van den Broecke R, Cocquyt V, Gespach C, Bracke M, De Wever O. Vacuolar H+ ATPase expression and activity is required for Rab27B-dependent invasive growth and metastasis of breast cancer. Int J Cancer 133: 843–854, 2013. [DOI] [PubMed] [Google Scholar]
- 60.Hermle T, Saltukoglu D, Grünewald J, Walz G, Simons M. Regulation of Frizzled-dependent planar polarity signaling by a V-ATPase subunit. Curr Biol 20: 1269–1276, 2010. [DOI] [PubMed] [Google Scholar]
- 61.Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, Verstreken P, Cao Y, Zhou Y, Kunz J, Bellen HJ. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell 121: 607–620, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hindenburg AA, Gervasoni JE, Krishna S, Stewart VJ, Rosado M, Lutzky J, Bhalla K, Baker MA, Taub RN. Intracellular distribution and pharmacokinetics of daunorubicin in anthracycline-sensitive and -resistant HL-60 cells. Cancer Res 49: 4607–4614, 1989. [PubMed] [Google Scholar]
- 63.Hinton A, Sennoune SR, Bond S, Fang M, Reuveni M, Sahagian GG, Jay D, Martinez-Zaguilan R, Forgac M. Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J Biol Chem 284: 16400–16408, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hirata R, Graham LA, Takatsuki A, Stevens TH, Anraku Y. VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+-ATPase. J Biol Chem 272: 4795–4803, 1997. [DOI] [PubMed] [Google Scholar]
- 65.Hirata T, Iwamoto-Kihara A, Sun-Wada GH, Okajima T, Wada Y, Futai M. Subunit rotation of vacuolar-type proton pumping ATPase: relative rotation of the G and C subunits. J Biol Chem 278: 23714–23719, 2003. [DOI] [PubMed] [Google Scholar]
- 66.Holliday LS, Lu M, Lee BS, Nelson RD, Solivan S, Zhang L, Gluck SL. The amino-terminal domain of the B subunit of vacuolar H+-ATPase contains a filamentous actin binding site. J Biol Chem 275: 32331–32337, 2000. [DOI] [PubMed] [Google Scholar]
- 67.Hong J, Nakano Y, Yokomakura A, Ishihara K, Kim S, Kang YS, Ohuchi K. Nitric oxide production by the vacuolar-type (H+)-ATPase inhibitors bafilomycin A1 and concanamycin A and its possible role in apoptosis in RAW 264.7 cells. J Pharmacol Exp Ther 319: 672–681, 2006. [DOI] [PubMed] [Google Scholar]
- 68.Hong J, Yokomakura A, Nakano Y, Ishihara K, Kaneda M, Onodera M, Nakahama K, Morita I, Niikura K, Ahn JW, Zee O, Ohuchi K. Inhibition of vacuolar-type (H+)-ATPase by the cytostatic macrolide apicularen A and its role in apicularen A-induced apoptosis in RAW 264.7 cells. FEBS Lett 580: 2723–2730, 2006. [DOI] [PubMed] [Google Scholar]
- 69.Horova V, Hradilova N, Jelinkova I, Koc M, Svadlenka J, Brazina J, Klima M, Slavik J, Hyrslova Vaculova A, Andera L. Inhibition of vacuolar ATPase attenuates the TRAIL-induced activation of caspase-8 and modulates the trafficking of TRAIL receptosomes. FEBS J 280: 3436–3450, 2013. [DOI] [PubMed] [Google Scholar]
- 70.Hrabeta J, Groh T, Khalil MA, Poljakova J, Adam V, Kizek R, Uhlik J, Doktorova H, Cerna T, Frei E, Stiborova M, Eckschlager T. Vacuolar-ATPase-mediated intracellular sequestration of ellipticine contributes to drug resistance in neuroblastoma cells. Int J Oncol 47: 971–980, 2015. [DOI] [PubMed] [Google Scholar]
- 71.Huang L, Lu Q, Han Y, Li Z, Zhang Z, Li X. ABCG2/V-ATPase was associated with the drug resistance and tumor metastasis of esophageal squamous cancer cells. Diagn Pathol 7: 180, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hurtado-Lorenzo A, Skinner M, El Annan J, Futai M, Sun-Wada GH, Bourgoin S, Casanova J, Wildeman A, Bechoua S, Ausiello DA, Brown D, Marshansky V. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat Cell Biol 8: 124–136, 2006. [DOI] [PubMed] [Google Scholar]
- 73.Ishisaki A, Hashimoto S, Amagasa T, Nishihara T. Caspase-3 activation during the process of apoptosis induced by a vacuolar type H+-ATPase inhibitor. Biol Cell 91: 507–513, 1999. [DOI] [PubMed] [Google Scholar]
- 74.Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, Guan KL. Differential regulation of mTORC1 by leucine and glutamine. Science 347: 194–198, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kallifatidis G, Hoepfner D, Jaeg T, Guzmán EA, Wright AE. The marine natural product manzamine A targets vacuolar ATPases and inhibits autophagy in pancreatic cancer cells. Mar Drugs 11: 3500–3516, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kallunki T, Olsen OD, Jäättelä M. Cancer-associated lysosomal changes: friends or foes? Oncogene 32: 1995–2004, 2013. [DOI] [PubMed] [Google Scholar]
- 77.Kane PM. Targeting reversible disassembly as a mechanism of controlling V-ATPase activity. Curr Protein Pept Sci 13: 117–123, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Karwatowska-Prokopczuk E, Nordberg JA, Li HL, Engler RL, Gottlieb RA. Effect of vacuolar proton ATPase on pHi, Ca2+, and apoptosis in neonatal cardiomyocytes during metabolic inhibition/recovery. Circ Res 82: 1139–1144, 1998. [DOI] [PubMed] [Google Scholar]
- 79.Kawai A, Uchiyama H, Takano S, Nakamura N, Ohkuma S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 3: 154–157, 2007. [DOI] [PubMed] [Google Scholar]
- 80.Kawasaki-Nishi S, Nishi T, Forgac M. Arg-735 of the 100-kDa subunit a of the yeast V-ATPase is essential for proton translocation. Proc Natl Acad Sci USA 98: 12397–12402, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Keizer HG, Joenje H. Increased cytosolic pH in multidrug-resistant human lung tumor cells: effect of verapamil. J Natl Cancer Inst 81: 706–709, 1989. [DOI] [PubMed] [Google Scholar]
- 82.Key L, Carnes D, Cole S, Holtrop M, Bar-Shavit Z, Shapiro F, Arceci R, Steinberg J, Gundberg C, Kahn A. Treatment of congenital osteopetrosis with high-dose calcitriol. N Engl J Med 310: 409–415, 1984. [DOI] [PubMed] [Google Scholar]
- 83.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10: 935–945, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim YC, Park HW, Sciarretta S, Mo JS, Jewell JL, Russell RC, Wu X, Sadoshima J, Guan KL. Rag GTPases are cardioprotective by regulating lysosomal function. Nat Commun 5: 4241, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kinouchi K, Ichihara A, Sano M, Sun-Wada GH, Wada Y, Kurauchi-Mito A, Bokuda K, Narita T, Oshima Y, Sakoda M, Tamai Y, Sato H, Fukuda K, Itoh H. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res 107: 30–34, 2010. [DOI] [PubMed] [Google Scholar]
- 86.Kobia F, Duchi S, Deflorian G, Vaccari T. Pharmacologic inhibition of vacuolar H+ ATPase reduces physiologic and oncogenic Notch signaling. Mol Oncol 8: 207–220, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kornfeld S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu Rev Biochem 61: 307–330, 1992. [DOI] [PubMed] [Google Scholar]
- 88.Kozik P, Hodson NA, Sahlender DA, Simecek N, Soromani C, Wu J, Collinson LM, Robinson MS. A human genome-wide screen for regulators of clathrin-coated vesicle formation reveals an unexpected role for the V-ATPase. Nat Cell Biol 15: 50–60, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kubisch R, Fröhlich T, Arnold GJ, Schreiner L, von Schwarzenberg K, Roidl A, Vollmar AM, Wagner E. V-ATPase inhibition by archazolid leads to lysosomal dysfunction resulting in impaired cathepsin B activation in vivo. Int J Cancer J Int Cancer 134: 2478–2488, 2014. [DOI] [PubMed] [Google Scholar]
- 90.Kulshrestha A, Katara GK, Ibrahim S, Pamarthy S, Jaiswal MK, Gilman Sachs A, Beaman KD. Vacuolar ATPase ”a2“ isoform exhibits distinct cell surface accumulation and modulates matrix metalloproteinase activity in ovarian cancer. Oncotarget 6: 3797–3810, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lakka SS, Gondi CS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, Rao JS. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 23: 4681–4689, 2004. [DOI] [PubMed] [Google Scholar]
- 92.Lange C, Prenninger S, Knuckles P, Taylor V, Levin M, Calegari F. The H(+) vacuolar ATPase maintains neural stem cells in the developing mouse cortex. Stem Cells Dev 20: 843–850, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 149: 274–293, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee BS, Holliday LS, Krits I, Gluck SL. Vacuolar H+-ATPase activity and expression in mouse bone marrow cultures. J Bone Miner Res 14: 2127–2136, 1999. [DOI] [PubMed] [Google Scholar]
- 95.Lee GH, Kim DS, Kim HT, Lee JW, Chung CH, Ahn T, Lim JM, Kim IK, Chae HJ, Kim HR. Enhanced lysosomal activity is involved in Bax inhibitor-1-induced regulation of the endoplasmic reticulum (ER) stress response and cell death against ER stress: involvement of vacuolar H+-ATPase (V-ATPase). J Biol Chem 286: 24743–24753, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee YY, Jeon HK, Hong JE, Cho YJ, Ryu JY, Choi JJ, Lee SH, Yoon G, Kim WY, Do IG, Kim MK, Kim TJ, Choi CH, Lee JW, Bae DS, Kim BG. Proton pump inhibitors enhance the effects of cytotoxic agents in chemoresistant epithelial ovarian carcinoma. Oncotarget 6: 35040–35050, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liang P, Jones CA, Bisgrove BW, Song L, Glenn ST, Yost HJ, Gross KW. Genomic characterization and expression analysis of the first nonmammalian renin genes from zebrafish and pufferfish. Physiol Genomics 16: 314–322, 2004. [DOI] [PubMed] [Google Scholar]
- 98.Liao C, Hu B, Arno MJ, Panaretou B. Genomic screening in vivo reveals the role played by vacuolar H+ ATPase and cytosolic acidification in sensitivity to DNA-damaging agents such as cisplatin. Mol Pharmacol 71: 416–425, 2007. [DOI] [PubMed] [Google Scholar]
- 99.Liberman R, Bond S, Shainheit MG, Stadecker MJ, Forgac M. Regulated assembly of vacuolar ATPase is increased during cluster disruption-induced maturation of dendritic cells through a phosphatidylinositol 3-kinase/mTOR-dependent pathway. J Biol Chem 289: 1355–1363, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liégeois S, Benedetto A, Garnier JM, Schwab Y, Labouesse M. The V0-ATPase mediates apical secretion of exosomes containing Hedgehog-related proteins in Caenorhabditis elegans. J Cell Biol 173: 949–961, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu P, Chen H, Han L, Zou X, Shen W. Expression and role of V1A subunit of V-ATPases in gastric cancer cells. Int J Clin Oncol 20: 725–735, 2015. [DOI] [PubMed] [Google Scholar]
- 102.Li YP, Chen W, Liang Y, Li E, Stashenko P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet 23: 447–451, 1999. [DOI] [PubMed] [Google Scholar]
- 103.Lozupone F, Borghi M, Marzoli F, Azzarito T, Matarrese P, Iessi E, Venturi G, Meschini S, Canitano A, Bona R, Cara A, Fais S. TM9SF4 is a novel V-ATPase-interacting protein that modulates tumor pH alterations associated with drug resistance and invasiveness of colon cancer cells. Oncogene 34: 5163–5174, 2015. [DOI] [PubMed] [Google Scholar]
- 104.Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, Marra M, Lugini L, Logozzi M, Lozupone F, Federici C, Iessi E, Parmiani G, Arancia G, Belardelli F, Fais S. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst 96: 1702–1713, 2004. [DOI] [PubMed] [Google Scholar]
- 105.Ludwig J, Kerscher S, Brandt U, Pfeiffer K, Getlawi F, Apps DK, Schägger H. Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. J Biol Chem 273: 10939–10947, 1998. [DOI] [PubMed] [Google Scholar]
- 106.Lu M, Ammar D, Ives H, Albrecht F, Gluck SL. Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump. J Biol Chem 282: 24495–24503, 2007. [DOI] [PubMed] [Google Scholar]
- 107.Lu Q, Lu S, Huang L, Wang T, Wan Y, Zhou CX, Zhang C, Zhang Z, Li X. The expression of V-ATPase is associated with drug resistance and pathology of non-small-cell lung cancer. Diagn Pathol 8: 145, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lu X, Qin W, Li J, Tan N, Pan D, Zhang H, Xie L, Yao G, Shu H, Yao M, Wan D, Gu J, Yang S. The growth and metastasis of human hepatocellular carcinoma xenografts are inhibited by small interfering RNA targeting to the subunit ATP6L of proton pump. Cancer Res 65: 6843–6849, 2005. [DOI] [PubMed] [Google Scholar]
- 109.Lu Y, Zhang R, Liu S, Zhao Y, Gao J, Zhu L. ZT-25, a new vacuolar H+-ATPase inhibitor, induces apoptosis and protective autophagy through ROS generation in HepG2 cells. Eur J Pharmacol 771: 130–138, 2016. [DOI] [PubMed] [Google Scholar]
- 110.MacLeod KJ, Vasilyeva E, Baleja JD, Forgac M. Mutational analysis of the nucleotide binding sites of the yeast vacuolar proton-translocating ATPase. J Biol Chem 273: 150–156, 1998. [DOI] [PubMed] [Google Scholar]
- 111.Ma L, Xu Y, Su J, Yu H, Kang J, Li H, Li X, Xie Q, Yu C, Sun L, Li Y. Autophagic flux promotes cisplatin resistance in human ovarian carcinoma cells through ATP-mediated lysosomal function. Int J Oncol 47: 1890–1900, 2015. [DOI] [PubMed] [Google Scholar]
- 112.Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F, Venturi G, Della Mina P, Pattingre S, Rivoltini L, Codogno P, De Milito A. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis 1: e87, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Marjuki H, Gornitzky A, Marathe BM, Ilyushina NA, Aldridge JR, Desai G, Webby RJ, Webster RG. Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell Microbiol 13: 587–601, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Marquardt D, Center MS. Involvement of vacuolar H+-adenosine triphosphatase activity in multidrug resistance in HL60 cells. J Natl Cancer Inst 83: 1098–1102, 1991. [DOI] [PubMed] [Google Scholar]
- 115.Marshansky V, Rubinstein JL, Grüber G. Eukaryotic V-ATPase: novel structural findings and functional insights. Biochim Biophys Acta 1837: 857–879, 2014. [DOI] [PubMed] [Google Scholar]
- 116.Martina JA, Diab HI, Li H, Puertollano R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell Mol Life Sci 71: 2483–2497, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martinez-Zaguilan R, Lynch RM, Martinez GM, Gillies RJ. Vacuolar-type H+-ATPases are functionally expressed in plasma membranes of human tumor cells Am J Physiol Cell Physiol 265: C1015–C1029, 1993. [DOI] [PubMed] [Google Scholar]
- 118.Martínez-Zaguilán R, Raghunand N, Lynch RM, Bellamy W, Martinez GM, Rojas B, Smith D, Dalton WS, Gillies RJ. pH and drug resistance. I. Functional expression of plasmalemmal V-type H+-ATPase in drug-resistant human breast carcinoma cell lines. Biochem Pharmacol 57: 1037–1046, 1999. [DOI] [PubMed] [Google Scholar]
- 119.Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol 21: 228–237, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell 137: 1062–1075, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mauvezin C, Nagy P, Juhász G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun 6: 7007, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 11: 1437–1438, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McHenry P, Wang WLW, Devitt E, Kluesner N, Davisson VJ, McKee E, Schweitzer D, Helquist P, Tenniswood M. Iejimalides A and B inhibit lysosomal vacuolar H+-ATPase (V-ATPase) activity and induce S-phase arrest and apoptosis in MCF-7 cells. J Cell Biochem 109: 634–642, 2010. [DOI] [PubMed] [Google Scholar]
- 124.Mei F, You J, Liu B, Zhang M, Liu J, Zhang B, Pei F. LASS2/TMSG1 inhibits growth and invasion of breast cancer cell in vitro through regulation of vacuolar ATPase activity. Tumour Biol 36: 2831–2844, 2015. [DOI] [PubMed] [Google Scholar]
- 125.Michel V, Licon-Munoz Y, Trujillo K, Bisoffi M, Parra KJ. Inhibitors of vacuolar ATPase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. Int J Cancer 132: E1–10, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mijaljica D, Prescott M, Devenish RJ. V-ATPase engagement in autophagic processes. Autophagy 7: 666–668, 2011. [DOI] [PubMed] [Google Scholar]
- 127.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer 6: 764–775, 2006. [DOI] [PubMed] [Google Scholar]
- 128.Morel N, Dedieu JC, Philippe JM. Specific sorting of the a1 isoform of the V-H+ATPase a subunit to nerve terminals where it associates with both synaptic vesicles and the presynaptic plasma membrane. J Cell Sci 116: 4751–4762, 2003. [DOI] [PubMed] [Google Scholar]
- 129.Morimura T, Fujita K, Akita M, Nagashima M, Satomi A. The proton pump inhibitor inhibits cell growth and induces apoptosis in human hepatoblastoma. Pediatr Surg Int 24: 1087–1094, 2008. [DOI] [PubMed] [Google Scholar]
- 130.Moriyama Y, Maeda M, Futai M. Energy coupling of l-glutamate transport and vacuolar H(+)-ATPase in brain synaptic vesicles. J Biochem 108: 689–693, 1990. [DOI] [PubMed] [Google Scholar]
- 131.Murakami T, Shibuya I, Ise T, Chen ZS, Akiyama S, Nakagawa M, Izumi H, Nakamura T, Matsuo K, Yamada Y, Kohno K. Elevated expression of vacuolar proton pump genes and cellular pH in cisplatin resistance. Int J Cancer 93: 869–874, 2001. [DOI] [PubMed] [Google Scholar]
- 132.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 11: 558–572, 2011. [DOI] [PubMed] [Google Scholar]
- 133.Namkoong S, Lee KI, Lee JI, Park R, Lee EJ, Jang IS, Park J. The integral membrane protein ITM2A, a transcriptional target of PKA-CREB, regulates autophagic flux via interaction with the vacuolar ATPase. Autophagy 11: 756–768, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 10: 767–777, 2011. [DOI] [PubMed] [Google Scholar]
- 135.Nishihara T, Akifusa S, Koseki T, Kato S, Muro M, Hanada N. Specific inhibitors of vacuolar type H+-ATPases induce apoptotic cell death. Biochem Biophys Res Commun 212: 255–262, 1995. [DOI] [PubMed] [Google Scholar]
- 136.Nishisho T, Hata K, Nakanishi M, Morita Y, Sun-Wada GH, Wada Y, Yasui N, Yoneda T. The a3 isoform vacuolar type H+-ATPase promotes distant metastasis in the mouse B16 melanoma cells. Mol Cancer Res 9: 845–855, 2011. [DOI] [PubMed] [Google Scholar]
- 137.O'Callaghan KM, Ayllon V, O'Keeffe J, Wang Y, Cox OT, Loughran G, Forgac M, O'Connor R. Heme-binding protein HRG-1 is induced by insulin-like growth factor I and associates with the vacuolar H+-ATPase to control endosomal pH and receptor trafficking. J Biol Chem 285: 381–391, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ochotny N, Flenniken AM, Owen C, Voronov I, Zirngibl RA, Osborne LR, Henderson JE, Adamson SL, Rossant J, Manolson MF, Aubin JE. The V-ATPase a3 subunit mutation R740S is dominant negative and results in osteopetrosis in mice. J Bone Miner 26: 1484–1493, 2011. [DOI] [PubMed] [Google Scholar]
- 139.Ohta T, Arakawa H, Futagami F, Fushida S, Kitagawa H, Kayahara M, Nagakawa T, Miwa K, Kurashima K, Numata M, Kitamura Y, Terada T, Ohkuma S. Bafilomycin A1 induces apoptosis in the human pancreatic cancer cell line Capan-1. J Pathol 185: 324–330, 1998. [DOI] [PubMed] [Google Scholar]
- 140.Oka T, Murata Y, Namba M, Yoshimizu T, Toyomura T, Yamamoto A, Sun-Wada GH, Hamasaki N, Wada Y, Futai M. a4, a unique kidney-specific isoform of mouse vacuolar H+-ATPase subunit a. J Biol Chem 276: 40050–40054, 2001. [DOI] [PubMed] [Google Scholar]
- 141.Otero-Rey EM, Somoza-Martín M, Barros-Angueira F, García-García A. Intracellular pH regulation in oral squamous cell carcinoma is mediated by increased V-ATPase activity via over-expression of the ATP6V1C1 gene. Oral Oncol 44: 193–199, 2008. [DOI] [PubMed] [Google Scholar]
- 142.Ouar Z, Bens M, Vignes C, Paulais M, Pringel C, Fleury J, Cluzeaud F, Lacave R, Vandewalle A. Inhibitors of vacuolar H+-ATPase impair the preferential accumulation of daunomycin in lysosomes and reverse the resistance to anthracyclines in drug-resistant renal epithelial cells. Biochem J 370: 185–193, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pamarthy S, Jaiswal MK, Kulshreshtha A, Katara GK, Gilman-Sachs A, Beaman KD. The vacuolar ATPase a2-subunit regulates Notch signaling in triple-negative breast cancer cells. Oncotarget 6: 34206–34220, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pascolo S. Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies. Eur J Pharmacol 771: 139–144, 2016. [DOI] [PubMed] [Google Scholar]
- 145.Pastor-Soler N, Beaulieu V, Litvin TN, Da Silva N, Chen Y, Brown D, Buck J, Levin LR, Breton S. Bicarbonate-regulated adenylyl cyclase (sAC) is a sensor that regulates pH-dependent V-ATPase recycling. J Biol Chem 278: 49523–49529, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Peña-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TAT, Zou L, Xie X-J, Corey DR, Brugarolas J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J 30: 3242–3258, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pérez-Sayáns M, Reboiras-López MD, Somoza-Martín JM, Barros-Angueira F, Diz PG, Rey JMG, García-García A. Measurement of ATP6V1C1 expression in brush cytology samples as a diagnostic and prognostic marker in oral squamous cell carcinoma. Cancer Biol Ther 9: 1057–1064, 2010. [DOI] [PubMed] [Google Scholar]
- 148.Peri F, Nüsslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133: 916–927, 2008. [DOI] [PubMed] [Google Scholar]
- 149.Peters C, Bayer MJ, Bühler S, Andersen JS, Mann M, Mayer A. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature 409: 581–588, 2001. [DOI] [PubMed] [Google Scholar]
- 150.Petzoldt AG, Gleixner EM, Fumagalli A, Vaccari T, Simons M. Elevated expression of the V-ATPase C subunit triggers JNK-dependent cell invasion and overgrowth in a Drosophila epithelium. Dis Model Mech 6: 689–700, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pisoni RL, Thoene JG, Christensen HN. Detection and characterization of carrier-mediated cationic amino acid transport in lysosomes of normal and cystinotic human fibroblasts. Role in therapeutic cystine removal? J Biol Chem 260: 4791–4798, 1985. [PubMed] [Google Scholar]
- 152.Powell B, Graham LA, Stevens TH. Molecular characterization of the yeast vacuolar H+-ATPase proton pore. J Biol Chem 275: 23654–23660, 2000. [DOI] [PubMed] [Google Scholar]
- 153.Raghunand N, Martínez-Zaguilán R, Wright SH, Gillies RJ. pH and drug resistance. II. Turnover of acidic vesicles and resistance to weakly basic chemotherapeutic drugs. Biochem Pharmacol 57: 1047–1058, 1999. [DOI] [PubMed] [Google Scholar]
- 154.Ramachandran N, Munteanu I, Wang P, Ruggieri A, Rilstone JJ, Israelian N, Naranian T, Paroutis P, Guo R, Ren ZP, Nishino I, Chabrol B, Pellissier JF, Minetti C, Udd B, Fardeau M, Tailor CS, Mahuran DJ, Kissel JT, Kalimo H, Levy N, Manolson MF, Ackerley CA, Minassian BA. VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol 125: 439–457, 2013. [DOI] [PubMed] [Google Scholar]
- 155.Rath S, Liebl J, Fürst R, Vollmar AM, Zahler S. Regulation of endothelial signaling and migration by v-ATPase. Angiogenesis 17: 587–601, 2014. [DOI] [PubMed] [Google Scholar]
- 156.Regad T. Targeting RTK signaling pathways in cancer. Cancers 7: 1758–1784, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rhodes CJ, Lucas CA, Mutkoski RL, Orci L, Halban PA. Stimulation by ATP of proinsulin to insulin conversion in isolated rat pancreatic islet secretory granules. Association with the ATP-dependent proton pump. J Biol Chem 262: 10712–10717, 1987. [PubMed] [Google Scholar]
- 158.Riediger F, Quack I, Qadri F, Hartleben B, Park JK, Potthoff SA, Sohn D, Sihn G, Rousselle A, Fokuhl V, Maschke U, Purfürst B, Schneider W, Rump LC, Luft FC, Dechend R, Bader M, Huber TB, Nguyen G, Muller DN. Prorenin receptor is essential for podocyte autophagy and survival. J Am Soc Nephrol 22: 2193–2202, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res 69: 2260–2268, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Robey IF, Nesbit LA. Investigating mechanisms of alkalinization for reducing primary breast tumor invasion. BioMed Res Int 2013: 485196, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol 13: 2160–2170, 2002. [DOI] [PubMed] [Google Scholar]
- 162.Rojas JD, Sennoune SR, Maiti D, Bakunts K, Reuveni M, Sanka SC, Martinez GM, Seftor EA, Meininger CJ, Wu G, Wesson DE, Hendrix MJC, Martínez-Zaguilán R. Vacuolar-type H+-ATPases at the plasma membrane regulate pH and cell migration in microvascular endothelial cells. Am J Physiol Heart Circ Physiol 291: H1147–H1157, 2006. [DOI] [PubMed] [Google Scholar]
- 163.Rothberg JM, Bailey KM, Wojtkowiak JW, Ben-Nun Y, Bogyo M, Weber E, Moin K, Blum G, Mattingly RR, Gillies RJ, Sloane BF. Acid-mediated tumor proteolysis: contribution of cysteine cathepsins. Neoplasia 15: 1125–1137, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rozhin J, Sameni M, Ziegler G, Sloane BF. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Res 54: 6517–6525, 1994. [PubMed] [Google Scholar]
- 165.Rudnick G, Clark J. From synapse to vesicle: the reuptake and storage of biogenic amine neurotransmitters. Biochim Biophys Acta 1144: 249–263, 1993. [DOI] [PubMed] [Google Scholar]
- 166.Rush JS, Quinalty LM, Engelman L, Sherry DM, Ceresa BP. Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis. J Biol Chem 287: 712–722, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Salerno M, Avnet S, Bonuccelli G, Hosogi S, Granchi D, Baldini N. Impairment of lysosomal activity as a therapeutic modality targeting cancer stem cells of embryonal rhabdomyosarcoma cell line RD. PloS One 9: e110340, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sautin YY, Lu M, Gaugler A, Zhang L, Gluck SL. Phosphatidylinositol 3-kinase-mediated effects of glucose on vacuolar H+-ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol Cell Biol 25: 575–589, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Schempp CM, Schwarzenberg von K, Schreiner L, Kubisch R, Müller R, Wagner E, Vollmar AM. V-ATPase inhibition regulates anoikis resistance and metastasis of cancer cells. Mol Cancer Ther 13: 926–937, 2014. [DOI] [PubMed] [Google Scholar]
- 171.Schneider LS, von Schwarzenberg K, Lehr T, Ulrich M, Kubisch-Dohmen R, Liebl J, Trauner D, Menche D, Vollmar AM. Vacuolar-ATPase inhibition blocks iron metabolism to mediate therapeutic effects in breast cancer. Cancer Res 75: 2863–2874, 2015. [DOI] [PubMed] [Google Scholar]
- 172.Schwab A, Fabian A, Hanley PJ, Stock C. Role of ion channels and transporters in cell migration. Physiol Rev 92: 1865–1913, 2012. [DOI] [PubMed] [Google Scholar]
- 173.Von Schwarzenberg K, Lajtos T, Simon L, Müller R, Vereb G, Vollmar AM. V-ATPase inhibition overcomes trastuzumab resistance in breast cancer. Mol Oncol 8: 9–19, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Von Schwarzenberg K, Wiedmann RM, Oak P, Schulz S, Zischka H, Wanner G, Efferth T, Trauner D, Vollmar AM. Mode of cell death induction by pharmacological vacuolar H+-ATPase (V-ATPase) inhibition. J Biol Chem 288: 1385–1396, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sennoune SR, Bakunts K, Martínez GM, Chua-Tuan JL, Kebir Y, Attaya MN, Martínez-Zaguilán R. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am J Physiol Cell Physiol 286: C1443–C1452, 2004. [DOI] [PubMed] [Google Scholar]
- 176.Sennoune SR, Luo D, Martínez-Zaguilán R. Plasmalemmal vacuolar-type H+-ATPase in cancer biology. Cell Biochem Biophys 40: 185–206, 2004. [DOI] [PubMed] [Google Scholar]
- 177.Seol JH, Shevchenko A, Shevchenko A, Deshaies RJ. Skp1 forms multiple protein complexes, including RAVE, a regulator of V-ATPase assembly. Nat Cell Biol 3: 384–391, 2001. [DOI] [PubMed] [Google Scholar]
- 178.Sethi N, Yan Y, Quek D, Schupbach T, Kang Y. Rabconnectin-3 is a functional regulator of mammalian Notch signaling. J Biol Chem 285: 34757–34764, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Seto ES, Bellen HJ. Internalization is required for proper Wingless signaling in Drosophila melanogaster. J Cell Biol 173: 95–106, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sharma M, Astekar M, Soi S, Manjunatha BS, Shetty DC, Radhakrishnan R. pH gradient reversal: an emerging hallmark of cancers. Recent Patents Anticancer Drug Discov 10: 244–258, 2015. [DOI] [PubMed] [Google Scholar]
- 182.Shen W, Zou X, Chen M, Liu P, Shen Y, Huang S, Guo H, Zhang L. Effects of diphyllin as a novel V-ATPase inhibitor on gastric adenocarcinoma. Eur J Pharmacol 667: 330–338, 2011. [DOI] [PubMed] [Google Scholar]
- 183.Simon S, Roy D, Schindler M. Intracellular pH and the control of multidrug resistance. Proc Natl Acad Sci USA 91: 1128–1132, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Smardon AM, Tarsio M, Kane PM. The RAVE complex is essential for stable assembly of the yeast V-ATPase. J Biol Chem 277: 13831–13839, 2002. [DOI] [PubMed] [Google Scholar]
- 185.Smith AN, Finberg KE, Wagner CA, Lifton RP, Devonald MA, Su Y, Karet FE. Molecular cloning and characterization of Atp6n1b: a novel fourth murine vacuolar H+-ATPase a-subunit gene. J Biol Chem 276: 42382–42388, 2001. [DOI] [PubMed] [Google Scholar]
- 186.Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet 26: 71–75, 2000. [DOI] [PubMed] [Google Scholar]
- 187.Staebler A, Pierce JH, Brazinski S, Heidaran MA, Li W, Schlegel R, Goldstein DJ. Mutational analysis of the beta-type platelet-derived growth factor receptor defines the site of interaction with the bovine papillomavirus type 1 E5 transforming protein. J Virol 69: 6507–6517, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Stock C, Schwab A. Protons make tumor cells move like clockwork. Pflügers Arch 458: 981–992, 2009. [DOI] [PubMed] [Google Scholar]
- 189.Stover EH, Borthwick KJ, Bavalia C, Eady N, Fritz DM, Rungroj N, Giersch ABS, Morton CC, Axon PR, Akil I, Al-Sabban EA, Baguley DM, Bianca S, Bakkaloglu A, Bircan Z, Chauveau D, Clermont MJ, Guala A, Hulton SA, Kroes H, Li Volti G, Mir S, Mocan H, Nayir A, Ozen S, Rodriguez Soriano J, Sanjad SA, Tasic V, Taylor CM, Topaloglu R, Smith AN, Karet FE. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet 39: 796–803, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Stransky LA, Forgac M. Amino acid availability modulates vacuolar H+-ATPase assembly. J Biol Chem 290: 27360–27369, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sun-Wada GH, Toyomura T, Murata Y, Yamamoto A, Futai M, Wada Y. The a3 isoform of V-ATPase regulates insulin secretion from pancreatic beta-cells. J Cell Sci 119: 4531–4540, 2006. [DOI] [PubMed] [Google Scholar]
- 192.Sun-Wada GH, Wada Y. Vacuolar-type proton pump ATPases: acidification and pathological relationships. Histol Histopathol 28: 805–815, 2013. [DOI] [PubMed] [Google Scholar]
- 193.Supek F, Supekova L, Mandiyan S, Pan YC, Nelson H, Nelson N. A novel accessory subunit for vacuolar H+-ATPase from chromaffin granules. J Biol Chem 269: 24102–24106, 1994. [PubMed] [Google Scholar]
- 194.Susani L, Pangrazio A, Sobacchi C, Taranta A, Mortier G, Savarirayan R, Villa A, Orchard P, Vezzoni P, Albertini A, Frattini A, Pagani F. TCIRG1-dependent recessive osteopetrosis: mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA. Hum Mutat 24: 225–235, 2004. [DOI] [PubMed] [Google Scholar]
- 195.Su Y, Zhou A, Al-Lamki RS, Karet FE. The a-subunit of the V-type H+-ATPase interacts with phosphofructokinase-1 in humans. J Biol Chem 278: 20013–20018, 2003. [DOI] [PubMed] [Google Scholar]
- 196.Takeuchi K, Ito F. Receptor tyrosine kinases and targeted cancer therapeutics. Biol Pharm Bull 34: 1774–1780, 2011. [DOI] [PubMed] [Google Scholar]
- 197.Toei M, Toei S, Forgac M. Definition of membrane topology and identification of residues important for transport in subunit a of the vacuolar ATPase. J Biol Chem 286: 35176–35186, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tognon E, Kobia F, Busi I, Fumagalli A, De Masi F, Vaccari T. Control of lysosomal biogenesis and Notch-dependent tissue patterning by components of the TFEB-V-ATPase axis in Drosophila melanogaster. Autophagy 12: 499–514, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Torigoe T, Izumi H, Ishiguchi H, Uramoto H, Murakami T, Ise T, Yoshida Y, Tanabe M, Nomoto M, Itoh H, Kohno K. Enhanced expression of the human vacuolar H+-ATPase c subunit gene (ATP6L) in response to anticancer agents. J Biol Chem 277: 36534–36543, 2002. [DOI] [PubMed] [Google Scholar]
- 200.Toyomura T, Murata Y, Yamamoto A, Oka T, Sun-Wada GH, Wada Y, Futai M. From lysosomes to the plasma membrane: localization of vacuolar-type H+-ATPase with the a3 isoform during osteoclast differentiation. J Biol Chem 278: 22023–22030, 2003. [DOI] [PubMed] [Google Scholar]
- 201.Toyomura T, Oka T, Yamaguchi C, Wada Y, Futai M. Three subunit a isoforms of mouse vacuolar H+-ATPase. Preferential expression of the a3 isoform during osteoclast differentiation. J Biol Chem 275: 8760–8765, 2000. [DOI] [PubMed] [Google Scholar]
- 202.Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science 299: 1400–1403, 2003. [DOI] [PubMed] [Google Scholar]
- 203.Tschan MP, Simon HU. The role of autophagy in anticancer therapy: promises and uncertainties. J Intern Med 268: 410–418, 2010. [DOI] [PubMed] [Google Scholar]
- 204.Turk V, Turk B, Guncar G, Turk D, Kos J. Lysosomal cathepsins: structure, role in antigen processing and presentation, and cancer. Adv Enzyme Regul 42: 285–303, 2002. [DOI] [PubMed] [Google Scholar]
- 205.Tuttle AM, Hoffman TL, Schilling TF. Rabconnectin-3a regulates vesicle endocytosis and canonical Wnt signaling in zebrafish neural crest migration. PLoS Biol 12: e1001852, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Vaccari T, Duchi S, Cortese K, Tacchetti C, Bilder D. The vacuolar ATPase is required for physiological as well as pathological activation of the Notch receptor. Development 137: 1825–1832, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Valapala M, Hose S, Gongora C, Dong L, Wawrousek EF, Samuel Zigler J, Sinha D. Impaired endolysosomal function disrupts Notch signalling in optic nerve astrocytes. Nat Commun 4: 1629, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Vargas-Poussou R, Houillier P, Le Pottier N, Strompf L, Loirat C, Baudouin V, Macher MA, Déchaux M, Ulinski T, Nobili F, Eckart P, Novo R, Cailliez M, Salomon R, Nivet H, Cochat P, Tack I, Fargeot A, Bouissou F, Kesler GR, Lorotte S, Godefroid N, Layet V, Morin G, Jeunemaître X, Blanchard A. Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol 17: 1437–1443, 2006. [DOI] [PubMed] [Google Scholar]
- 209.Victor BC, Anbalagan A, Mohamed MM, Sloane BF, Cavallo-Medved D. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res 13: R115, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Vitavska O, Wieczorek H, Merzendorfer H. A novel role for subunit C in mediating binding of the H+-V-ATPase to the actin cytoskeleton. J Biol Chem 278: 18499–18505, 2003. [DOI] [PubMed] [Google Scholar]
- 211.Vukovic V, Tannock IF. Influence of low pH on cytotoxicity of paclitaxel, mitoxantrone and topotecan. Br J Cancer 75: 1167–1172, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wang Y, Toei M, Forgac M. Analysis of the membrane topology of transmembrane segments in the C-terminal hydrophobic domain of the yeast vacuolar ATPase subunit a (Vph1p) by chemical modification. J Biol Chem 283: 20696–20702, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wang Z, Liu S, Kakizaki M, Hirose Y, Ishikawa Y, Funato H, Yanagisawa M, Yu Y, Liu Q. Orexin/hypocretin activates mTOR complex 1 (mTORC1) via an Erk/Akt-independent and calcium-stimulated lysosome v-ATPase pathway. J Biol Chem 289: 31950–31959, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 11: 671–677, 2011. [DOI] [PubMed] [Google Scholar]
- 215.Wiedmann RM, von Schwarzenberg K, Palamidessi A, Schreiner L, Kubisch R, Liebl J, Schempp C, Trauner D, Vereb G, Zahler S, Wagner E, Müller R, Scita G, Vollmar AM. The V-ATPase-inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho-GTPase Rac1. Cancer Res 72: 5976–5987, 2012. [DOI] [PubMed] [Google Scholar]
- 216.Xu J, Xie R, Liu X, Wen G, Jin H, Yu Z, Jiang Y, Zhao Z, Yang Y, Ji B, Dong H, Tuo B. Expression and functional role of vacuolar H+-ATPase in human hepatocellular carcinoma. Carcinogenesis 33: 2432–2440, 2012. [DOI] [PubMed] [Google Scholar]
- 217.Xu X, Liu B, Zou P, Zhang Y, You J, Pei F. Silencing of LASS2/TMSG1 enhances invasion and metastasis capacity of prostate cancer cell. J Cell Biochem 115: 731–743, 2014. [DOI] [PubMed] [Google Scholar]
- 218.Xu X, You J, Pei F. Silencing of a novel tumor metastasis suppressor gene LASS2/TMSG1 promotes invasion of prostate cancer cell in vitro through increase of vacuolar ATPase activity. J Cell Biochem 113: 2356–2363, 2012. [DOI] [PubMed] [Google Scholar]
- 219.Xu Y, Parmar A, Roux E, Balbis A, Dumas V, Chevalier S, Posner BI. Epidermal growth factor-induced vacuolar H+-ATPase assembly: a role in signaling via mTORC1 activation. J Biol Chem 287: 26409–26422, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23: 33–42, 1998. [DOI] [PubMed] [Google Scholar]
- 221.Yamamoto H, Komekado H, Kikuchi A. Caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of beta-catenin. Dev Cell 11: 213–223, 2006. [DOI] [PubMed] [Google Scholar]
- 222.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 10: 1533–1541, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yan Y, Denef N, Schüpbach T. The vacuolar proton pump, V-ATPase, is required for Notch signaling and endosomal trafficking in Drosophila. Dev Cell 17: 387–402, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yin JJ, Pollock CB, Kelly K. Mechanisms of cancer metastasis to the bone. Cell Res 15: 57–62, 2005. [DOI] [PubMed] [Google Scholar]
- 225.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266: 17707–17712, 1991. [PubMed] [Google Scholar]
- 226.Yoshimoto Y, Imoto M. Induction of EGF-dependent apoptosis by vacuolar-type H+-ATPase inhibitors in A431 cells overexpressing the EGF receptor. Exp Cell Res 279: 118–127, 2002. [DOI] [PubMed] [Google Scholar]
- 227.You H, Jin J, Shu H, Yu B, De Milito A, Lozupone F, Deng Y, Tang N, Yao G, Fais S, Gu J, Qin W. Small interfering RNA targeting the subunit ATP6L of proton pump V-ATPase overcomes chemoresistance of breast cancer cells. Cancer Lett 280: 110–119, 2009. [DOI] [PubMed] [Google Scholar]
- 228.Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, Wu GS, Wu K. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett 369: 20–27, 2015. [DOI] [PubMed] [Google Scholar]
- 229.Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL, Wu YQ, Li TY, Liang Y, Lu Z, Lian G, Liu Q, Guo H, Yin Z, Ye Z, Han J, Wu JW, Yin H, Lin SY, Lin SC. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab 20: 526–540, 2014. [DOI] [PubMed] [Google Scholar]
- 230.Zhang T, Zhou Q, Ogmundsdottir MH, Möller K, Siddaway R, Larue L, Hsing M, Kong SW, Goding CR, Palsson A, Steingrimsson E, Pignoni F. Mitf is a master regulator of the v-ATPase, forming a control module for cellular homeostasis with v-ATPase and TORC1. J Cell Sci 128: 2938–2950, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Zhang Z, Zheng Y, Mazon H, Milgrom E, Kitagawa N, Kish-Trier E, Heck AJR, Kane PM, Wilkens S. Structure of the yeast vacuolar ATPase. J Biol Chem 283: 35983–35995, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zhao J, Benlekbir S, Rubinstein JL. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 521: 241–245, 2015. [DOI] [PubMed] [Google Scholar]
- 233.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334: 678–683, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zuo J, Vergara S, Kohno S, Holliday LS. Biochemical and functional characterization of the actin-binding activity of the B subunit of yeast vacuolar H+-ATPase. J Exp Biol 211: 1102–1108, 2008. [DOI] [PubMed] [Google Scholar]