

Abstract
The liposome dialyzer is a small-volume equilibrium dialysis device, built from commercially available materials, that is designed for rapid exchange of small volumes of an extraliposomal reagent pool against a liposome preparation. The dialyzer is prepared by modification of commercially available dialysis cartridges and consists of a reactor with two 300 µL chambers and a 1.56 cm2 dialysis surface area. The dialyzer is prepared in three stages: 1) disassembly of dialysis cartridges to obtain required parts; 2) assembly of the dialyzer; and 3) sealing the dialyzer with epoxy. Preparation of the dialyser takes about 1.5 h, not including overnight epoxy curing. Each round of dialysis takes 1–24 h, depending on the analyte and membrane employed. We previously used the dialyzer for small-volume nonenzymatic RNA synthesis reactions inside fatty acid vesicles. In this protocol, we demonstrate other applications, including removal of unencapsulated calcein from vesicles, remote loading, and vesicle microscopy.
Introduction
General description
The liposome dialyzer (Figure 1) allows the equilibrium dialysis of small volume samples, enabled by a large dialysis membrane surface area. It is prepared by the modification of inexpensive commercially available Thermo Scientific Slide-A-Lyzer Dialysis Cassettes (ca. US $25 total cost). The physical construction of the dialyzer is ideal for reactions taking place within liposomes. It is small and lightweight, allowing it to be placed on a shaker or rotator for extended tumbling, or in an incubator for temperature cycling. The device consists of two chambers, each of which can hold 250–300 µL. The effective dialysis area is 1.56 cm2, corresponding to a surface area to volume ratio of ca. 500–600 mm2/mL. These characteristics make the dialyzer ideal for liposome formulations, including remote loading, preparation of liposomes containing high-value encapsulation solutions, and general biophysical studies of liposomes. Additionally, given the correct choice of dialysis membrane, we anticipate the dialyzer would be suitable for dialysis of macromolecules (e.g., proteins or DNA/RNA) against solutions of small molecules.
Figure 1. Diagram of the liposome dialyzer.
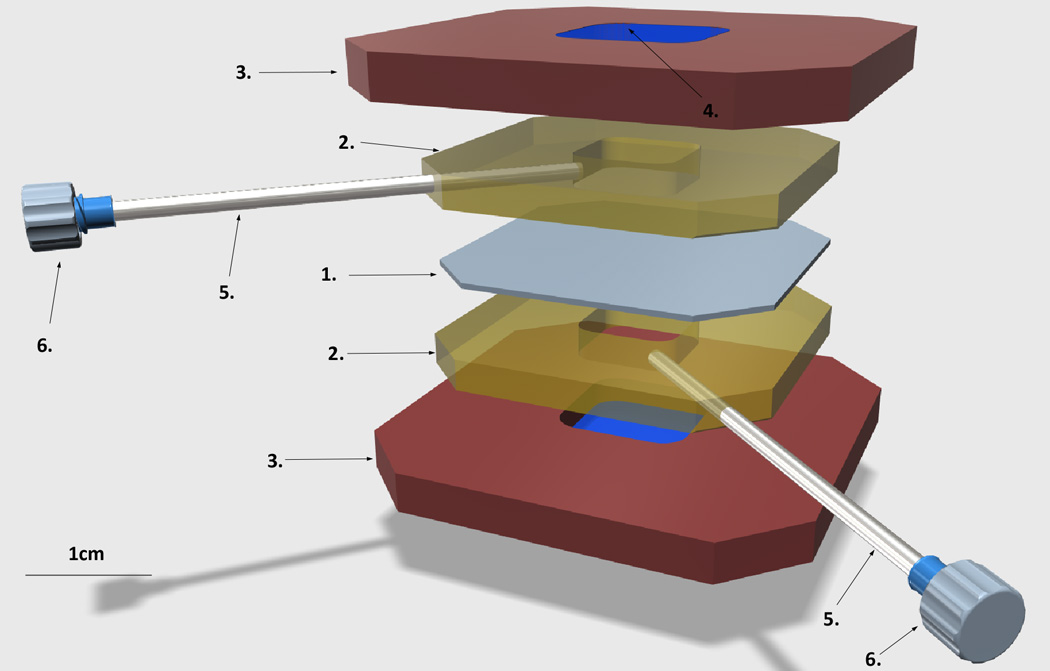
1. Dialysis membrane, 20K MWCO
2. Silicone gaskets
3. Plastic cassette, top and bottom
4. The window on either side of the plastic cassette, sealed with the PCR film
5. Needles for loading sample and wash buffers
6. Needle luer caps
Sample chambers are colored yellow.
Development
We originally developed the dialyzer to facilitate our experiments in non-enzymatic template-directed RNA polymerization within lipid vesicles, as part of our research program targeted towards the study of the emergence of the earliest cellular life. In particular, the dialyzer was first used to facilitate the first non-enzymatic RNA template copying system capable of functioning inside fatty acid model protocells.1 In a typical experiment, the RNA template and primers are encapsulated inside fatty acid vesicles, and 2-methyl imidazole activated nucleotide monomers are added to the outside of the vesicles. We required a means of removing hydrolyzed activated RNA nucleotide monomers from the liposome sample (which inhibit polymerization) and to deliver fresh portions of these monomers. Activated RNA nucleotides are prepared in-house by labor-intensive processes and have a limited shelf life; accordingly, we sought to conserve the use of these monomers to the maximum extent possible.
Typical commercially available dialysis systems lacked one or more of the following aspects, which we required and are common to many liposome applications: 1) the means to dialyze a small volume of valuable sample (containing liposomes with encapsulated RNA) against small volumes of equally valuable solution (containing activated monomers), 2) a large surface area to volume ratio (for rapid equilibration of small volume samples), 3) the ability to tumble the sample, (liposomes, especially fatty acid ones, aggregate rapidly if not constantly tumbled), 4) the ability to easily add and remove solutions from one or both chambers repeatedly, and 5) a means to view the samples easily to monitor liposome integrity (aggregation of liposomes, resulting in a precipitate, can be observed with the naked eye). The sample is then dialyzed against buffer containing fresh activated monomers. During dialysis, the hydrolyzed nucleotide monomers and 2-methyl imidazole (non-enzymatic RNA primer extension reaction inhibitors) are removed, and new activated monomer is delivered to the vesicles.
The general principle of removing hydrolyzed activated monomer is extensible to a wide range of nonenzymatic primer extension chemistries, including OAt esters, as employed by Richert et al in a similar system requiring immobilization of RNA for hydrolysis product removal.2 After optimizing the construction of the dialyzer, we developed other uses for the dialyzer in liposome studies performed in our research (see example uses). Numerous uses for dialysis exist in liposome studies. We have previously shown that bulk dialysis (i.e., against excess buffer) can be employed to prepare uniformly sized large liposomes 3,4. Many other groups have employed liposomes as bioreactors; almost all such experiments require a purification step after liposome preparation to remove unencapsulated solutes. Several examples of recent work in this field include cell-free transcription 5 and protein synthesis 6,7, as well as employing liposomes to add sensory capabilities to cells 8 and protein selection methods 9. Liposomes have also attracted broad interest as drug delivery platforms for delivering chemical therapeutics 10,11 and nanoparticles 12. One of the most widely known examples of liposomal drug delivery successfully applied in cancer treatment is Doxil, a liposomal doxorubicin formulation prepared by remote loading of the drug into liposomes 13. All these applications require the use of low-abundance materials encapsulated within liposomes.
The bulk of our work with the dialyzer employs the Slide-A-Lyzer (original) device, which is optimal for preparing the dialyzer. A newer version of the Slide-A-Lyzer exists, called the Slide-A-Lyzer G2. This device has two principal new features: 1) an integrated air pocket, negating the need for the use of foam floats when dialyzing and 2) the introduction of stoppered ports in the silicone gasket, allowing for sample introduction through the use of a pipette instead of a syringe and needle. The first feature is not relevant to the use of the dialyzer described in this method. While the second feature is desirable, the construction of the Slide-A-Lyzer G2 differs from that of the original device in that the plastic frames are of two different shapes. The plastic frame that contains the port in the gasket has a small lip on the bottom. This does not preclude the preparation of a functional dialyzer, but it requires careful alignment of the two plastic frames when assembling the dialyzer and clamping it during epoxy curing. In contrast, the original Slide-A-Lyzer can be assembled more easily. We present a description of the use of the Slide-A-Lyzer G2 in Box 1. The data obtained in Figure 3 were collected using dialyzers prepared by this method. This formulation of the dialyzer will be optimal for samples highly sensitive to shear, such as giant vesicles, that are better introduced by a pipette than a needle. Additionally, in the event that the original Slide-A-Lyzer is discontinued, this represents an alternate method of dialyzer preparation.
Box 1. Alternate method for dialyzer preparation using Slide-A-Lyzer G2.
TIMING 24h
In the main protocol, we have presented the dialyzer assembly using the first generation Slide-A-Lyzer. However, it is also possible to prepare a liposome dialyzer using Slide-A-Lyzer G2 dialysis cassettes.
The main difference between the classical design and the G2 is the presence of the sample port in the G2, which eliminates the need to use a needle to introduce and remove samples.
The basic assembly workflow is similar to the one described in the main part of the protocol. The most important differences are highlighted below.
-
1
(Related to the step 1 in the main protocol) Disassembling the G2 Slide-a-Lyzer requires the removal of the sample port plug prior to the separation of the plastic frame.
*CRITICAL STEP Take special care not to puncture the dialysis membrane. Avoid handling the dialyzer by the membrane.
-
2
(Related to step 4 in the main protocol) Attaching the PCR film to create the transparent sample viewing window has to be done with great care, adjusting the PCR film to the angle of the inside of the G2 plastic frame. It is helpful to use a pipette tip or the tip of a pencil to push the PCR film into the grooves inside the plastic frame.
-
3
(Related to step 5 in the main protocol) The presence of sample ports makes needle insertion optional with G2-based dialyzers. The sample and dialysis buffer can be successfully dispensed through the sample ports in the gasket, so there is no need to perform step 5 of the original protocol.
-
4
(Related to step 7 in the main protocol) Before clamping the G4 assembly, make sure the sample port plugs are properly aligned – parallel to each other and parallel to the top of the dialyzer frame. (Figures S16 and S17)
*CRITICAL STEP The gasket of the G2 Slide-a-Lyzer is uneven in thickness (wider on the top, slightly narrower on the bottom) It is important to adjust the tension of the bar clamp very carefully, so that the dialyzer remains firmly squeezed during the hardening of the epoxy, but the sample port plugs are not pushed into each other.
-
5
(Related to steps 9 and 10 in the main protocol) To avoid leaks, epoxy needs to be applied firmly to the whole surface of the gasket. The sample port plugs make the access to the top part of the gasket very difficult; the application to the top of the gasket in G2 assembly can be achieved with a narrow spatula or pipette tip.
*CRITICAL STEP Do not apply epoxy to the sample port plugs. The plugs need to remain removable for the proper use of the dialyzer.
*TROUBLESHOOTING We recommend applying epoxy to the top of the G2 assembly first, before covering the sides. This allows for inspection of the sample port plugs before proceeding with epoxy application to the rest of the gasket. If, despite great care, the sample port plug is glued in place, the dialyzer assembly can be repaired by inserting a needle into the side of the silicone gasket, as described in the step 5 of the main protocol. The needle will then be used, instead of the sample port, to deliver and recover sample and/or dialysis buffer.
-
6
After the epoxy cures, the sample plugs need to be removed and the sample ports inspected to make sure both ports are clear. (Figure S18)
After the epoxy cures, the G2 dialyzer can be used for the same purposes as the standard Slide-a-Lyzer based dialyzer, with one difference: sample and dialysis buffer is inserted and removed through sample ports instead of the side needles. (Figure S19)
Figure 3. Removal of unencapsulated small molecule from liposome preparations.
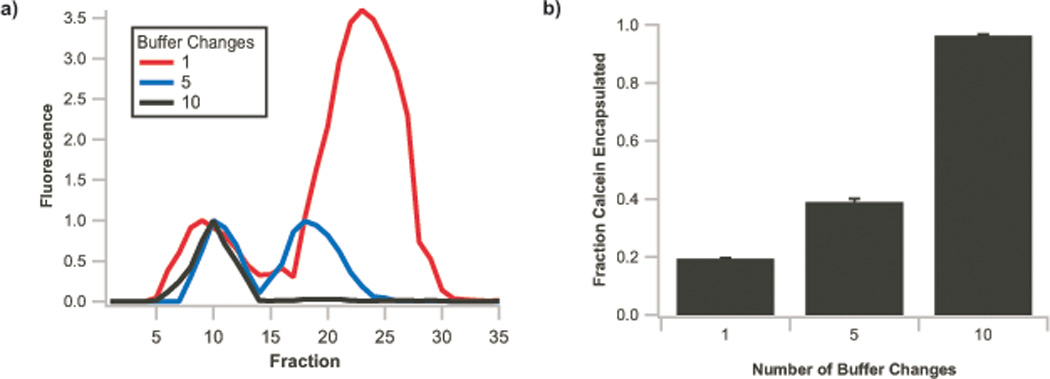
a) Repurification traces of vesicles (100 mM oleic acid) prepared with 1 mM calcein, dialyzed one (red trace), five (blue trace), or ten times (black trace) against a dye-free vesicle solution. b) Fraction of calcein encapsulated within vesicles after dialysis, calculated by integration of fluorescence signals from a), demonstrating removal of ca. 90% of free small molecules. Error bars indicate S.E.M., N=3.
The methods for preparation of fatty acid vesicles with encapsulated small molecule solutes and purification of vesicles are described in our previous work.1,21
Comparison with Other Methods
Dialysis devices can be described in two broad classes: all devices employ a small-volume chamber containing the high-molecular weight analyte of interest, such as a protein, nucleic acid, or liposome. This chamber is allowed to equilibrate through the dialysis membrane with another solution containing the small molecule of interest. In some, dialysis against a large excess of bulk solution is performed (this is the intended application of the parent Slide-a-Lyzer device employed in this protocol). In other dialysis experiments, typically termed equilibrium dialysis, compartments of equal or near-equal volume are used and the small molecule of interest is allowed to equilibrate between the two chambers; we required such a dialyzer for our experiments. A number of devices exist to perform such experiments, such as those available from Spectra/Por and Harvard Apparatus, but of those we found, each lacked one or more of 1) a large surface area-to-volume ratio, 2) compatibility with tumbling, and 3) viewing windows to view liposome samples during incubation to monitor their integrity. The combination of these features makes our dialyzer ideal for liposome experiments.
Limitations
Due to the construction of the device, it is not suitable for high-throughput applications (e.g., where a 96-well plate dialyzer would be more appropriate). We have found the dialyzer’s internal volume of 250–300 µL/chamber, coupled with its large surface area (500–600 mm2/mL) to be an ideal combination for the scale of reactions described in this work that employ a high-value small molecule. Larger dialyzers from the same product family exist (up to 30 mL) of the same design that should be readily adaptable to the technique we have described. Much larger or smaller applications, or those employing readily-available small molecules, may be better-suited to other devices (such as devices intended for dialysis against bulk solution).
Experimental Design
Calcein dialysis
One small molecule we employed to test the system is the fluorescent dye calcein; we show results for this in the Anticipated Results section. In typical experiments, vesicles are prepared from thin films dried from chloroform or dichloromethane solution, or, in the case of liquid fatty acids, neat lipids. Due to the high solubility of fatty acids relative to diacylphospholipids, fatty acid vesicles should be included in both dialysis chambers to avoid lipid depletion; for phospholipid vesicles, they are required in only one chamber.
Remote Loading of Phospholipid Vesicles
The equal volume chambers of the dialyzer are ideal for remote loading procedures, wherein an ion gradient is employed to concentrate weakly basic or acidic small molecules within phospholipid vesicles.14 Typical remote loading experiments employ an ammonium ion gradient and a weakly basic substrate for loading, such as acridine orange (employed in the study described below) or doxorubicin (remotely loaded into vesicles in the manufacture of the liposomal formulation of this molecule sold as Doxil).15 As a result of the high membrane permeability of neutral ammonia, this molecule can leave the liposome and exchange with extraliposomal small molecule, actively loading it into the liposome. Similarly, acetate gradients have been employed to load weak acids, such as diclofenac and insulin, into liposomes16. As remote loading is a method of concentrating the vesicle payload, typical applications demand both the maximum possible concentration of payload, both within the liposome, as well as in the bulk solution containing the liposomes (i.e., a high overall concentration of lipid). Loading of the liposomes with the dialyzer enables both minimal dilution, in contrast to non-dialysis techniques, such as adding a dilute solution of intended payload to the vesicle preparation. Additionally, in contrast to bulk dialysis, the equal-volume inner chambers of the dialyzer allow for loading a minimal volume of relatively concentrated solution of intended payload. Additionally, the ability to incorporate up to four needles (one in each corner of the dialyzer, two in each gasket) allows for loading vesicles under flow, facilitating the loading of low-solubility payloads while maintaining a constant reservoir of fresh small molecules.
In these experiments, a thin film of the desired lipid or lipid mixture is rehydrated to the desired lipid concentration in a remote-loading compatible salt solution.
Staining of Phospholipid Vesicles
The dialyzer allows for the incorporation of stains into vesicles for analysis by microscopy. The equal-volume chambers allow for maintaining isoosmotic conditions throughout staining without the addition of the lytic concentrations of the solvents necessary to solubilize high concentrations of many dyes (e.g., ethanol and DMSO). In these experiments, vesicles are prepared by thin film hydration of a thin film of the desired lipid or lipid mixture.
Dynamics of Single-Chain Amphiphile Assemblies
Other applications of the dialyzer relate to the study of lipid dynamics. One unique characteristic of liposomes formed from single-chain amphiphiles is their dynamic nature. As a result of the substantially higher solubility of single-chain amphiphiles (cf. oleate, with ca. 100 µM solubility, as opposed to dilauroylphosphatidylcholine (DLPC), with ca. 25 nM solubility), vesicles formed from these lipids are more prone to undergo lipid exchange via the soluble lipid fraction. The dialyzer enables a number of experiments relating to observation of these dynamics, examples of which are described below.
Size Characterization of Lipid During Transfer Between Vesicle Populations
While it is well-understood that lipid exchange occurs between vesicles formed from single-chain amphiphiles via the soluble lipid fraction, it is less clear what lipid aggregation state (free lipid, or larger, likely micellar aggregates) is responsible for this exchange. The analysis of populations of liposomes and lipid micelles and free lipids is difficult; the size range of the population of lipid assemblies in these systems ranges from on the order of 1 nm to several microns – over three orders of magnitude. This marked polydispersity complicates analysis by optical techniques, such as DLS, and the large size of liposomes results in slow tumbling, making analysis by NMR difficult or impossible. Dialysis affords one means of characterizing mass transfer between liposome systems. While single-chain amphiphiles are of low molecular weight (e.g., oleic acid, MW 282), even micelles, the smallest assemblies typically associated with these compounds, typically have an aggregation number of ca. 50. This corresponds to a mass of over 10,000 Da for these assemblies, even neglecting associated water and counterions. As a result, assemblies of these lipids have dramatically different permeability to dialysis membranes than free lipid. Thus, the size of the lipid species exchanged between vesicles can be probed by using the dialyzer, for which membranes spanning this size range (2K-20K MWCO) exist.
Dynamics of Fatty Acid Vesicle Formation
Phospholipid vesicles are typically formed in laboratory conditions either by rehydration of a thin film of amphiphiles, or by injection of a concentrated solution (typically ethanolic) of the lipid into buffered solution.17 While fatty acids can form vesicles by these methods as well, another method exists, wherein a solution of fatty acids at high pH (containing micelles) is subjected to a drop in pH. As a result, the micelles are partially protonated, micelles dissociate, and vesicles are formed. One unique characteristic of such “pH drop” experiments is that vesicle formation proceeds autocatalytically. In these experiments, an induction phase occurs, in which minimal vesicle formation is observed. As vesicles begin to form, the rate of vesicle formation increases. Consistent with an autocatalytic reaction, “seeding” of these solutions with vesicles results in an increase in the rate of vesicle formation.18 The dialyzer is suitable for monitoring of vesicle formation kinetics by transfer of lipid from existing vesicles. For example, it can be used in experiments where one chamber initially contains a high concentration of single-chain amphiphiles vesicles, the other initially contains none, and vesicle formation is monitored by absorbance measurements of the second chamber.
Materials
Equipment
Building the dialyzer
-
Slide-A-Lyzer Dialysis Cassettes, 20K MWCO (molecular weight cut-off), 0.5 mL (Thermo Fischer, cat. no. 66005). Other volumes employ the same general design and should be adaptable to the technique, but we have not used them. The MWCO of the cassette should be selected according to the application, but this MWCO is the highest available and thus gives the fastest dialysis rates.
*CRITICAL The design of this dialysis cassette is integral to that of the Dialyzer, so this cassette must be used. Lower MWCOs from the same family can be used if desired, but the dialysis rate will be slowed.
<USEFUL TIP> For most applications, the original Slide-A-Lyzer dialysis cassettes, not the self-floating Slide-A-Lyzer G2 cassettes, are optimal. However, G2 cassettes can be used, and their needle-free port design may be useful in applications employing shear-sensitive samples, such as giant vesicles. Instructions for the assembly of dialyzers employing these cassettes differ slightly and are presented in Box 1.
Gel knife (Invitrogen, cat. no. EI9010). Any wide flat metal spatula or common putty knife, approximately 3 to 6 inches long, should be suitable.
25 ga (gauge) regular bevel needles, 1.5 in. (Becton Dickinson, cat. no 305127). Most needles from 23 to 27 ga should be suitable.
25 ga. blunt needles, 1.5 in. (Sai-Infusion, cat. no. B25–150 100B). The blunt needle gauge should match the above sharp needle diameter or be slightly larger (−2 to 0 ga. difference).
Male Luer integral lock ring cap, closed at grip (Value Plastics, cat. no. LP4–6)
Clear PCR sealing film (Applied Biosystems MicroAmp Clear Adhesive Film, cat. no. 4306311). Any clear adhesive PCR sealing film should be suitable.
Bar clamp (Irwin, cat. no. 512QC). Any 12 in. quick-grip type clamp with pads of sufficient size to span the Dialyzer windows (Figure S10) should be suitable. We have also employed binder clips on each side of the dialyzer, but the bar clamp method is preferred.
-
Epoxy (Hardman DOUBLE-BUBBLE Extra Fast Set Non-sag Epoxy, cat. no. 04001). Any waterproof epoxy should be suitable.
*CRITICAL Note that Step 10 is timed for the 30 minute cure time required for this epoxy to reach handling strength; cure time will vary for other epoxies.
Tube rotator (Bibby/Techne, cat. no. SB3/120V/60 or similar)
Vesicle experiments
Liposome extruder (Avanti Polar Lipids, cat. no. 610023).
10 mm filter supports (Avanti Polar Lipids, cat. no. 610014)
19 mm 100 nm track-etched membranes (Whatman, cat. no. WHA800309)
Reagents
Oleic Acid (Nu-Chek, cat. no. U-46-A) is typically used in fatty acid encapsulations. Technical grade oleic acid (Sigma-Aldrich, cat. no. 364525) has proven to be satisfactory when used in high-volume applications (e.g., in running buffer for purifications).
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, cat. no. 850457)
Chloroform (Sigma-Aldrich, cat. no. C2432 or similar) or dichloromethane (Sigma-Aldrich, cat. no. 270997 or similar).
Vybrant DiI (Life Technologies, cat. no. V-22885)
Calcein (Sigma-Aldrich, cat. no. C0875)
Guanosine monophosphate, 2-methylimidazolide (prepared as described previously)19
1M Tris-HCl pH 8 (Life Technologies, included in cat. no. AM9010 or available separately as cat. no. AM9855G)
1M MgCl2 (Life Technologies, included in cat. no. AM9010 or available separately as cat. no. AM9530G)
Nuclease-free water, non-DEPC-treated (Life Technologies, cat. no. AM9939)
Sepharose 4B (Sigma-Aldrich, cat. no. 4B200)
Reagent Setup
Liposome formulations
In all cases, liposomes must be prepared at or above the CAC (critical aggregation concentration) of the lipid employed. This can vary from the nanomolar range (for diacylphospholipids, in which case CAC is effectively irrelevant) to tens to hundreds of millimolar (for short-chain fatty acids, such as decanoic acid). Liposomes are typically prepared from a thin film of lipid dried from chloroform or dichloromethane solution, when using mixed-lipid vesicles of any type or vesicles employing solid lipids, such as phospholipid or saturated fatty acid vesicles. In all cases, denaturing detergents, such as Triton X-100, should be avoided in liposome preparations. In the case of liquid single-amphiphile vesicles (e.g., oleic acid), neat lipid can be used. We typically incubate vesicles with tumbling and use them within >12 but <72 h after preparation. Generally speaking, phospholipid (diacyl) vesicles are more robust than single chain amphiphile (e.g., fatty acid) vesicles. As most of our work employs at least one single-chain amphiphile in liposome formulations, our guidelines are more stringent than those required by a laboratory employing only diacylphospholipid liposomes.
Oleic Acid Vesicles
Oleate vesicles are prepared by addition of neat oleic acid to buffered solution (typically 100–250 mM Tris-HCl pH 8) containing the desired encapsulation substrates and ½ equivalent of NaOH or KOH (relative to oleic acid) and overnight tumbling at room temperature (22±2 °C).
POPC Vesicles
POPC vesicles are prepared by hydration of a thin film prepared by evaporation of a CHCl3 or CH2Cl2 of lipid under a stream of nitrogen. The hydration solution consists of buffered solution (typically 100–250 mM Tris-HCl pH 8) containing the desired encapsulation substrates. Hydration is performed with overnight tumbling for vesicles to be extruded or is conducted with minimum agitation for the preparation of giant vesicles.
Dialysis Buffer Composition
Typically, the dialysis buffer used is of identical composition to that in the chamber containing the liposomes of interest, with a few exceptions. When unencapsulated solutes are being removed, the desired solute to be removed is omitted from the buffer. When remote loading experiments are being performed, the desired payload is added to the dialysis buffer. Finally, when fatty acid vesicles are used, vesicles comprised of identical lipid composition and concentration are added to the dialysis buffer, so as to avoid depletion of the soluble lipid in the vesicle preparation.
Procedure
Assembly of the Liposome dialyzer
-
1
Open both Slide-A-Lyzer cassettes, inserting the spatula between the edges of the plastic cassette and applying sideways pressure to crack the plastic frame at the seams along the long axes. It is important to avoid damaging the soft silicone gasket inside the plastic cassette and avoid damaging at least one of the dialysis membranes.
The silicone gaskets from both cassettes will be used. Only one plastic frame will be used (two parts of one full frame, with and without the plastic middle ridge) (Figure S1).
-
2
Detach all but one of the dialysis membranes from the silicone gasket (remove both membranes from one of the gaskets and one membrane from another gasket). It is best achieved by bending the gasket slightly; the membranes are attached fairly loosely and should detach easily. It is important not to damage the membrane that remains attached to one of the gaskets.
*CRITICAL STEP Take special care not to puncture the remaining dialysis membrane.
-
3
Cut out two pieces of PCR film in the shape of the membranes. It is helpful to use one of the removed dialysis membranes as a template. (Figure S2)
-
4
Attach one of the pieces of PCR film to the inside of one side of the plastic frame, sticky side out, completely covering the opening in the frame. The film is best sealed by placing the PCR film loosely on the inside of the plastic frame and, using a disposable 200 µL pipette tip or the tip of a soft pencil, pushing the PCR film into the grooves on the inside of the frame, starting from the groove closest to the opening. After tracing all the grooves, apply gentle pressure to the whole film to set it firmly in place. Repeat with the other side of the plastic frame and second piece of PCR film (Figures S3 and S4).
*CRITICAL STEP Make sure no holes remain and the PCR film is sealed all around.
-
5
With a beveled needle, pre-puncture one corner of the silicone gasket. Insert the blunt needle into the pre-punctured hole, with the end of the blunt needle almost flush with the inner edge of the gasket. The needle must penetrate the full thickness of the gasket, but the end should not stick out more than a half of a millimeter on the inside of the gasket. Penetrating the gasket further will impair removal of the contents of the assembled dialyzer. Repeat with the other gasket, paying special attention to the still-attached dialysis membrane. Puncturing the membrane at this stage is a common error; avoid the tip of either needle touching the dialysis membrane at any point (Figures S5 and S6).
*CRITICAL STEP It is important to center the needle in the thickness of the gasket to avoid leaks.
-
6
Align the silicone gasket on the plastic cassette. Then, align the second gasket on top of the first, with the attached dialysis membrane pointing down, so that the membrane is between the gaskets (Figures S7 and S8). Cover the assembly with the other half of the plastic cassette. The finished assembly should look similar to the original Slide-A-Lyzer, except thicker (because there are two silicone gaskets instead of one) (Figure S9).
-
7
Place the assembly on the bar clamp and gently tighten the clamp until the dialyzer assembly is evenly squeezed.
*CRITICAL STEP Applying too much pressure might damage the dialysis membrane, but not pressing the clamp tight enough will not compress the gaskets, resulting in a leaky dialyzer.
Ensure the dialyzer is centered over the rubber ends of the clamps, with these ends fully covering the sides of the dialyzer.
It is important to transport the assembly gently from the bench top to the clamp, so that it doesn’t misalign and the needles won’t punch the dialysis membrane (Figure S10).
*USEFUL TIP Wrapping both arms of the bar clamp in plastic wrap (e.g., Saran Wrap) will prevent the dialyzer assembly from being permanently glued to the clamps, and will help keep the clamp clean of overflowing epoxy. Do not use aluminum foil for this, as the epoxy adheres well to the foil, resulting in pieces of foil becoming stuck to the dialyzer.
-
8
Inspect the whole assembly in the bar clamp. The gaskets should be parallel and centered within the plastic frame (Figure S11). Both needles should penetrate the gasket fully but not protrude more than half of a millimeter into the chambers of the dialyzer.
<CRITICAL STEP> If the assembly is not correctly aligned, with both silicone gaskets flush with one another and aligned with the grooves in the plastic frame, the dialyzer will leak.
-
9
Prepare the epoxy for use, according to the manufacturer’s instructions. Apply the epoxy to the full circumference of the dialyzer assembly using a disposable 200 µL pipette tip (Figure S12).
*CRITICAL STEP Be sure not to contaminate the inside of the dialyzer, particularly the dialysis membrane, with epoxy.
*CRITICAL STEP It is important to cover the full circumference of the dialyzer with epoxy and to glue the needles in place securely to avoid possible shifting of the needles during use of the dialyzer. It is also important to ensure the epoxy thoroughly covers the sides of the silicone gaskets (under the plastic ridge on the side of the plastic cassette). We typically use two 4 g packets of Hardman DOUBLE-BUBBLE fast-setting epoxy to cover the full circumference of the dialyzer. Because the working time of the epoxy is only a few minutes, we recommend applying the contents of one packet, then mixing and using the other packet.
-
10
Leave the dialyzer in the bar clamp until the epoxy glue is fully cured. We typically allow an overnight cure for even fast-setting epoxy (30 minutes until handling strength is reached) to ensure a full cure.
*CRITICAL STEP Removing the dialyzer from the bar clamp before the epoxy is fully cured could result in leaks in the dialyzer.
-
11
Cap the needles with luer caps to avoid dust entering the dialyzer (Figures S13 and S14). Take care to support the dialyzer and needles when manipulating the luered ends of the needles to avoid bending the needles.
-
12
After the epoxy is cured, remove the dialyzer form the bar clamps and carefully inspect the completed device.
Dialysis of Liposomes
-
13
Pre-wet the dialysis membrane with the buffer that will be used for dialysis by adding an equal volume of dialysis buffer to both dialysis chambers and gently shaking manually for about a minute. This also serves to remove glycerol from the membrane.
-
14
Load the first change of dialysis buffer into one dialyzer chamber, then load your liposome sample into the other chamber. The duration of dialysis and frequency of buffer changes is dictated by the application and liposome composition (Table 1).
When loading or removing samples from the dialyzer, use a syringe larger than the volume of sample you wish to load. To load samples, remove air from the dialyzer chamber of ca. equal volume to the sample you intend to load prior to loading. To remove samples, load air prior to dispensing sample into the dialyzer. By doing this, the dialyzer will achieve ca. atmospheric pressure after sample loading or removal.
*CRITICAL STEP It is very helpful to mark which chamber contains the liposomes of interest and which contains buffer, particularly with samples where liposomes are in both chambers, such as fatty acid vesicle experiments. We typically mark a dot on the plastic casing on the sample side with a lab marker.
-
15
Always tumble the dialyzer when using fatty acid vesicles.
We have found that attaching the dialyzer to a rotary tumbler with paper clips, tape, or elastic bands is the best method. With tumbling, vesicles are dialyzed according to their composition and desired application (Table 1).
*CRITICAL STEP The needles can easily be bent, which will impair sample retrieval. Ensure the needles will not collide with anything when rotating. For example, we have noticed that one sometimes attaches the dialyzer assembly at the top of the stopped rotor, with the needles sticking outside the edges of the rotor. When the tumbler is turned on and moves the dialyzer to the position closest to the bottom, the needles then hit the bench and bend or break.
Table 1.
Duration of Liposome Dialysis by Composition and Application
| Liposome type |
Experiment type | Typical timing |
|---|---|---|
| Fatty acid vesicles |
Non-enzymatic template directed RNA synthesis |
4–6 hours for each change of activated monomer Typically 8–10 monomer changes Total reaction time 48–96 hours |
| Phospholipid or fatty acid |
Removal of unencapsulated solutes |
10 buffer changes 1 hour intervals for the first five exchanges 2 hour intervals for the next three 12 hour intervals for the last two |
| Phospholipid or fatty acid |
Staining of liposomes | Single change of buffer Staining complete in 1 hour |
| Phospholipids | Remote loading of weakly basic small molecules driven by an ammonium ion gradient |
Single change of buffer Overnight tumbling (12–24 hours) |
Reuse of the dialyzer
-
16
The dialyzer can be reused, especially to dialyze another portion of the same type of sample. Wash the dialyzer at least once with a volume of water sufficient to fill at least one-half the internal volume (i.e., ca. 200 µL for the dialyzer employed in most experiments here), then again at least twice with a buffer of identical composition to that used in the experiment,
-
17
It is necessary to confirm the integrity of the inner dialysis membrane before using the dialyzer again. To do this, fill one chamber of the dialyzer with buffer and monitor the second chamber for leakage. The pressure from having one chamber filled against an empty chamber is typically enough to observe leakage through any small tears in the dialysis membrane. If no significant leakage is observed after few minutes, the membrane is deemed intact.
Timing
Steps 1–9 1–2 h
Step 10 30 min-1 h to reach handling strength, overnight (12 h) for full cure
*CRITICAL STEP the timing of this step depends on the type of the epoxy glue used
Liposome preparation: typically overnight tumbling to pre-form liposomes
Steps 16–17 ca. 30 min.
Liposome dialysis: times vary depending on the application (see Table 1)
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting
| Step | Problem | |
|---|---|---|
| Problem could occur at any assembly or usage step |
Dialyzer is leaking buffer or sample outside of the plastic case |
The outside seal film has been punctured. This can happen at any assembly step, or after completing the dialyzer. Inspect the dialyzer, the tears in the PCR thermal seal are usually easy to spot by eye. If the seal is punctured, either the assembly process needs to be repeated, or, in some cases, the tear can be repaired by applying another layer of the protective film on the outside of the dialyzer. Repair by applying another layer of PCR film works best. |
| 6 | Dialyzer has not been properly aligned. If the leak is discovered after the epoxy has cured, the assembly needs to be repeated with new parts. Make sure to properly align both gaskets with the grooves on the inside of the plastic casing. |
|
| 7, 8, 9 | Dialyzer has not been properly sealed. If the bar clamps are too loose, the dialyzer will be glued with insufficient pressure on the gaskets to create the proper seal. If the leak is discovered after the epoxy has cured, the dialyzer assembly needs to be repeated with new parts. |
|
| 10 | The epoxy has not cured sufficiently. Insert the dialyzer back into the bar clamp and leave for longer period of time. Note that an overnight cure is typically employed, even with fast-setting (30 minutes working time epoxy) to ensure a tight seal. Consult the instructions for the specific epoxy glue used. |
|
| 1, 2, 4 | Dialyzer is leaking inside, between the compartments |
The inside membrane has been punctured. This could happen at any stage of the assembly; in our experience, we most commonly observe this to happen during step 1, 2 or 4. The dialyzer assembly needs to be repeated with new parts. |
| 6 | The dialyzer has not been properly aligned If the leak is discovered after the epoxy has cured, the dialyzer assembly needs to be repeated with new parts. Be sure to properly align both gaskets with the grooves on the inside of the plastic casing. |
|
| 10 | The epoxy has not cured enough. Reassemble the dialyzer-bar clamp assembly and allow to cure for a longer period of time. Consult the specific manual of the epoxy glue used. In our experience, overnight curing time is enough for most fast-curing epoxy glues. |
|
| 14–15 | Very low dialysis rate |
Wrong MWCO membrane used. If the dialysis rate is too low, new dialyzer needs to be assembled, using higher MWCO Slide-A-Lyzer as a starting material. |
| Insufficient dialysis buffer changes. Change dialysis buffer volume more often. | ||
| Insufficient time to equilibrate with the dialysis buffer Allow to equilibrate longer. | ||
| It is possible that perceived low dialysis rate, measured as removal of a compounds from inside or delivery into the liposomes, is not due to the wrong size of the dialyzer membrane, but due to the low permeability of the particular liposome membrane used. To diagnose this problem, perform a size exclusion purification-based leakage experiment, to establish whether the dialyzed solute crosses the vesicle membrane freely. |
||
| 13–15 | Sample or buffer leaks through the needle port |
Damaged luer stopper. Replace the luer stoppers on the ends of the needle. |
| 5 | Sample or buffer leaks through the hole around the needle |
The beveled needle used to puncture the gasket for blunt needle insertion was of too large a diameter. In our experience, in that case no amount of epoxy added afterwards will seal the gasket properly. The dialyzer needs to be assembled with new gaskets, with a beveled needle of +2 to 0 ga size difference relative to the blunt needle. |
| 9 | The gasket around the needle is leaking. This can sometimes be fixed by adding more epoxy around the needle ports. |
|
| 14–16 | Liposomes aggregate during dialysis |
The dialyzer was not tumbled vigorously enough during the dialysis. In our experience, especially in case of fatty acid vesicles, vigorous tumbling is critical to prevent aggregation of liposomes. Multilamellar vesicles are particularly susceptible to this. |
| 14–15 | Liposomes leak inside dialyzer (lose encapsulated contents) |
Liposomes aggregated and subsequently leaked – see troubleshooting above. |
| 1–9 | Trace detergent contamination of the inner surfaces of the dialyzer. Make sure the clear PCR film, gloves and all surfaces that the inner parts of the dialyzer touch during assembly process are free of detergents. |
|
| Liposomes precipitate inside the dialyzer |
Fatty acid vesicles will precipitate in presence of divalent cations. Make sure the dialysis buffer does not contain excess divalent cations. If vesicles precipitated, remove the sample and dialysis buffer completely, then wash both chambers of the dialyzer with several changes of buffer. The dialyzer can then be re-used. |
|
| Liposomes can precipitate in the presence of excess salt. Make sure the salt concentration of the dialysis buffer matches (or does not exceed) the salinity of the vesicle buffer. If liposomes precipitated in the dialyzer, remove all remaining sample and dialysis buffer and wash both chambers of the dialyzer with several changes of buffer. The dialyzer can then be re-used. |
Anticipated Results
Nonenzymatic RNA Primer Extension
Figure 2 shows results of an experiment where the dialyser was used to enable nonenzymatic primer extension of mixed-sequence RNA. The sequence of the template used in the experiments shown was 5′-AGC CGC CAG UCA GUC UAC GC-3′, and the primer sequence was 5′-Cy3-GCG UAG ACU GAC UGG −3′. Reactions contained 2 µM primer, 10 µM template, 50 mM MgCl2, 200mM citric acid (made pH 8 with NaOH or KOH), 50 mM 2MeImpG, 50 mM 2MeImpC, and 0.25 M Tris HCl buffer, pH 8, as well as 100 mM oleic acid.1,19 Vesicles were downsized by extrusion (Box 2).
Figure 2. Nonenzymatic copying of mixed (G+C) RNA templates.

Primer, template, and monomers were encapsulated inside oleic acid vesicles as described in the experimental design. Primer extension was conducted with either no dialysis (lane 1), five exchanges of activated monomer (lane 2), or ten exchanges of activated monomer (lane 3) over 48 hours. The methods for preparation of fatty acid vesicles and encapsulated RNA primer extension are described in our previous work.1
Box 2. Liposome Extrusion.
Downsizing vesicles and making them unilamellar is required for most experiments. As a general guideline, we typically extrude fatty acid vesicles to 100 nm for most applications. For imaging, large liposomes can be prepared according to the method described earlier 4.
<USEFUL TIP> When downsizing vesicles, an initial coarse downsizing step, such as freeze-thaw cycling, brief (5 minute) bath sonication, or a single passage through a 0.2 µM syringe filter is often helpful, particularly with high-concentration (> 50 mM lipid) samples.
Procedure
-
1
Assemble the liposome extruder according to the instructions from Avanti Polar Lipids. If using different extruder, follow the manufacturer’s guidelines.
-
2
Pre-wet the extruder with one syringe full of buffer. Perform a cycle of extrusion (i.e., pushing the buffer into the extruder from both sides). Discard the buffer.
<CRITICAL STEP> It is important to perform the pre-wetting step not only to condition the extrusion membrane, but also to ensure the assembled extruder does not leak and the extrusion membrane has not ruptured.
-
3
Inspect the liposome samples visually for traces of precipitate or aggregation. We do not recommend using samples that already show traces of aggregation.
-
4
Load the sample into one extruder syringe, extrude, using a syringe pump, and collect the sample in syringe on the opposite side. We typically extrude vesicles 7, 9, or 11 times.
<CRITICAL STEP> Perform an odd number of passages through the extruder. By doing this, the extruded vesicles are collected from the opposite end of the extruder in which they were loaded. This ensures the collected vesicles have all transited through the filter membrane, and the small dead volume in the syringe used to load the sample into the extruder is not collected (which may contain large multilamellar vesicles).
-
5
Tumble the vesicles for at least 10 minutes (preferably 1 hour) after extrusion before loading into the dialyzer.
After 48 hours, primer extension samples were removed from the dialyzer and purified by gel filtration (Sepharose 4B) using a mobile phase containing vesicles and buffer of identical composition to the reaction, lacking only RNA oligonucleotides and monomers. Vesicles were then lysed, and primer extension efficiency was analyzed by polyacrylamide gel electrophoresis (PAGE) (Figure 2). Only dialyzed samples exhibit full-length copying, due to the removal of inhibitors generated by monomer hydrolysis and replenishment of activated monomer.
Exchange of Unencapsulated Solutes
Here, liquid oleic acid sufficient to give 500 µL of a 100mM solution was rehydrated in 250 mM tris-HCl, pH 8.0, with 50 mM NaOH (i.e., ½ equivalent relative to fatty acid) containing 1 mM calcein, producing multilamellar vesicles. Alternately, these vesicles can be downsized by extrusion (Box 2). This solution was dialyzed against 500 µL of a dye-free solution containing vesicles and buffer. The dialyzer is tumbled with regular changes of buffer (at one-hour intervals for the first five exchanges, two-hour intervals for the next three, and 12-hour intervals for the last two). Representative repurification traces for one, five, and ten buffer changes (normalized to encapsulated fluorescence intensity) are shown in Figure 3. In this experiment, a dialyzer prepared from Slide-A-Lyzer G2 cassettes was used, as described in Box 1.
Remote loading of phospholipid vesicles
Figure 4 shows results of experiments where remote loading of phospholipid vesicles was performed. In these experiments, a thin film of the desired lipid or lipid mixture is rehydrated to the desired lipid concentration in a remote-loading compatible salt solution. Here, 100 mM POPC was rehydrated in 100 mM aqueous (NH4)2SO4 with overnight tumbling. The resulting multilamellar vesicles were then downsized and made unilamellar by seven passages through an extruder (Box 2). Extraliposomal ammonium salts were removed on a Sepharose 4B column with an isoosmotic mobile phase lacking ammonium ion (150 mM NaCl). 250 µL of the purified unilamellar vesicles were loaded into one chamber of the dialyzer, and 250 µL of the desired small molecule to be loaded (here, 100 µM acridine orange hydrochloride in 150 mM NaCl) were loaded into the other. After overnight tumbling of the Dialyzer, the dialyzer contents were removed and analyzed visually (Figure 4A), by absorbance spectroscopy (Figure 4B and 4C), and by repurification, followed by absorbance and fluorescence spectroscopy (Figure 4D), verifying remote loading had occurred.
Figure 4. Remote loading of liposomes with small molecules.
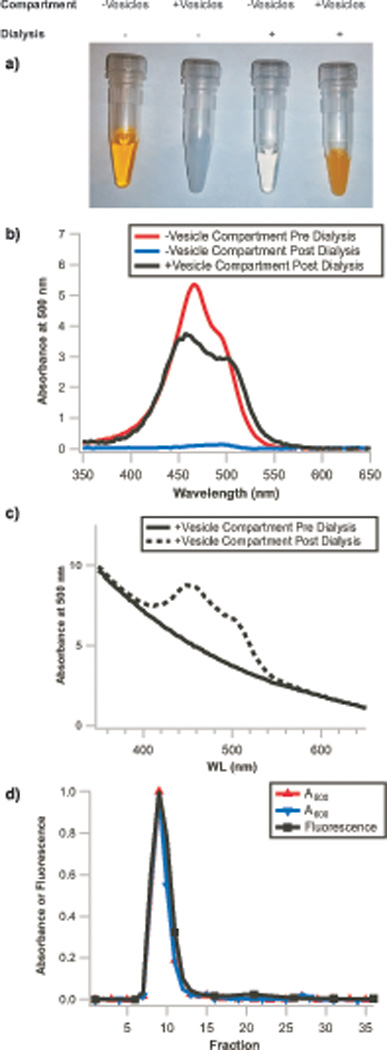
a) Images of vesicle-free and vesicle-containing solutions before and after dialysis. Selective accumulation of acridine orange occurs in the compartment containing vesicles as dye molecules are depleted from the vesicle-free compartment, demonstrating remote loading occurs. Similarly, b) The vesicle-free solution from the dialyzer, initially containing 100 µM acridine orange (red trace), is >95% depleted of dye (blue trace) after dialysis due to accumulation of dye within the vesicles (black trace, background corrected for scattering by subtraction of a vesicle blank). c) The non-background corrected post-dialysis vesicle sample (dashed line) exhibits the same A600 (only vesicle scattering contributes to absorbance at this wavelength) as the pre-dialysis vesicle sample (solid line), demonstrating that the vesicles are not diluted by this protocol. Absorbance signals are from NanoDrop measurements and are normalized to 10mm path length. d) Repurification of these vesicles results in superimposable normalized A500 (acridine orange absorbance), A600 (vesicle scattering), and acridine orange fluorescence traces (λex=500 nm, λem=530 nm), demonstrating that the dye has both crossed the dialysis membrane and accumulated within the vesicle lumen. Integration of fluorescence signal in the encapsulated dye fractions (fractions 6–16) and free dye fractions (fractions 17–27) indicates >95% encapsulation efficiency.
The methods for preparation of vesicles are described in our previous work1,21; the remote loading protocol (without the use of the dialyzer) was previously described elsewhere.14
Staining of Phospholipid Vesicles
In these experiments, vesicles were prepared by thin film hydration of a thin film of POPC. A 1mM chloroform solution of POPC was dried on the surface of a roughened PTFE strip for > 24 h, as described previously.20 The resulting film was hydrated with a sucrose solution (200 mM) to a final concentration of 20 mM lipid. This assembly was heated at 60 °C for 12 h and briefly vortexed. 300 µL of the resulting vesicles were added to one compartment of the dialyzer that had been pre-wetted in an isosmotic buffer (125 mM tris-HCl, pH 8). 300 µL of this buffer was added to the second compartment. To initiate dye insertion, 2.5 µL of a Vybrant DiI Cell-labeling solution was added to the vesicle-free compartment of the dialyzer. The dialyzer was then tumbled, and samples were removed at subsequent time points and imaged on a Nikon A1R MP Confocal microscope (Figure 5).
Figure 5. Staining of giant phospholipid vesicles.

Overlay of the confocal fluorescence and DIC bright field images of POPC vesicles incubated with Vybrant DiI Cell labeling solution for the specified time intervals. Scale bars are 15 µm.
The methods for preparation of liposomes were described in our previous work.1,21
Supplementary Material
Acknowledgments
This work was supported in part by NASA Exobiology grant NNX07AJ09G to J.W.S. and a grant from the Simons Collaboration on the Origin of Life to J.W.S. A.E.E. and N.P.K. were supported by appointments to the NASA Postdoctoral Program, administered by Oak Ridge Associated Universities through a contract with NASA. A.E.E. was supported by a Tosteson Fellowship from the Massachusetts General Hospital Executive Committee on Research. J.W.S. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions
K.A., A.E.E., N.P.K., and L.J. performed experiments. K.A., A.E.E., N.P.K., L.J., and J.W.S. wrote the manuscript. J.W.S. supervised the research. K.A. and A.E.E. contributed equally to this work.
Supplementary Figures S1-S14, showing assembly of the liposome dialyzer.
Supplementary Figures S15-S19, showing assembly of the G2 version of the liposome dialyzer.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Adamala K, Szostak JW. Nonenzymatic template-directed RNA synthesis inside model protocells. Science. 2013;342:1098–1100. doi: 10.1126/science.1241888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deck C, Jauker M, Richert C. Efficient enzyme-free copying of all four nucleobases templated by immobilized RNA. Nat. Chem. 2011;3:603–608. doi: 10.1038/nchem.1086. [DOI] [PubMed] [Google Scholar]
- 3.Zhu TF, Szostak JW. Coupled growth and division of model protocell membranes. Journal of the American Chemical Society. 2009;131:5705–5713. doi: 10.1021/ja900919c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu TF, Szostak JW. Preparation of large monodisperse vesicles. PloS one. 2009;4:e5009. doi: 10.1371/journal.pone.0005009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsumoto K, Nomura S-iM, Nakatani Y, Yoshikawa K. Giant Liposome as a Biochemical Reactor: Transcription of DNA and Transportation by Laser Tweezers. Langmuir : the ACS journal of surfaces and colloids. 2001;17:7225–7228. [Google Scholar]
- 6.Murtas G, Kuruma Y, Bianchini P, Diaspro A, Luisi PL. Protein synthesis in liposomes with a minimal set of enzymes. Biochemical and biophysical research communications. 2007;363:12–17. doi: 10.1016/j.bbrc.2007.07.201. [DOI] [PubMed] [Google Scholar]
- 7.Nishikawa T, Sunami T, Matsuura T, Yomo T. Directed Evolution of Proteins through In Vitro Protein Synthesis in Liposomes. Journal of nucleic acids. 2012;2012:923214. doi: 10.1155/2012/923214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lentini R, et al. Integrating artificial with natural cells to translate chemical messages that direct E. coli behaviour. Nature communications. 2014;5:4012. doi: 10.1038/ncomms5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujii S, et al. Liposome display for in vitro selection and evolution of membrane proteins. Nature protocols. 2014;9:1578–1591. doi: 10.1038/nprot.2014.107. [DOI] [PubMed] [Google Scholar]
- 10.Tan ML, Choong PF, Dass CR. Recent developments in liposomes, microparticles and nanoparticles for protein and peptide drug delivery. Peptides. 2010;31:184–193. doi: 10.1016/j.peptides.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Advanced drug delivery reviews. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 12.Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends in pharmacological sciences. 2009;30:592–599. doi: 10.1016/j.tips.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Gabizon A, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer research. 1994;54:987–992. [PubMed] [Google Scholar]
- 14.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochimica et biophysica acta. 1993;1151:201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 15.Barenholz Y. Doxil(R)--the first FDA-approved nano-drug: lessons learned. Journal of controlled release : official journal of the Controlled Release Society. 2012;160:117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 16.Hwang SH, Maitani Y, Qi XR, Takayama K, Nagai T. Remote loading of diclofenac, insulin and fluorescein isothiocyanate labeled insulin into liposomes by pH and acetate gradient methods. International journal of pharmaceutics. 1999;179:85–95. doi: 10.1016/s0378-5173(98)00392-5. [DOI] [PubMed] [Google Scholar]
- 17.Stano P, et al. Novel camptothecin analogue (gimatecan)-containing liposomes prepared by the ethanol injection method. Journal of liposome research. 2004;14:87–109. doi: 10.1081/lpr-120039794. [DOI] [PubMed] [Google Scholar]
- 18.Stano P, Luisi PL. Achievements and open questions in the self-reproduction of vesicles and synthetic minimal cells. Chem Commun (Camb) 2010;46:3639–3653. doi: 10.1039/b913997d. [DOI] [PubMed] [Google Scholar]
- 19.Joyce GF, Inoue T, Orgel LE. Non-enzymatic template-directed synthesis on RNA random copolymers. Poly(C, U) templates. J Mol Biol. 1984;176:279–306. doi: 10.1016/0022-2836(84)90425-x. [DOI] [PubMed] [Google Scholar]
- 20.Kamat NP, et al. A Generalized System for Photo-Responsive Membrane Rupture in Polymersomes. Advanced functional materials. 2010;20:2588–2596. doi: 10.1002/adfm.201000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adamala K, Szostak JW. Competition between model protocells driven by an encapsulated catalyst. Nat Chem. 2013;5:495–501. doi: 10.1038/nchem.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.