

 This work is licensed under a
This work is licensed under a Abstract
Preterm labour (PTL) is commonly associated with infection and/or inflammation. Lipopolysaccharide (LPS) from different bacteria can be used to independently or mutually activate Jun N-terminal kinase (JNK)/AP1- or NF-κB-driven inflammatory pathways that lead to PTL. Previous studies using Salmonella abortus LPS, which activates both JNK/AP-1 and NF-κB, showed that selective inhibition of NF-κB delays labour and improves pup outcome. Where labour is induced using Escherichia coli LPS (O111), which upregulates JNK/AP-1 but not NF-κB, inhibition of JNK/AP-1 activation also delays labour. In this study, to determine the potential role of JNK as a therapeutic target in PTL, we investigated the specific contribution of JNK signalling to S. Abortus LPS-induced PTL in mice. Intrauterine administration of S. Abortus LPS to pregnant mice resulted in the activation of JNK in the maternal uterus and fetal brain, upregulation of pro-inflammatory proteins COX-2, CXCL1, and CCL2, phosphorylation of cPLA2 in myometrium, and induction of PTL. Specific inhibition of JNK by co-administration of specific D-JNK inhibitory peptide (D-JNKI) delayed LPS-induced preterm delivery and reduced fetal mortality. This is associated with inhibition of myometrial cPLA2 phosphorylation and proinflammatory proteins synthesis. In addition, we report that D-JNKI inhibits the activation of JNK/JNK3 and caspase-3, which are important mediators of neural cell death in the neonatal brain. Our data demonstrate that specific inhibition of TLR4-activated JNK signalling pathways has potential as a therapeutic approach in the management of infection/inflammation-associated PTL and prevention of the associated detrimental effects to the neonatal brain.
Introduction
Globally, more than 15 million babies are born preterm each year (Blencowe et al. 2013), of which around 1 million die from associated complications. Many survivors experience lifetime learning disabilities and visual and hearing problems. The risk of cerebral palsy (CP) is increased 70-fold in infants born <28 weeks gestation and some 90% of infants born before 30 weeks gestation show brain abnormalities on MRI when imaged at term-corrected age (Romero et al. 2006). Intrauterine infection and/or inflammation plays an important aetiological role in early preterm delivery and is a risk factor for subsequent CP in both term and preterm infants (Wu & Colford 2000).
Inflammation represents a common biochemical pathway critically involved in both term and preterm labour (PTL; Romero et al. 2006). To study the underlying biochemistry of inflammation-associated PTL, several investigators have developed mouse models based on intrauterine or systemic injection of bacteria or bacterial lipopolysaccharide (Hirsch et al. 1995, Kaga et al. 1996, Elovitz et al. 2003, Elovitz & Mrinalini 2004). Bacterial products induce PTL through interaction with toll-like receptor-4 (TLR4) and subsequent activation of pro-inflammatory and pro-contractile pathways within the uterus. Studies using TLR4 mutant mice show that TLR4 plays an essential role in LPS-induced PTL (Elovitz et al. 2003, Wang & Hirsch 2003). Activation of the TLR4 signalling pathway by LPS leads to the upregulation of prostaglandin (PG) synthesis and production of proinflammatory cytokines (Gravett et al. 1994, Pollard & Mitchell 1996, Elovitz et al. 2006). PG E2 and PGF2 and cytokines induce cervical ripening (Kelly 2002) and stimulate uterine contractions (Baggia et al. 1996). There is now good evidence that in addition to inducing preterm delivery, proinflammatory cytokines also mediate antenatal brain injury (Bell et al. 2004, Elovitz et al. 2006). In animal models of prenatal brain injury, injection of LPS or intact bacteria causes lesions in white matter of the neonatal brain (Debillon et al. 2000), including both periventricular leukomalacia (PVL), a pathology associated with the development of CP (Drougia et al. 2007), and diffuse white matter injury, a brain abnormality more commonly associated with adverse neurodevelopmental outcome (Dyet et al. 2006). Due to the association between inflammation, the onset of labour and the risk of perinatal brain injury, attention is being focused on anti-inflammatory agents as novel therapeutic options to prevent PTL (Rinaldi et al. 2011, MacIntyre et al. 2012, Sykes et al. 2014).
Animal studies have demonstrated that several anti-inflammatory agents delay preterm delivery and improve pup survival, including cytokine IL-10 (Terrone et al. 2001, Rodts-Palenik et al. 2004), short-chain fatty acids (Voltolini et al. 2012) and lipoxins (Macdonald et al. 2011). The transcription factor nuclear factor kappa B (NF-κB) plays a pivotal role in the upregulation of pro-labour and pro-inflammatory genes associated with parturition (Choi et al. 2007) and is proposed to inhibit progesterone receptor function and thus block uterine quiescence (Condon et al. 2006). We, and others, have shown that NF-κB inhibition leads to decreased synthesis of cytokines that trigger both preterm delivery and neonatal brain injury, making it an attractive therapeutic target in the management of PTL (Adams Waldorf et al. 2008, Pirianov et al. 2009, Li et al. 2010, Nath et al. 2010).
We have recently shown that normal labour onset in the mouse involves the sequential activation of the transcription factors NF-κB and AP-1 within the uterus (MacIntyre et al. 2014). Additionally, we have identified differential activation of NF-κB and Jun N-terminal kinase (JNK) in two mouse models of LPS-induced PTL (Pirianov et al. 2009, MacIntyre et al. 2014). PTL induced using highly TRL4-specific Salmonella abortus LPS activates both NF-κB and JNK and leads to the upregulation of cPLA2 and COX-2, both central to prostaglandin synthesis, as well as the stimulation of labour-associated cytokines, CCL-2 and CXCL-1, in the myometrium (Pirianov et al. 2009). Inhibition of NF-κB activity, JNK activity and cytokine synthesis by the anti-inflammatory cyclopentenone prostaglandin 15-deoxy-δ12, 14-prostaglandin J2 (15d-PGJ2), delays preterm birth and improves pup survival. In contrast, induction of PTL by Escherichia coli-derived LPS involves AP-1 activation via JNK, but does not involve NF-κB activation (MacIntyre et al. 2014). In this model, inhibition of JNK using SP600125 delays PTL. Where labour is induced using the PR/GR antagonist RU486, no sequential activation of NF-κB and AP-1 is detected in the uterus, but labour itself is associated with increased JNK activity. Thus NF-κB activation appears to be a feature of normal labour and of labour induced by some LPS serotypes, but it is not a universal feature of all models of inflammation- or non-inflammation-induced PTL.
JNK-mediated activation of AP-1 appears to be common to both inflammation- or non-inflammation-induced PTL. Further, brain activation of JNK has been shown to play a key role in perinatal brain injury. Three mammalian JNK genes (JNK1, JNK2 and JNK3) have been identified of which JNK3 is the form most predominantly active in the brain. In a mouse model where JNK3 expression was ablated, pup brain levels of c-Jun were reduced compared to wild-type (WT) animals and led to partial protection against hypoxic–ischaemic injury through a reduction of caspase-3 activation (Pirianov et al. 2007).
Specific inhibition of JNK, in the context of inflammation-induced PTL, has the potential to both delay delivery and improve pup outcome. However, where inflammation activates both NF-κB and JNK, specific inhibition of JNK may be insufficient to either delay preterm birth or improve pup outcome. In this study S. abortus LPS was used to activate both uterine NF-κB and JNK activation and cause preterm birth. Animals were then treated with a highly specific JNK inhibitor, D-JNK inhibitory peptide (D-JNKI), to determine if in vivo inhibition of myometrial JNK-activation and downstream inflammatory mediators such as COX-2, cPLA and cytokines delays LPS-induced preterm delivery, improves neonatal mortality and reduces JNK3 activation and thus damaging the neonatal brain.
Materials and methods
Reagents and antibodies
Antibodies against serine 505-phosphorylated cPLA2, HRP-conjugated secondary antibodies and JNK in vitro kinase kit and cleaved caspase-3 antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). COX-2, CCL2, CXCL1 and JNK2 antibodies were purchased from Santa Cruz Biotechnology. JNK1 and JNK1/2 antibodies were obtained from Pharmingen (San Jose, CA, USA). The antibody against β-actin was from Abcam (Cambridge, UK). LPS (TLR-4 grade S form from Salmonella abortus) was purchased from Enzo Biosciences (Nottingham, UK). D-JNKI was kindly provided by Dr H Mehmet (Merck, Kenilworth, NJ, USA).
Mouse model of intrauterine inflammation
All animal experiments were approved by the Imperial College London Ethical Review Board and conformed to the British Home Office regulations. CD-1 outbred mice were used in this study. This strain is commonly used in LPS-mediated models of PTL and inflammation more generally and does not exhibit any known LPS resistance. CD-1 outbred, timed-pregnant mice were obtained from Harlan Laboratories (Bicester, UK) on gestation day 13 after mating and were acclimatised for 3 days before being used in experiments. Surgery was performed on day 16 of gestation. Dams were anaesthetised by isofluorane, the uterine horns exteriorised following laparotomy and kept moist with sterile PBS. The uterine horn containing the greatest number of viable fetuses was selected for injection. A 25 μl volume LPS (1.0 μg) alone, 5 μg D-JNKI, or 5 μg LPS plus 5 μg D-JNKI was injected into the lumen of the uterus between the first and second anterior fetuses using a 33-gauge Hamilton syringe, taking care not to enter the amniotic cavity. The wound was closed in two layers. Mice received Vetalar analgesia (Parke Davis & Co. Ltd., Eastleigh, UK) and were recovered in a warm environment. For observations, each mouse was housed separately. Control animals received no anaesthesia or surgery, and sham animals received anaesthesia and an intrauterine injection of 25 μl of saline. The time from surgery to delivery was recorded, with delivery of the newborn pups and their survival rate monitored every 6 h. Animals studies were in accordance with UK Home Office licence conditions.
Tissue harvesting and processing
Maternal uterine tissue (myometrium) and foetal brain were collected close to the injection site in the uterine horn 1 and 6 h post-injection with LPS ±D-JNKI (1, 2 or 5 μg) or vehicle control. Samples were flash frozen in liquid nitrogen and stored at −80 °C. Tissues were lysed by sonication in a non-denaturing phosphate lysis buffer consisting of 20 mM sodium phosphate, 137 mM NaCl, 25 mM sodium β-glycerophosphate, 2 mM sodium pyrophosphate, 2 mM EDTA, 10% glycerol, 1% Triton X-100 and protease inhibitor cocktail (Sigma–Aldrich). Cell lysates were incubated on ice for 20 min and centrifuged for 20 min at 12 000 g at 4 °C. Protein concentration was determined by the bicinchoninic acid method (Pierce, Rockford, IL, USA).
RT-PCR
Total RNA was isolated using RNA STAT-60 (Tel-Test, Inc., Friendswood, TX, USA) according to the manufacturer's instructions. A total of 1 μg RNA was used as a template for reverse transcription. Expression of OTR, COX-1, COX-2, connexin 26 and 43, CCL2, CXCL-1 and GAPDH were assessed by real-time RT-PCR using an ABI PRISM 7700 Sequence Detection System according to the manufacturer's protocol (Applied Biosystems/Life Technologies). Taqman primers and probes were designed using the primer express programme (Applied Biosystems/Life Technologies). The data were analysed using Sequence Detector version 1.7 Software (Applied Biosystems/Life Technologies) and were normalised to GAPDH.
SDS–PAGE and immunoblotting
Tissue lysates (50 μg) were separated on a 10% SDS–PAGE gel and transferred to PVDF membranes (Millipore, Billerica, MA, USA) and blocked using 5% (w/v) skimmed milk in Tris-buffered saline (TBS) supplemented with 0.1% (v/v) Tween-20 (TBST) for 1 h at room temperature. Blots were incubated overnight at 4 °C with primary antibody (1:1000 dilutions in TBS, 1% milk). After washing in TBST, blots were incubated with HRP-conjugated goat anti-rabbit antibody or rabbit anti-mouse antibody at room temperature for 1 h in 5% milk prepared in TBST. Following the final wash, immune-reactive bands were visualized on film using a chemiluminescent substrate (ECL Plus, GE Healthcare and Buckinghamshire, UK). Densitometric analysis was performed using 1D Kodak Digital Science Software (Kodak). The levels of cellular β-actin were used as a loading control.
Immunoprecipitation of cleaved caspase-3
Tissue lysates (200 μg protein) were incubated overnight at 4 °C with G agarose beads (GE Healthcare) pre-bound with cleaved p17 caspase-3 antibody. After washing, the beads were resuspended in a sample loading buffer and heated for 5 min at 95 °C and spun down. Samples (50 μl) were separated by electrophoresis on a 14% SDS–PAGE gel, transferred to a PVDF membrane and finally incubated with cleaved caspase-3 antibody (1:1000) overnight at 4 °C following the western blotting procedure previously described.
In vitro kinase assay for JNK
JNK activity was measured using a specific kit (Cell Signaling Technology) following the manufacture's instructions and using GST-Jun (1–79) fusion peptide as the specific substrate for JNK. In brief, tissue lysates (100 μg protein) were incubated overnight at 4 °C with GST-Jun fusion protein beads. After washing, the beads were resuspended in kinase buffer containing ATP and the kinase reaction was allowed to proceed for 30 min at 30 °C. The reaction was stopped by the addition of a sample loading buffer. Proteins were separated by electrophoresis on a 10% SDS–PAGE gel, transferred to a PVDF membrane and finally incubated with phospho-c-Jun (Ser63) and c-Jun antibodies. Finally, blots were subjected to enhanced chemiluminescence and kinase activity determined by densitometric analysis.
In vitro kinase assay for JNK3
Currently, specific non-cross-reactive antibodies for the major JNK isoforms 1, 2 and 3 are not available. Therefore we used an approach for capturing JNK3 following immunodepletion of JNK1 and JNK2 isoforms. To specifically determine the presence of the active JNK3 isoform, cell lysates were first immunoprecipitated with a mixture of JNK1 (cross-reacts with JNK1 and 2 but not with JNK3; data not shown) and JNK2 (cross-reacts with JNK2 and 1 but not with JNK3; data not shown) antibodies already pre-bound to Protein-G beads to remove both JNK1 and JNK2 from the lysates. This process was repeated twice to ensure that JNK1 and JNK2 were completely removed from the supernatant. Depletion of JNK1 and JNK2 was confirmed by western blot analysis of cell lysates before and after immunodepletion using a JNK1/2 antibody that recognizes both the JNK1 and JNK2 isoforms. The residual active JNK3 isoform in the supernatant was subjected to in vitro kinase assay to measure JNK3 activity, as described above.
Statistical analysis
Biochemical and molecular data are reported as mean±s.d. and analysed with one-way ANOVA followed by the Bonferroni post-test for multiple comparisons using GraphPad Prism version 4.0. The differences between multiple groups in PTL model were analysed by Kruskal-Wallis equality-of-population rank test.
Results
D-JNKI supresses LPS-induced JNK activation in mouse myometrium and fetal brain
We have previously shown that the administration of S. abortus LPS activates TLR4/JNK signalling in an experimental model of PTL (Pirianov et al. 2009). Here, the effects of D-JNKI (a specific inhibitor of JNK) on PTL were investigated. In the first series of experiments we determined the ability of co-administration of D-JNKI (0–5 μg) to block S. abortus LPS-induced TLR4/JNK activation in the myometrium and in the fetal brain in this mouse model of PTL. Animals were sacrificed 1 h post-injection of LPS. Because D-JNKI prevents JNK/substrate interactions but not JNK phosphorylation, we utilised an in vitro kinase assay to monitor the inhibitory effects of D-JNKI in tissue lysates. Kinase assay data showed that LPS injection induced JNK activity at 1 h post-injection in both the maternal myometrium and the neonatal brain. Inhibition of LPS-induced JNK activity was achieved by D-JNKI at an injection dose of 5 μg (Fig. 1A). Although this single western blot gives the impression that JNK is activated at a higher level in the D-JNKI 5 μg treated samples compared to controls and LPS+D-JNKI treated samples, quantification of three separate experiments (Fig. 2A and B) shows that D-JNKI at 5 μg has no effect on JNK activity. Based on this result, JNK activity assays were performed using 5 μg of D-JNKI as shown in Fig. 2. This dose of D-JNKI was sufficient to inhibit JNK activity up to 6 h post-injection in both the myometrium and neonatal brain following LPS injection (Fig. 2A and B).
Figure 1.
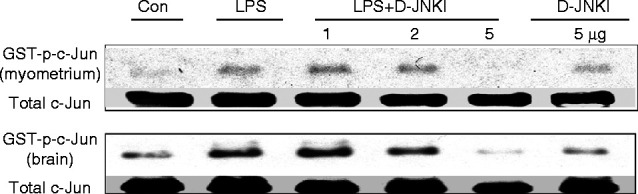
D-JNKI inhibits LPS-induced JNK activation in myometrium and neonatal brain. Timed pregnant CD-1 mice were injected on day 16 with Salmonella abortus LPS (1.0 μg) alone, LPS together with (1, 2 or 5 μg) of D-JNKI or with D-JNKI (5 μg) alone. Tissue samples from myometrium and fetal brain were isolated at 1 h post-treated and examined for JNK activity using an in vitro kinase assay. Maximal inhibition of LPS-induced JNK activation was achieved with 5 μg D-JNKI.
Figure 2.
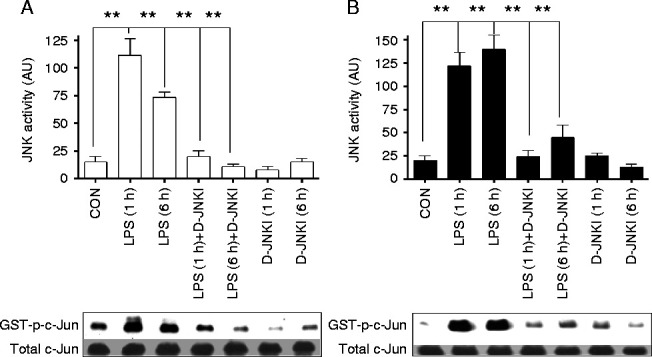
D-JNKI inhibits LPS-induced JNK activation in myometrium and neonatal brain at 1 and 6 h post-injection. Timed pregnant CD-1 mice were injected on gestation day 16 with Salmonella abortus LPS (1.0 μg) alone, LPS together with (5 μg) of D-JNKI or with D-JNKI (5 μg) alone. (A) Tissue samples from myometrium and (B) fetal brain were isolated at 1 and 6 h post-injection and analysed for JNK activity using an in vitro kinase assay. (n=3 **P<0.001 compared to LPS alone at each time point ANOVA).
D-JNKI delays LPS-induced preterm delivery
In the next experiments we determined whether specific inhibition of JNK by D-JNKI would lead to delayed S. abortus LPS-induced PTL in this animal model. Animals were injected on gestation day 16 with S. abortus alone or together with 5 μg of D-JNKI or with 5 μg of D-JNKI alone. The pregnancy was monitored at 6 h intervals until delivery. S. abortus LPS alone caused preterm delivery after ∼20 h (±s.e.m.) (Fig. 3A) with 70% pup mortality (Fig. 3B). Co-injection of 5 μg of D-JNKI delayed preterm delivery by an average of 10 h (to a mean of 32 h; Fig. 3A) and reduced pup mortality to 10% (Fig. 3B). Injection of 5 μg of D-JNKI alone was associated with later preterm delivery at 40 h, with no pup mortality. These data show that the specific inhibition of JNK delays S. abortus LPS-induced PTL and increases pup survival.
Figure 3.
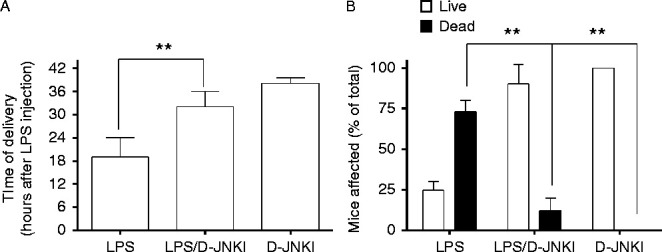
D-JNKI delays LPS-induced preterm delivery and increases pup survival. Timed pregnant CD-1 mice were injected on gestation day 16 with 1.0 μg Salmonella abortus LPS (n=7) or LPS together with 5 μg of D-JNKI (n=8) or with 5 μg of D-JNKI alone (n=8). Delivery of the newborn pups and their survival rate was monitored every 6 h. D-JNKI led to a significant increase in time to delivery as well as increased pup survival. Data represent the mean (±s.e.m.) of n animals at each group. **P<0.01, Kruskal-Wallis equality-of-population rank test.
D-JNKI inhibits both LPS-induced cPLA2 phosphorylation and upregulation of proinflammatory proteins COX-2, CCL2 and CXCL1 in mouse myometrium
We have previously reported that PTL induced in a mouse model using S. abortus LPS has no effect on OTR, connexins 23 and 46 or COX-1 but significantly upregulates mRNA levels of COX-2, CCL2 and CXCL1 and increases phosphorylation of cPLA2 in the myometrium 6 h post-injection. We therefore studied the effect of D-JNKI on S. abortus LPS-induced expression of COX-2, CCL2 and CXCL1 and cPLA2 phosphorylation. Upregulation of COX-2, CCL2 and CXCL1 were significantly suppressed by co-administration of D-JNKI (Fig. 4A, B and C). Using western blotting analysis, we validated the mRNA data at the protein level COX-2, CCL2 and CXCL1 in mouse myometrium (Fig. 4D and E). S. abortus LPS injection induced cPLA2 phosphorylation in maternal myometrium, which was inhibited by D-JNKI at 1 h but not after 6 h following injection (Fig. 5).
Figure 4.

D-JNKI inhibits LPS-induced cPLA2 phosphorylation and production of COX-2, CCL-2 and CXCL-1 in myometrium. Timed pregnant CD-1 mice were injected on day 16 with 1.0 μg of Salmonella abortus LPS alone, LPS together with 5 μg of D-JNKI or with 5 μg of D-JNKI alone. Tissue samples from the myometrium were collected at 6 h and analysed for mRNA and protein levels of COX-2 (A and D), CCL-2 (B and E) and CXCL-1 (C and F) expression and normalised to loading controls (GAPDH and actin) as arbitrary units (AU). Data represent the mean (±s.e.m.) of three animals at each data point. The difference between LPS alone or together with D-JNKI was statistically significant by ANOVA *P<0.05 and **P<0.01.
Figure 5.
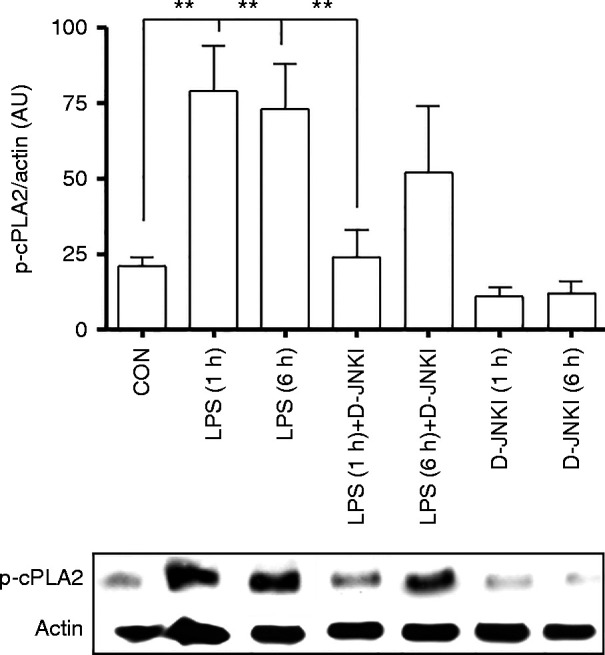
D-JNKI inhibits LPS-induced cPLA2 phosphorylation. Timed pregnant CD-1 mice were treated on gestation day 16 with Salmonella abortus LPS (1.0 μg) alone, LPS together with 5 μg of D-JNKI, or with 5 μg of D-JNKI alone. Tissue samples from the myometrium were prepared at 6 h and analysed for cPLA2 phosphorylation and normalised to loading control actin as arbitrary units (AU). Data represent the mean (±s.e.m.) of three animals at each data point. The difference between LPS alone or together with D-JNKI was statistically significant by ANOVA (**P<0.01).
D-JNKI supresses LPS-induced JNK3 and caspase-3 activation in the fetal brain
We have previously shown that the JNK3 isoform plays a critical role in hypoxia-ischaemia-induced neural cell death and neonatal brain injury (Pirianov et al. 2007). We therefore examined the effect of D-JNKI on S. abortus LPS-driven JNK3 and caspase-3 activation in the fetal brain. Using an in vitro immunodepletion kinase assay (Pirianov et al. 2006) to measure JNK/JNK3 activity, we found that co-injection of D-JNKI downregulates JNK activation and completely inhibits JNK3 activity in the fetal brain (Fig. 6A). This effect of D-JNKI is associated with a decrease in S. abortus LPS-induced caspase-3 activity, measured as a production of cleaved p17 caspase-3 active fragment (Fig. 6B).
Figure 6.
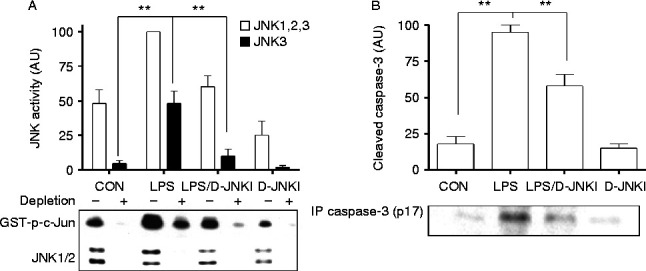
D-JNKI inhibits LPS-induced JNK/JNK3 and caspase-3 activation in the fetal brain. Pup brain extracts collected at 6 h following injection with 1.0 μg of Salmonella abortus LPS alone, LPS together with 5 μg of D-JNKI, or with 5 μg of D-JNKI alone were analysed for (A) JNK/JNK3 and (B) cleaved caspase-3 activation. Data represent the mean (±s.e.m.) of three animals at each data point. The difference between LPS alone or together with D-JNKI was statistically significant by ANOVA (**P<0.05).
Discussion
There exists a spectrum of severity of inflammation associated with human PTL. Overt clinical signs of chorioamnionitis are rarely seen during pregnancy in women who go on to experience an infection-associated PTL. In most cases, evidence of chorioamnionitis is only obtained following histological examination of the placenta and membranes following delivery. We originally developed the mouse model of inflammation-associated preterm delivery described in this study to model the clinical presentation in humans. The model is based on those developed by Hirsch et al. (1995) and Elovitz et al. (2003, 2006), in which sonicated bacteria or E. coli-derived LPS are used to induce preterm birth. However, these models report high variations in the LPS dose required, time to delivery and level of pup mortality. Our S. abortus LPS model has the advantage of reliable preterm delivery rates and controllable rates of pup mortality thus permitting in vivo study more representative of human presentation.
LPS derived from different bacterial species are known to differentially induce TLR4 signalling pathways (Park & Lee 2013). However, it is increasingly recognised that LPS molecules from the same bacterial species belonging to different serotypes also differentially interact and stimulate the TLR4 receptor (Chessa et al. 2014). We have previously reported that LPS isolated from either S. abortus or E. coli will activate PTL in the mouse, yet time to delivery and pup survival rates vary significantly (Pirianov et al. 2009, MacIntyre et al. 2014). Moreover, we have shown that E. coli-derived LPS serotypes differentially activate inflammatory pathways in the mouse myometrial and pup brain leading to major differences in maternal and fetal outcomes (unpublished data). While all four of the E. coli LPS serotypes tested led to preterm birth, time to preterm delivery onset was strongly correlated with the level of JNK/AP1 activation but not correlated with NF-κB activation. Recent studies using E. coli serotype O111 (B4) alone have shown that preterm delivery in the mouse involves myometrial activation of AP-1 via JNK without upregulation of NF-κB activity and that inhibition of JNK can delay PTL (MacIntyre et al. 2014). In contrast, S. abortus LPS leads to preterm delivery that is associated with myometrial activation of both NF-κB and JNK/AP-1 and that inhibition of NF-κB by the cyclo-pentenone 15d-PGJ2 delays preterm delivery, improves pup survival and inhibits myometrial and brain inflammation in the mouse PTL model primarily (Pirianov et al. 2009). The relative contribution of NF-κB and JNK/AP-1 to both PTL and fetal outcomes is yet to be fully elucidated. Thus, in this study we set out to determine whether specific inhibition of JNK would also be effective in a model in which both NF-κB and JNK/AP-1 are activated. We selected the D-JNKI rather than SP600125 because the latter is a comparably weak and non-specific inhibitor of JNK (Jin et al. 2009). The D-JNKI used in this study is a small peptide pharmacological of JNK activity based on specific interaction with substrate binding domain of JNK (Bonny et al. 2001, Barr et al. 2002). D-JNKI is a highly cell permeable, decoy substrate, which potently and selectively inhibits all JNK isoforms and has the potential to be used as a small molecule drug.
In the initial dose response studies, animals were sacrificed at 1 h after injection of LPS. This time point was chosen for the harvesting of uterine and brain samples because JNK is thought to be involved in the early signal transduction pathways leading to labour onset and neonatal brain injury, and previous work has shown that cytokine levels in the mouse uterus and brain are elevated within 1–8 h of LPS administration (Hirsch et al. 1995, Elovitz et al. 2006). Accordingly, effective and clinically relevant strategies for JNK inhibition should target the early phase of inflammatory pathway induction. Once the suitable dose of D-JNKI had been determined, the effect of that dose at early (1 h) and later (6 h) times points was confirmed (Fig. 1B). As D-JNKI prevents JNK/substrate interactions but not JNK phosphorylation, we then utilised an in vitro kinase assay, rather than a marker of JNK phosphorylation (indirect JNK activation), to monitor the inhibitory effects of D-JNKI in myometrium and brain tissue lysates (Fig. 2A and B). Significant inhibition of JNK activity by D-JNKI was also achieved at both 1 and 6 h time points indicating that D-JNKI possesses a rapid and direct blockage of JNK/AP1 activity that is sustained for at least 6 h in an in vivo model of infection-induced preterm birth.
Consistent with our previous findings, S. abortus LPS was shown to induce PTL onset 18–24 h post-treatment with 70% pup mortality (Pirianov et al. 2009) (Fig. 3). Co-injection of 5 μg of D-JNKI delayed preterm delivery until 32 h post-injection and reduced pup mortality to 10%. Therefore, inhibition of JNK/AP1 by D-JNKI delays preterm delivery and improves pup mortality to a similar extent as inhibition of NF-κB by the cyclo-pentenone 15d-PGJ2, when tested in the same mouse model of preterm birth (Pirianov et al. 2009). There were no major differences between the ability of D-JNKI or 15d-PGJ2 to delay labour or improve pup outcome suggesting that a strategy of directly targeting JNK rather than NF-κB will be effective whether the inflammatory stimulus does or does not activate NF-κB.
In addition to inhibition of inflammatory transcription activation, we also showed that D-JNKI significantly reduced mRNA and protein levels of various downstream pro-labour and pro-contractile mediators including CCL2, CXCL1 and COX-2 when assessed at 6 h post-LPS injection (Fig. 4). Moreover, inhibition of enzyme cPLA2, which catalyses the release of arachidonic acid from membrane phospholipids, was achieved by D-JNKI at 1 h compared with LPS-alone treated animals. While the ERK1/2 (p44/p42) or p38 isoforms of MAPK have historically been considered as the primary kinases responsible for cPLA2 serine 505 phosphorylation (Leslie 1997, Ghosh et al. 2006), JNK has recently been identified as the key mediator of cPLA2 serine 505 phosphorylation and subsequent translocation to the membrane in human macrophages (Casas et al. 2009), suggesting that these events are likely to be cell type and activation stimuli dependent. In support of the latter, we show that D-JNKI inhibits early phosphorylation of cPLA2 at 1 h post-treatment but not at the later 6 h time point. This raises the possibility that cPLA2 phosphorylation in the myometrium is modulated in a phasic manner by multiple kinases.
The typical length of gestation in these animals is 18.5 days. We found that D-JNKI alone had a modest impact on reducing gestation length. It did not, however, have any effect on CCL-2, CXCL-1 or COX-2 expression. This suggests that D-JNKI alone did not lead to early changes in the inflammatory response. Why D-JNKI alone caused slight preterm delivery is unclear, but, importantly, pup mortality rates were similar in the D-JNKI and injected control group. D-JNKI does inhibit all three isoforms of JNK and we cannot exclude the possibility that one of these isoforms is involved in the regulation of parturition at term. JNK has been shown to play a key role in neonatal hypoxic–ischaemic brain injury (Dreskin et al. 2001). While JNK1 and JNK2 are widely expressed in a range of tissues, expression of JNK3 is largely confined to the brain and testis (Kyriakis et al. 1994). Gene ablation studies have shown that JNK1/JNK2 are essential for normal development while deletion of JNK3 results in apparently normal animals (Kuan et al. 2003). However, JNK3 knockout mice are resistant to excitotoxic injury (Yang et al. 1997) and partially resistant to experimental hypoxic–ischaemic brain injury (Kuan et al. 2003). This resistance to hypoxic–ischaemic brain injury applies to JNK3 knockout neonatal mice in which absence of JNK3 significantly truncates hypoxic–ischaemic induction of caspase-3 and the associated apoptosis (Pirianov et al. 2007). Fetal LPS exposure via the intrauterine route alone has been shown to cause a reduction in oligodendrocyte or myelin markers without macroscopic lesions being evident. However, fetal LPS exposure via the intrauterine route sensitises the brain to subsequent hypoxic–ischaemic brain injury. We therefore hypothesised that JNK3 activation may be unregulated in the brain following LPS exposure via the intrauterine route. We found that S. abortus LPS leads to the upregulation of overall JNK activity in the brain. Using an immunodepletion assay in which JNK1 and JNK2 are removed, we showed that LPS injection causes a large increase in JNK3 activity, increasing its contribution to total JNK activity to 50%. This is associated with increased caspase-3 activity. Observed increases in both JNK3 and caspase-3 activity in response to LPS were significantly inhibited by D-JNKI (Fig. 6), suggesting that this small molecule has a neuroprotective function.
In mouse models of infection/inflammation-induced PTL, activation of JNK is a common final pathway leading to parturition independent of the serotype of LPS used to induce labour. Specific inhibition of JNK prolongs pregnancy, improves neonatal pup death rates and reduces the expression of inflammatory mediators within the myometrium. LPS exposure of the fetus via the intrauterine injection route activates JNK3 and caspase-3, known mediators of neuronal cell death. This activation is also inhibited by D-JNKI. The data that we present here shows that inhibition of JNK improves immediate pup survival rates and reduces JNK-3 neurotoxic activity in the brain. Whether this will in fact lead to measurable improvements in outcomes of PTL and neonatal cerebral injury requires further investigation. However, a small molecule inhibition of JNK represents an attractive anti-inflammatory strategy in the management of PTL and its associated neonatal cerebral injury.
Declaration of interests
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by Wellcome Trust (DSRR-P24225 to P R Bennett) and SPARKS (04 IMP 04 to P R Bennett).
References
- Adams Waldorf KM, Persing D, Novy MJ, Sadowsky DW, Gravett MG. Pretreatment with toll-like receptor 4 antagonist inhibits lipopolysaccharide-induced preterm uterine contractility, cytokines, and prostaglandins in rhesus monkeys. Reproductive Sciences. 2008;15:121–127. doi: 10.1177/1933719107310992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggia S, Gravett MG, Witkin SS, Haluska GJ, Novy MJ. Interleukin-1 β intra-amniotic infusion induces tumor necrosis factor-α, prostaglandin production, and preterm contractions in pregnant rhesus monkeys. Journal of the Society for Gynecologic Investigation. 1996;3:121–126. doi: 10.1016/1071-5576(96)00002-0. [DOI] [PubMed] [Google Scholar]
- Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. Journal of Biological Chemistry. 2002;277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- Bell MJ, Hallenbeck JM, Gallo V. Determining the fetal inflammatory response in an experimental model of intrauterine inflammation in rats. Pediatric Research. 2004;56:541–546. doi: 10.1203/01.PDR.0000139407.89883.6B. [DOI] [PubMed] [Google Scholar]
- Blencowe H, Cousens S, Chou D, Oestergaard M, Say L, Moller A-B, Kinney M, Lawn J. Born too soon: the global epidemiology of 15 million preterm births. Reproductive Health. 2013;10(Suppl 1):S2. doi: 10.1186/1742-4755-10-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of β-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- Casas J, Meana C, Esquinas E, Valdearcos M, Pindado J, Balsinde J, Balboa MA. Requirement of JNK-mediated phosphorylation for translocation of group IVA phospholipase A2 to phagosomes in human macrophages. Journal of Immunology. 2009;183:2767–2774. doi: 10.4049/jimmunol.0901530. [DOI] [PubMed] [Google Scholar]
- Chessa D, Spiga L, De Riu N, Delaconi P, Mazzarello V, Ganau G, Rubino S. Lipopolysaccharides belonging to different Salmonella serovars are differentially capable of activating Toll-like receptor 4. Infection and Immunity. 2014;82:4553–4562. doi: 10.1128/IAI.02297-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SJ, Oh Sy, Kim JH, Roh CR. Changes of nuclear factor κ B (NF-κB), cyclooxygenase-2 (COX-2) and matrix metalloproteinase-9 (MMP-9) in human myometrium before and during term labor. European Journal of Obstetrics, Gynecology, and Reproductive Biology. 2007;132:182–188. doi: 10.1016/j.ejogrb.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Condon JC, Hardy DB, Kovaric K, Mendelson CR. Up-regulation of the progesterone receptor (PR)-C isoform in laboring myometrium by activation of nuclear factor-κB may contribute to the onset of labor through inhibition of PR function. Molecular Endocrinology. 2006;20:764–775. doi: 10.1210/me.2005-0242. [DOI] [PubMed] [Google Scholar]
- Debillon T, Gras-Leguen C, Vérielle V, Winer N, Caillon J, Rozé JC, Gressens P. Intrauterine infection induces programmed cell death in rabbit periventricular white matter. Pediatric Research. 2000;47:736–742. doi: 10.1203/00006450-200006000-00009. [DOI] [PubMed] [Google Scholar]
- Dreskin SC, Thomas GW, Dale SN, Heasley LE. Isoforms of Jun kinase are differentially expressed and activated in human monocyte/macrophage (THP-1) cells. Journal of Immunology. 2001;166:5646–5653. doi: 10.4049/jimmunol.166.9.5646. [DOI] [PubMed] [Google Scholar]
- Drougia A, Giapros V, Krallis N, Theocharis P, Nikaki A, Tzoufi M, Andronikou S. Incidence and risk factors for cerebral palsy in infants with perinatal problems: a 15-year review. Early Human Development. 2007;83:541–547. doi: 10.1016/j.earlhumdev.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Dyet LE, Kennea N, Counsell SJ, Maalouf EF, Ajayi-Obe M, Duggan PJ, Harrison M, Allsop JM, Hajnal J, Herlihy AH, et al. Natural history of brain lesions in extremely preterm infants studied with serial magnetic resonance imaging from birth and neurodevelopmental assessment. Pediatrics. 2006;118:536–548. doi: 10.1542/peds.2005-1866. [DOI] [PubMed] [Google Scholar]
- Elovitz MA, Mrinalini C. Animal models of preterm birth. Trends in Endocrinology and Metabolism. 2004;15:479–487. doi: 10.1016/j.tem.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Elovitz MA, Wang Z, Chien EK, Rychlik DF, Phillippe M. A new model for inflammation-induced preterm birth: the role of platelet-activating factor and Toll-like receptor-4. American Journal of Pathology. 2003;163:2103–2111. doi: 10.1016/S0002-9440(10)63567-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elovitz MA, Mrinalini C, Sammel MD. Elucidating the early signal transduction pathways leading to fetal brain injury in preterm birth. Pediatric Research. 2006;59:50–55. doi: 10.1203/01.pdr.0000191141.21932.b6. [DOI] [PubMed] [Google Scholar]
- Ghosh M, Tucker DE, Burchett SA, Leslie CC. Properties of the Group IV phospholipase A2 family. Progress in Lipid Research. 2006;45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Gravett MG, Witkin SS, Haluska GJ, Edwards JL, Cook MJ, Novy MJ. An experimental model for intraamniotic infection and preterm labor in rhesus monkeys. American Journal of Obstetrics and Gynecology. 1994;171:1660–1667. doi: 10.1016/0002-9378(94)90418-9. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Saotome I, Hirsh D. A model of intrauterine infection and preterm delivery in mice. American Journal of Obstetrics and Gynecology. 1995;172:1598–1603. doi: 10.1016/0002-9378(95)90503-0. [DOI] [PubMed] [Google Scholar]
- Jin H-O, Seo S-K, Woo S-H, Kim E-S, Lee H-C, Yoo D-H, Choe T-B, Hong S-I, Kim J-I, Park I-C. SP600125 negatively regulates the mammalian target of rapamycin via ATF4-induced Redd1 expression. FEBS Letters. 2009;583:123–127. doi: 10.1016/j.febslet.2008.11.035. [DOI] [PubMed] [Google Scholar]
- Kaga N, Katsuki Y, Obata M, Shibutani Y. Repeated administration of low-dose lipopolysaccharide induces preterm delivery in mice: a model for human preterm parturition and for assessment of the therapeutic ability of drugs against preterm delivery. American Journal of Obstetrics and Gynecology. 1996;174:754–759. doi: 10.1016/S0002-9378(96)70460-X. [DOI] [PubMed] [Google Scholar]
- Kelly RW. Inflammatory mediators and cervical ripening. Journal of Reproductive Immunology. 2002;57:217–224. doi: 10.1016/S0165-0378(02)00007-4. [DOI] [PubMed] [Google Scholar]
- Kuan C-Y, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, et al. A critical role of neural-specific JNK3 for ischemic apoptosis. PNAS. 2003;100:15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- Leslie CC. Properties and regulation of cytosolic phospholipase A2. Journal of Biological Chemistry. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- Li L, Kang J, Lei W. Role of toll-like receptor 4 in inflammation-induced preterm delivery. Molecular Human Reproduction. 2010;16:267–272. doi: 10.1093/molehr/gap106. [DOI] [PubMed] [Google Scholar]
- Macdonald LJ, Boddy SC, Denison FC, Sales KJ, Jabbour HN. A role for lipoxin A4 as an anti-inflammatory mediator in the human endometrium. Reproduction. 2011;142:345–352. doi: 10.1530/REP-11-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIntyre DA, Sykes L, Teoh TG, Bennett PR. Prevention of preterm labour via the modulation of inflammatory pathways. Journal of Maternal-Fetal & Neonatal Medicine. 2012;1:17–20. doi: 10.3109/14767058.2012.666114. [DOI] [PubMed] [Google Scholar]
- MacIntyre DA, Lee YS, Migale R, Herbert BR, Waddington SN, Peebles D, Hagberg H, Johnson MR, Bennett PR. Activator protein 1 is a key terminal mediator of inflammation-induced preterm labor in mice. FASEB Journal. 2014;28:2358–2368. doi: 10.1096/fj.13-247783. [DOI] [PubMed] [Google Scholar]
- Nath CA, Ananth CV, Smulian JC, Peltier MR. Can sulfasalazine prevent infection-mediated pre-term birth in a murine model? American Journal of Reproductive Immunology. 2010;63:144–149. doi: 10.1111/j.1600-0897.2009.00773.x. [DOI] [PubMed] [Google Scholar]
- Park BS, Lee J-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Experimental & Molecular Medicine. 2013;45:e66. doi: 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirianov G, Jesurasa A, Mehmet H. Developmentally regulated changes in c-Jun N-terminal kinase signalling determine the apoptotic response of oligodendrocyte lineage cells. Cell Death and Differentiation. 2006;13:531–533. doi: 10.1038/sj.cdd.4401805. [DOI] [PubMed] [Google Scholar]
- Pirianov G, Brywe KG, Mallard C, Edwards AD, Flavell RA, Hagberg H, Mehmet H. Deletion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hypoxic–ischaemic injury. Journal of Cerebral Blood Flow and Metabolism. 2007;27:1022–1032. doi: 10.1038/sj.jcbfm.9600413. [DOI] [PubMed] [Google Scholar]
- Pirianov G, Waddington SN, Lindström TM, Terzidou V, Mehmet H, Bennett PR. The cyclopentenone 15-deoxy-δ 12,14-prostaglandin J(2) delays lipopolysaccharide-induced preterm delivery and reduces mortality in the newborn mouse. Endocrinology. 2009;150:699–706. doi: 10.1210/en.2008-1178. [DOI] [PubMed] [Google Scholar]
- Pollard JK, Mitchell MD. Intrauterine infection and the effects of inflammatory mediators on prostaglandin production by myometrial cells from pregnant women. American Journal of Obstetrics and Gynecology. 1996;174:682–686. doi: 10.1016/S0002-9378(96)70450-7. [DOI] [PubMed] [Google Scholar]
- Rinaldi SF, Hutchinson JL, Rossi AG, Norman JE. Anti-inflammatory mediators as physiological and pharmacological regulators of parturition. Expert Review of Clinical Immunology. 2011;7:675–696. doi: 10.1586/eci.11.58. [DOI] [PubMed] [Google Scholar]
- Rodts-Palenik S, Wyatt-Ashmead J, Pang Y, Thigpen B, Cai Z, Rhodes P, Martin JN, Granger J, Bennett WA. Maternal infection-induced white matter injury is reduced by treatment with interleukin-10. American Journal of Obstetrics and Gynecology. 2004;191:1387–1392. doi: 10.1016/j.ajog.2004.06.093. [DOI] [PubMed] [Google Scholar]
- Romero R, Espinoza J, Kusanovic JP, Gotsch F, Hassan S, Erez O, Chaiworapongsa T, Mazor M. The preterm parturition syndrome. BJOG. 2006;113(Suppl):17–42. doi: 10.1111/j.1471-0528.2006.01120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes L, MacIntyre DA, Teoh TG, Bennett PR. Anti-inflammatory prostaglandins for the prevention of preterm labour. Reproduction. 2014;148:R29–R40. doi: 10.1530/REP-13-0587. [DOI] [PubMed] [Google Scholar]
- Terrone DA, Rinehart BK, Granger JP, Barrilleaux PS, Martin JN, Bennett WA. Interleukin-10 administration and bacterial endotoxin-induced preterm birth in a rat model. Obstetrics and gynecology. 2001;98:476–480. doi: 10.1016/S0029-7844(01)01424-7. [DOI] [PubMed] [Google Scholar]
- Voltolini C, Battersby S, Etherington SL, Petraglia F, Norman JE, Jabbour HN. A novel antiinflammatory role for the short-chain fatty acids in human labor. Endocrinology. 2012;153:395–403. doi: 10.1210/en.2011-1457. [DOI] [PubMed] [Google Scholar]
- Wang H, Hirsch E. Bacterially-induced preterm labor and regulation of prostaglandin-metabolizing enzyme expression in mice: the role of toll-like receptor 4. Biology of Reproduction. 2003;69:1957–1963. doi: 10.1095/biolreprod.103.019620. [DOI] [PubMed] [Google Scholar]
- Wu YW, Colford JM. Chorioamnionitis as a risk factor for cerebral palsy: a meta-analysis. Journal of the American Medical Association. 2000;284:1417–1424. doi: 10.1001/jama.284.11.1417. [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincón M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]