

Abstract
Fungal maleidrides are an important family of bioactive secondary metabolites that consist of 7, 8, or 9‐membered carbocycles with one or two fused maleic anhydride moieties. The biosynthesis of byssochlamic acid (a nonadride) and agnestadride A (a heptadride) was investigated through gene disruption and heterologous expression experiments. The results reveal that the precursors for cyclization are formed by an iterative highly reducing fungal polyketide synthase supported by a hydrolase, together with two citrate‐processing enzymes. The enigmatic ring formation is catalyzed by two proteins with homology to ketosteroid isomerases, and assisted by two proteins with homology to phosphatidylethanolamine‐binding proteins.
Keywords: biosynthesis, cyclization, enzymes, maleidride, polyketides
Maleidrides are bioactive medium‐ring carbocyclic compounds with one or two maleic anhydride moieties and are produced by fungi. Byssochlamic acid (1; Figure 1), discovered by Raistrick and Smith in the 1930s,1 was one of the first maleidrides to have its structure elucidated in the 1960s.2, 3, 4 The structures of its isomers glaucanic acid (2)3 and heveadride (3)5 were also solved at that time. More recent examples include phomoidride A (4, an inhibitor of squalene synthase and Ras farnesyl transferase),6 rubratoxins such as 5 (recently shown to suppress cancer metastasis),7 cornexistin (6, which shows potent and selective herbicidal activity),8 and zopfiellin (7).9 Heptadrides are also known, for example, agnestadride A (8), which was isolated along with byssochlamic acid (1) from Byssochlamys fulva.10
Figure 1.
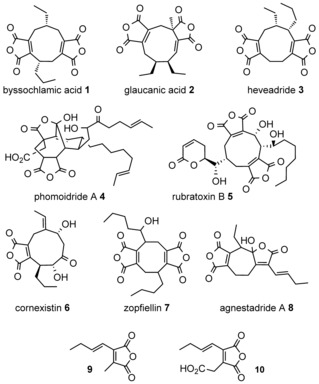
Examples of maleidrides and the hypothesized precursor.
In 1965, Barton and Sutherland proposed a scheme for the biosynthesis of 1, 2, and 3.3 They envisaged biosynthesis from the C9‐maleic anhydride monomer 9, with the differences in the position of the substituents explained by either “head‐to‐tail” or “head‐to‐head” dimerization. However the molecular basis for this dimerization has remained unknown until now.
A reinvestigation of the metabolites produced by B. fulva led to the identification of the probable monomer 10, which undergoes facile decarboxylation to 9. Decarboxylation is also proposed to produce the exo‐methylene putative intermediate 11 (Scheme 1),10 which can then undergo cyclization with a second molecule of 10 to produce 1. Another mode of dimerization of 10 (head‐to‐side dimerization) explains the biosynthesis of agnestadride A (8).10
Scheme 1.
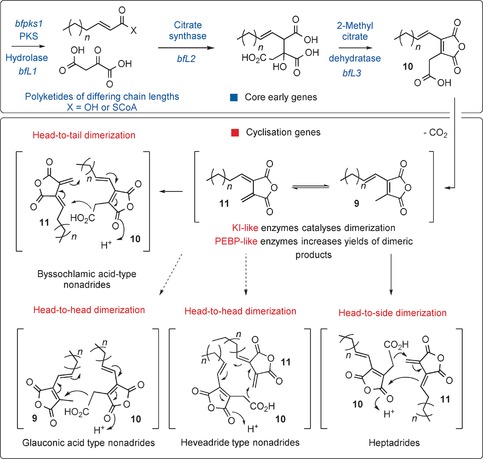
Proposed general pathway to maleidrides.
Barton and Sutherland also hypothesized that monomer 9 is a modified product of the citric acid cycle, produced from a polyketide‐derived hexanoate with oxaloacetate.3 Isotopic feeding studies supported this hypothesis11 and suggested that a candidate maleidride biosynthetic gene cluster (BGC) should contain a highly reducing polyketide synthase (hrPKS),12 as well as a gene encoding a citrate‐synthase‐like enzyme.13
Based on this hypothesis, we sequenced the genomes of two B. fulva strains, IMI 40021 and IMI 58422, both of which are linked to the original strain investigated by Raistrick and Smith.1 The former reliably produces byssochlamic acid, but the latter is an unreliable and generally poor producer. BLAST14 analyses were utilized to search the genomes of both B. fulva strains for likely maleidride BGCs, which led to the identification of a highly homologous maleidride‐type BGC in each genome (Figure 2, Genbank accession KU928136). Each BGC contains an hrPKS, a citrate‐synthase‐like enzyme, and several other interesting genes, including a methylcitrate dehydratase. Transcriptomic analysis of B. fulva IMI 40021 under byssochlamic acid producing and non‐producing conditions confirmed that the putative BGC is highly differentially expressed (Figure 2). The two BGCs are more than 97 % identical but show differences in the bfR4 promoter region, which might explain the low productivity we observed in the IMI 58422 strain (see the Supporting Information).
Figure 2.
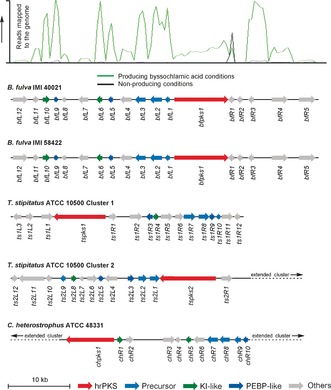
Top: transcriptome analysis of the byssochlamic acid (1) biosynthetic gene cluster in B. fulva IMI 40021 under producing (green) and non‐producing (black) conditions. Bottom: comparative genomic analysis of other putative maleidride BGCs.
BLAST14 searches of genome sequences deposited at the NCBI, utilizing bfpks1 and the citrate synthase‐like gene (bfL2) as queries, revealed several further likely maleidride BGCs in different fungi. Two BGCs were identified from Talaromyces stipitatus. Many Talaromyces species are known to produce glaucanic acid (2), as well as the more complex rubratoxin B (5).15 A third BGC was identified in a Cochliobolus species. Cochliobolus species are synonymous with Helminthosporium, which are the original producers of heveadride (3).5, 16
The bfpks1 gene that encodes the PKS was knocked out by using the bipartite method17 to produce B. fulva bfΔpks1 strains, which no longer produced byssochlamic acid (1) or agnestadride A (8; Figure 3 A and 3 B). This is consistent with heptadride 8 being formed from the same pathway as byssochlamic acid.
Figure 3.
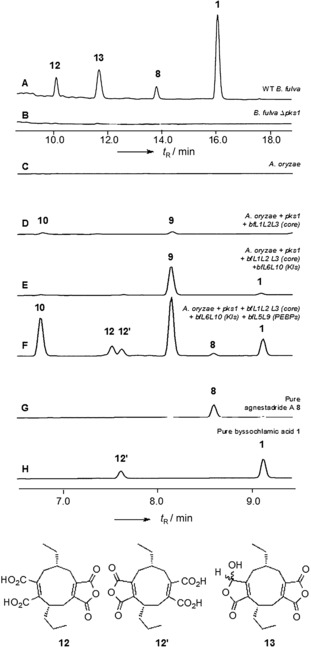
HPLC (DAD, 210–600 nm) chromatograms. A) Extract from WT B. fulva; B) extract from B. fulva Δpks1; C) extract from untransformed A. oryzae NSAR1; D) extract from A. oryzae transformant BF‐PMCH; E) extract from A. oryzae transformant BF‐PMCH+KIs; F) extract from A. oryzae transformant BF‐PMCH+KIs+PEBPs; G) Purified agnestadride A (8); H) Purified byssochlamic acid (1). Compounds 12, 12′, and 13 are cometabolites of 1 and have been previously identified and characterized.10
In a recent investigation of the biosynthesis of phomoidride A (4), Oikawa and co‐workers also identified a BGC containing an hrPKS, a citrate‐synthase‐like enzyme, and a methylcitrate dehydratase, as well as the two maleidride‐type BGCs from Talaromyces stipitatus.18 Heterologous expression experiments utilizing the three genes from one of the T. stipitatus BGCs allowed the isolation of a longer‐chain decarboxylated monomer. These experiments confirm the polyketide/oxaloacetate origin of maleidride monomers.18 However the proteins responsible for the key cyclisation reaction necessary for maleidride biosynthesis remain unkown.
From comparison of the two B. fulva BGCs and the other BGCs identified above, four highly conserved genes were chosen for heterologous expression experiments in Aspergillus oryzae NSAR1:19, 20 the hrPKS bfpks1; the citrate‐synthase‐like gene bfL2; the methylcitrate dehydratase bfL3; and the hydrolase 341 bfL1, which encodes a protein homologous to Type II thiolesterases such as RifR,21 which are involved in the release of abberant ACP‐bound polyketide intermediates during modular polyketide biosynthesis, as well as to LovG, which releases the lovastatin nonaketide from its PKS.22
Expression of these four core genes in A. oryzae NSAR1 (strains AO‐BF‐PMCH 1–7) led to the production of both 9 and 10 (Figure 3 D). Compound 9 is already known to be the decarboxylation product of the highly volatile and unstable 10.10 We also expressed bfpks1, bfL2, and bfL3 in the absence of bfL1, which encodes the hydrolase. Under these conditions, 9 and 10 were not produced (see the Supporting Information), thus strongly suggesting that the hydrolase is essential in the byssochlamic acid system.
Oikawa and co‐workers suggested that the longer‐chain decarboxylated monomer isolated from their heterologous expression experiments is produced by an adventitious native decarboxylase.18 Our experiments have previously identified the natural product 10 by comparing its LCMS characteristics with data collected for the synthetic homologue, which was also observed to decarboxylate spontaneously to give 9. This demonstrates that, at least for the shorter‐chain maleic anhydrides, no enzymatic decarboxylation is necessary.10
To investigate the dimerization process, several further genes were selected for heterologous expression. Analysis of the BGC comparisons from several organisms (Figure 2) revealed that in addition to the core genes, two further types of genes are common to each BGC, namely either one or two genes encoding putative ketosteroid isomerase (KI)‐like proteins and either one or two putative phosphatidylethanolamine‐binding proteins (PEBP).
Co‐expression of the two KI‐like genes (bfL6 and bfL10) with the core genes (bfpks1, bfL1, bfL2, and bfL3) in the heterologous host (strains AO‐BF‐PMCH+KIs 1–21) led to the production of byssochlamic acid (1) and the decarboxylated intermediate 9 in several transformants (Figure 3 E). Agnestadride A (8) and the intermediate 10 were also detected, but in low titer (see the Supporting Information). This result shows that in the A. oryzae NSAR1 background, the four core “monomer” genes and the KI‐like genes are sufficient to form both nonadrides and heptadrides.
To determine whether the presence of both KI‐like genes is necessary for dimerization, or alternatively, whether each KI‐like gene controls a different dimerization mode (nonadride/ heptadride), the core “monomer” genes were co‐expressed with either KI1 (bfL6) or KI2 (bfL10) alone to give strains AO‐BF‐PMCH+KI1 1–7 and AO‐BF‐PMCH+KI2 1–7. Both sets of strains only produced 9 and 10, with no evidence for dimerized products (see the Supporting Information). Reverse‐transcription PCR (RT‐PCR) confirmed that both KI1 and KI2 genes were expressed in each experiment, therefore it can be concluded that within the A. oryzae NSAR1 background, the core “monomer” genes and KI1 or KI2 alone are not sufficient to catalyze dimerization.
The BGC comparisons also revealed that genes encoding proteins similar to PEBPs are common to all of the maleidride BGCs (Figure 2). The two PEBP genes (bfL5 and bfL9) were thus co‐expressed with the core genes and the KI‐like genes in A. oryzae NSAR1 to create strains AO‐BF‐PMCH+KIs+PEBPs 1–8. No new compounds were identified from extracts from these strains. However, direct quantitative comparisons between strains containing the PEBP genes in addition to KI genes showed an over 20‐fold increase in the biosynthesis of byssochlamic acid (to 44 mg L−1, Figure 3 F), as well as a corresponding increase in the production of the heptadride 8. Byssochlamic acid (1) isolated from these experiments was chromatographically and spectroscopically identical to 1 isolated from wild‐type (WT) B. fulva (see the Supporting Information). Compounds 12 and 12′ were also observed in these experiments: these are most likely formed in vitro through partial hydrolysis of 1 during extraction and analysis. However, the reduction product 13 is not formed in A. oryzae, thus suggesting that 13 arises in B. fulva through the activity of an adventitious enzyme.
The biosynthetic pathway to byssochlamic acid and agnestadride A has thus been fully elucidated through genome and transcriptome sequencing, gene disruption, and heterologous expression. Putative maleidride BGCs were identified from the genomes of Talaromyces and Cochliobolus species. Both genera are known to produce nonadrides. Comparison of the byssochlamic acid cluster to the putative maleidride gene clusters identified a core set of genes that are common to all five BGCs. Four genes were identified (bfpks1, bfL1, bfL2, and bfL3) that, when expressed in the heterologous host A. oryzae NSAR1, produce the monomer 10, as well as its spontaneous decarboxylation product 9, both of which have been previously identified from WT B. fulva extracts.10 Experiments to determine the genes involved in dimerization demonstrated that both KI‐like genes co‐expressed with the core genes are necessary. Furthermore, co‐expression of the core genes with both KI‐like genes and both PEBPs gave increased yields of dimerized products. The mechanism for the dimerization still needs to be established through in vitro experiments.
Based on previous biosynthetic investigations, our discovery of the byssochlamic acid pathway, and research conducted by Oikawa and co‐workers,18 we now propose a general biosynthetic route to maleidrides (Scheme 1). In each maleidride pathway, the initial steps are common, with potential differences including polyketide chain length and pattern of reduction. For example, the biosynthesis of 1–3, 6, and 8 requires triketides; 5 requires pentaketides; and 4 requires hexaketides. The polyketide is presumably released from the PKS by the BfL1 hydrolase, although it is not yet known whether this is as the free acid or as a CoA‐thiolester. This result contrasts with the recently reported work of Oikawa, who showed that the hydrolase did not appear to be required in the case of a Talaromyces PKS.18 However it is known that some iterative fungal PKSs, for example the squalestatin tetraketide synthase,23 can release polyketides without the requirement for a dedicated thiolesterase. Reaction between the polyketide and oxaloacetate, catalyzed by the citrate‐synthase‐like enzyme, is followed by dehydration by the methylcitrate dehydratase homologue, which then sets up the synthesis of the maleic anhydride decarboxylated monomers 9 and 11. It is interesting to note that fungi have evolved at least two distinct routes to maleic anhydride moieties. Other organisms oxidize vicinal aromatic methyl groups to form this biologically important motif.24, 25
The KI‐like enzyme(s) can then react 10 with either 9 or 11. These differences, and the orientation of the reacting species, determine the size, substitution pattern, and stereochemistry of the central carbocyclic ring. Previous in vitro work by Baldwin and others26 has shown that such reactions are chemically feasible, although in the absence of enzymes, strong bases are required and low yields are observed. Our experiments suggest that the KI‐like proteins from B. fulva act together to form heterodimeric enzymes that show “flexibility” in that they create both nonadrides and heptadrides. To our knowledge, B. fulva IMI 40021 is the first fungus known to produce cross‐class maleidrides (i.e., nonadride and heptadride), so this “flexibility” may be specific to the byssochlamic acid/agnestadride A pathway. Other known maleidride producers appear to have pathways that only involve one class of dimerization, although it is possible that reinvestigation of the metabolites produced may identify other minor compounds with different dimerization modes. The PEBP‐like enzymes also appear to be involved in the dimerization, and although their catalytic role is not yet clear, it is possible their known anionic binding ability may be involved.27 One possibility may be the chaperoning of highly unstable carboxylates such as 10 in order to prevent premature decarboxylation prior to dimerization. However further understanding of the role of the KI and PEBP proteins must await in vitro experiments. Gene clusters encoding the biosynthesis of octadrides such as zopfiellin (7) have not yet been reported, but our results suggest that similar KI‐like and PEBP‐like proteins may be involved in their biosynthesis.
In conclusion, our experiments have revealed for the first time that maleidride biosynthesis can be recreated in a heterologous host and the observations open the way for future experiments to create new compounds in this class with potentially interesting and useful biological properties through pathway engineering. Further experiments are currently underway in our laboratories to understand the mechanisms and selectivity of the ring‐forming enzymes.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank BBSRC (KW, BB/J006289/1) and Syngenta (AS) for funding. Analytical and preparative LCMS were provide by EPSRC (EP/F066104/1) and DFG (INST 187/621). 500 MHz NMR (EP/L011999/1) was provided by EPSRC. We thank Dr Kate de Mattos‐Shipley (BB/K002341/1) for helpful discussions and bioinformatic assistance with maleidride clusters. We thank Dr Sian Deller, Jo Mattocks and Dr Dianne Irwin at Syngenta for culturing B. fulva isolates and preliminary profiling of byssochlamic acid production, and for preparation of samples for genomic‐ and RNA‐sequencing, and Dr John Clough and Dr William Whittingham at Syngenta for valuable scientific input. B. fulva IMI 58422 was sequenced and assembled at The Genome Analysis Centre (Norwich, UK) under contract to Syngenta through Genome Enterprises Ltd. B. fulva IMI 40021 was sequenced and assembled at the University of Bristol Genomics Facility.
K. Williams, A. J. Szwalbe, N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis, T. J. Simpson, R. J. Cox, Angew. Chem. Int. Ed. 2016, 55, 6784.
Contributor Information
Dr. Katherine Williams, Email: katherine.williams@oci.uni-hannover.de
Prof. Russell J. Cox, Email: russell.cox@oci.uni-hannover.de.
References
- 1. Raistrick H., Smith G., Biochem. J. 1933, 27, 1814–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baldwin J. E., Barton D. H., Bloomer J. L., Jackman L. M., Rodriguez-Hahn L., Sutherland J. K., Experientia 1962, 18, 345–352. [DOI] [PubMed] [Google Scholar]
- 3. Barton D. H. R., Sutherland J. K., J. Chem. Soc. 1965, 1769–1772. [Google Scholar]
- 4. Baldwin J. E., Barton D. H. R., Sutherland J. K., J. Chem. Soc. 1965, 1787–1798. [Google Scholar]
- 5. Crane R. I., Hedden P., Macmillan J., Turner W. B., J. Chem. Soc. Perkin Trans. 1 1973, 194–200. [DOI] [PubMed] [Google Scholar]
- 6. Dabrah T. T., Harwood H. J., Huang L. H., Jankovich N. D., Kaneko T., Li J. C., Lindsey S., Moshier P. M., Subashi T. A., Therrien M., Watts P. C., J. Antibiot. 1997, 50, 1–7. [DOI] [PubMed] [Google Scholar]
- 7. Wada S.-I., Usami I., Umezawa Y., Inoue H., Ohba S.-I., Someno T., Kawada M., Ikeda D., Cancer Sci. 2010, 101, 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakajima M., Itoi K., Takamatsu Y., Sato S., Furukawa Y., Furuya K., Honma T., Kadotani J., Kozasa M., Haneishi T., J. Antibiot. 1991, 44, 1065–1072. [DOI] [PubMed] [Google Scholar]
- 9. Futagawa M., Wedge D. E., Dayan F. E., Pestic. Biochem. Physiol. 2002, 73, 87–93. [Google Scholar]
- 10. Szwalbe A. J., Williams K., O'Flynn D. E., Bailey A. M., Mulholland N. P., Vincent J. L., Willis C. L., Cox R. J., Simpson T. J., Chem. Commun. 2015, 51, 17088–17091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bloomer J. L., Moppett C. E., Sutherland J. K., J. Chem. Soc. C 1968, 588–591. [Google Scholar]
- 12. Cox R. J., Org. Biomol. Chem. 2007, 5, 2010–2026. [DOI] [PubMed] [Google Scholar]
- 13. Wiegand G., Remington S. J., Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 97–117. [DOI] [PubMed] [Google Scholar]
- 14. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., J. Mol. Biol. 1990, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 15. Yilmaz N., Visagie C. M., Houbraken J., Frisvad J. C., Samson R. A., Stud. Mycol. 2014, 78, 175–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hosoe T., Fukushima K., Itabashi T., Nozawa K., Takizawa K., Kawai K. I., Heterocycles 2004, 63, 2581–2589. [Google Scholar]
- 17. Nielsen M. L., Albertsen L., Lettier G., Nielsen J. B., Mortensen U. H., Fungal Genet. Biol. 2006, 43, 54–64. [DOI] [PubMed] [Google Scholar]
- 18. Fujii R., Matsu Y., Minami A., Nagamine S., Takeuchi I., Gomi K., Oikawa H., Org. Lett. 2015, 17, 5658–5661. [DOI] [PubMed] [Google Scholar]
- 19. Pahirulzaman K. A. K., Williams K., Lazarus C. M., Methods Enzymol. 2012, 517, 241–260. [DOI] [PubMed] [Google Scholar]
- 20. Jin F. H., Maruyama J.-I., Juvvadi P. R., Arioka M., Kitamoto K., FEMS Microbiol. Lett. 2004, 239, 79–85. [DOI] [PubMed] [Google Scholar]
- 21. Claxton H. B., Akey D. L., Silver M. K., Admiraal S. J., Smith J. L., J. Biol. Chem. 2009, 284, 5021–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu W., Chooi Y.-H., Choi J. W., Li S., Vederas J. C., Da Silva N. A., Tang Y., Angew. Chem. Int. Ed. 2013, 52, 6472–6475; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6600–6603. [Google Scholar]
- 23. Cox R. J., Glod F., Hurley D., Lazarus C. M., Nicholson T. P., Rudd B. A. M., Simpson T. J., Wilkinson B., Zhang Y., Chem. Commun. 2004, 2260–2261. [DOI] [PubMed] [Google Scholar]
- 24. Davison J., al Fahad A., Cai M., Song Z., Yehia S. Y., Lazarus C. M., Bailey A. M., Simpson T. J., Cox R. J., Proc. Natl. Acad. Sci. USA 2012, 109, 7642–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. al Fahad A., Abood A., Simpson T. J., Cox R. J., Angew. Chem. Int. Ed. 2014, 53, 7519–7523; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7649–7653. [Google Scholar]
- 26. Baldwin J. E., Beyeler A., Cox R. J., Keats C., Pritchard G. J., Adlington R. M., Watkin D. J., Tetrahedron 1999, 55, 7363–7374. [Google Scholar]
- 27. Serre L., Vallée B., Bureaud N., Schoentgen F., Zelwer C., Structure 1998, 6, 1255–1265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary