

Abstract
Purpose of review
LVADs have markedly improved the survival for patients with advanced heart failure but are plagued with significant morbidity, including pump thrombosis and bleeding. Better understanding of the platelet, and their role in the balance of bleeding and thrombosis, stands to impact the frequency and treatment of these significant complications.
Recent findings
In patients with LVADs, there is little consistency linking traditional biomarkers of platelet activation and clinical events. A number of innovative methods of assessing platelet functionality, including shedding of platelet receptors and formation of microparticle complexes as well as measuring mitochondrial membrane potentials, exist and appear to be clinically relevant. Acquired von Willebrand syndrome, while not explaining all bleeding events, is a central feature of mechanical support and offers a target for innovative therapies
Summary
Although the platelet is only one component of impacting thrombosis and bleeding in patients supported with LVADs, it plays a central role in mediating these two opposing forces. Innovations in both understanding platelet physiology as well as manipulating genomic and receptor interactions for an individual patient will be critical if we are to decrease these serious adverse events in the future.
Keywords: platelets, bleeding, thrombosis, LVAD, heart failure, coagulation
Introduction
Left ventricular assist devices (LVAD) are an invaluable part of the therapeutic armamentarium for patients suffering from advanced heart failure. When used either as a bridge to transplant, to promote myocardial recovery, or as lifetime use, LVADs have proven to prolong survival and improve quality of life.1–3 Unfortunately, mechanical circulatory support carries significant morbidity that imparts a significant burden to the patient and socioeconomically – including multiple readmissions and reoperations.
One of the more vexing issues for patients and providers is the physiologic toggling between thrombosis and bleeding. While the overall rate of adverse events has decreased in the continuous flow era, adverse events including hemolysis, stroke, renal dysfunction, and respiratory failure are more common.4 For those taking care of chronic LVAD patients, the threat of gastrointestinal bleeding, stroke, and pump thrombosis is a routine part of care for this patient population. Pump thrombosis, in particular, has become a mainstream concern following a report suggesting an increase incidence in this event.5 This dreaded complication, and the need for pump exchange, portends markedly reduced life-expectancy with one-year survivals of 65% after a second implant, and only 50% after a third implant.4
Blood moving though a mechanical circulatory support pump is subject to altered rheologic conditions and contact with a foreign surface. This unnatural state results in activation of platelets and the clotting cascade, activation of endothelial cells and leucocytes, fibrinolysis, and the development of acquired von Willebrand syndrome. Cumulatively, these forces conspire to create an environment that may breed hemolysis, bleeding, or clotting. In the middle of this storm is the platelet. In this report, we will focus on the role of the platelet in regulating these events and its central role in balancing the yin and yang of bleeding and thrombosis.
The birth of the platelet
The origin of platelets from bone marrow megakaryocytes was recognized early in the 20th century.6 Platelets are generated by megakaryocytes cytoplasmic extensions termed proplatelets. Apoptotic stimulation through mitochondrial caspase activation regulates proplatelet differentiation and platelet shedding. In addition, megakaryocyte development and platelet formation are regulated by a multitude of cytokines, principally thrombopoietin and interleukins (IL)-3, -6, and -11. Thrombopoietin drives megakaryocyte maturation resulting in the accumulation of distinctive cytoplasmic components: platelet-specific proteins, organelles, and the demarcation membrane system – an elaborate network of membrane channels composed of flattened cisternae and tubules.7
The biogenesis of secretory granules is a cardinal feature of thrombopoiesis. The most abundant are α-granules that contain an electron dense nucleoid within a granular matrix. α-granules contain proteins essential for platelet adhesion, including fibrinogen, fibronectin, von Willebrand factor (vWF), and thrombospondin. Dense granules contain small molecules such as serotonin, epinephrine and ADP. Both types of granules originate from the Golgi complex, are present in mature megakaryocytes, and acquire their contents through a combination of endogenous synthesis and uptake of plasma components by receptor-mediated endocytosis and pinocytosis. Transformation of the megakaryocyte cytoplasm initiates erosion at one pole of the cell and subsequently concentrates cellular contents into proplatelet extension. Mature megakaryocytes migrate into close proximity to sinus endothelial cells, thereby enabling projected proplatelets to enter the vascular space. Proplatelet formation represents the terminal phase in the megakaryocyte lifecycle (Figure 1).8
Figure 1.
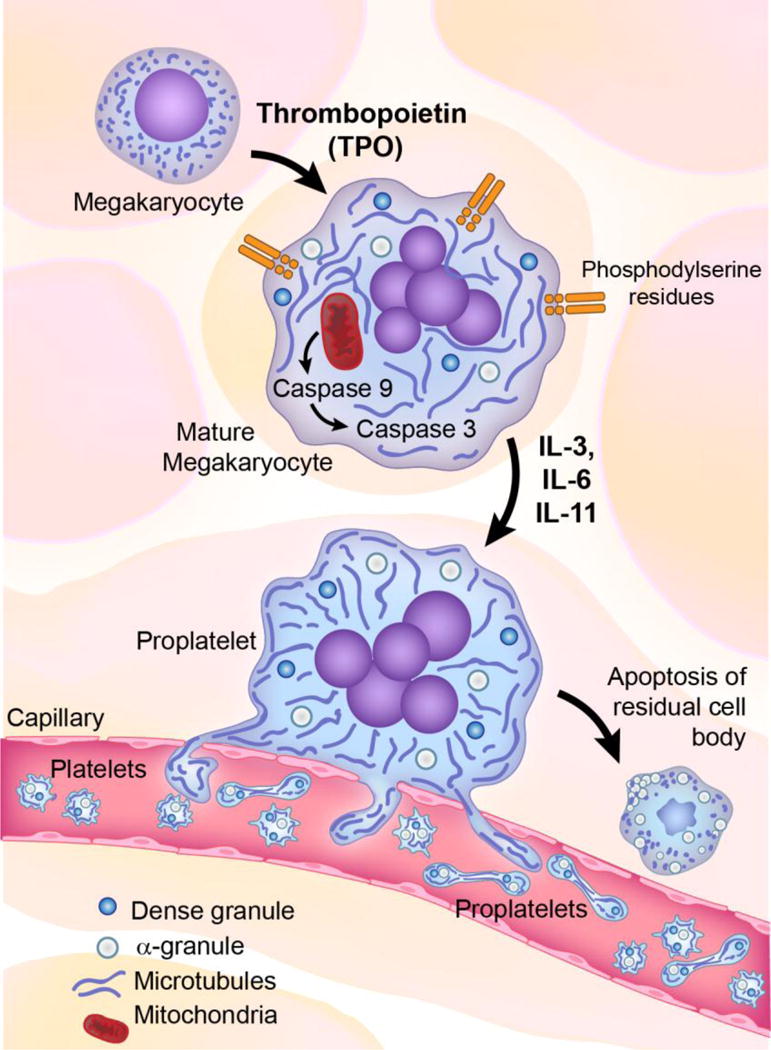
Birth of the platelet. Platelets formation involves mitochondria apoptotic activation. Tpo drives mononuclear MKs precursors to undergo endomitosis and become polyploidy. Further MK maturation results in the accumulation of cytoplasmic components as platelet-specific proteins, organelles and the demarcation membrane system composed of flattened cisternae and tubules. Next occurs the biogenesis of α-granules and dense granules by receptor mediated endocytosis and pinocytosis. Transformation of the MK cytoplasm initiates with erosion at one pole of the cell. Pseudopodial proplatelets extend into the bone marrow sinusoids and project proplatelets into the vascular space. Proplatelet formation is the terminal phase of the MK cycle of life. It is unclear whether MKs release fully formed individual platelets or proplatelet chains within the bloodstream. (Tpo, thrombopoietin; IL, interleukin; MK, megacaryocytes)
Platelet function
Hemostasis is the process of clot formation at the site of vessel injury. The hemostatic response must be quick, localized and carefully regulated. The initiation and the formation of the platelet plug is the first step of the hemostatic process. The functional response of activated platelets at the site of vascular injury involves three distinct processes:
Adhesion: With exposure of the subendothelial space after vascular injury, platelets adhere to exposed basement membrane proteins (proteogylcans, collagen, fibulin, and laminin) and molecules secreted locally (von Willebrand Facor), through their membrane glycoprotein receptors (GP1a). This process is energy dependent, requiring ATP. Platelet interaction with collagen not only provides a surface for additional platelets adhesion but also serves as a strong stimulus for platelets activation.
Aggregation: After adhering, platelets change their shape, spreading along the collagen fibrils. Secretion of thromboxane A2 and ADP synergistically attracts and stimulates neighboring platelets. These activated platelets directly bind to the circulating coagulation protein fibrinogen. Fibrinogen binds to platelet integrin GPIIb/IIIa receptors, thereby cross-linking adjacent platelets.
Secretion: Once activated, platelets release the contents of their α- and dense granules that further promote activation and adherence of additional platelets.
As mentioned, platelet activation is not an isolated physiologic event. It is intimately associated with the natural clotting cascade. Intrinsic activation from the exposure of procoagulant phospholipids on the platelet surface, as well as extrinsic activation by tissue factor at the site of injury, allow for convergence and activation of factor X. The resultant conversion of prothromin to thrombin not only leads to fibrin formation but also serves to perpetuate the continued activation of additional platelets. Consequently, platelet activation and fibrin deposition are intimately linked, maximizing the growth and strength of the hemostatic plug.
Platelets and hemocompatability
Thrombotic complications associated with LVADs have multiple etiologies (Figure 2).9 Of these, hemocompatibility – that subset of biocompatibility related to the effects of blood elements and a mechanical device – and shear stress play significant role. Virchow’s triad (endothelial injury, hypercoagulability, and stasis) can apply, by analogy, to LVAD patients. The bioreactive material of the device, circulating activated platelets, and aberrant flows create an environment for thrombogenesis.10 Shear stress and hemolysis are complicit to this process as ADP leakage from damaged red cells is a potent platelet activator.
Figure 2.
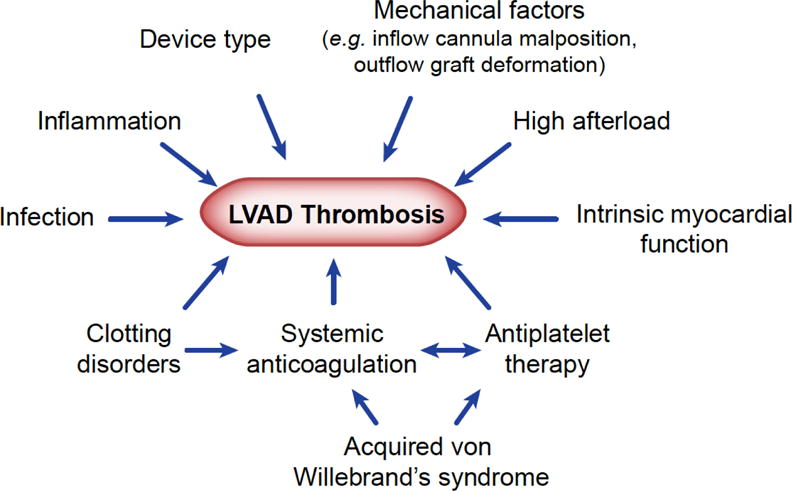
Clinical factors influencing LVAD-associated thrombosis (reproduced from9)
Historically, hemocompatibility has been the Achilles heel of support pumps. When plasma proteins (including but not limited to factor XII, prekallikrein, factor XI and kininogen) interact with the negatively charged foreign surface in a circulatory pump, the intrinisic cascade is activated. Despite current efforts to make uniformly smooth metallic surfaces, irregularities on the pump surface may promote platelet adhesion.11 When blood is exposed to this rough surface, fibrinogen and vWF are adsorbed to the metallic surface, providing a nidus for platelet adhesion. Once contact adhesion occurs, spreading of platelets onto the surface and release of their secretory granules further drives platelet activation and subsequent deposition of fibrin, platelet aggregates, leucocytes and erythrocytes (Figure 3).
Figure 3.
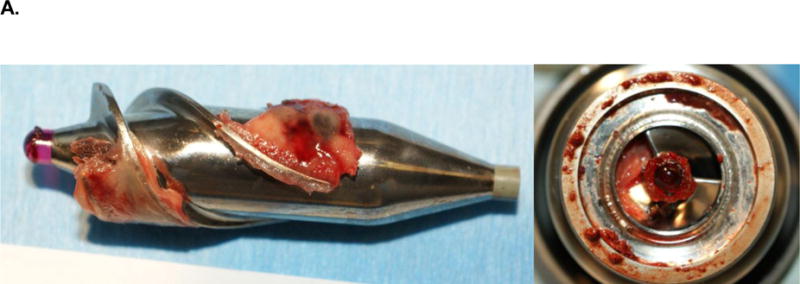
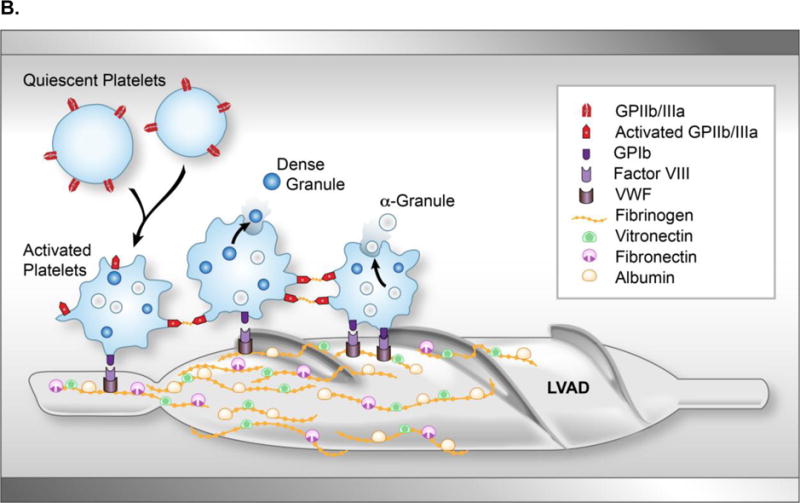
Clot formation on an axial flow pump. (A) Explanted HeartMate II LVAD with evidence of red- and white-clot around inflow as well as in main rotor. (B) Upon contact with the blood, the titanium bioreactive surface of the LVAD adsorbs several plasma proteins, including albumin, fibrinogen, fibronectin and vitronectin forming a protein layer covering the LVAD surface. With contact, activated platelets change their discoid shape by putting out pseudopods and release their dense and α-granules. Fibrinogen, vWF, and factor VIII form a complex with the GP1b receptor site of the activated platelet to cause adherence. This complex produces a conformational change in the GPIIb/IIIa receptor allowing the platelet to bind with fibrinogen. As this occurs, the newly recruited platelets release their granule content, which accelerate the thrombus development by increasing linkages between the platelets with fibrinogen/fibrin bridges. Subsequently thrombin is generated and stabilizes the local thrombus. (vWF, von Willebrand factor; GP, glycoprotein; LVAD, left ventricular assist device)
Platelet activation and LVADs
With activation, platelets expose several cell surface glycoproteins and release contents of their secretory granules, including β-thromboglobulin, platelet factor-4, P-selectin (CD62), thrombospondin, and integrins (GPαIIbβ3, GPIbα and CD63).12 Of these, P-selectin and thrombospondin have been most studied. P-selectin on the surface of platelets mediates homotypic platelet-platelet aggregation as well as heterotypic platelet-leucocyte aggregate formation (PLAs). Thrombospondin serves as a stabilizing ligand between glycoprotein IIb/IIIa and fibrinogen during clot formation.
There are multiple studies examining platelet activation in patients with LVADs. Some of this evidence remains controversal, likely due in part to differences in patient populations, pre-analytical variables, and the timing of measurements as well as biological aspects of these various activation indices Table 1. For example, in patients supported with pulsatile LVADs (Novacor and BerlinHeart), P-selectin and thrombospondin peaked on post-operative day 3, and, although decreased after 3 weeks, remained above normal levels.13 Similarly, CD63 expression was initially increased and subsequently decreased after 4 weeks, but not to normal levels. To-date, altered expression of platelet activation markers have not been correlated with bleeding time or clinical events. In a group of patients supported with external pulsatile VADs, persistent membrane expression of P-selectin, but not integrin αIIbβ3 and Ibα expression was observed, suggesting only moderate persistent platelet activation.14
Table 1.
Biomarkers activated in continuous flow LVADs.
| Immediate | Long-term | |
|---|---|---|
| Platelet activation markers | ||
| CD62 (P-selectin)) | ━ | ━ |
| CD40 | ━ | ━ |
| CD41 | ━ | ━ |
| CD42b | ━ | ━ |
| CD31 | ━ | ━ |
| Micriparticles (MP) | ||
| Platelet MP | ⬆ | ⬆ |
| Leukocyte MP | ⬆ | ⬆ |
| Endothelial MP | ⬆ | ⬆ |
| Endothelial activation | ||
| ICAM-1 | ⬆ | ⬆ |
| VCAM-1 | ⬆ | ⬆ |
| E-selectin | ⬆ | ⬆ |
| IL-6 | ⬆ | ⬆ |
| IL-8 | ⬆ | ⬆ |
| TF | ⬆ | ⬆ |
| Monocyte activation | ||
| CD14 | ⬆ | ⬆ |
| Fibrinolytic activation | ||
| Fibrinogen | ⬆ | ↓ |
| D-Dimers | ⬆ | ↓ |
Several studies have examined platelet activation in second-generation axial-flow devices. In patients receiving the MicroMed pump, increased expression of P-selectin, CD63, and CD31 was observed in the early postoperative period that returned to baseline several weeks post-implant.15 In the Jarvik 2000 LVAD, sustained expression of P-selectin, GP IIb/IIIa, and soluble CD40L was not observed.16 Similar lack of platelet biomarker expression was also observed in patients supported with the HeartMate II LVAD – independent of length of LVAD support, patient age, or antiplatelet regiment.17, 18
A recent study compared platelet activation in patients supported with the HeartMate II and the 3rd generation centrifugal HeartWare LVAD. No differences were observed in expression of platelet markers CD41, CD42b, or CD62. Significant differences in other markers of hemolysis and coagulation were identified – higher levels of LDH in HeartMate II patients and higher levels of fibrin-split products (D-dimers) in HeartWare patients.19 Taken together, these studies demonstrate that simply measuring standard biomarkers of platelet activation might not tell the whole story.
Role of platelet microparticles
When blood traverses the high shear environment of mechanical pumps, hemolysis and complement activation are involved with generation of microparticles. In particular, LVAD patients express increased levels of platelet-derived microparticles that may contribute to thrombus formation.20, 21
When activated, platelets and leukocyte microparticles bind to endothelial cells and induce inflammatory gene expression with release of IL-6 and IL-8 and upregulation of cellular adhesion molecules including intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin. In patients supported with LVADs, these markers peaked in the early post-operative and remained elevated for 6 months.22 Microparticles may be involved in the perpetuation of a vicious cycle of endothelial damage that propogate vascular dysfunction and increase the risk for thromboembolic complications.23
Increased monocyte activation and CD14 expression has been implicated in thrombotic events and inflammation in patients with advanced heart failure. Following LVAD implantation, increased density of monocyte membrane expression of CD14 is associated with increased monocyte-platelet complexes (MPCs). These aggregates induce expression of monocyte tissue factor witch contribute to procoagulant responses, thrombogenesis and the proinflammatory state. In 15 patients, no differences in standard platelet biomarkers were seen after 6 months, but 40% had persistent elevation of MPCs, for which 2 of these patients had major events (embolic stroke and multi-organ failure).24 Summarily, the influence of platelets likely involves more than its release of its stored granules and the paracrine effect of those substances, but also its central role in interacting with other vascular and blood stream components.
LVADs, platelets, and bleeding
Non-surgical bleeding among LVAD-support patients is a major problem and is responsible for hospital readmissions, blood products transfusion, and impaired quality of life. While dysregulated platelet function likely contributes to non-surgical bleeding, few studies have examined this issue in chronically supported continuous flow patients. The Maryland group studied 25 LVAD patients and demonstrated that platelet reactive oxygen species (ROS), mitochondrial damage, and apoptosis is increased in advanced heart failure patients at baseline and is further increased after LVAD insertion. In addition, when comparing patients with and without bleeding, no differences in the levels of ROS were observed, but patients with bleeding had a 2-fold and 1.5-fold rise in depolarized and apoptotic platelets, respectively. Post-implant, these platelet abnormalities remained elevated in the bleeding group, while decreasing in the non-bleeders, thereby suggesting a potential, albeit incompletely understood, role of platelet dysfunction on bleeding complications.25
The same group of investigators then compared patients supported with three different devices – HeartMate II, HeartWare HVAD, and the Jarvik 2000. Flow cytometric analysis showed significantly higher intraplatelet ROS, an increase in mitochondrial membrane potential (ΔΨm), and more platelet apoptosis in HeartWare supported patients compared to the other two devices. These laboratory abnormalities correlated with more observed bleeding complications (nearly 50%) in the HeartWare patients.25
The concept of platelet dysfunction as manifest by altered mitochondrial membrane potential is intriguing. An association between platelet dysfunction and the systemic inflammatory response has been observed in multiple patient populations.26 In CF-LVAD patients that develop the systemic inflammatory response (SIRS), there is a positive association between platelet function as assessed by PFA collagen, ADP, and epinephrine closure times and the percentages of depolarized ΔΨm platelets. While the increased incidence of major clinical events could be associated with the broader cycle of SIRS, mitochondrial energy status, quantified by ΔΨm, is associated with qualitative platelet defects and thrombocytopathies that could be a useful marker for post-LVAD adverse events.27
LVADs and platelet shedding
Amongst other effects, exposure of blood to elevated shear stress results in shedding of platelet surface receptors, including the glycoprotein Ibα (GPIbα) receptor. Platelet GPIbα is expressed as the ligand-binding unit of the GPIb-IX-V complex with vWF. The interaction produces transmembrane signaling and platelet activation. Increased plasma GPIbα level is associated with decreased ristocetin-induced platelet aggregation and an increased risk of bleeding.28 In LVAD supported patients, higher levels of GPIbα ectodomain shedding (in the first 2 weeks after implant) were observed in patients with clinical bleeding events. Assaying this shedded receptor might provide a diagnostic tool for predicting bleeding in LVAD patients.29 Several in vitro studies have also examined the influence of shear stress and shedding of platelet receptors, including GP1bα, GPVI, and GP IIb/IIIa.30, 31
GPIIb/IIIa polymorphism
In addition to upregulation and shedding of platelet glycoprotein receptors, genomic changes can also contribute to altered platelet function. The Berlin group examined the influence of genotypes that code for the GPIIa/IIIb receptor on clinical events. They observed that patients with the A1A1 genotype had significantly more bleeding complications (and less thrombotic) than did patients with the A1A2 genotype. Conversely, A1A2 patients had more thromboembolic events.32
These finding have significant clinical implications with regard to oral platelet inhibitors, as A1A1 subjects are more sensitive to antiplatelet therapy. In A1 homozygotes, aspirin results in a reduction in the speed of thrombin formation, prothrombin consumption, and fibrinogen removal, while the presence of the A2 allele is associated with impaired antithrombotic action.32 While others have not confirmed genotype-associated complications, LVAD patients with the A1A1 polymorphism did express higher levels of CD62P, CD63, thrombospondin, and monocyte-platelet complexes.24
Acquired Von Willebrand syndrome and LVAD
Von Willebrand Factor (vWF) is a multifunctional plasma protein that plays an important role in the events that lead to hemostasis and thrombosis. vWF is synthesized and stored in the Weibel-Palade bodies of endothelial cells and in the α-granules of platelets. Platelet vWF constitutes about 15–20% of the total vWF present in the blood. In the plasma, vWF functions as a stabilizing chaperone for coagulation factor VIII. It has binding sites for collagen and for platelet membrane glycoprotein GPIb and GPIIb-IIIa, mediating the adhesion and aggregation of platelets at sites of vascular injury by acting as a bridging element to structures in the subendothelial matrix.
Von Willebrand factor circulates in the blood as a large soluble multimeric protein that is cleaved by a metalloprotease (ADAMTS13). When exposed to conditions of high shear stress, structural changes occur in the von Willebrand molecule thereby exposing the bond between amino acids 842 and 843(6) to the specific von Willebrand protease.33 This results in acquired type 2 von Willebrand Syndrome that is characterized by the loss of the largest von Willebrand multimers, which are critical for effective platelet-mediated hemostasis.
In patients supported with the HeartMate II, ristocetin-induced platelet aggregation is significantly impaired, especially in those with minor or major bleeding events. The specific activity of vWF is also reduced due to the lack of high molecular weight vWF multimers. As such, HeartMate II patients develop an acquired form of vWF deficiency that appears similar to that observed in patients with aortic stenosis and other cardiac defects.34 Another study demonstrated that all 26 HeartMate II patients examined had severe impairment of platelet aggregation as well as loss of large vWF multimers.35 This acquired von Willebrand syndrome reverses after transplantation and device removal.
When comparing HeartMate II patients to those supported with the third generation HeartWare HVAD centrifugal flow pump, vWF profiles did not discriminate between patients with and without bleeding and thromboembolic events. The investigators did observe, however, that the speed of the HVAD pump correlated to a lower percentage of high molecular weight multimers, whereas vWF breakdown in HeartMate II patients was not related to pump speed.36
Despite significant reduction in vWF multimers, not all patient have bleeding events.37 Indeed, vWF loss does not fully account for bleeding complication in CF-LVAD patients. Angiodysplastic lesions, either present prior to implant or developed after implant, are important pathologic features of bleeding in these patients. While their relationship to nonpulsatile blood flow remains hypothetical, some data suggests that enhanced pulsatility can decrease the incidence of bleeding complication.38 Summarily, acquired vWF is practically ubiquitous with mechanical circulatory support, but it is likely only one part of the story related to bleeding and thrombotic events in LVAD patients.
Therapeutic considerations
Manipulating factors that alter the balance of bleeding and thrombosis appear conceptually attractive. The morbidity and socioeconomic burden of their related complications mandate the community to try to find ways to decrease these adverse events. Because of its central role in this paradox, the platelet is an obvious target. Antiplatelet therapy with aspirin is a core therapy for all patients on LVADs. Some manufacturers suggest dual antiplatelet therapy (dipyridamole) as well (i.e. Jarvik, Inc.). When bleeding occurs, many will decrease aspirin from 325mg to the lower 81mg dose. However, with the HeartWare HVAD, lower aspirin dosing was correlated with increased embolic events.39
Several other mechanistically driven platelet specific therapies have been investigated. Desmopressin administration has long been used for the treatment of von Willebrand disease and uremic bleeding. It liberates intact vWF from platelets and endothelial cells. While anecdotally utilized for LVAD non-surgical bleeding, no formal study has investigated its use. Alternatively, a recent case report described utilizing purified vWF concentrate to treat a patient with intractable gastrointestinal bleeding.40 Replacing the disrupted vWF multimers with “new” concentrated vWF has potential for acute bleeding events, but its long-term therapeutic efficacy remains to be determined.
In the setting of enhanced shear stress with LVADs, acquired von Willebrand syndrome appears to be significantly related to upregulation of ADAMTS-13 which correspondingly increases vWF proteolysis.41 Manipulating the natural regulation of vWF proteolysis by ADAMTS-13 represents a potential therapeutic option for nonsurgical LVAD bleeding. Direct inhibition of vWF degradation could also be achieved with monoclonal antibodies targeting the vWF D4 domain. This approach partially inhibited vWF-ADAMTS13 binding and reduced but not fully inhibited loss of HMW multimers under condition of high shear stress.42 This ex-vivo proof-of-concept of an antibody-based therapy for the treatment of vWF degradation induced by circulatory support has the potential provide more sustained stabilization of vWF.
Antibody-directed therapy could also target other platelet components. An interesting in vitro study examined the effect of the local delivery of the GP IIb/IIIa receptor inhibitor, TAK-029. When the circuit was primed with albumin or albumin-bound TAK-029, the amount of adsorbed fibrinogen was significantly reduced with a beneficial effect on platelet adhesion at the interface.43 A controlled and local drug release from the pump itself could deter thrombus formation. This biomedical engineering solution holds promise but will take much work to identify suitable chemical and surface bonding that will appropriately balance the antiplatelet effect and potential for bleeding complications.
Conclusions
While no single factor can take responsibility for toggling the predisposition to bleed or clot, the platelet certainly plays a central role in both activities. Advanced heart failure patients and those that are subsequently implanted with a durable LVAD have multiple markers that suggest platelet activation. Yet, chronic LVAD support does not result in consistent changes in traditional platelet biomarkers that can than be linked to clinical events. Platelet functionality and the formation of the hemostatic plug are likely more relevant than pure biomarker tests. While all mechanical pumps create a disruptive environment, some patients might be genetically predisposed to more aggressive platelet dysfunction. Acquired von Willebrand syndrome has the potentially to be mitigated with some magnetically levitated pumps, but targeting abnormalities in vWF biology offers promising therapeutic targets. Insights related to interactions with blood-formed microparticles as well as platelet receptor shedding provide new areas of both mechanistic and therapeutic research.
Summarily, the yin-yang of bleeding and thrombosis remains a vexing problem in patients supported with LVADs. Platelet biology is transforming beyond simple markers of aggregation and release of secretory granules. Future directions will be based on manipulating their transcriptomic and proteomic molecular signatures and more targeted approaches to membrane and protein-protein interactions. Although the LVAD guarantees platelets a rough environment, how an individual’s platelet responds to that stress is likely different. Over the next 5–10 years, platelet-related therapies for LVAD patients will become more personalized, accounting for genetic differences in the patient and genomic differences in the platelet.
Key Points.
Linking clinical events to standard biomarkers of platelet activation is inadequate
Shedding of platelet receptors and formation of microparticle complexes offer more sophisticated approaches for LVAD-induced platelet activation
Novel methods of platelet functionality, including altered mitochondrial membrane potentials, exist
Acquired von Willebrand syndrome, while not explaining all bleeding events, is a central feature of mechanical support and offers a target for innovative therapies
Acknowledgments
none
Financial support: This work was funded in part by grants from the International Society for Heart and Lung Transplantation (AK, CHS), National Institute of Health RO1 HL089592 (CHS), the American College of Surgeons and Association for Academic Thoracic Surgery (SHM), the NHLBI (HL112311 and HL126547 to MTR), and the NIA (AG048022 to MTR).
Footnotes
Conflict of interest: none
References
- 1.Slaughter MS, Rogers JG, Milano CA, Russell SD, Conte JV, Feldman D, Sun B, Tatooles AJ, Delgado RM, 3rd, Long JW, Wozniak TC, Ghumman W, Farrar DJ, Frazier OH, HeartMate III. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med. 2009;361:2241–2251. doi: 10.1056/NEJMoa0909938. [DOI] [PubMed] [Google Scholar]
- 2.Aaronson KD, Slaughter MS, Miller LW, McGee EC, Cotts WG, Acker MA, Jessup ML, Gregoric ID, Loyalka P, Frazier OH, Jeevanandam V, Anderson AS, Kormos RL, Teuteberg JJ, Levy WC, Naftel DC, Bittman RM, Pagani FD, Hathaway DR, Boyce SW. Use of an intrapericardial, continuous-flow, centrifugal pump in patients awaiting heart transplantation. Circulation. 2012;125:3191–3200. doi: 10.1161/CIRCULATIONAHA.111.058412. [DOI] [PubMed] [Google Scholar]
- 3.Estep JD, Starling RC, Horstmanshof DA, Milano CA, Selzman CH, Shah KB, Loebe M, Moazami N, Long JW, Stehlik J, Kasirajan V, Haas DC, O’Connell JB, Boyle AJ, Farrar DJ, Rogers JG. Risk Assessment and Comparative Effectiveness of Left Ventricular Assist Device and Medical Management in Ambulatory Heart Failure Patients: Results From the ROADMAP Study. J Am Coll Cardiol. 2015;66:1747–1761. doi: 10.1016/j.jacc.2015.07.075. [DOI] [PubMed] [Google Scholar]
- 4.Kirklin JK, Naftel DC, Pagani FD, Kormos RL, Stevenson LW, Blume ED, Myers SL, Miller MA, Baldwin JT, Young JB. Seventh INTERMACS annual report: 15,000 patients and counting. J Heart Lung Transplant. 2015 doi: 10.1016/j.healun.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Starling RC, Moazami N, Silvestry SC, Ewald G, Rogers JG, Milano CA, Rame JE, Acker MA, Blackstone EH, Ehrlinger J, Thuita L, Mountis MM, Soltesz EG, Lytle BW, Smedira NG. Unexpected abrupt increase in left ventricular assist device thrombosis. N Engl J Med. 2014;370:33–40. doi: 10.1056/NEJMoa1313385. [DOI] [PubMed] [Google Scholar]
- 6.Wright J. The origin and nature of blood platelets. Boston Med Surg J. 1906;154:643–645. [Google Scholar]
- 7.Schulze H, Shivdasani RA. Mechanisms of thrombopoiesis. Journal of thrombosis and haemostasis: JTH. 2005;3:1717–1724. doi: 10.1111/j.1538-7836.2005.01426.x. [DOI] [PubMed] [Google Scholar]
- 8.Italiano JE, Jr, Shivdasani RA. Megakaryocytes and beyond: the birth of platelets. Journal of thrombosis and haemostasis: JTH. 2003;1:1174–1182. doi: 10.1046/j.1538-7836.2003.00290.x. [DOI] [PubMed] [Google Scholar]
- 9.Stehlik J, Johnson SA, Selzman CH. Gold standard in anticoagulation assessment of left ventricular assist device patients?: how about bronze. JACC. Heart failure. 2015;3:323–326. doi: 10.1016/j.jchf.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 10**.de Biasi AR, Manning KB, Salemi A. Science for surgeons: understanding pump thrombogenesis in continuous-flow left ventricular assist devices. J Thorac Cardiovasc Surg. 2015;149:667–673. doi: 10.1016/j.jtcvs.2014.11.041. A timely review of the major issues surrounding thrombogenesis of LVADs. [DOI] [PubMed] [Google Scholar]
- 11.Linneweber J, Dohmen PM, Kertzscher U, Affeld K, Nose Y, Konertz W. The effect of surface roughness on activation of the coagulation system and platelet adhesion in rotary blood pumps. Artif Organs. 2007;31:345–351. doi: 10.1111/j.1525-1594.2007.00391.x. [DOI] [PubMed] [Google Scholar]
- 12.Kawahito K, Mohara J, Misawa Y, Fuse K. Platelet damage caused by the centrifugal pump: in vitro evaluation by measuring the release of alpha-granule packing proteins. Artif Organs. 1997;21:1105–1109. doi: 10.1111/j.1525-1594.1997.tb00450.x. [DOI] [PubMed] [Google Scholar]
- 13.Dewald O, Schmitz C, Diem H, Goehring P, Vetter HO, Roell W, Goedje O, Tschoepe D, Reichart B. Platelet activation markers in patients with heart assist device. Artif Organs. 2005;29:292–299. doi: 10.1111/j.1525-1594.2005.29050.x. [DOI] [PubMed] [Google Scholar]
- 14.Houel R, Mazoyer E, Boval B, Kirsch M, Vermes E, Drouet L, Loisance DY. Platelet activation and aggregation profile in prolonged external ventricular support. J Thorac Cardiovasc Surg. 2004;128:197–202. doi: 10.1016/j.jtcvs.2003.11.059. [DOI] [PubMed] [Google Scholar]
- 15.Bonaros N, Mueller MR, Salat A, Schima H, Roethy W, Kocher AA, Wolner E, Wieselthaler GM. Extensive coagulation monitoring in patients after implantation of the MicroMed Debakey continuous flow axial pump. Asaio J. 2004;50:424–431. doi: 10.1097/01.mat.0000136515.97686.a2. [DOI] [PubMed] [Google Scholar]
- 16.Loffler C, Straub A, Bassler N, Pernice K, Beyersdorf F, Bode C, Siegenthaler MP, Peter K. Evaluation of platelet activation in patients supported by the Jarvik 2000* high-rotational speed impeller ventricular assist device. J Thorac Cardiovasc Surg. 2009;137:736–741. doi: 10.1016/j.jtcvs.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 17.Ashbrook M, Walenga JM, Schwartz J, Heroux A, Jeske WP, Escalante V, Bakhos M. Left ventricular assist device-induced coagulation and platelet activation and effect of the current anticoagulant therapy regimen. Clin Appl Thromb Hemost. 2013;19:249–255. doi: 10.1177/1076029612464900. [DOI] [PubMed] [Google Scholar]
- 18.Slaughter MS, Sobieski MA, 2nd, Graham JD, Pappas PS, Tatooles AJ, Koenig SC. Platelet activation in heart failure patients supported by the HeartMate II ventricular assist device. Int J Artif Organs. 2011;34:461–468. doi: 10.5301/IJAO.2011.8459. [DOI] [PubMed] [Google Scholar]
- 19**.Birschmann I, Dittrich M, Eller T, Wiegmann B, Reininger AJ, Budde U, Struber M. Ambient hemolysis and activation of coagulation is different between HeartMate II and HeartWare left ventricular assist devices. J Heart Lung Transplant. 2014;33:80–87. doi: 10.1016/j.healun.2013.11.010. A clinical study that compares differences in acquired von Willebrand syndrome and activation of coagulation pathways between the two most frequently used LVADs. [DOI] [PubMed] [Google Scholar]
- 20.Hugel B, Martinez MC, Kunzelmann C, Freyssinet JM. Membrane microparticles: two sides of the coin. Physiology (Bethesda) 2005;20:22–27. doi: 10.1152/physiol.00029.2004. [DOI] [PubMed] [Google Scholar]
- 21.Mackman N. On the trail of microparticles. Circulation research. 2009;104:925–927. doi: 10.1161/CIRCRESAHA.109.196840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.John R, Panch S, Hrabe J, Wei P, Solovey A, Joyce L, Hebbel R. Activation of endothelial and coagulation systems in left ventricular assist device recipients. Ann Thorac Surg. 2009;88:1171–1179. doi: 10.1016/j.athoracsur.2009.06.095. [DOI] [PubMed] [Google Scholar]
- 23.Diehl P, Aleker M, Helbing T, Sossong V, Beyersdorf F, Olschewski M, Bode C, Moser M. Enhanced microparticles in ventricular assist device patients predict platelet, leukocyte and endothelial cell activation. Interactive cardiovascular and thoracic surgery. 2010;11:133–137. doi: 10.1510/icvts.2010.232603. [DOI] [PubMed] [Google Scholar]
- 24.Radovancevic R, Matijevic N, Bracey AW, Radovancevic B, Elayda M, Gregoric ID, Frazier OH. Increased leukocyte-platelet interactions during circulatory support with left ventricular assist devices. Asaio J. 2009;55:459–464. doi: 10.1097/MAT.0b013e3181b235af. [DOI] [PubMed] [Google Scholar]
- 25**.Mondal NK, Sorensen EN, Hiivala NJ, Feller ED, Pham SM, Griffith BP, Wu ZJ. Intraplatelet reactive oxygen species, mitochondrial damage and platelet apoptosis augment non-surgical bleeding in heart failure patients supported by continuous-flow left ventricular assist device. Platelets. 2015;26:536–544. doi: 10.3109/09537104.2014.948840. Study focusing on more novel forms of platelet function and clinical events. [DOI] [PubMed] [Google Scholar]
- 26.Rondina MT, Garraud O. Emerging evidence for platelets as immune and inflammatory effector cells. Frontiers in immunology. 2014;5:653. doi: 10.3389/fimmu.2014.00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Mondal NK, Sorensen EN, Feller ED, Pham SM, Griffith BP, Wu ZJ. Systemic Inflammatory Response Syndrome After Contentious-Flow Left Ventricular Assist Device Implantation and Change in Platelet Mitochondrial Membrane Potential. Journal of cardiac failure. 2015;21:564–571. doi: 10.1016/j.cardfail.2015.04.007. First study to focus on innovative concepts of platelet function assessed by mitochondrial damage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Himmelfarb J, Nelson S, McMonagle E, Holbrook D, Benoit SE, Michelson AD, Ault K. Elevated plasma glycocalicin levels and decreased ristocetin-induced platelet agglutination in hemodialysis patients. American journal of kidney diseases: the official journal of the National Kidney Foundation. 1998;32:132–138. doi: 10.1053/ajkd.1998.v32.pm9669434. [DOI] [PubMed] [Google Scholar]
- 29**.Hu J, Mondal NK, Sorensen EN, Cai L, Fang HB, Griffith BP, Wu ZJ. Platelet glycoprotein Ibalpha ectodomain shedding and non-surgical bleeding in heart failure patients supported by continuous-flow left ventricular assist devices. J Heart Lung Transplant. 2014;33:71–79. doi: 10.1016/j.healun.2013.08.013. Demonstrates relationship between glycoprotein receptor shedding and non-surgical bleeding and implicates a novel biomarker of platelet function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Z, Mondal NK, Ding J, Gao J, Griffith BP, Wu ZJ. Shear-induced platelet receptor shedding by non-physiological high shear stress with short exposure time: glycoprotein Ibalpha and glycoprotein VI. Thrombosis research. 2015;135:692–698. doi: 10.1016/j.thromres.2015.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Z, Mondal NK, Ding J, Koenig SC, Slaughter MS, Griffith BP, Wu ZJ. Activation and shedding of platelet glycoprotein IIb/IIIa under non-physiological shear stress. Molecular and cellular biochemistry. 2015;409:93–101. doi: 10.1007/s11010-015-2515-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potapov EV, Ignatenko S, Nasseri BA, Loebe M, Harke C, Bettmann M, Doller A, Regitz-Zagrosek V, Hetzer R. Clinical significance of PlA polymorphism of platelet GP IIb/IIIa receptors during long-term VAD support. Ann Thorac Surg. 2004;77:869–874. doi: 10.1016/j.athoracsur.2003.08.013. discussion 874. [DOI] [PubMed] [Google Scholar]
- 33.Schmugge M, Rand ML, Freedman J. Platelets and von Willebrand factor. Transfus Apher Sci. 2003;28:269–277. doi: 10.1016/S1473-0502(03)00046-6. [DOI] [PubMed] [Google Scholar]
- 34.Klovaite J, Gustafsson F, Mortensen SA, Sander K, Nielsen LB. Severely impaired von Willebrand factor-dependent platelet aggregation in patients with a continuous-flow left ventricular assist device (HeartMate II) J Am Coll Cardiol. 2009;53:2162–2167. doi: 10.1016/j.jacc.2009.02.048. [DOI] [PubMed] [Google Scholar]
- 35.Meyer AL, Malehsa D, Bara C, Budde U, Slaughter MS, Haverich A, Strueber M. Acquired von Willebrand syndrome in patients with an axial flow left ventricular assist device. Circulation. Heart failure. 2010;3:675–681. doi: 10.1161/CIRCHEARTFAILURE.109.877597. [DOI] [PubMed] [Google Scholar]
- 36*.Meyer AL, Malehsa D, Budde U, Bara C, Haverich A, Strueber M. Acquired von Willebrand syndrome in patients with a centrifugal or axial continuous flow left ventricular assist device. JACC. Heart failure. 2014;2:141–145. doi: 10.1016/j.jchf.2013.10.008. A retrospective study highlighting that all continuous flow pumps develop acquired von Willebrand syndrome with similar complication rates. This study recognizes that this acquired syndrome is an important, but not the only, contributing factor for bleeding. [DOI] [PubMed] [Google Scholar]
- 37.Crow S, Chen D, Milano C, Thomas W, Joyce L, Piacentino V, 3rd, Sharma R, Wu J, Arepally G, Bowles D, Rogers J, Villamizar-Ortiz N. Acquired von Willebrand syndrome in continuous-flow ventricular assist device recipients. Ann Thorac Surg. 2010;90:1263–1269. doi: 10.1016/j.athoracsur.2010.04.099. discussion 1269. [DOI] [PubMed] [Google Scholar]
- 38.Wever-Pinzon O, Selzman CH, Drakos SG, Saidi A, Stoddard GJ, Gilbert EM, Labedi M, Reid BB, Davis ES, Kfoury AG, Li DY, Stehlik J, Bader F. Pulsatility and the risk of nonsurgical bleeding in patients supported with the continuous-flow left ventricular assist device HeartMate II. Circulation. Heart failure. 2013;6:517–526. doi: 10.1161/CIRCHEARTFAILURE.112.000206. [DOI] [PubMed] [Google Scholar]
- 39.Teuteberg JJ, Slaughter MS, Rogers JG, McGee EC, Pagani FD, Gordon R, Rame E, Acker M, Kormos RL, Salerno C, Schleeter TP, Goldstein DJ, Shin J, Starling RC, Wozniak T, Malik AS, Silvestry S, Ewald GA, Jorde UP, Naka Y, Birks E, Najarian KB, Hathaway DR, Aaronson KD. The HVAD Left Ventricular Assist Device: Risk Factors for Neurological Events and Risk Mitigation Strategies. JACC. Heart failure. 2015;3:818–828. doi: 10.1016/j.jchf.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 40.Fischer Q, Huisse MG, Voiriot G, Caron C, Lepage L, Dilly MP, Nataf P, Ajzenberg N, Kirsch M. Von Willebrand factor, a versatile player in gastrointestinal bleeding in left ventricular assist device recipients? Transfusion. 2015;55:51–54. doi: 10.1111/trf.12788. [DOI] [PubMed] [Google Scholar]
- 41.Bartoli CR, Restle DJ, Zhang DM, Acker MA, Atluri P. Pathologic von Willebrand factor degradation with a left ventricular assist device occurs via two distinct mechanisms: mechanical demolition and enzymatic cleavage. J Thorac Cardiovasc Surg. 2015;149:281–289. doi: 10.1016/j.jtcvs.2014.09.031. [DOI] [PubMed] [Google Scholar]
- 42.Rauch A, Legendre P, Christophe OD, Goudemand J, van Belle E, Vincentelli A, Denis CV, Susen S, Lenting PJ. Antibody-based prevention of von Willebrand factor degradation mediated by circulatory assist devices. Thromb Haemost. 2014;112:1014–1023. doi: 10.1160/TH14-02-0148. [DOI] [PubMed] [Google Scholar]
- 43.Linneweber J, Dohmen PM, Kertzscher U, Affeld K, Konertz W. Local glycoprotein IIb/IIIa receptor inhibitor delivery from the pump surface attenuates platelet adhesion in continuous flow ventricular assist devices. Artif Organs. 2008;32:792–799. doi: 10.1111/j.1525-1594.2008.00632.x. [DOI] [PubMed] [Google Scholar]