

Abstract
Objective
To identify circulating proteins that distinguish between active anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) and remission in a manner complementary to markers of systemic inflammation.
Methods
Twenty-eight serum proteins representing diverse aspects of the biology of AAV were measured before and 6 months after treatment in a large clinical trial of AAV. Subjects (n=186) enrolled in the Rituximab in ANCA-Associated Vasculitis (RAVE) trial were studied. Erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels were available for comparison. The primary outcome was the ability of markers to distinguish severe AAV (Birmingham Vasculitis Activity Score for Wegener’s granulomatosis (BVAS/WG)≥3 at screening) from remission (BVAS/WG=0 at month 6), using areas under receiver operating characteristic (ROC) curve (AUC).
Results
All subjects had severe active vasculitis (median BVAS/WG=8) at screening. In the 137 subjects in remission at month 6, 24 of the 28 markers showed significant declines. ROC analysis indicated that levels of CXCL13 (BCA-1), matrix metalloproteinase-3 (MMP-3) and tissue inhibitor of metalloproteinases-1 (TIMP-1) best discriminated active AAV from remission (AUC>0.8) and from healthy controls (AUC>0.9). Correlations among these markers and with ESR or CRP were low.
Conclusions
Many markers are elevated in severe active AAV and decline with treatment, but CXCL13, MMP-3 and TIMP-1 distinguish active AAV from remission better than the other markers studied, including ESR and CRP. These proteins are particularly promising candidates for future studies to address unmet needs in the assessment of patients with AAV.
INTRODUCTION
Granulomatosis with polyangiitis (Wegener’s, GPA) and microscopic polyangiitis (MPA) are severe inflammatory diseases that share the features of necrotising vasculitis of small vessels in multiple organ systems and characteristic autoantibodies (anti-neutrophil cytoplasmic antibodies (ANCAs), with specificity for proteinase-3 (PR3) or myeloperoxidase (MPO)), and thus can be described together as ANCA-associated vasculitis (AAV). In addition, GPA features necrotising granulomatous inflammation. Thus there are many plausible avenues for discovery of new biomarkers in these diseases, based on activation of T and B cells, acute and chronic inflammation, damage to the microvasculature, and tissue damage and repair considered either generally or in an organ-specific manner. New biomarkers are needed for AAV because the course of disease after initial treatment is highly variable, and existing markers such as ANCA titres and non-specific markers of inflammation such as C-reactive protein (CRP) and the erythrocyte sedimentation rate (ESR) have limited value.1–8
As the first step in a programme to identify markers that are useful in staging vasculitis activity, distinguishing vasculitis from other inflammatory diseases such as infections, and predicting response to treatment and risk of relapse, we assessed the ability of 28 proteins to distinguish severe AAV from remission and from healthy controls. We measured these proteins simultaneously in a large number of patients followed under standardised conditions in order to compare their ability to discriminate between disease states.
The proteins were chosen from a list of 108 proteins previously validated for assay on our custom microarray platform, in order to reflect a range of disease-related processes distinct from autoantibody specificity or systemic inflammation—broadly categorised as cytokines, chemokines, soluble receptors, markers of microvascular damage, and markers of tissue damage and repair. Some of these markers— such as ACE,9 CCL5,10 CCL17,11 CXCL8 (interleukin-8, IL-8),9,10,12,13 intercellular adhesion molecule-1 (ICAM-1),9,14–17 interferon-γ (IFNγ),9 interleukin-6 (IL-6),12,15,18 interleukin-18 (IL-18),19,20 IL-18 binding protein (IL-18BP),20 soluble interleukin-2 receptor (sIL-2R),21–24 soluble IL-6 receptor (sIL-6R),25 matrix metalloproteinase-3 (MMP-3),26,27 neutrophil gelatinase-associated lipocalin (NGAL, lipocalin-2),28 osteopontin,29 tissue inhibitor of metalloproteinases-1 (TIMP-1),26,27 soluble tumour necrosis factor receptors I or II (sTNF-RI/II),22,25,28,30 and vascular cell adhesion molecule-1 (VCAM-1)9,14,16,31—have shown promise previously in smaller studies of AAV. The remaining proteins—clusterin, CXCL10 (IP-10), CXCL13 (BCA-1), basic fibroblast growth factor (bFGF), granulocyte colony-stimulating factor (G-CSF), granulocyte–monocyte colony-stimulating factor (GM-CSF), interleukin-15 (IL-15), kidney injury molecule-1 (KIM-1, TIM-1), nerve growth factor β (NGFβ), plasminogen activator inhibitor-1 (PAI-1) and platelet-derived growth factor, A and B subunits (PDGF-AB)—have not to our knowledge been studied previously in AAV.
METHODS
Study design
Subjects for this study were enrolled in the Rituximab in ANCA-Associated Vasculitis (RAVE) trial. Of the 197 subjects in RAVE, 11 were excluded on the basis of withdrawal from the study by month 4 (n=7), absence of serum collection at screening (n=2), or frequent absence of serum collection at other times throughout the trial (n=2). Of the remaining 186 subjects, all samples at screening were analysed, but data from the month 6 visit were analysed only from the 162 subjects who had completed 6 months without blinded cross-over to the other treatment and had serum collected at month 6.
The primary outcome of this study was the difference in marker level between active AAV (at screening) and remission (at month 6) in the same patients (n=137), as determined by analysis of the absolute changes in marker levels and the areas under receiver operating characteristic (ROC) curves (see below under Statistical analysis). Secondary outcomes included differences in marker levels in groups defined by treatment at the time of sample collection, comparison of active AAV with healthy controls, and comparison of remission with recurrent active disease (n=25) at month 6.
Summary of clinical trial and clinical outcome measures
RAVE was a randomised, double-blinded, multicentre clinical trial that compared standard remission-induction therapy using oral cyclophosphamide (CYC) and glucocorticoids with experimental treatment using the B-cell-depleting agent, rituximab (RTX), and glucocorticoids, in 197 patients with new or recurrent, severe AAV (GPA or MPA).32 All patients tested positive for antibodies to either PR3 or MPO. Subjects randomised to receive CYC were switched to maintenance therapy with azathioprine (AZA) if they were clinically in remission between months 4 and 6. Subjects in the RTX arm were not placed on a maintenance agent, but the great majority still had no detectable B cells at month 6 and were therefore still considered to be ‘on treatment’ at this time point. Ongoing or recurrent severe disease during the first 6 months led to blinded cross-over to the other treatment arm for re-induction therapy or withdrawal from blinded treatment. Per protocol, glucocorticoid (prednisone) treatment was completely withdrawn before 6 months, although investigators had the option to restart prednisone at no more than 10 mg/day to control recurrent symptoms of mild disease. The rates of achievement of remission were equivalent in the two treatment arms.32
Activity of vasculitis was assessed using the Birmingham Vasculitis Activity Score for Wegener’s granulomatosis (BVAS/WG),33 in which each severe disease manifestation is given 3 points and each non-severe (mild) manifestation is given 1 point.33 Remission was defined as BVAS/WG=0. Every patient had a BVAS/WG of at least 3 at screening.
All subjects were enrolled using a protocol and informed consent documents approved by institutional review boards at all participating sites.
Healthy controls
Sixty-eight subjects self-identified as being in good health were recruited separately at Boston University School of Medicine under a protocol approved by the institutional review board. Specifically, these persons denied having any autoimmune or inflammatory disease or cardiovascular disease, or taking immune-suppressive, antiplatelet or anticoagulant drugs, based on responses to a questionnaire.
Processing of serum samples
Serum was processed and stored at each study site, then shipped to a central repository, then shipped to the study laboratory. All samples remained frozen at −80°C until the day the assays were performed.
Biomarker assays
Antibody arrays
Additional laboratory data
Westergren ESR and CRP were assayed at the participating sites at the time of each study visit, and data were retrieved from the clinical report forms.
Statistical analysis
Distributions, correlations and adjustment for multiple testing
Distributions of marker values were evaluated for normality using Shapiro–Wilk and Kolmogorov–Smirnov tests, as well as visual inspection of histograms. Correlation between pairs of markers was measured using Spearman correlation coefficients. Findings in all analyses were considered significant at p≤0.05 after adjustment for multiple comparisons (30 markers tested in each analysis) by calculating the false discovery rate as described by Benjamini and Hochberg.34 All analyses were performed using SAS V.9.1 or InStat.
Distinguishing active AAV from remission or controls
Since very few of the markers showed normally distributed values among subjects with active vasculitis (online supplementary figure S1), data are reported as medians and inter-quartile range (IQR) and were analysed using non-parametric statistics (Wilcoxon signed rank test for univariate analyses and Wilcoxon rank sum test for bivariate analyses).
To further assess the ability of markers to distinguish active AAV from remission, ROC curves were constructed for each marker using logistic regression, with marker level as the predictor variable and one of two dichotomous outcomes: active AAV versus remission, or active AAV versus healthy controls. The area under each ROC curve (AUC or C-statistic) was calculated. The standard errors (SEs) of the AUCs for selected markers were calculated and used to determine whether one AUC was significantly greater than another.35,36 An optimal cut-off point for each analysis was defined using the Youden Index37: the maximum of the sum of sensitivity (percentage of subjects with active AAV in whom marker level was elevated) and specificity (percentage of subjects in remission, or healthy controls, in whom marker level was not elevated). Positive likelihood ratios (LRs) (sensitivity/(1–specificity)) for active AAV versus remission or controls were calculated at these optimal cut-off points.
In analyses in which active AAV (at screening) was compared with AAV in remission, the dataset contained repeated measurements from the same subjects and thus had the potential to require adjustment. We reasoned that the balanced nature of the study (one active and one remission sample from each subject) obviated problems with repeated measures, but, to be certain, we performed a simulation in which each subject was randomly assigned to one of two groups, with one group used to provide the active AAV data and the other used to provide the remission data. This process was repeated 20 times, and the resulting ROC curves were averaged. Results were nearly identical (ie, within the error range expected from only 20 trials) with those obtained with a single analysis of the full dataset (data not shown).
RESULTS
Patient characteristics at screening and follow-up
The 186 subjects included 91 male and 95 female patients with median age 52 (IQR 44–66), all of whom had severe disease: median BVAS/WG 8, IQR 6–10, range 3–16. Of the 186 subjects evaluated at screening, 139 had been diagnosed with GPA and 46 with MPA; 124 were positive for anti-PR3 and 62 for anti-MPO; 90 had a new diagnosis of AAV, whereas 96 had established diagnoses and were experiencing relapses. At screening, 92 patients were receiving glucocorticoids, and 104 were receiving some immune-suppressive drug (glucocorticoids, other drugs, or both).
Samples from month 6 were also available for 162 patients who had not undergone re-induction therapy. Of these 162 subjects, 137 (85%) were in remission, and data from these 137 subjects were used for the primary analysis. The remaining 25 patients (15%) had active disease at month 6, and disease was usually mild (BVAS/WG 1 or 2 in 21/25 subjects). Of the 137 subjects in remission, 20 (15%) were receiving prednisone ≤10 mg/day, and the remaining subjects were off prednisone.
The 68 healthy controls included 28 men and 40 women, median age 41 (IQR 28–57).
Marker levels in active AAV, remission and healthy controls
Twenty-four of the 28 experimental markers, as well as ESR and CRP, were significantly different at times of active AAV compared with remission in the same subjects (table 1 and online supplementary figure S1). Levels of 20 proteins declined and four increased after successful treatment. Twenty-four of the 28 markers were also significantly different (22 higher and two lower) in active AAV compared with healthy controls (table 1).
Table 1.
Marker levels in severe active ANCA-associated vasculitis (screening) and remission among the patients who were in remission at month 6
| Marker | Screening (n=137) | Month 6 remission (n=137) | Difference (n=137) | p Value* | Healthy controls (n=68) | p Value** |
|---|---|---|---|---|---|---|
| Cytokines | ||||||
| G-CSF (pg/ml) | 20.4 (8.01;45.9) | 10.5 (5.63;23.7) | 5.78 (−0.74;26.0) | <0.0001* | 7.58 (4.89;12.7) | <0.0001** |
| GM-CSF (pg/ml) | 27.6 (2.28;269) | 1.17 (<0.98;4.99) | 16.8 (0.00;241) | <0.0001* | 1.39 (<0.98;7.27) | <0.0001** |
| IFNγ (pg/ml) | <0.49 (<0.49;2.01) | <0.49 (<0.49;<0.49) | 0.00 (0.00;0.95) | <0.0001* | <0.49 (<0.49;<0.49) | 0.011** |
| IL-6 (pg/ml) | 2.14 (<0.69;19.8) | <0.49 (<0.49;0.77) | 1.36 (0.00;18.3) | <0.0001* | <0.49 (<0.49;<0.49) | <0.0001** |
| IL-15 (pg/ml) | 21.6 (7.65;109) | 5.69 (2.60;13.5) | 14.6 (1.65;92.4) | <0.0001* | 2.87 (2.18;4.31) | <0.0001** |
| IL-18 (pg/ml) | 57.4 (37.2;101) | 51.9 (31.0;85.8) | 8.49 (−23.4;37.7) | 0.023* | 36.2 (20.1;60.8) | <0.0001** |
| Osteopontin (ng/ml) | 65.0 (38.8;101) | 54.4 (37.8;80.9) | 5.48 (−15.9;37.8) | 0.013* | 36.2 (29.5;42.4) | <0.0001** |
| Chemokines | ||||||
| BCA-1 (pg/ml) | 170 (74.2;489) | 32.0 (18.2;55.6) | 116 (27.6;416) | <0.0001* | 29.6 (19.8;44.8) | <0.0001** |
| IL-8 (pg/ml) | 19.5 (7.33;51.2) | 7.09 (3.59;15.3) | 7.60 (−2.26;40.4) | <0.0001* | 3.00 (1.30;5.19) | <0.0001** |
| IP-10 (pg/ml) | 11.1 (6.01;23.7) | 13.2 (7.68;25.0) | −0.87 (−7.95;7.95) | 0.73 | 3.28 (2.19;5.30) | <0.0001** |
| RANTES (ng/ml) | 60.3 (33.4;107) | 52.3 (30.8;90.0) | 0.00 (−16.6;31.3) | 0.43 | 58.3 (27.7;91.4) | 0.11 |
| TARC (pg/ml) | 520 (234;1535) | 655 (347;>2500) | −62.7 (−439;92.4) | 0.0046* | 177 (115;367) | <0.0001** |
| Soluble receptors | ||||||
| IL-18BP (pg/ml) | 116 (21.6;768) | 14.6 (<6.11;55.1) | 87.7 (1.46;537) | <0.0001* | 13.9 (6.11;46.5) | <0.0001** |
| sIL-2R (pg/ml) | <2.44 (<2.44;153) | <2.44 (<2.44;<2.44) | 0.00 (0.00;120) | <0.0001* | <2.44 (<2.44;<2.44) | 0.0005** |
| sIL-6R (ng/ml) | 27.4 (21.1;43.1) | 21.9 (15.4;33.0) | 4.27 (−3.12;12.5) | 0.0002* | 15.8 (11.7;20.0) | <0.0001** |
| sTNF-RII (pg/ml) | 2671 (1306;4855) | 2417 (1350;5808) | 122 (−2068;1606) | 0.86 | 499 (295;687) | <0.0001** |
| Tissue damage and repair | ||||||
| ACE (ng/ml) | 105 (73.8;144) | 178 (130;252) | −70.9 (−137; −24.6) | <0.0001* | 96.7 (81.4;115) | 0.076 |
| bFGF (pg/ml) | 3.05 (<0.98;34.4) | <0.98 (<0.98;9.77) | 0.00 (0.00;28.5) | <0.0001* | 2.39 (<0.98;11.0) | 0.42 |
| KIM-1 (pg/ml) | 242 (73.4;744) | 45.6 (17.2;127) | 134 (27.1;691) | <0.0001* | 19.8 (8.08;128) | <0.0001** |
| MMP-3 (ng/ml) | 96.6 (46.6;148) | 15.6 (11.8;29.1) | 79.1 (27.4;126) | <0.0001* | 10.2 (7.02;15.5) | <0.0001** |
| NGFβ (pg/ml) | 9.11 (3.15;37.0) | 2.48 (1.25;4.32) | 5.11 (0.67;31.4) | <0.0001* | 2.11 (0.77;4.53) | <0.0001** |
| PDGF-AB (pg/ml) | 4298 (1573;6585) | 3260 (879;5374) | 798 (−617;2980) | 0.0003* | 8877 (5812;11510) | <0.0001** |
| TIMP-1 (ng/ml) | 477 (302;862) | 166 (125;233) | 269 (67.1;638) | <0.0001* | 117 (65.4;163) | <0.0001** |
| Inflammation and vascular injury | ||||||
| Clusterin (μg/ml) | 77.1 (65.3;89.3) | 73.0 (59.4;85.9) | 5.52 (−8.12;18.2) | 0.0079* | 71.9 (61.2;83.0) | 0.0085** |
| CRP (mg/dl) | 1.2 (0.5;4.0) | 0.5 (0.3;1.2) | 0.7 (0.0;3.3) | <0.0001* | ND | ND |
| ESR (mm/h) | 37 (16;60) | 14 (7;22) | 19 (2;39) | <0.0001* | ND | ND |
| ICAM-1 (ng/ml) | 463 (307;933) | 537 (345;882) | −61.3 (−198;103) | 0.004* | 281 (226;337) | <0.0001** |
| NGAL (ng/ml) | 271 (176;399) | 172 (129;237) | 80.3 (6.96 ;214) | <0.0001* | 117 (92.4;150) | <0.0001** |
| PAI-1 (pg/ml) | 1491 (<977;5650) | 1202 (<977;4719) | 0.00 (−1449;2325) | 0.39 | 3307 (1083;7236) | 0.0008** |
| VCAM-1 (ng/ml) | 133 (95.4;174) | 148 (108;224) | −7.75 (−84.4;26.9) | 0.0035* | 139 (84.3;188) | 0.9 |
Values are median (IQR).
p<0.05 by Wilcoxon signed rank test, after adjustment for multiple comparisons.
p<0.05 by Wilcoxon rank sum test comparing screening to healthy controls, after adjustment for multiple comparisons.
BCA-1, CXCL13; bFGF, basic fibroblast growth factor; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–monocyte colony-stimulating factor; ICAM-1, intercellular adhesion molecule-1; IFN, interferon; IL, interleukin; IL-8, CXCL8; IL-18BP, interleukin 18 binding protein; IP-10, CXCL10; KIM-1, kidney injury molecule-1; MMP-3, matrix metalloproteinase-3; NGAL, neutrophil gelatinase-associated lipocalin; NGFβ, nerve growth factor β; PAI-1, plasminogen activator inhibitor-1; PDGF-AB, platelet-derived growth factor, A and B subunits; RANTES, CCL5; sIL-2R, soluble IL 2 receptor; sIL-6R, soluble IL 6 receptor; sTNF-RII, soluble TNF receptor II; TARC, CCL17; TIMP-1, tissue inhibitor of metalloproteinases-1; VCAM-1, vascular cell adhesion molecule-1.
The ability of each marker to distinguish active AAV from remission or from healthy controls was quantified by measuring the AUC and calculating LR. Eleven proteins had AUC>0.7 for both comparisons, and three (CXCL13/BCA-1, MMP-3 and TIMP-1) stood out as the best-performing markers, with AUC>0.8 and positive LR 4.3–6.8 comparing active AAV with remission (p<0.05 compared with p=0.76 for ESR), and AUC>0.9 and positive LR 10.7–12.4 comparing active AAV with controls (table 2 and figure 1).
Table 2.
Distinction of active ANCA-associated vasculitis (AAV) from remission or from healthy controls: receiver operating characteristic (ROC) analyses and likelihood ratios
| Active AAV versus remission
|
Active AAV versus controls
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Marker | AUC* | OCP† | Sensitivity‡ | Specificity‡ | LR+§ | AUC* | OCP† | Sensitivity‡ | Specificity‡ | LR+§ |
| BCA-1 | 0.86 | 70 | 77 (69–83) | 85 (78–90) | 5.13 | 0.91 | 70 | 75 (68–81) | 93 (84–98) | 10.7 |
| ESR | 0.76 | 23 | 68 (59–76) | 76 (68–83) | 2.83 | ND | ||||
| GM-CSF | 0.72 | 10 | 60 (51–68) | 82 (74–88) | 3.33 | 0.72 | 11 | 59 (52–66) | 81 (70–89) | 3.11 |
| IL-6 | 0.75 | 0.68 | 75 (67–82) | 71 (62–78) | 2.59 | 0.77 | 0.51 | 77 (70–83) | 78 (66–87) | 3.50 |
| IL-15 | 0.74 | 17 | 58 (50–67) | 82 (75–88) | 3.22 | 0.86 | 4.5 | 87 (81–92) | 78 (66–87) | 3.95 |
| IL-18BP | 0.74 | 93 | 54 (45–63) | 85 (78–91) | 3.60 | 0.73 | 22.5 | 73 (66–79) | 69 (57–80) | 2.35 |
| KIM-1 | 0.76 | 90 | 72 (64–80) | 69 (59–75) | 2.32 | 0.74 | 43 | 86 (80–91) | 63 (51–75) | 2.32 |
| MMP-3 | 0.89 | 38 | 82 (74–88) | 88 (81–93) | 6.83 | 0.94 | 29.5 | 87 (81–92) | 93 (84–98) | 12.4 |
| NGAL | 0.73 | 193 | 74 (66–81) | 64 (55–72) | 2.06 | 0.89 | 190 | 72 (65–78) | 91 (82–97) | 8.00 |
| NGFβ | 0.74 | 4.5 | 67 (58–74) | 77 (69–84) | 2.91 | 0.74 | 5.7 | 61 (53–68) | 82 (71–91) | 3.39 |
| TIMP-1 | 0.83 | 270 | 78 (70–85) | 82 (74–88) | 4.33 | 0.93 | 237 | 80 (73–85) | 93 (84–98) | 11.4 |
AUC, area under the ROC curve. An AUC of 1 indicates perfect discrimination between groups; an AUC of 0.5 indicates no discrimination.
OCP, optimal cut-off point, which is the maximum sum of sensitivity and specificity; for units, see table 1.
Sensitivity and specificity, at OCP, determined by values greater than OCP in active AAV and less than OCP in remission or controls unless otherwise noted. 95% confidence intervals (asymptotic method) are shown. ND, not done.
LR+, positive likelihood ratio at the OCP, which equals sensitivity/(1–specificity).
BCA-1, CXCL13; ESR, erythrocyte sedimentation rate; GM-CSF, granulocyte–monocyte colony-stimulating factor; IL, interleukin; IL-18BP, interleukin 18 binding protein; KIM-1, kidney injury molecule-1; MMP-3, matrix metalloproteinase-3; NGAL, neutrophil gelatinase-associated lipocalin; NGFβ, nerve growth factor β; TIMP-1, tissue inhibitor of metalloproteinases-1.
Figure 1.
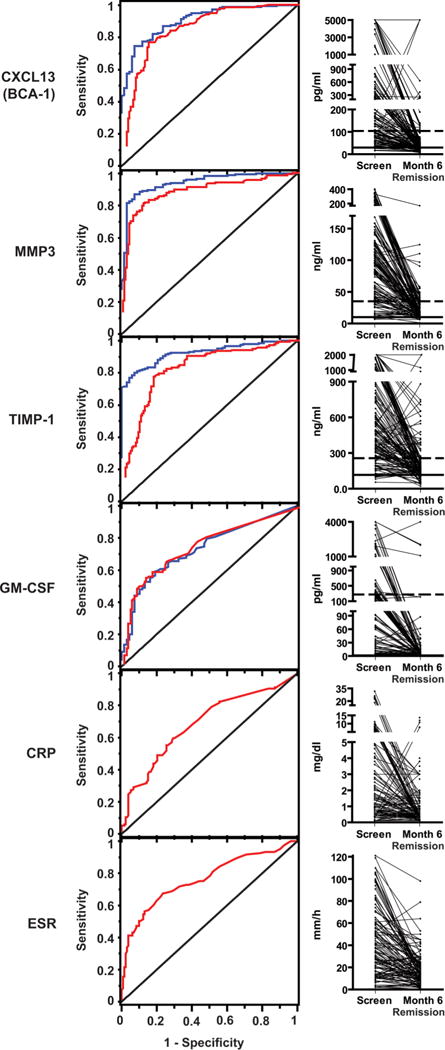
Levels of selected markers in severe active ANCA-associated vasculitis (AAV), AAV in remission, and in healthy controls. Left panels: receiver operating characteristic curves showing the ability of selected markers to distinguish severe active AAV (at screening) from remission at month 6 (blue curves), and severe active AAV from healthy controls (red curves). The diagonal line indicates what would be expected with no discrimination between groups. Erythrocyte sedimentation rate and C-reactive protein were not measured in healthy controls. Right panels: levels in individual patients. All subjects had severe active AAV at screening and were in remission at month 6. The medians and 95th centiles among healthy controls are shown with horizontal solid and dotted lines, respectively. CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; GM-CSF, granulocyte–monocyte colony-stimulating factor; MMP3, matrix metalloproteinase-3; TIMP-1, tissue inhibitor of metalloproteinases-1.
Four markers (CXCL13/BCA-1, G-CSF, IL-15 and TIMP-1) were significantly higher at month 6 in the 25 subjects with active, usually very mild, disease than in the 137 subjects in remission. Discrimination between mild disease and remission at month 6 was limited, with all AUC<0.7 (online supplementary table S1). However, power for detecting differences was low, and determining disease activity (versus consequences of damage) in such cases is challenging clinically.
Correlations among markers that distinguish active vasculitis from remission
Correlation coefficients between all pairs of markers are shown in figure 2. ESR and CRP correlated only weakly with all 28 experimental proteins. The three markers of greatest interest based on discrimination of active disease, remission and healthy controls also correlated weakly with each other: 0.20 for CXCL13/BCA-1 with MMP-3; 0.13 for CXCL13/BCA-1 with TIMP-1; and 0.17 for MMP-3 with TIMP-1.
Figure 2.
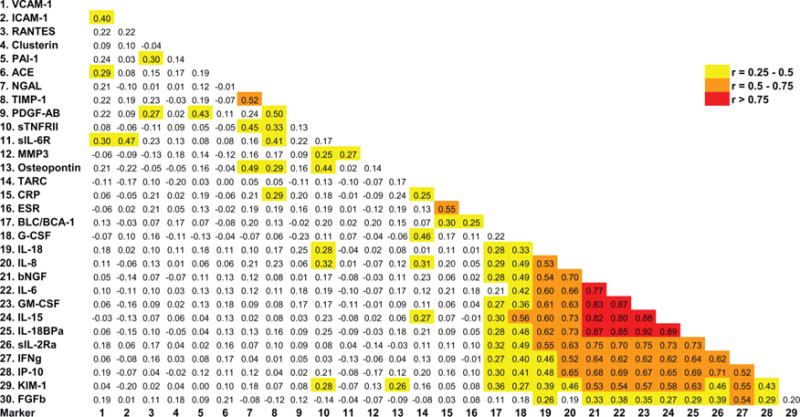
Correlations between marker levels. Spearman correlation coefficients for all pairs of experimental markers as well as erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are shown. Background colour indicates strength of association (red, >0.75; orange, 0.5–0.75; yellow, 0.25–0.5; white, <0.25). BCA-1, CXCL13; FGFb, basic fibroblast growth factor; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–monocyte colony-stimulating factor; ICAM-1, intercellular adhesion molecule-1; IFN, interferon; IL, interleukin; IL-8, CXCL8; IL-18BP, interleukin 18 binding protein; IP-10, CXCL10; KIM-1, kidney injury molecule-1; MMP-3, matrix metalloproteinase-3; NGAL, neutrophil gelatinase-associated lipocalin; NGFβ, nerve growth factor β; PAI-1, plasminogen activator inhibitor-1; PDGF-AB, platelet-derived growth factor, A and B subunits; RANTES, CCL5; sIL-2R, soluble IL 2 receptor; sIL-6R, soluble IL 6 receptor; sTNF-RII, soluble TNF receptor II; TARC, CCL17; TIMP-1, tissue inhibitor of metalloproteinases-1; VCAM-1, vascular cell adhesion molecule-1.
In contrast, a group of 12 proteins correlated highly with each other: GM-CSF, IFNγ, IL-15, IL-18, IL-18BP, sIL-2Rα, IL-6, CXCL8/IL-8, CXCL10/IP-10, KIM-1, bFGF and G-CSF. Most of these markers had shown moderate ability to distinguish active AAV from remission or from healthy controls (tables 1 and 2). We confirmed the validity of the results for these 12 markers using individual ELISAs on 20 patients and 12 controls selected to include low, mid-range and extremely high values (data not shown).
Effects of treatment on marker levels
Table 3 shows marker levels in groups at screening defined by newly diagnosed AAV versus relapse or by use of prednisone at the time of screening. Several markers were significantly higher in subjects with newly diagnosed AAV than in subjects with relapsing disease; all of these markers were also nominally higher in subjects not taking prednisone than in those already taking prednisone, although the differences often did not reach statistical significance.
Table 3.
Marker levels in patients with newly diagnosed or relapsing ANCA-associated vasculitis (AAV), and in patients treated or not treated with glucocorticoids at the time of screening
| Newly diagnosed or relapsing AAV
|
Glucocorticoid treatment at screening
|
|||||
|---|---|---|---|---|---|---|
| Marker | Relapsing (n=96) | New (n=90) | p Value | Yes (n=92) | No (n=94) | p Value |
| Cytokines | ||||||
| G-CSF | 23.4 (11.5;56.3) | 17.2 (6.60;41.2) | 0.032 | 21.5 (10.2;47.6) | 22.7 (8.79;49.2) | 0.81 |
| GM-CSF | 39.0 (2.78;243) | 18.1 (1.72;269) | 0.4 | 26.7 (1.92;236) | 23.3 (2.66;269) | 0.85 |
| IFNγ | <0.49 (<0.49;2.06) | <0.49 (<0.49;2.11) | 0.87 | <0.49 (<0.49;0.79) | <0.49 (<0.49;3.56) | 0.4 |
| IL-6 | 2.52 (0.53;19.9) | 1.93 (0.69;20.0) | 0.92 | 2.04 (<0.49;20.4) | 2.12 (0.63;19.8) | 0.87 |
| IL-15 | 30.2 (10.4;110) | 17.9 (7.23;99.3) | 0.31 | 26.8 (9.11;102) | 20.6 (8.28;116) | 0.4 |
| IL-18 | 58.0 (34.5;113) | 52.1 (30.7;101) | 0.31 | 56.5 (38.6;145) | 56.6 (30.7;99.4) | 0.22 |
| Osteopontin | 49.0 (34.0;75.5) | 76.5 (45.7;116) | 0.0004* | 50.7 (32.2;81.4) | 68.9 (46.3;107) | 0.0021 |
| Chemokines | ||||||
| BCA-1 | 125 (53.4;320) | 232 (84.5;771) | 0.011* | 153 (62.9;474) | 199 (74.5;603) | 0.65 |
| IL-8 | 19.1 (7.11;44.8) | 17.9 (5.71;53.3) | 0.94 | 17.4 (5.82;44.8) | 19.9 (7.18;51.2) | 0.48 |
| IP-10 | 12.4 (6.55;23.2) | 9.92 (5.21;23.4) | 0.54 | 13.3 (6.63;21.8) | 9.85 (5.09;23.7) | 0.35 |
| RANTES | 59.2 (34.0;113) | 64.2 (36.0;93.4) | 0.66 | 56.5 (35.4;89.5) | 74.7 (36.5;117) | 0.19 |
| TARC | 869 (238;1901) | 444 (227;999) | 0.042 | 504 (187;1582) | 595 (287;1841) | 0.23 |
| Soluble receptors | ||||||
| IL-18BP | 160 (22.7;828) | 80.3 (20.2;969) | 0.73 | 119 (14.5;834) | 134 (22.6;825) | 0.74 |
| sIL-2Rα | <2.44 (<2.44;206) | <2.44 (<2.44;250) | 0.98 | <2.44 (<2.44;206) | <2.44 (<2.44;250) | 0.91 |
| sIL-6R | 25.6 (19.5;33.0) | 29.0 (22.7;44.8) | 0.021 | 26.4 (20.2;37.0) | 26.5 (21.5;43.3) | 0.71 |
| sTNF-RII | 1912 (1167;3885) | 3110 (1646;5392) | 0.012* | 2039 (1123;4532) | 2886 (1481;4661) | 0.054 |
| Tissue damage and repair | ||||||
| ACE | 113 (76.2;166) | 106 (70.1;145) | 0.38 | 113 (73.2;156) | 105 (72.0;150) | 0.96 |
| bFGF | 3.86 (<0.98;50.6) | 2.86 (<0.98;37.4) | 0.54 | 3.20 (<0.98;55.1) | 2.86 (<0.98;37.4) | 0.38 |
| KIM-1 | 190 (49.5;709) | 274 (89.6;1267) | 0.08 | 157 (48.5;698) | 298 (113;1550) | 0.011 |
| MMP3 | 83.3 (48.0;156) | 101 (47.1;146) | 0.77 | 94.6 (54.8;160) | 88.7 (37.0;136) | 0.077 |
| NGFβ | 9.57 (4.08;43.9) | 6.68 (2.05;35.6) | 0.17 | 9.19 (3.69;40.6) | 8.85 (2.61;37.9) | 0.76 |
| PDGF-AB | 2769 (968;5002) | 4682 (2322;7392) | 0.0072* | 3541 (1261;5940) | 4136 (1440;7338) | 0.2 |
| TIMP-1 | 389 (236;673) | 525 (311;1149) | 0.01* | 473 (290;730) | 430 (253;1055) | 0.78 |
| Inflammation and vascular injury | ||||||
| Clusterin | 76.7 (65.3;87.0) | 80.9 (66.4;96.1) | 0.24 | 79.0 (66.5;94.8) | 75.6 (65.7;88.8) | 0.17 |
| CRP | 0.8 (0.39;2.45) | 1.96 (0.6;6.3) | 0.0026* | 0.9 (0.3;2.3) | 1.9 (0.6;4.9) | 0.003 |
| ESR | 29 (13;52) | 49 (26;80) | 0.0002* | 29 (13;53) | 48 (25;70) | 0.005 |
| ICAM-1 | 413 (292;775) | 555 (346;1005) | 0.02 | 450 (305;974) | 484 (317;819) | 0.96 |
| NGAL | 223 (153;342) | 291 (222;431) | 0.0057* | 224 (161;340) | 283 (204;428) | 0.045 |
| PAI-1 | 1089 (<977;4761) | 1002 (<977;6481) | 0.77 | 1887 (<977;5817) | <977 (<977;4509) | 0.28 |
| VCAM-1 | 120 (87.8;168) | 144 (106;177) | 0.026 | 127 (95.3;179) | 138 (95.5;77.9) | 0.52 |
Values are median (IQR). For units, see table 1.
p<0.05 comparing newly diagnosed with relapsing AAV, still significant after adjustment for multiple comparisons. No marker met this standard comparing subjects treated or not treated with glucocorticoids, although several unadjusted p values were<0.05.
BCA-1, CXCL13; bFGF, basic fibroblast growth factor; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–monocyte colony-stimulating factor; ICAM-1, intercellular adhesion molecule-1; IFN, interferon; IL, interleukin; IL-8, CXCL8; IL-18BP, interleukin 18 binding protein; IP-10, CXCL10; KIM-1, kidney injury molecule-1; MMP-3, matrix metalloproteinase-3; NGAL, neutrophil gelatinase-associated lipocalin; NGFβ, nerve growth factor β; PAI-1, plasminogen activator inhibitor-1; PDGF-AB, platelet-derived growth factor, A and B subunits; RANTES, CCL5; sIL-2R, soluble IL 2 receptor; sIL-6R, soluble IL 6 receptor; sTNF-RII, soluble TNF receptor II; TARC, CCL17; TIMP-1, tissue inhibitor of metalloproteinases-1; VCAM-1, vascular cell adhesion molecule-1.
The effects of different treatments in the first 6 months were examined using subjects who were in remission at month 6 and had remained in their original treatment groups. As shown in table 4, after adjustment for multiple comparisons, only one marker (IL-15) was significantly different in patients who received CYC/AZA versus RTX, and only one marker (CXCL13/BCA) was significantly different comparing patients on and off prednisone at month 6, although the sample size for the latter analysis was small.
Table 4.
Marker levels in patients in remission at month 6, stratified by treatment
| Treatment group
|
Glucocorticoids at month 6
|
|||||
|---|---|---|---|---|---|---|
| Marker | CYC/AZA (n=60) | RTX (n=72) | p Value | Yes (n=20) | No (n=117) | p Value |
| Cytokines | ||||||
| G-CSF | 9.56 (4.67;19.6) | 11.9 (6.81;29.9) | 0.19 | 21.9 (4.03;36.6) | 10.2 (6.14;20.7) | 0.24 |
| GM-CSF | 1.38 (<0.98;4.84) | 1.08 (<0.98;8.33) | 0.72 | 1.18 (<0.98;7.66) | 1.17 (<0.98;4.68) | 0.88 |
| IFNγ | <0.49 (<0.49;<0.49) | <0.49 (<0.49;<0.49) | 0.15 | <0.49 (<0.49;<0.49) | <0.49 (<0.49;<0.49) | 0.84 |
| IL-6 | <0.49 (<0.49;0.73) | <0.49 (<0.49;0.97) | 0.35 | 0.59 (<0.49;1.18) | <0.49 (<0.49;0.73) | 0.2 |
| IL-15 | 9.93 (3.80;16.4) | 4.11 (1.77;11.1) | 0.0014* | 4.93 (2.11;13.8) | 5.69 (2.60;13.5) | 0.69 |
| IL-18 | 49.7 (27.2;83.3) | 45.7 (31.1;88.0) | 0.49 | 45.2 (28.0;128) | 52.8 (33.1;84.8) | 0.99 |
| Osteopontin | 60.4 (37.7;98.4) | 50.5 (37.2;72.9) | 0.072 | 54.4 (38.6;76.4) | 54.5 (37.6;81.9) | 0.82 |
| Chemokines | ||||||
| BCA-1 | 38.4 (23.5;60.0) | 23.9 (16.8;49.2) | 0.0053 | 64.3 (35.1;191) | 26.9 (17.1;48.2) | <0.0001* |
| IL-8 | 6.40 (3.78;13.0) | 8.33 (3.49;22.1) | 0.41 | 6.65 (1.76;24.5) | 7.23 (3.74;14.8) | 0.72 |
| IP-10 | 12.6 (7.23;25.4) | 12.9 (7.68;23.5) | 0.98 | 16.6 (6.89;30.0) | 13.1 (7.68;24.2) | 0.71 |
| RANTES | 67.0 (35.5;100) | 45.3 (29.2;84.2) | 0.08 | 62.6 (42.8;200) | 51.1 (30.2;89.5) | 0.16 |
| TARC | 555 (247;2500) | 700 (393;2500) | 0.36 | 653 (491;2161) | 655 (322;2500) | 0.58 |
| Soluble receptors | ||||||
| IL-18BP | 13.0 (6.11;42.4) | 18.4 (<6.11;65.2) | 0.5 | 12.0 (<6.11;49.3) | 16.4 (<6.11;55.7) | 0.51 |
| sIL-2Rα | <2.44 (<2.44;<2.44) | <2.44 (<2.44;<2.44) | 0.19 | <2.44 (<2.44;<2.44) | <2.44 (<2.44;<2.44) | 0.49 |
| sIL-6R | 21.2 (16.0;33.9) | 22.5 (15.2;32.8) | 0.89 | 23.0 (15.2;35.7) | 21.7 (15.5;32.4) | 0.81 |
| sTNF-RII | 2039 (1137;4009) | 2902 (1470;6569) | 0.087 | 1528 (978;3285) | 2524 (1445;5985) | 0.079 |
| Tissue damage and repair | ||||||
| ACE | 190 (127;253) | 170 (132;260) | 0.83 | 165 (142;253) | 180 (129;251) | 0.91 |
| bFGF | <0.98 (<0.98;11.6) | <0.98 (<0.98;4.53) | 0.88 | 3.15 (<0.98;21.0) | <0.98 (<0.98;3.75) | 0.16 |
| KIM-1 | 40.6 (17.3;111) | 53.9 (15.9;134) | 0.67 | 28.2 (12.8;119) | 49.3 (18.5;127) | 0.3 |
| MMP-3 | 14.9 (11.0;26.0) | 16.6 (12.9;30.5) | 0.39 | 29.3 (16.1;43.0) | 14.9 (11.6;24.0) | 0.0039 |
| NGFβ | 2.30 (0.94;4.21) | 2.71 (1.35;4.29) | 0.24 | 2.83 (0.75;7.17) | 2.35 (1.26;4.03) | 0.58 |
| PDGF-AB | 2437 (701;4814) | 3807 (1463;5864) | 0.018 | 3049 (748;3967) | 3295 (886;5610) | 0.34 |
| TIMP-1 | 169 (125;233) | 162 (129;263) | 0.9 | 212 (144;355) | 160 (125;224) | 0.078 |
| Inflammation and vascular injury | ||||||
| Clusterin | 78.3 (64.6;91.3) | 68.2 (57.5;83.8) | 0.024 | 69.4 (57.2;82.6) | 73.0 (60.0;86.8) | 0.26 |
| CRP | 0.4 (0.3;1.2) | 0.5 (0.3;1.0) | 0.78 | 0.5 (0.3;0.8) | 0.5 (0.3;1.3) | 0.77 |
| ESR | 17 (9;26) | 11 (7;21) | 0.034 | 19 (7;28) | 12 (7;22) | 0.15 |
| ICAM-1 | 461 (318;905) | 607 (356;895) | 0.25 | 786 (458;1088) | 490 (329;791) | 0.14 |
| NGAL | 169 (99.9;215) | 179 (137;287) | 0.028 | 157 (119;222) | 174 (130;243) | 0.61 |
| PAI-1 | 1200 (<977;3733) | 1645 (<977;5217) | 0.49 | 2545 (<977;5358) | 1202 (<977;3969) | 0.38 |
| VCAM-1 | 155 (116;230) | 138 (102;216) | 0.36 | 208 (126;455) | 145 (107;209) | 0.027 |
Values are median (IQR). For units, see table 1.
p<0.05 comparing groups, still significant after adjustment for multiple comparisons.
BCA-1, CXCL13; bFGF, basic fibroblast growth factor; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–monocyte colony-stimulating factor; ICAM-1, intercellular adhesion molecule-1; IFN, interferon; IL, interleukin; IL-8, CXCL8; IL-18BP, interleukin 18 binding protein; IP-10, CXCL10; KIM-1, kidney injury molecule-1; MMP-3, matrix metalloproteinase-3; NGAL, neutrophil gelatinase-associated lipocalin; NGFβ, nerve growth factor β; PAI-1, plasminogen activator inhibitor-1; PDGF-AB, platelet-derived growth factor, A and B subunits; RANTES, CCL5; sIL-2R, soluble IL 2 receptor; sIL-6R, soluble IL 6 receptor; sTNF-RII, soluble TNF receptor II; TARC, CCL17; TIMP-1, tissue inhibitor of metalloproteinases-1; VCAM-1, vascular cell adhesion molecule-1.
Overall, these results suggest that recent treatment did not have a dramatic effect on marker levels at screening, and that the changes seen in marker levels from screening to month 6 were not driven by effects of particular treatments.
DISCUSSION
This study of markers of active AAV identifies several novel markers (CXCL13/BCA-1, NGFβ, bFGF, GM-CSF, G-CSF, IL-15, IL-18BP, sIL-6R, KIM-1 and NGAL), confirms several (IFNγ, sIL-2R, IL-6, CXCL8/IL-8, MMP-3, TIMP-1), and suggests that some previously studied markers may not be valuable (ICAM-1, VCAM-1). Multiple markers appeared to distinguish between active AAV and remission better than did ESR or CRP, but, more importantly, provided complementary information to ESR and CRP based on low correlation coefficients.38
Markers that distinguish between clinically obvious disease states are the most promising ones for further studying the more important and challenging goals of biomarker development. These goals have not been enumerated for vasculitis; we propose that they include staging of current internal organ involvement, distinguishing active vasculitis from infections or other conditions, distinguishing mildly active vasculitis from remission, predicting permanent and/or progressive damage to organ systems, predicting response to treatment or risk of relapse, and assessing efficacy of treatment early in its course. These goals are thematically similar to what have been proposed by expert panels in other rheumatic diseases.39,40
The strengths of this study include the large study population, standardised prospective collection of detailed clinical information, inclusion of enough healthy controls to provide good estimates of normal ranges, and testing and comparison of many novel markers drawn from diverse aspects of autoimmune inflammatory disease processes. As above, the main limitation of the study is that it represents only a first step in identifying clinically useful biomarkers. Clinical use will require studies with longer follow-up, many samples associated with mildly active disease, and comparison with other disease states, as well as assessment of marker variability within and between individuals,40,41 technical validation of assay methodologies,42 and evaluation of pre-analytical factors such as posture, time of day, effects of food or exercise, and stability during processing and storage. The three markers of greatest interest in distinguishing active disease from remission in this study clearly have limited capacity to distinguish active AAV from other inflammatory conditions and infections.
MMP-3 has previously been identified by our research group using the same cohort,43 and the current study provides a useful technical validation of that finding, since different assay methodologies were used. Multiple cell types and tissues could contribute to circulating MMP-3 levels, and elevated levels have been detected in inflammatory diseases presumed to be pathophysiologically distinct, such as GPA,26,27 Takayasu’s arteritis44 and multiple forms of inflammatory arthritis.45
TIMP-1 (in plasma rather than serum) has been previously reported to be elevated in active GPA compared with remission or healthy controls.26 This marker is not specific for AAV, since it circulates at high levels in sepsis.46 The tissue source in AAV of this widespread antiproteinase is not clear but is probably not circulating leucocytes, since transcription of TIMP-1 was found to be much higher in peripheral blood mononuclear cells from patients in remission than in patients with active GPA.26
Elevation of CXCL13/BCA-1 in AAV is a particularly interesting finding in considering pathophysiology, although there are clearly limits to what can be inferred about mechanisms of disease from circulating levels of a locally produced, locally acting protein. This chemokine is important in the development of secondary and ectopic lymphoid tissues and is critical in B cell homing via the receptor, CXCR5.47 The importance of B cells in AAV was shown in the RAVE Trial.32 Circulating CXCL13/BCA-1 has not been measured in AAV, but levels have been reported to be elevated in patients with cryoglobulinaema,48 systemic lupus erythematosus,49–51 multiple sclerosis,52,53 HIV infection54 and sepsis.49 Although elevation of CXCL13 in all of these diseases, including AAV, implies a connection with abnormalities in B cell function, CXCR5 is also expressed on subpopulations of CD4 Tcells and dendritic cells47; similarly, although follicular dendritic cells are the main producers of CXCL13 in lymph nodes,47 it is not clear which cells are the source of elevated circulating levels in disease states.
Finally, the poor correlation between the best-performing markers and ESR and CRP indicates that the use of multiple markers will improve on the performance of any individual marker, which will be important in approaching the outcomes of greatest need for biomarker development in AAV: distinguishing active vasculitis from infections or other inflammatory diseases, distinguishing mild disease from remission, staging of current organ-system involvement, and prediction of response to treatment, future relapse, and other long-term outcomes. Large longitudinal studies such as the RAVE trial will provide the essential framework for development and validation of models in pursuit of these clinically relevant goals.
Supplementary Material
Acknowledgments
Funding This work was sponsored by the Vasculitis Clinical Research Consortium which has received support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (U54AR057319, RC1 AR058303 and P60 AR047785), the National Center for Research Resources (U54 RR019497), the National Institute of Neurological Disorders and Stroke (NS064808), and the Office of Rare Diseases Research. The RAVE Trial was performed with the support of the Immune Tolerance Network (NIH Contract N01 AI15416), an international clinical research consortium supported by the National Institute of Allergy and Infectious Diseases and the Juvenile Diabetes Research Foundation (see online appendix for the list of all members of the RAVE-ITN Research Group). Genentech and Biogen Idec provided the study medications and partial funding. At the Mayo Clinic and Foundation, the trial was supported by a Clinical and Translational Science Award from the National Center for Research Resources (NCRR) (RR024150-01), at Johns Hopkins University, by grants from the NCRR (RR025005) and career development awards (K24 AR049185 to JHS, and K23 AR052820 to PS), and at Boston University, by a Clinical and Translational Science Award (RR 025771), grants from the National Institutes of Health (M01 RR00533) and a career development award (K24 AR02224 to Dr Peter A Merkel). Dr Paul A Monach was supported by an Arthritis Investigator Award from the Arthritis Foundation.
Footnotes
Handling editor Tore K Kvien
Contributors All authors meet the requirements for authorship as per BMJ/ARD policies.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Kerr GS, Fleisher TA, Hallahan CW, et al. Limited prognostic value of changes in antineutrophil cytoplasmic antibody titer in patients with Wegener’s granulomatosis. Arthritis Rheum. 1993;36:365–71. doi: 10.1002/art.1780360312. [DOI] [PubMed] [Google Scholar]
- 2.Kyndt X, Reumaux D, Bridoux F, et al. Serial measurements of antineutrophil cytoplasmic autoantibodies in patients with systemic vasculitis. Am J Med. 1999;106:527–33. doi: 10.1016/s0002-9343(99)00064-9. [DOI] [PubMed] [Google Scholar]
- 3.Boomsma MM, Stegeman CA, van der Leij MJ, et al. Prediction of relapses in Wegener’s granulomatosis by measurement of antineutrophil cytoplasmic antibody levels: a prospective study. Arthritis Rheum. 2000;43:2025–33. doi: 10.1002/1529-0131(200009)43:9<2025::AID-ANR13>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 4.Girard T, Mahr A, Noel LH, et al. Are antineutrophil cytoplasmic antibodies a marker predictive of relapse in Wegener’s granulomatosis? A prospective study. Rheumatology (Oxford) 2001;40:147–51. doi: 10.1093/rheumatology/40.2.147. [DOI] [PubMed] [Google Scholar]
- 5.Finkielman JD, Merkel PA, Schroeder D, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med. 2007;147:611–19. doi: 10.7326/0003-4819-147-9-200711060-00005. [DOI] [PubMed] [Google Scholar]
- 6.Kalsch AI, Csernok E, Munch D, et al. Use of highly sensitive C-reactive protein for followup of Wegener’s granulomatosis. J Rheumatol. 2010;37:2319–25. doi: 10.3899/jrheum.100302. [DOI] [PubMed] [Google Scholar]
- 7.Hind CR, Winearls CG, Lockwood CM, et al. Objective monitoring of activity in Wegener’s granulomatosis by measurement of serum C-reactive protein concentration. Clin Nephrol. 1984;21:341–5. [PubMed] [Google Scholar]
- 8.Sproson EL, Jones NS, Al-Deiri M, et al. Lessons learnt in the management of Wegener’s Granulomatosis: long-term follow-up of 60 patients. Rhinology. 2007;45:63–7. [PubMed] [Google Scholar]
- 9.Olle EW, Deogracias MP, Messamore JE, et al. Screening of serum samples for Wegener’s granulomatosis patients using antibody microarrays. Proteomics Clinical Applications. 2007;1:1212–20. doi: 10.1002/prca.200600906. [DOI] [PubMed] [Google Scholar]
- 10.Torheim EA, Yndestad A, Bjerkeli V, et al. Increased expression of chemokines in patients with Wegener’s granulomatosis—modulating effects of methylprednisolone in vitro. Clin Exp Immunol. 2005;140:376–83. doi: 10.1111/j.1365-2249.2005.02770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dallos T, Heiland GR, Strehl J, et al. CCL17/thymus and activation-related chemokine in Churg-Strauss syndrome. Arthritis Rheum. 2010;62:3496–503. doi: 10.1002/art.27678. [DOI] [PubMed] [Google Scholar]
- 12.Ohlsson S, Wieslander J, Segelmark M. Circulating cytokine profile in anti-neutrophilic cytoplasmatic autoantibody-associated vasculitis: prediction of outcome? Mediators Inflamm. 2004;13:275–83. doi: 10.1080/09629350400003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomasson G, Lavalley M, Tanriverdi K, et al. Relationship between markers of platelet activation and inflammation with disease activity in Wegener’s granulomatosis. J Rheumatol. 2011;38:1048–54. doi: 10.3899/jrheum.100735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mrowka C, Sieberth HG. Circulating adhesion molecules ICAM-1, VCAM-1 and E-selectin in systemic vasculitis: marked differences between Wegener’s granulomatosis and systemic lupus erythematosus. Clin Investig. 1994;72:762–8. doi: 10.1007/BF00180543. [DOI] [PubMed] [Google Scholar]
- 15.Ohta N, Fukase S, Aoyagi M. Serum levels of soluble adhesion molecules ICAM-1, VCAM-1 and E-selectin in patients with Wegener’s granulomatosis. Auris Nasus Larynx. 2001;28:311–14. doi: 10.1016/s0385-8146(01)00097-9. [DOI] [PubMed] [Google Scholar]
- 16.Ara J, Mirapeix E, Arrizabalaga P, et al. Circulating soluble adhesion molecules in ANCA-associated vasculitis. Nephrol Dial Transplant. 2001;16:276–85. doi: 10.1093/ndt/16.2.276. [DOI] [PubMed] [Google Scholar]
- 17.Stegeman CA, Tervaert JW, Huitema MG, et al. Serum levels of soluble adhesion molecules intercellular adhesion molecule 1, vascular cell adhesion molecule 1, and E-selectin in patients with Wegener’s granulomatosis. Relationship to disease activity and relevance during followup. Arthritis Rheum. 1994;37:1228–35. doi: 10.1002/art.1780370818. [DOI] [PubMed] [Google Scholar]
- 18.Muller Kobold AC, Kallenberg CG, Tervaert JW. Monocyte activation in patients with Wegener’s granulomatosis. Ann Rheum Dis. 1999;58:237–45. doi: 10.1136/ard.58.4.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hultgren O, Andersson B, Hahn-Zoric M, et al. Serum concentration of interleukin-18 is up-regulated in patients with ANCA-associated vasculitis. Autoimmunity. 2007;40:529–31. doi: 10.1080/08916930701622783. [DOI] [PubMed] [Google Scholar]
- 20.Novick D, Elbirt D, Dinarello CA, et al. Interleukin-18 binding protein in the sera of patients with Wegener’s granulomatosis. J Clin Immunol. 2009;29:38–45. doi: 10.1007/s10875-008-9217-0. [DOI] [PubMed] [Google Scholar]
- 21.Schmitt WH, Heesen C, Csernok E, et al. Elevated serum levels of soluble interleukin-2 receptor in patients with Wegener’s granulomatosis. Association with disease activity. Arthritis Rheum. 1992;35:1088–96. doi: 10.1002/art.1780350914. [DOI] [PubMed] [Google Scholar]
- 22.Nassonov EL, Samsonov MY, Tilz GP, et al. Serum concentrations of neopterin, soluble interleukin 2 receptor, and soluble tumor necrosis factor receptor in Wegener’s granulomatosis. J Rheumatol. 1997;24:666–70. [PubMed] [Google Scholar]
- 23.D’Cruz D, Direskeneli H, Khamashta M, et al. Lymphocyte activation markers and von Willebrand factor antigen in Wegener’s granulomatosis: potential markers for disease activity. J Rheumatol. 1999;26:103–9. [PubMed] [Google Scholar]
- 24.Sanders JS, Huitma MG, Kallenberg CG, et al. Plasma levels of soluble interleukin 2 receptor, soluble CD30, interleukin 10 and B cell activator of the tumour necrosis factor family during follow-up in vasculitis associated with proteinase 3-antineutrophil cytoplasmic antibodies: associations with disease activity and relapse. Ann Rheum Dis. 2006;65:1484–9. doi: 10.1136/ard.2005.046219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tesar V, Jirsa M, Jr, Zima T, et al. Soluble cytokine receptors in renal vasculitis and lupus nephritis. Med Sci Monit. 2002;8:BR24–9. [PubMed] [Google Scholar]
- 26.Bjerkeli V, Halvorsen B, Damas JK, et al. Expression of matrix metalloproteinases in patients with Wegener’s granulomatosis. Ann Rheum Dis. 2004;63:1659–63. doi: 10.1136/ard.2003.017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Leeuw K, Sanders JS, Stegeman C, et al. Accelerated atherosclerosis in patients with Wegener’s granulomatosis. Ann Rheum Dis. 2005;64:753–9. doi: 10.1136/ard.2004.029033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohlsson S, Wieslander J, Segelmark M. Increased circulating levels of proteinase 3 in patients with anti-neutrophilic cytoplasmic autoantibodies-associated systemic vasculitis in remission. Clin Exp Immunol. 2003;131:528–35. doi: 10.1046/j.1365-2249.2003.02083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenzen J, Lovric S, Kramer R, et al. Osteopontin in antineutrophil cytoplasmic autoantibody-associated vasculitis: relation to disease activity, organ manifestation and immunosuppressive therapy. Ann Rheum Dis. 2010;69:1169–71. doi: 10.1136/ard.2009.113621. [DOI] [PubMed] [Google Scholar]
- 30.Jonasdottir O, Petersen J, Bendtzen K. Tumour necrosis factor-alpha (TNF), lymphotoxin and TNF receptor levels in serum from patients with Wegener’s granulomatosis. APMIS. 2001;109:781–6. doi: 10.1034/j.1600-0463.2001.d01-146.x. [DOI] [PubMed] [Google Scholar]
- 31.Schneeweis C, Rafalowicz M, Feist E, et al. Increased levels of BLyS and sVCAM-1 in anti-neutrophil cytoplasmatic antibody (ANCA)-associated vasculitides (AAV) Clin Exp Rheumatol. 2010;28(1 Suppl 57):62–6. [PubMed] [Google Scholar]
- 32.Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–32. doi: 10.1056/NEJMoa0909905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham vasculitis activity score. International Network for the Study of the Systemic Vasculitides (INSSYS) Arthritis Rheum. 2001;44:912–20. doi: 10.1002/1529-0131(200104)44:4<912::AID-ANR148>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 34.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc B. 1995;57:289–300. [Google Scholar]
- 35.Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- 36.Hanley JA, McNeil BJ. A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology. 1983;148:839–43. doi: 10.1148/radiology.148.3.6878708. [DOI] [PubMed] [Google Scholar]
- 37.Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3:32–5. doi: 10.1002/1097-0142(1950)3:1<32::aid-cncr2820030106>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 38.Pinsky PF, Zhu CS. Building multi-marker algorithms for disease prediction-the role of correlations among markers. Biomark Insights. 2011;6:83–93. doi: 10.4137/BMI.S7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bauer DC, Hunter DJ, Abramson SB, et al. Classification of osteoarthritis biomarkers: a proposed approach. Osteoarthritis Cartilage. 2006;14:723–7. doi: 10.1016/j.joca.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Maksymowych WP, Landewe R, Tak PP, et al. Reappraisal of OMERACT 8 draft validation criteria for a soluble biomarker reflecting structural damage endpoints in rheumatoid arthritis, psoriatic arthritis, and spondyloarthritis: the OMERACT 9 v2 criteria. J Rheumatol. 2009;36:1785–91. doi: 10.3899/jrheum.090346. [DOI] [PubMed] [Google Scholar]
- 41.Fraser CG. Test result variation and the quality of evidence-based clinical guidelines. Clin Chim Acta. 2004;346:19–24. doi: 10.1016/j.cccn.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 42.Cummings J, Ward TH, Greystoke A, et al. Biomarker method validation in anticancer drug development. Br J Pharmacol. 2008;153:646–56. doi: 10.1038/sj.bjp.0707441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monach PA, Tomasson G, Specks U, et al. Circulating markers of vascular injury and angiogenesis in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2011;63:3988–97. doi: 10.1002/art.30615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuyama A, Sakai N, Ishigami M, et al. Matrix metalloproteinases as novel disease markers in Takayasu arteritis. Circulation. 2003;108:1469–73. doi: 10.1161/01.CIR.0000090689.69973.B1. [DOI] [PubMed] [Google Scholar]
- 45.Ribbens C, Martin y Porras M, Franchimont N, et al. Increased matrix metalloproteinase-3 serum levels in rheumatic diseases: relationship with synovitis and steroid treatment. Ann Rheum Dis. 2002;61:161–6. doi: 10.1136/ard.61.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lauhio A, Hastbacka J, Pettila V, et al. Serum MMP-8, -9 and TIMP-1 in sepsis: high serum levels of MMP-8 and TIMP-1 are associated with fatal outcome in a multicentre, prospective cohort study. Hypothetical impact of tetracyclines. Pharmacol Res. 2011;64:590–4. doi: 10.1016/j.phrs.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 47.Muller G, Hopken UE, Lipp M. The impact of CCR7 and CXCR5 on lymphoid organ development and systemic immunity. Immunol Rev. 2003;195:117–35. doi: 10.1034/j.1600-065x.2003.00073.x. [DOI] [PubMed] [Google Scholar]
- 48.Sansonno D, Tucci FA, Troiani L, et al. Increased serum levels of the chemokine CXCL13 and up-regulation of its gene expression are distinctive features of HCV-related cryoglobulinemia and correlate with active cutaneous vasculitis. Blood. 2008;112:1620–7. doi: 10.1182/blood-2008-02-137455. [DOI] [PubMed] [Google Scholar]
- 49.Schiffer L, Kumpers P, Davalos-Misslitz AM, et al. B-cell-attracting chemokine CXCL13 as a marker of disease activity and renal involvement in systemic lupus erythematosus (SLE) Nephrol Dial Transplant. 2009;24:3708–12. doi: 10.1093/ndt/gfp343. [DOI] [PubMed] [Google Scholar]
- 50.Wong CK, Wong PT, Tam LS, et al. Elevated production of B cell chemokine CXCL13 is correlated with systemic lupus erythematosus disease activity. J Clin Immunol. 2010;30:45–52. doi: 10.1007/s10875-009-9325-5. [DOI] [PubMed] [Google Scholar]
- 51.Ezzat M, El-Gammasy T, Shaheen K, et al. Elevated production of serum B-cell-attracting chemokine-1 (BCA-1/CXCL13) is correlated with childhood-onset lupus disease activity, severity, and renal involvement. Lupus. 2011;20:845–54. doi: 10.1177/0961203311398513. [DOI] [PubMed] [Google Scholar]
- 52.Michalowska-Wender G, Losy J, Biernacka-Lukanty J, et al. Impact of methylprednisolone treatment on the expression of macrophage inflammatory protein 3alpha and B lymphocyte chemoattractant in serum of multiple sclerosis patients. Pharmacol Rep. 2008;60:549–54. [PubMed] [Google Scholar]
- 53.Festa ED, Hankiewicz K, Kim S, et al. Serum levels of CXCL13 are elevated in active multiple sclerosis. Mult Scler. 2009;15:1271–9. doi: 10.1177/1352458509107017. [DOI] [PubMed] [Google Scholar]
- 54.Widney DP, Breen EC, Boscardin WJ, et al. Serum levels of the homeostatic B cell chemokine, CXCL13, are elevated during HIV infection. J Interferon Cytokine Res. 2005;25:702–6. doi: 10.1089/jir.2005.25.702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.