

Abstract
Background
Long noncoding RNAs (lncRNAs) represent a class of RNA molecules that are implicated in regulation of gene expression in both mammals and plants. While much progress has been made in determining the biological functions of lncRNAs in mammals, the functional roles of lncRNAs in plants are still poorly understood. Specifically, the roles of long intergenic nocoding RNAs (lincRNAs) in plant defence responses are yet to be fully explored.
Results
In this study, we used strand-specific RNA sequencing to identify 1113 lincRNAs in potato (Solanum tuberosum) from stem tissues. The lincRNAs are expressed from all 12 potato chromosomes and generally smaller in size compared to protein-coding genes. Like in other plants, most potato lincRNAs possess single exons. A time-course RNA-seq analysis between a tolerant and a susceptible potato cultivar showed that 559 lincRNAs are responsive to Pectobacterium carotovorum subsp. brasiliense challenge compared to mock-inoculated controls. Moreover, coexpression analysis revealed that 17 of these lincRNAs are highly associated with 12 potato defence-related genes.
Conclusions
Together, these results suggest that lincRNAs have potential functional roles in potato defence responses. Furthermore, this work provides the first library of potato lincRNAs and a set of novel lincRNAs implicated in potato defences against P. carotovorum subsp. brasiliense, a member of the soft rot Enterobacteriaceae phytopathogens.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2967-9) contains supplementary material, which is available to authorized users.
Keywords: Noncoding RNA, Potato, Pectobacterium, lincRNA, Plant defence, Soft rot bacteria, Solanum tuberosum, Entereobacteria
Background
Advances in transcriptome profiling techniques especially with the advent of deep sequencing approaches (RNA-sequencing) have revealed that transcription in eukaryotes is much more complex than previously anticipated. It is now apparent that the bulk of eukaryotic genomes is pervasively transcribed giving rise to noncoding RNAs (ncRNAs) which exert pivotal effects on gene regulation [1]. Noncoding RNAs can be grouped based on their lengths, into either (1) short ncRNAs (<200 bp) which have been extensively studied and generally include microRNAs (miRNAs), small nucleolar RNAs (snoRNAs), small nuclear RNA (snRNAs), and small interfering RNAs (siRNAs); and (2) long ncRNAs which are generally greater than 200 bp in length. Like mRNAs, lncRNAs have a 5’ cap and a 3’ poly-A tail; are mostly localized within the nucleus [2, 3], and can be multi-exonic [4]. LncRNAs can exhibit cell or tissue specific expression patterns and have been observed to show poor conservation across different species [4]. Based on their genomic location and context, lncRNAs are classified into intergenic (long intergenic noncoding RNA; lincRNA), long intronic noncoding RNA, and natural antisense transcripts (NATs). Natural antisense transcripts are RNA molecules with complementarity to other transcripts and can be grouped into cis-NATs (NATs fully antisense to protein coding genes on opposite strand) and trans-NATs (NATs with partial complementarity and transcribed from different loci) [1]. Some lincRNAs can be located in close proximity to protein-coding genes (CDS), thus, may be referred to as adjacent-lncRNAs, and usually associated with CDS promoter and terminator regions. Furthermore, lincRNAs on one strand can partially overlap with CDS regions on the opposite strand and such lincRNAs may be termed antisense-lncRNAs.
In the past decade, much progress has been made towards understanding the roles of small non coding RNAs in plants [5]. However, unlike small RNAs, the regulatory roles of lncRNAs remain poorly understood. Furthermore, compared to human and animal species, genome-wide discovery of lncRNAs in plants is still in its infancy [6]. Consequently, lncRNAs in plants constitute a class of ncRNAs that is less well-characterized. Nonetheless, regulatory roles of plant lncRNAs are now beginning to be recognized in diverse plant species through employing whole genome tilling arrays, in silico predictions and RNA-seq approaches [7–11]. These emerging evidences demonstrate that lncRNAs play important roles in diverse biological processes in plants ranging from plant reproductive development, and responses to biotic and abiotic stresses [9, 12, 13].
The functional mechanisms of lncRNAs in many plant species are not yet fully understood with only a few lncRNAs having been fully characterized. In Arabidopsis, lncRNAs such as COLDAIR (cold-assisted intronic non-coding RNA) and COOLAIR (cold induced long antisense intragenic RNA) have been demonstrated to mediate chromatin modifying activities in transcriptional silencing of FLC during vernalization [14, 15]. Another antisense lncRNA, ASL, a non-polyadenylated transcript, was recently discovered, and is implicated in epigenetic silencing of FLC [16]. Additional regulatory functions of some lincRNAs such as AT4 and IPS1 (INDUCED BY PHOSPHATE STARVATION1) involve acting as decoys of miRNAs by a target mimicry mechanism, thus sequestering the regulatory roles of miRNAs away from their intended target genes [17–19]. It has recently been suggested that the Alternative Splicing Competitor long noncoding RNA (ASCO-lncRNA) also acts as a decoy, regulating gene expression in Arabidopsis during development [20]. The ASCO-lncRNA acts by competing to bind alternative splicing (AS) regulators, thus, diverting them from their AS mRNA targets [20]. Furthermore, plant lincRNAs have been implicated in important biological roles in responses to external stimuli [18, 19, 21, 22]. In plants, genome-wide analysis of lncRNAs using deep sequencing transcriptomic data (mainly from RNA-seq approaches) have been performed on only a few plant species including Arabidopsis thaliana [9, 13, 23], Triticum aestivum [8], Medicago truncatula [24], Oryza sativa [12] tomato [25] and Zea mays [10, 26]. Recently, a computational genome scale investigation of lncRNAs associated with annotated gene models was performed on 37 plant species including identification of 6788 potato (Solanum tuberosum) lncRNAs [11]. However, to date, investigation of the pervasive transcription in intergenic regions in potato and identification of lincRNAs have not yet been done on a genome-wide scale.
Potato is an important staple crop ranking fourth in global production after maize, rice and wheat. It can be severely affected by soft rot Enterobacteriaceae (SRE) species, in particular, Pectobacterium carotovorum subsp brasiliense (Pcb1692), an emerging member of the SRE, which is the most important causal agent of potato blackleg and soft rot globally including South Africa. Consequently, this pathogen poses a major threat to the potato industry in terms of yield, tuber quality and tuber seed exports [27]. Pathogen-responsive lincRNAs have been implicated in defence responses against Fusarium oxysporum infection in Arabidopsis [9], and powdery mildew infection responses in wheat [8]. Given the importance, albeit not well characterized, of lincRNAs in plant response to these pathogens, it would be interesting to unravel the repertoire of lincRNAs in potato and identify those responsive to this important emerging soft rot bacterium.
We identified 1113 potato candidate lincRNAs present in two potato cultivars that are susceptible (S. tuberosum cv. Valor) and tolerant (S. tuberosum cv. BP1) to Pcb1692. Using potato time-course RNA-seq data following infection with Pcb1692, we identified 559 potato lincRNA candidates that showed significant differential expression in the stems of the resistant and susceptible cultivars, compared to the mock-inoculated samples. Of these, six were validated using RT-qPCR. Importantly, expression of 17 lincRNAs was highly correlated with potato defence-related genes. Thus, our results suggest that lincRNAs are involved in potato defence mechanisms.
Methods
Plant material and growth conditions
Seed tubers of two potato cultivars, susceptible (Solanum tuberosum cv. Valor) and tolerant (S. tuberosum cv. BP1) to Pectobacterium carotovorum subsp brasiliense strain 1692 (Pcb1692) infection were grown in the greenhouse under standard conditions (22 to 26 °C, 16 h light/ 8 h dark photoperiod and 70 % relative humidity). Stem inoculations were done as previously described in Kubheka et al. [28], except that we used wild-type Pcb1692 for the inoculations and inoculated plants were assessed and sampled at 0, 6, 12, 24, and 72 h post inoculation (hpi) in triplicates (three plants were pooled together for each biological replicate).
Total RNA preparation
Total RNA was extracted from potato stems using the QIAGEN RNeasy plant mini kit (Qiagen) including DNAse treatment (Qiagen). RNA was quantified using the NanoDrop (Thermo Scientific, Sugarland, TX, USA) and the quality and integrity checked using Agilent 2100 BioAnalyzer system (Agilent, Santa Clara, CA, USA).
Whole transcriptome library construction and sequencing
The construction of whole transcriptome libraries and sequencing were carried out at the Beijing Genomics Institute (BGI-Shenzhen, China). For the preparation of strand-specific libraries, total RNA was pooled from five time-points (0, 6, 12, 24, 72 hpi) for BP1 and Valor. Whole transcriptome libraries were constructed using the TruSeq Stranded RNA Sample Prep Kit v2 (Illumina, San Diego, CA), according to the manufacturer’s instructions. For the time-course experiment, standard (normal) transcriptome libraries were constructed using RNA samples from individual biological replicates (n = 3) from each time-point using the TruSeq RNA sample Prep Kit v2 (Illumina, San Diego, CA) following manufacturer’s instructions. The libraries were quality checked and quantified using Agilent BioAnalyzer 2100 system and qPCR. Finally, the libraries were sequenced on the Illumina HiSeq 2000 system generating 90 bp paired-end reads. The data have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through the GEO accession number, GSE74871.
Assembly of RNA transcripts
Strand-specific sequencing reads for each cultivar were quality checked using FASTQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc) and mapped to the potato reference genome (Genome assembly: PGSC_DM_v4.03; http://solanaceae.plantbiology.msu.edu/pgsc_download.shtml) using TopHat2 (version 2.0.13) [−−library-type fr-firststrand –G] [29]. For the alignments, the minimum (−i) and maximum (−l) intron sizes were obtained at http://solanaceae.plantbiology.msu.edu/pgsc_download.shtml, and set at 10 bp and 15,000 bp, respectively. Transcript assembly was performed using Cufflinks (version 2.2.1) [-g -u --library-type fr-firststrand] [30].
Bioinformatics identification of lincRNAs
The assembled potato transcripts were compared with annotated potato protein sequences (http://potato.plantbiology.msu.edu/data/PGSC_DM_V403_representative_genes.gff.zip) using IntersectBed (v2.22.1) [31]. All assembled transcripts overlapping with potato coding sequences and less than 200 bp from protein coding regions were removed. For size selection, java scripts were used to filter out all transcripts less than 200 nucleotides in length. For the sequencing depth filter, HTSeq-count [python -m HTSeq.scripts.count -f bam -s reverse] [32] was used and only transcripts with at least two reads were considered. Following sequencing depth filter, IntersectBed (v2.22.1) [31] and stringent Blastn (Evalue: 1.0E-100) was used to extract novel transcripts present in both BP1 and Valor. Since lncRNA transcripts are generally known not to have any coding capacity, all the transcripts common to BP1 and Valor were tested for protein-coding potential using the Coding Potential Calculator (CPC) [33]. Following the coding potential filter, only transcripts with a negative CPC score were retained as potential novel lincRNA candidates.
Distribution of lincRNAs and protein-coding genes in the potato genome
A circular representation of the distribution of lincRNAs and mRNAs was constructed using Circos [34] for comparative visualizations among the 12 chromosomes.
Classification of lincRNAs
Potato lincRNAs were classified into three categories based on their genomic location and distance from protein-coding genes nearest to each lincRNA transcript using IntersectBed (v2.22.1) [31] and java scripts. The lincRNAs were grouped into: 1) intergenic-lncRNA, without any overlaps with protein-coding genes on both strands and at least 1 kb away from the nearest CDS 2) adjacent-lncRNA, which are in close proximity to protein coding genes but without any overlaps and 3) antisense-lncRNA, which partially overlap with genes on the opposite strand.
Differential expression analysis of lincRNAs between the tolerant and susceptible potato cultivars
Time course RNA-seq data from stems of BP1 and Valor was used to identify lincRNAs responsive to P. carotovorum subsp. brasiliense infection. Briefly, to identify differentially expressed lincRNAs between Valor and BP1, RNA-seq reads were quality checked using FASTQC and mapped to the potato reference genome using TopHat2 [29]. HTSeq-count was used to make read counts mapped to lincRNA transcripts and DeSeq2 [35] was used to determine the differential expression with a false discovery rate threshold of 10 %.
Quantitative reverse transcription PCR (RT-qPCR)
For RT-qPCR, first-strand cDNA synthesis was done from total RNA using Superscript III First-Strand cDNA Synthesis SuperMix kit (Invitrogen, USA) following manufacturer’s instructions. Quantitative real-time PCR using Applied Biosystems SYBR Green Master Mix was performed in the QuantStudio 12 K Flex Real-Time PCR system (Life Technologies, Carlsbad, CA, USA). For RT-qPCR, 2 μl of sample was added to 8 μl of Applied Biosystems SYBR Green Master Mix and primers at a concentration of 0.4 μM. The cycling conditions were as follows: an initial denaturation at 50 °C for 5 min and 95 °C for 2 min followed by 45 cycles of 95 °C for 15 s and 60 °C for 1 min. Each sample was run in triplicate and two biological replicates were employed. The samples were normalized to 18S rRNA and elongation factor 1-α (ef1α) as the reference genes [36] and the mock treated samples used as calibrators. The comparative CT (ΔΔct) method was used to measure relative expression [37]. Primers used were designed online using Primer3Plus (http://primer3plus.com/cgi-bin/dev/primer3plus.cgi) and are listed in Additional file 1: Table S5.
LincRNA-mRNA coexpression analysis
To investigate correlations of expression between lincRNA and differentially expressed mRNA transcripts under the same conditions, an all-against-all hierarchical clustering analysis was performed based on log2 fold changes using Cluster 3.0 software [38]. Briefly, LincRNA and mRNA datasets were filtered so that only transcripts with an expression of at least 2-fold at any of the 5 time-points tested were considered. Clustering was performed using the Spearman Rank Correlation similarity metric (rrho > |0.8|) and the complete linkage clustering method. Visualization was done using TreeView program [39]. In order to predict potential lincRNA functions, mRNA transcripts grouped together with lincRNAs in various clusters were used to perform Gene Ontology (GO) analysis based on the Panther Classification System (version 10.0) web server [40]. Corresponding orthologs in Arabidopsis of the differentially expressed potato mRNA genes were used for the GO enrichment analysis, based on BLASTp (e-value: 1.0E-05), implemented in ProteinOrtho program [41]. Lastly, pairwise Spearman correlation coefficient was calculated by cor.test() in R, between CDS genes and lincRNAs grouped within each cluster associated with response to stimulus GO biological process terms. To assign putative functional annotations to the lincRNAs, GO terms of CDS genes significantly correlated with lincRNAs were mapped to the lincRNAs.
RT-PCR validation of lincRNA transcripts
First-strand cDNA was synthesized as described above for RT-qPCR and the PCR was performed on Bio-RAD T100TM Thermal Cycler conventional PCR (Bio-RAD, USA). The lincRNA primers were designed online using Primer3plus (Additional file 2: Table S6). PCR was performed in a 25 μl reaction mix containing 1 μl of template cDNA (~40 ng), Taq DNA Polymerase, 10x Taq Buffer (New England Biolabs, UK), 2.5 mM dNTPs each and 0. 5 μM of forward and reverse primer each. Thermal cycling conditions were: 95 °C for 2 min; 30 cycles of 95 °C for 30 s, 57 °C for 30 s, 72 °C for 60 s, and the final extension at 72 °C for 5 min. The PCR products were analysed on 1.5 % agarose gel including 1 kb DNA molecular weight ladder 470 (NEB, UK). To check for genomic DNA contamination, a non reverse-transcriptase control was included.
Prediction of lincRNA and miRNA interactions
Potato lincRNAs targeted by miRNAs were predicted using the psRNATarget [42] server by using default parameters.
Results
Genome-wide identification of lincRNAs in potato
In order to identify long intergenic noncoding RNA (lincRNAs) related to potato defence networks, we employed a computational approach using strand-specific RNA-seq (ssRNA-seq) data derived from stems of Solanum tuberosum cultivars Valor and BP1 (Fig. 1). Samples of potato stems of each cultivar were harvested from six time-points post inoculation with Pcb1692, RNA isolated and pooled together. Sequencing was conducted on representative RNA pools of the susceptible and tolerant cultivars. The ssRNA-seq generated approximately 36 million (33 million uniquely mapped) and 38 million (35.3 million uniquely mapped) paired-end reads in S. tuberosum cvs Valor and BP1, respectively. From these data, a computational strategy was used that enabled the identification of lincRNAs after read mapping and transcript abundance assembly using Tophat2 (v2.0.13) and Cufflinks (v2.2.1), respectively [29, 30]. As an initial step, all transcript loci, from the potato genome annotation without strand information were removed prior to performing read alignments and transcript assembly. Subsequently, 59,681 and 60,292 transcripts were reconstructed for S. tuberosum cvs Valor and BP1, respectively. The majority of the assembled transcripts (84.7 %) represented annotated genes and allelic isoforms in the potato reference genome assembly (PGSC_DM_v4.03) for both S. tuberosum cvs Valor and BP1.
Fig. 1.
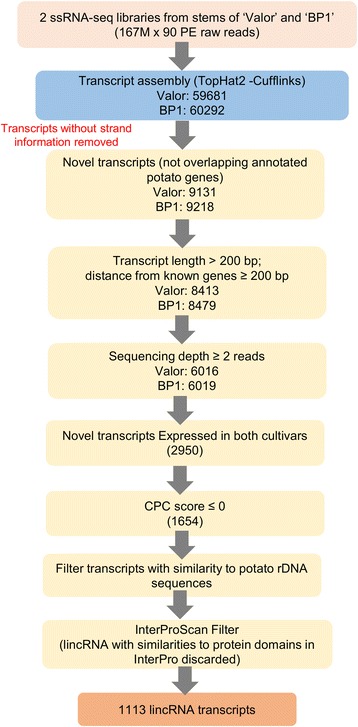
Schematic diagram of the bioinformatics approach used for identification of potato lincRNAs
Identification of novel transcriptionally active regions
To identify transcripts representing novel transcriptionally active regions (TARs) from the Cufflinks assembled transcripts, we first eliminated all the transcripts that overlapped with annotated potato features on the same strand. Our main focus in this study was particularly to identify novel noncoding RNA transcription; thus, only transcripts at a distance of more than 200 nucleotides from known genes on the same strand were considered, with lengths above 200 bp. Furthermore, to eliminate the possibility of genomic DNA contamination, only transcripts with a sequencing depth of at least two reads per transcript were retained (Fig. 1). Additionally, since we were interested in measuring and comparing the variation in transcript abundances of the TARs between S. tuberosum cvs Valor and BP1 using time-course RNA-seq data in our downstream analysis, we started by first determining novel transcripts that were common between both the cultivars based on strand-specific RNA-seq data. Using the IntersectBed tool (v2.22.1) [31] and Blastn (Evalue: 1.0E-100) we identified 2950 novel transcripts that were present in the two cultivars. To determine a set of long intergenic noncoding RNA transcripts (lincRNAs) that is novel, the coding capability of these transcripts was then assessed using the Coding Potential Calculator (CPC) [33]. CPC evaluates the protein-coding potential of transcripts based on prediction and assessment of potential open reading frames (ORFs) features and BLASTX (E-value cut-off 1.0E-10) homology searches against the non-redundant Uniprot Reference Clusters (UniRef90) protein database. Based on the extracted feature information, CPC algorithm, uses a score to classify transcripts into either protein-coding or noncoding. In this regard, all the transcripts showing evidence for protein-coding (CPC score > 0) were eliminated. Consequently, we obtained 1654 lincRNAs expressed in both potato cultivars with CPC scores less than zero. The 1654 lincRNAs obtained were further filtered to remove any lincRNAs with similarity to potato ribosomal DNA sequences (obtained from EnsemblPlants SolTub_3.0 Assembly (Blastn: E-value 1.0E-2)) resulting in 1649 lincRNA candidates. Of these, 1177 were high-confidence novel lincRNAs (CPC score < −1) and 472 were weak-novel lincRNA based on the CPC scores (Additional file 3: Table S1). However, because CPC uses a stringent Blastx cutoff (E-value: 1.0E-10), and only performs similarity analysis against the UniRef90 protein database, it is possible that some mRNA transcripts with relatively weak protein signatures could be falsely classified as potential lincRNA transcripts. Thus, we further screened the 1649 lincRNA candidates against InterPro [43], using InterProScan5 [44]. LincRNA sequences with similarities to protein families and domains from any of the databases within the InterPro consortium were considered as protein-coding and eliminated. Finally, following the additional filters, a list of 1113 transcripts was regarded as novel lincRNA potato transcripts expressed in stems of S. tuberosum cvs Valor and BP1 (Additional file 3: Table S1). Semi-quantitative reverse transcription (RT)-PCR confirmed nine of the RNA-seq identified lincRNAs, thus validating the assembly quality and identification pipeline (Fig. 2).
Fig. 2.
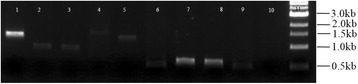
RT-PCR validation of nine lincRNA transcripts. Agarose gel electrophoresis of the PCR amplicon fragments representing each lincRNA. Lane 1. LincRNA9, Lane 2. LincRNA10, Lane 3. LincRNA12, Lane 4. LincRNA13, Lane 5. LincRNA20, Lane 6. LincRNA178, Lane 7. LincRNA1405, Lane 8. LincRNA24, Lane 9. LincRNA1102, Lane 10. No reverse transcriptase control, Lane 11. 1 kb DNA ladder
Characterization and classification of potato lincRNAs
Using basic features of the identified lincRNAs in a genomic context, we found that the lincRNAs ranged from 200 to 17,256 bp in size and were transcribed from all the 12 potato chromosomes (Potato Genome Assembly: PGSC_DM_v4.03) (Additional file 4: Figure S1A). The highest and least numbers of lincRNAs were transcribed from chromosome one (183 lincRNAs) and chromosome 12 (nine lincRNAs), respectively. As with mRNA transcripts, lincRNAs appeared to be distributed uniformly across all chromosomes, with the exception of chromosome 12, were lincRNAs were only concentrated within the region up to 5 Mbp (Additional file 4: Figure S1A). In addition, based on length distribution, lincRNAs can be divided into three groups, namely, short-length, medium-length and long-length lincRNAs [4]. Thus, the majority of potato lincRNAs (71 %) are short-length lincRNA (200–1000 bp), 26 % are medium-length lincRNA (1–5 kb) and only 3 % are long-length lincRNA (>5 kb). In contrast, most of the protein-coding transcripts, 54 % comprise medium transcripts (Additional file 4: Figure S1B). Comparing the number of exons between annotated potato genes and lincRNAs showed that on average, lincRNAs possess fewer exons (Table 1). Furthermore, we assessed the repeat content (including presence of transposons) of potato lincRNAs using RepeatMasker (http://www.repeatmasker.org) and the TIGR Solanum Repeat Database v3.2 (plantrepeats.plantbiology.msu.edu/downloads.html). Almost half of the lincRNAs (42.3 %) contain repetitive sequences.
Table 1.
Exon numbers of lincRNA and protein-coding genes in Solanum tubersoum cvs Valor and BP1
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ≥10 | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Valor | 5 | 791 | 145 | 93 | 32 | 11 | 9 | 4 | 6 | 6 | 10 | 1113 |
| BP1 | 17 | 779 | 158 | 74 | 33 | 11 | 14 | 5 | 9 | 3 | 10 | 1113 |
| amRNA | 16883 | 6937 | 3561 | 2753 | 1960 | 1599 | 1144 | 937 | 709 | 621 | 1924 | 39 028 |
amRNA exons obtained from the potato genome assembly (PGSC_DM_v4.03)
Even though it is still not yet clear how classification of lncRNAs based on their proximity to coding genes reflects biological function, knowing where lncRNAs are located in the genome and their expression profiles provides useful insights into their biological significance and primary mechanisms of action [45]. Thus, we classified the identified lincRNAs into three types, based on their genomic location and proximity with respect to their closest protein-coding genes, namely: intergenic (distance > 1 kb; without any overlaps with CDSs on both strands), adjacent (distance < 1 kb) and antisense-lncRNAs (those partially overlapping protein-coding genes on the opposite strand) (Fig. 3a). Most of the lincRNAs (87 %) are located at least 1 kb away from annotated potato gene models on either strand, 8 % of lincRNA are adjacent-lncRNA, located in close proximity to protein-coding genes, and only a small proportion (5 %) constituted antisense-lncRNAs (Fig. 3b and Additional file 3: Table S1). The percentage difference observed between lincRNAs at distance > 1 kb from CDS regions and antisense-lncRNA is consistent with previous observations made in maize [10] and tomato [25].
Fig. 3.
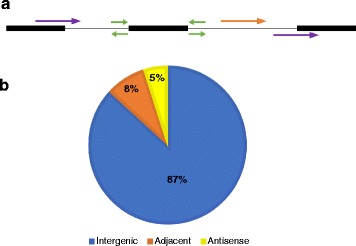
Classification of potato lincRNAs relative to protein-coding transcripts. a A schematic diagram showing the location of lincRNAs in relation to adjacent protein-coding genes (black rectangles). Purple arrows represent antisense-lncRNAs which overlap annotated genes on the opposite strand; Green arrows show adjacent-lncRNAs which are positioned in close proximity to annotated genes; and the Orange arrow represents long intergenic noncoding RNAs (lincRNAs). b Percentage and distribution of lincRNAs in three classes
Furthermore, the identified lincRNA sequences were compared with lncRNA sequences from tomato [25], Populus [22], and Arabidopsis [21, 46, 47] to determine the set of potato lincRNAs with similarity to these plant species (BLASTn e-value < 1.0E-10). As expected, the lincRNAs displayed poor conservation. Only 13 % lincRNAs showed multiple homologous regions (>80 % identity and alignment length > 100 bp) with 231 lncRNAs from tomato and two from Arabidopsis (Additional file 5: Table S2). Thus, unlike most mRNAs which are highly conserved across organisms, lincRNAs tend to evolve rapidly resulting in poor conservation [48]. Lastly, to check the novelty of our set of lincRNAs, we checked for overlaps against potato lncRNAs reported by Gallart et al. [11]. Comparisons using IntersectBed tool (v2.22.1), showed that only nine lincRNAs out of the 1113 lincRNAs from our set overlapped with and were similar to nine previously reported potato lncRNAs (Additional file 6: Figure S2).
Quantitative analysis of potato lncRNAs responsive to Pectobacterium carotovorum subspecies brasiliense infection
From our previous work [28], we showed that S. tuberosum cv. Valor is highly susceptible to Pcb1692 infection showing typical blackleg symptoms upon infection. On the other hand, S. tuberosum cv. BP1 was shown to be tolerant to Pcb1692. Furthermore, gene expression analysis performed in our lab between Valor and BP1 revealed differentially expressed protein-coding genes involved in potato defence responses to Pcb1692 infection (unpublished data). However, it remains to be investigated whether lincRNA expression is activated in response to Pcb1692 infection in potato. Thus, we hypothesized that lincRNAs could be involved in potato defence mechanisms and therefore differentially expressed in the tolerant compared to susceptible cultivar. Consequently, in the present work, we sought to determine novel lincRNA transcripts that were differentially expressed between the two cultivars and could thus be implicated in potato defences against the necrotrophic plant pathogen, Pcb1692. Our bioinformatics analysis showed that a total of 1113 lincRNAs were expressed in both cultivars. Thus, to identify defence-related lincRNAs, the expression levels of these commonly expressed 1113 lincRNAs were compared between the two cultivars, S. tuberosum cvs Valor and BP1. To do this, each cultivar was inoculated with Pcb1692 (1 × 109 cfu.ml−1) and samples obtained at five different time points (0 (Buffer inoculated control), 6, 12, 24, 72 h post inoculation; hpi). RNA was isolated from each time point and three biological replications per time point were prepared and sequenced independently. Using the resulting time-course RNA-seq data, the differential expression patterns of the 1113 lincRNAs was evaluated. In total, 485, 416, 539, 364, and 449, lincRNAs were differentially expressed (DE) at 0, 6, 12, 24, and 72 hpi, respectively, between S. tuberosum cvs Valor and BP1, at 10 % false discovery rate (FDR) (Additional file 7: Table S3). Compared with S. tuberosum cv Valor, an average of 51 % lincRNAs were upregulated in S. tuberosum cv BP1, throughout the time-course. Numbers of up-regulated lincRNAs in the tolerant cultivar were slightly higher at 6, 12 and 72 hpi, with the most up-regulated lincRNAs observed at 6 hpi (54 %) (Fig 4a). Furthermore, in order to determine the expression profiles of these differentially expressed lincRNAs showing cultivar-specific differences, DE lincRNAs at each time-point (post inoculation) were compared to mock-inoculated samples (0 hpi) in each cultivar. In total, 173 DE lincRNAs were present in both cultivars and 267 and 119 were only significantly expressed in S. tuberosum cv Valor and S. tuberosum cv BP1, respectively (Fig. 4b and c).
Fig. 4.
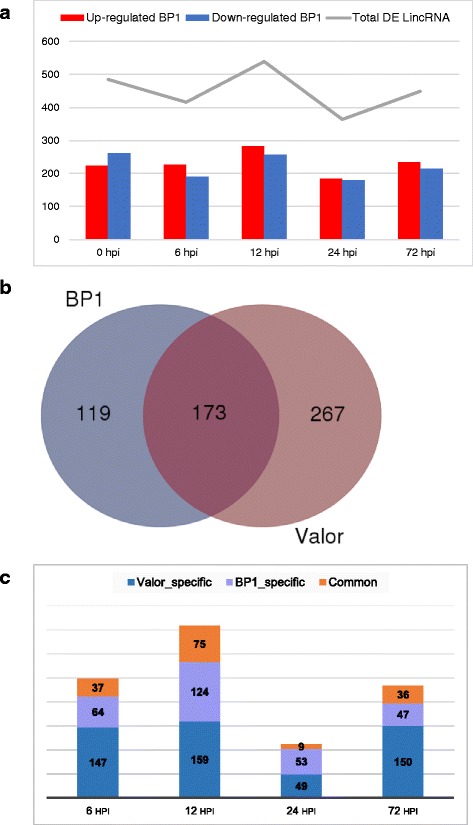
LincRNAs significantly expressed over time between Valor and BP1 following infection with Pcb1692. a Pairwise comparisons between S. tuberosum cv Valor and S. tuberosum cv BP1 at each time-point. Red represent significantly upregulated and blue represent significantly downregulated. b Comparison of DE lincRNAs specific to each cultivar in relation to the mock-inoculated samples (0 hpi). c Numbers of DE lincRNAs common or specific to each cultivar at individual time-points in relation to mock inoculated samples
To confirm RNA-seq expression patterns and determine whether the differentially expressed lincRNAs are involved in potato defence responses, six of these lincRNAs were arbitrarily selected representing lincRNAs that were up or down regulated at one or more time-points and validated experimentally using reverse-transcription quantitative PCR (RT-qPCR) (Fig. 5). The RT-qPCR results were in concordance with the RNA-seq data, thus, implicating the DE lincRNAs in potato defence responses.
Fig. 5.
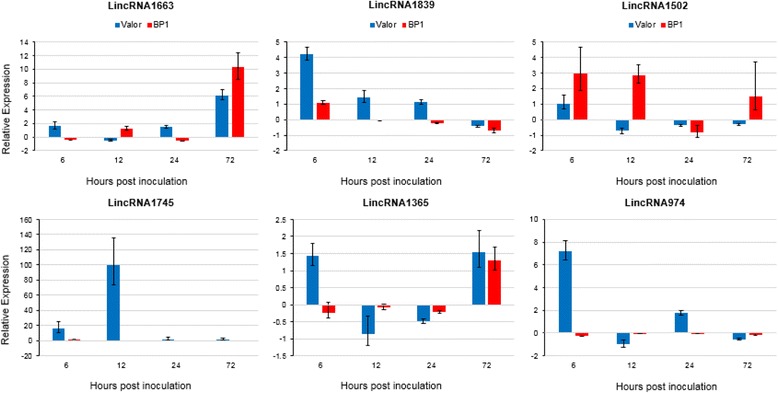
RT-qPCR validation of time-course RNA-seq data using six selected lincRNAs differentially expressed over time. 18S rRNA and elongation factor 1-α (ef1α) were used as the reference genes. The relative expression levels of lincRNAs at each time point were calculated relative to calibrator (control sample; 0 hpi). Error bars represent the range of relative expression (fold change) calculated by 2-(ΔΔCt±SD). Two biological replicates were used in triplicate
LincRNA/mRNA genes expression correlation
In order to understand the possible biological roles of the differentially expressed (DE) lincRNAs in relation to potato defence responses, we investigated all-against-all coexpression patterns between lincRNA transcripts and DE mRNA genes within the time-course using hierarchical clustering. In total, 179 lincRNAs were highly correlated with 3573 mRNA genes (Spearman rank correlation (rrho) > |0.8| and were clustered into 62 different groups. Interestingly, Gene Ontology (GO) enrichment analysis using the Panther Classification System web server [40], showed that 32 clusters contained CDS genes enriched in “response to stimulus”, including secondary GO terms such as “defence response to bacterium”, “response to stress” and “response to endogenous stimulus”. Therefore, to highlight potential lincRNA functions and/ or interactions with CDS genes involved in potato defence mechanisms, we further performed pairwise correlations between CDS genes and lincRNAs within each cluster associated with response to stimulus GO terms. Overall, 17 lincRNAs exhibited extremely high positive correlation (rrho ≥ 0.9) with 12 potato defence-related CDS genes (Table 2). These results suggest that these highly correlated lincRNAs could be involved in potato defence responses against Pcb1692 infection.
Table 2.
LincRNA transcripts highly coexpressed with defence-related CDS genes
| LincRNA ID | LincRNA class | r rho a | Potato gene ID | Arabidopsis ortholog | Gene description | Gene Ontology classification (Biological process) |
|
|---|---|---|---|---|---|---|---|
| LincRNA739 | Intergenic | 1 | PGSC0003DMG400014801 | AT4G17760 | rad1-like | response to stimulus (GO:0050896) | response to stress (GO:0006950) |
| LincRNA1304 | Intergenic | 1 | PGSC0003DMG400020345 | AT2G38080 | Laccase-4 | response to stimulus (GO:0050896) | response to toxic substance (GO:0009636) |
| LincRNA127 | Intergenic | 1 | PGSC0003DMG400011631 | AT3G46230 | 17.4 kDa class I heat shock protein (HSP17.4A) | response to stimulus (GO:0050896) | response to stress (GO:0006950) |
| LincRNA803 | Intergenic | 1 | AT1G53540 | 17.6 kDa class I heat shock protein 3 (HSP17.6C) | response to stimulus (GO:0050896) | response to stress (GO:0006950) | |
| LincRNA127 | Intergenic | 1 | PGSC0003DMG400000996 | AT3G47570 | Probable LRR receptor-like serine/threonine-protein kinase | response to stimulus (GO:0050896) | defence response to bacterium (GO:0042742) |
| AT3G47580 | Leucine-rich repeat protein kinase family protein | ||||||
| AT3G47090 | Leucine-rich repeat protein kinase-like protein | ||||||
| LincRNA1464 | Intergenic | 1 | PGSC0003DMG400012994 | AT5G21950 | Hydrolase, alpha/beta fold family protein | response to stimulus (GO:0050896) | response to toxic substance (GO:0009636) |
| AT4G33180 | |||||||
| LincRNA907 | Intergenic | 1 | PGSC0003DMG400000757 | AT2G23620 | Methylesterase 1 | response to stimulus (GO:0050896) | response to toxic substance (GO:0009636) |
| LincRNA1118 | Intergenic | 0.9 | |||||
| LincRNA258 | Intergenic | 0.9 | |||||
| LincRNA632 | Intergenic | 1 | PGSC0003DMG400025635 | AT3G45920 | Protein kinase family protein | defence response to bacterium (GO:0042742) | |
| PGSC0003DMG400004885 | AT2G24370 | Adenine nucleotide alpha hydrolase domain-containing protein kinase | defence response to bacterium (GO:0042742) | ||||
| LincRNA758 | Intergenic | 1 | |||||
| LincRNA749 | Intergenic | 1 | |||||
| AT4G09570 | Calcium-dependent protein kinase 4 | response to endogenous stimulus (GO:0009719) | |||||
| PGSC0003DMG400026077 | AT1G35670 | Calcium-dependent protein kinase 11 | |||||
| LincRNA758 | Intergenic | 1 | |||||
| LincRNA749 | Intergenic | 1 | |||||
| PGSC0003DMG400013679 | AT1G30270 | CBL-interacting serine/threonine-protein kinase 23 | response to stimulus (GO:0050896) | ||||
| LincRNA1112 | Intergenic | 1 | |||||
| LincRNA908 | Intergenic | 0.9 | |||||
| LincRNA1712 | Intergenic | 0.9 | |||||
| PGSC0003DMG400030755 | AT1G77110 | Probable auxin efflux carrier component 6 | response to stimulus (GO:0050896) | response to endogenous stimulus (GO:0009719) | |||
| LincRNA178 | Intergenic | 0.9 | |||||
| LincRNA1583 | Intergenic | 1 | |||||
| PGSC0003DMG400021008 | AT4G20940 | Probable LRR receptor-like serine/threonine-protein kinase | response to stimulus (GO:0050896) | defense response to bacterium (GO:0042742) | |||
| LincRNA379 | Intergenic | 0.9 | |||||
aSpearman correlation coefficient
Prediction of interactions between lincRNAs and miRNAs
Long noncoding RNAs can be involved in diverse cellular molecular functions depending on their mode of action [4]. Because lincRNAs are functional RNA molecules, they can be targeted and regulated by miRNAs post transcriptionally, triggering degradation of the targeted lincRNAs. To investigate whether the identified potato lincRNAs are targeted by miRNAs, we analyzed the 1113 lincRNAs using psRNATarget [42]. A total of 57 lincRNAs were predicted to be targeted by 98 potato miRNAs (Additional file 8: Table S4). Of these lincRNAs, four were targeted by six miRNAs implicated in plant immune defences [49] (Additional file 8: Table S4). Interestingly, none of these four lincRNAs were differentially expressed in the time-course following inoculation with Pcb1692 possibly reflecting their miRNA mediated cleavage and degradation. RT-qPCR analysis confirmed expression of these defence-related miRNAs under the same experimental conditions, adding credence to their possible interaction or regulation of their target lincRNAs (Additional file 9: Figure S3). An additional 10 lincRNAs were targets of various members of stu-miR5303 family which is part of nine miRNA families unique to solanaceous plants [50] (Additional file 8: Table S4). Previously identified potential targets of stu-miRNA5303 family include proteins responsive to abiotic stress, metabolic enzymes and proteins of unknown function [50]. The large numbers of stu-miRNA5303 members implies its biological importance. Consequently, it is plausible to assume that their target lincRNA transcripts play important biological roles in potato.
Discussion
The regulatory roles of lincRNAs are increasingly being unraveled in plants, as indicated by the number of various reports on the identification of lncRNAs in plant species including maize, millet, rice, Populus and Arabidopsis [7, 10, 12, 21–23, 26]. However, most of these reports have focused on lncRNAs involved in plant development, reproduction and abiotic stress responses [3, 10, 12, 13, 51]. In contrast, reports about lincRNAs involved in defence regulatory mechanisms against pathogens are just beginning to emerge [8, 9]. In potato, previous studies on noncoding RNA have predominantly focused on miRNA identification and functional analysis [49, 52–54], but no data have been reported for lincRNAs, especially in association with potato defence responses. In this study, we conducted a genome-wide analysis of potato lincRNAs, by integrating strand-specific RNA sequencing with time-course RNA-seq data. We identified novel candidate lincRNAs potentially associated with potato defence response mechanisms during challenge by Pcb1692. Hence, this present work provides an important resource of potato lincRNAs that can be useful to other researchers.
To facilitate the identification of lincRNAs, a strand-specific RNA-seq (ssRNA-seq) approach was employed which made it possible to determine the strand from which the lincRNAs were produced. This is in contrast to some previous lincRNA identification reports in plants, which had the limitation of RNA-seq data lacking strand information [10, 22]. Knowledge of the strand information of lncRNAs is important in localizing their genome context and position since lincRNAs are transcribed from intergenic regions with some being adjacent or antisense to protein-coding regions [55]. Thus, strand-specific RNA-seq allowed us to classify the identified lincRNAs into three categories, based on their proximity to protein-coding genes (Fig. 3b). Classification of lincRNAs based on their genomic location can be a useful preliminary step in determining potential functional roles of lincRNAs [4]. In addition, our present work revealed that, like protein-coding genes, lincRNAs are distributed throughout the potato genome (Additional file 4: Figure S1A). Thus, the pervasive expression of lincRNAs in the entire 12 potato chromosomes suggests that they are common RNA molecules representing a functional component of the potato genome.
Additionally, by aligning the ssRNA-seq reads, using Tophat2, a splice-aware aligner and performing transcript reconstruction using the software tool Cufflinks [30], a parsimonious representation of exon boundaries for the lincRNAs was obtained. As a result, the structure of lincRNA transcripts was resolved. Furthermore, since consideration was only given to lincRNA candidates conserved in S. tuberosum cvs. Valor and BP1, the identified 1113 lincRNAs constitute a reliable list of lincRNAs from potato stems, extending the current understanding of the potato transcriptome landscape.
In general, functional characterization of lincRNAs in plants is still in its infancy. Moreover, little is known about regulatory functions of lincRNAs in biotic stress responses in plants. Currently, the function of lncRNAs cannot be inferred directly from primary sequence or structure as is the case with miRNAs and protein-coding mRNA [56]. However, key insights into biological roles of lncRNAs can be derived from the conditions in which they are expressed [45]. In the present study, we identified 559 differentially expressed (DE) lincRNAs at different time-points (up to 72 hpi) in S. tubersom cvs. Valor and BP1, compared to mock-inoculated samples (Fig 4b). Meanwhile, the responsiveness of six DE lincRNAs to Pcb1692 infection was also confirmed using RT-qPCR, further alluding to the potential functional activity of these lincRNAs.
From a systems biology perspective, the guilt-by-association principle has been applied successfully for the functional characterization of various genes in humans and other mammals assuming functional relationships between co-expressed genes [57, 58]. Thus, in the present study, a hierarchical clustering strategy was employed in order to infer potential functional roles of DE lincRNAs in the time-course, and enriched functions of CDS genes within individual clusters were identified. Only clusters with genes enriched for biological process GO terms under the “response to stimulus” category were considered. Generally, coexpression analysis is used to predict biological processes and infer novel members (genes, ncRNA transcripts etc.) of known processes and/or pathways [59]. In addition, identifying lincRNAs associated with the “response to stimulus” category was particularly relevant in this study due to the fact that most defence-related genes are often overrepresented within this category. Therefore, GO terms of CDS genes that showed significantly high pairwise coexpression with lincRNAs (rrho > 0.9) within the clusters, were mapped to lincRNAs (Table 2). Based on this analysis, 17 lincRNAs were co-expressed with genes associated with defence-related GO terms such as response to stimulus, defence response to bacterium, response to endogenous stimulus, response to stress and response to toxic substance (Table 2). Most of these CDS genes belong to the Leucine-rich repeat protein kinase family, which mainly function as pattern recognition receptors in plant innate immune responses. Thus, our results implicate these co-expressed lincRNAs in defense responses against Pcb1692 in the tolerant cultivar, making them key candidates for future experimental validations.
Interestingly, these 17 lincRNAs are located more than 100 kb from their correlated CDS genes, and some of them are interchromosomal with regards to CDS genes they are co-expressed with. Thus, these lincRNAs are possibly trans-acting, functioning as transcriptional regulators that interact with genes at distal locations across multiple chromosomes. However, there is a paucity of information regarding molecular mechanisms employed by lincRNAs in regulating their distal gene targets. Nonetheless, we anticipate that these mechanisms will become more apparent in the near future.
Conclusions
This study focused on the genome-wide discovery of lincRNAs using strand-specific RNA-seq and resulted in the first catalogue of potato lincRNAs, comprising 1113 transcripts, including 1104 novel lincRNA candidates, derived from stem tissue. In addition, we identified 559 lincRNAs that were responsive to P. carotovorum subsp. brasiliense infection in S. tuberosum cvs Valor and BP1. Importantly, 17 differentially expressed lincRNAs were highly associated with defence-related CDS genes, thus representing key candidates for future functional studies.
Abbreviations
CDS, protein-coding genes; LincRNA, long intergenic noncoding RNA; lncRNAs, long noncoding RNAs; miRNAs, microRNAs; NATs, Natural antisense transcripts; ncRNAs, noncoding RNAs; Pcb1692, Pectobacterium carotovorum subsp. brasiliense; rrho, Spearman Rank correlation coefficient; RT-PCR, reverse transcription PCR; RT-qPCR, quantitative reverse transcription PCR; siRNAs, small interfering RNAs; snoRNAs, small nucleolar RNAs; snRNAs, small nuclear RNA; SRE, soft rot Enterobacteriaceae; ssRNA-seq, strand-specific RNA sequencing
Acknowledgements
We thank the National Research Foundation (NRF), South Africa for funding this work through Thuthuka grant number 69362; Research Development Grant for Y-Rated Researchers 93357; Bioinformatics and Functional Genomics (BFG 93685) and The Genomics Research Institute, University of Pretoria.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI’s Gene Expression Omnibus (GEO) repository [GEO accession number, GSE74871; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=czatweashdgvdyl&acc=GSE74871].
Authors’ contributions
SK and LNM conceived the study. SK performed bioinformatics analysis and experimental validations. SK, PRJB, and LNM wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional files
List of RT-qPCR primers used in this study. (XLS 26 kb)
List of RT-PCR primers used in this study. (XLSX 8 kb)
List of the identified 1113 lincRNA candidates. (XLS 166 kb)
A) Comparison of the genomic distribution of lincRNAs and protein-coding genes across the 12 potato chromosomes. The outer grey track represents the 12 potato chromosomes, with a scale (Mb) showing the length of each chromosome. The red histograms (second track with an outer orientation) and blue histograms (third track with inner orientation) represent the abundance and distribution of mRNA and lincRNAs, respectively, throughout the potato genome. The bin size (histogram width) = 5 Mbp). B) Comparison of LincRNA lengths to protein-coding mRNA transcripts in potato (PGSC_DM_v4.03 genome assembly). (PDF 258 kb)
LincRNA conservation analysis. (XLSX 32 kb)
Comparison of the 1113 lincRNA transcripts identified in the present study with potato lncRNAs available in the GreenC database. (PDF 105 kb)
Differentially expressed lincRNA transcripts. (XLSX 231 kb)
LincRNAs targeted by potato miRNAs. (XLS 30 kb)
RT-qPCR confirmation of five potato defense-related miRNAs in S. tuberosum cv BP1, computationally predicted to target some of the lincRNA transcripts. U6 snRNA was used as the reference gene. The fold changes of miRNAs at each time point were calculated relative to calibrator (control sample; 0 hpi). The experiments were done in triplicate. Error bars represent the fold change range calculated by 2-(ΔΔCt±SD). (PDF 86 kb)
Contributor Information
Stanford Kwenda, Email: stan.kwenda@fabi.up.ac.za.
Paul R. J. Birch, Email: Paul.Birch@hutton.ac.uk
Lucy N. Moleleki, Email: Lucy.Moleleki@up.ac.za
References
- 1.Bai Y, Dai X, Harrison AP, Chen M. RNA regulatory networks in animals and plants: a long noncoding RNA perspective. Brief Funct Genomics 2015;14(2):91-101. [DOI] [PubMed]
- 2.Wierzbicki AT. The role of long non-coding RNA in transcriptional gene silencing. Curr Opin Plant Biol. 2012;15(5):517–522. doi: 10.1016/j.pbi.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y-C, Chen Y-Q. Long noncoding RNAs: new regulators in plant development. Biochem Biophys Res Commun. 2013;436(2):111–114. doi: 10.1016/j.bbrc.2013.05.086. [DOI] [PubMed] [Google Scholar]
- 4.Ma L, Bajic VB, Zhang Z. On the classification of long non-coding RNAs. RNA Biol. 2013;10(6):924–933. doi: 10.4161/rna.24604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X, Hao L, Li D, Zhu L, Hu S. Long Non-coding RNAs and their biological roles in plants. Genomics Proteomics Bioinformatics. 2015;13(3):137–47. doi: 10.1016/j.gpb.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim E-D, Sung S. Long noncoding RNA: unveiling hidden layer of gene regulatory networks. Trends Plant Sci. 2012;17(1):16–21. doi: 10.1016/j.tplants.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 7.Qi X, Xie S, Liu Y, Yi F, Yu J. Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol Biol. 2013;83(4-5):459–473. [DOI] [PubMed]
- 8.Xin M, Wang Y, Yao Y, Song N, Hu Z, Qin D, Xie C, Peng H, Ni Z, Sun Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol. 2011;11(1):61. doi: 10.1186/1471-2229-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu QH, Stephen S, Taylor J, Helliwell CA, Wang MB. Long noncoding RNAs responsive to Fusarium oxysporum infection in Arabidopsis thaliana. New Phytol. 2014;201(2):574–584. doi: 10.1111/nph.12537. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Eichten SR, Shimizu R, Petsch K, Yeh C-T, Wu W, Chettoor AM, Givan SA, Cole RA, Fowler JE. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014;15(2):R40. doi: 10.1186/gb-2014-15-2-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallart AP, Pulido AH, de Lagrán IAM, Sanseverino W, Cigliano RA. GREENC: a wiki-based database of plant lncRNAs. Nucleic Acids Res. 2016;44(D1):D1161–D1166. doi: 10.1093/nar/gkv1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y-C, Liao J-Y, Li Z-Y, Yu Y, Zhang J-P, Li Q-F, Qu L-H, Shu W-S, Chen Y-Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014;15(12):512. doi: 10.1186/s13059-014-0512-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amor BB, Wirth S, Merchan F, Laporte P, d’Aubenton-Carafa Y, Hirsch J, Maizel A, Mallory A, Lucas A, Deragon JM. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2009;19(1):57–69. doi: 10.1101/gr.080275.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swiezewski S, Liu F, Magusin A, Dean C. Cold-induced silencing by long antisense transcripts of an Arabidopsis polycomb target. Nature. 2009;462(7274):799–802. doi: 10.1038/nature08618. [DOI] [PubMed] [Google Scholar]
- 15.Heo JB, Sung S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science. 2011;331(6013):76–79. doi: 10.1126/science.1197349. [DOI] [PubMed] [Google Scholar]
- 16.Shin JH, Chekanova JA. Arabidopsis RRP6L1 and RRP6L2 function in FLOWERING LOCUS C silencing via regulation of antisense RNA synthesis. PLoS Genet. 2014;10(9):e1004612. doi: 10.1371/journal.pgen.1004612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu H-J, Wang Z-M, Wang M, Wang X-J. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 2013;161(4):1875–1884. doi: 10.1104/pp.113.215962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, García JA, Paz-Ares J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 2007;39(8):1033–1037. doi: 10.1038/ng2079. [DOI] [PubMed] [Google Scholar]
- 19.Shin H, Shin HS, Chen R, Harrison MJ. Loss of At4 function impacts phosphate distribution between the roots and the shoots during phosphate starvation. Plant J. 2006;45(5):712–726. doi: 10.1111/j.1365-313X.2005.02629.x. [DOI] [PubMed] [Google Scholar]
- 20.Bardou F, Ariel F, Simpson CG, Romero-Barrios N, Laporte P, Balzergue S, Brown JW, Crespi M. Long noncoding RNA modulates alternative splicing regulators in Arabidopsis. Dev Cell. 2014;30(2):166–176. doi: 10.1016/j.devcel.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Jung C, Xu J, Wang H, Deng S, Bernad L, Arenas-Huertero C, Chua N-H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell. 2012;24(11):4333–4345. doi: 10.1105/tpc.112.102855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shuai P, Liang D, Tang S, Zhang Z, Ye C-Y, Su Y, Xia X, Yin W. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J Exp Bot. 2014;65(17):4975–83. doi: 10.1093/jxb/eru256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song D, Yang Y, Yu B, Zheng B, Deng Z, Lu B-L, Chen X, Jiang T. Computational prediction of novel non-coding RNAs in Arabidopsis thaliana. BMC Bioinf. 2009;10(Suppl 1):S36. doi: 10.1186/1471-2105-10-S1-S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen J, Parker BJ, Weiller GF. In silico identification and characterization of mRNA-like noncoding transcripts in Medicago truncatula. In Silico Biol. 2007;7(4):485–505. [PubMed] [Google Scholar]
- 25.Zhu B, Yang Y, Li R, Fu D, Wen L, Luo Y, Zhu H. RNA sequencing and functional analysis implicate the regulatory role of long non-coding RNAs in tomato fruit ripening. J Exp Bot. 2015;66(15):4483–95. doi: 10.1093/jxb/erv203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boerner S, McGinnis KM. Computational identification and functional predictions of long noncoding RNA in Zea mays. PLoS One. 2012;7(8):e43047. doi: 10.1371/journal.pone.0043047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van der Merwe JJ, Coutinho TA, Korsten L, van der Waals JE. Pectobacterium carotovorum subsp. brasiliensis causing blackleg on potatoes in South Africa. Eur J Plant Pathol. 2010;126(2):175–185. doi: 10.1007/s10658-009-9531-2. [DOI] [Google Scholar]
- 28.Kubheka GC, Coutinho TA, Moleleki N, Moleleki LN. Colonization patterns of an mCherry-tagged pectobacterium carotovorum subsp. Brasiliense strain in potato plants. Phytopathology. 2013;103(12):1268–1279. doi: 10.1094/PHYTO-02-13-0049-R. [DOI] [PubMed] [Google Scholar]
- 29.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anders S, Pyl PT, Huber W. HTSeq–A Python framework to work with high-throughput sequencing data. Bioinforma.2014;btu638. [DOI] [PMC free article] [PubMed]
- 33.Kong L, Zhang Y, Ye Z-Q, Liu X-Q, Zhao S-Q, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(suppl 2):W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicot N, Hausman J-F, Hoffmann L, Evers D. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J Exp Bot. 2005;56(421):2907–2914. doi: 10.1093/jxb/eri285. [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics 2004;20(17):3246-248. [DOI] [PubMed]
- 40.Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44(D1):D336–D342. doi: 10.1093/nar/gkv1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lechner M, Findeiß S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (Co-) orthologs in large-scale analysis. BMC Bioinf. 2011;12(1):124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39(2):W155–W159. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitchell A, Chang H-Y, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res.2014;gku1243. [DOI] [PMC free article] [PubMed]
- 44.Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atkinson SR, Marguerat S, Bahler J: Exploring long non-coding RNAs through sequencing. Seminars in cell & developmental biology. 2012;23(2):200-5. [DOI] [PubMed]
- 46.Jin J, Liu J, Wang H, Wong L, Chua N-H. PLncDB: plant long noncoding RNA database. Bioinformatics. 2013;29(8):1068–71. doi: 10.1093/bioinformatics/btt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yi X, Zhang Z, Ling Y, Xu W, Su Z. PNRD: a plant non-coding RNA database. Nucleic Acids Res. 2015;43(D1):D982–D989. doi: 10.1093/nar/gku1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li F, Pignatta D, Bendix C, Brunkard JO, Cohn MM, Tung J, Sun H, Kumar P, Baker B. MicroRNA regulation of plant innate immune receptors. Proc Natl Acad Sci. 2012;109(5):1790–1795. doi: 10.1073/pnas.1118282109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu M, Liu W, Meng Q, Zhang W, Chen A, Sun S, Xu G. Identification of microRNAs in six solanaceous plants and their potential link with phosphate and mycorrhizal signaling. J Integr Plant Biol. 2014;56(12):1164–1178. doi: 10.1111/jipb.12233. [DOI] [PubMed] [Google Scholar]
- 51.Wang M, Yuan D, Tu L, Gao W, He Y, Hu H, Wang P, Liu N, Lindsey K, Zhang X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.) New Phytol. 2015;207(4):1181–97. doi: 10.1111/nph.13429. [DOI] [PubMed] [Google Scholar]
- 52.Xie F, Frazier TP, Zhang B. Identification, characterization and expression analysis of MicroRNAs and their targets in the potato (Solanum tuberosum) Gene. 2011;473(1):8–22. doi: 10.1016/j.gene.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 53.Zhang R, Marshall D, Bryan GJ, Hornyik C. Identification and characterization of miRNA transcriptome in potato by high-throughput sequencing. PLoS One. 2013;8(2):e57233. doi: 10.1371/journal.pone.0057233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang W, Luo Y, Gong X, Zeng W, Li S. Computational identification of 48 potato microRNAs and their targets. Comput Biol Chem. 2009;33(1):84–93. doi: 10.1016/j.compbiolchem.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 55.Clark BS, Blackshaw S. Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front Genet. 2014;5:164. doi: 10.3389/fgene.2014.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 57.Saito K, Hirai MY, Yonekura-Sakakibara K. Decoding genes with coexpression networks and metabolomics–‘majority report by precogs’. Trends Plant Sci. 2008;13(1):36–43. doi: 10.1016/j.tplants.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 58.Wolfe C, Kohane I, Butte A. Systematic survey reveals general applicability of “guilt-by-association” within gene coexpression networks. BMC Bioinf. 2005;6(1):227. doi: 10.1186/1471-2105-6-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rhee SY, Mutwil M. Towards revealing the functions of all genes in plants. Trends Plant Sci. 2014;19(4):212–221. doi: 10.1016/j.tplants.2013.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are available in the NCBI’s Gene Expression Omnibus (GEO) repository [GEO accession number, GSE74871; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=czatweashdgvdyl&acc=GSE74871].