

Abstract
Adult T‐cell leukemia/lymphoma (ATL) is an aggressive T‐cell malignancy caused by human T‐cell lymphotropic virus 1. Treatment options for acute ATL patients include chemotherapy, stem cell transplantation, and recently the anti‐chemokine (C‐C motif) receptor 4 antibody, although most patients still have a poor prognosis and there is a clear need for additional options. HBI‐8000 is a novel oral histone deacetylase inhibitor with proven efficacy for treatment of T‐cell lymphomas that recently received approval in China. In the present study, we evaluated the effects of HBI‐8000 on ATL‐derived cell lines and primary cells obtained from Japanese ATL patients. In most cases HBI‐8000 induced apoptosis in both primary ATL cells and cell lines. In addition, findings obtained with DNA microarray suggested Bim activation and, interestingly, the contribution of the NLR family, pyrin domain containing 3 (NLRP3) inflammasome pathway in HBI‐8000‐induced ATL cell death. Further investigations using siRNAs confirmed that Bim contributes to HBI‐8000‐induced apoptosis. Our results provide a rationale for a clinical investigation of the efficacy of HBI‐8000 in patients with ATL. Although the role of NLRP3 inflammasome activation in ATL cell death remains to be verified, HBI‐8000 may be part of a novel therapeutic strategy for cancer based on the NLRP3 pathway.
Keywords: Adult T‐cell leukemia lymphoma, apoptosis, Bim, histone deacetylase inhibitors, NLRP3
Adult T‐cell leukemia/lymphoma (ATL) is an aggressive malignancy of mature T‐lymphocytes that result from infection with human T‐cell lymphotropic virus 1 (HTLV‐1). The disease can be divided into four stages: indolent, chronic, lymphoma, and acute, with no accepted curative therapy presently available.1, 2, 3 Outside of clinical trials, the predominant treatment option for acute, lymphoma, and unfavorable chronic type is sequential chemotherapy, whereas additional options for aggressive disease include azidothymidine and interferon‐α and allogenic hematopoietic stem cell transplantation.2, 3 Development of additional therapies has been slow, with the anti‐chemokine (C‐C motif) receptor 4 (CCR4) antibody mogamulizumab the only novel agent introduced in recent years. Although approximately 90% of ATL patients with aggressive disease express CCR4,4, 5 not all patients eligible to receive mogamulizumab respond to treatment; those who do often show relapse or become refractory soon thereafter.6 Thus, the prognosis for most affected patients remains poor and there is a clear unmet need for novel therapeutic options.
Histone deacetylases (HDACs) are epigenetic modulators that regulate key cellular processes, including proliferation and survival, by modulating gene expression and protein activity. Histone deacetylase inhibitors (HDACi) induce re‐expression of genes repressed during oncogenesis, including those involved in growth arrest, DNA damage repair, and apoptosis. As a consequence, HDACi have been shown to be potent inducers of growth inhibition and apoptosis in a variety of transformed cells, including those related to lymphoid malignancies.7, 8 At present, four different HDACi have been approved for treatment of T‐cell lymphoma, including romidepsin and belinostat for peripheral T‐cell lymphoma (PTCL), vorinostat and romidepsin for cutaneous T‐cell lymphoma, and more recently the orally administered HBI‐8000 (chidamide) for PTCL was approved in China. Even though there are similarities between PTCL, cutaneous T‐cell lymphoma, and ATL, and while previously published data indicate that HDACi cause apoptosis of ATL cells and HTLV‐1‐transformed cell lines,9, 10 none of the approved HDACi have been studied in regard to clinical efficacy for ATL.
HBI‐8000 is an orally bioavailable new chemical entity from the benzamide class of HDACi rationally designed to specifically block the catalytic pocket of class I HDACs.11, 12 HBI‐8000 has shown direct antitumor activity and is also known to enhance immune cell‐mediated tumor cytotoxicity by both natural killer cells and tumor‐specific CTLs.12, 13, 14, 15, 16, 17 In addition, its efficacy as monotherapy for T‐cell lymphomas has been proven.18
In the present study, we evaluated the effects of HBI‐8000 in ATL‐derived cell lines and fresh primary ATL cells obtained from ATL patients. Our results indicate that HBI‐8000 inhibits cell viability in both primary ATL cells and ATL‐derived cell lines. The results provide evidence to support further investigation of the potential utility of HBI‐8000 for ATL patients.
Materials and Methods
Sample preparation and cell cultures
Eleven samples were taken from 10 patients diagnosed with aggressive type ATL, based on Shimoyama's diagnostic criteria (Table S1).1 The diagnosis of ATL was confirmed by monoclonal integration of HTLV‐1 proviral DNA (Southern blotting; data not shown) in all cases.19 Peripheral blood mononuclear cells were obtained from ATL patients and healthy adult volunteers by density gradient centrifugation using a Lympho‐prep (Axis Shield, Oslo, Norway). CD4+ lymphocytes from healthy donors were purified from PBMCs by the magnetic bead method (CD4+ T Cell Isolation Kit; Miltenyi Biotec, Auburn, CA, USA). The PBMCs from ATL patients contained more than 85% leukemic cells and were maintained in medium supplemented with 10 ng/mL interleukin‐2 (IL2). Among 10 patients with ATL, ATL cases 5, 7, 9, and 10 had cells of sufficient quantity to carry out Western blotting or protein expression array. Following approval by the Ethics Committee of Nagasaki University Hospital (No. 13102878; Nagasaki, Japan), all patient samples were obtained after receiving informed consent.
Cell lines and cell cultures
The ATL‐derived lines KOB, KK1, ST1, SO4, LMY1, and LMY2 were established from patients with aggressive type ATL and kept in our laboratory.20, 21 LMWT5 is a newly established cell line from PBMCs from a patient with acute type ATL. The primary ATL origin of the cells was confirmed by concordance of the results of Southern blotting (data not shown). Human T‐cell leukemia cell line Jurkat and human monocytic leukemia cell line THP‐1 were obtained from ATCC (Manassas, VA, USA). ST1, Jurkat, and THP‐1 cells were maintained in medium (RPMI‐1640 supplemented with 10% FBS) in an IL2‐independent manner. Other cell lines were maintained in medium supplemented with 10 ng/mL IL2. In a previous study, we clarified that KOB, LMY1, LMY2, and ST1 cells have wild‐type p53.22
Chemicals and assays of cell viability, apoptosis, and cell cycle
HBI‐8000 (HUYA Bioscience International, San Diego, CA, USA) was used in this study. The MTS cell viability assay was carried out using a Cell Titer 96 Aqueous Cell Proliferation Assay kit (Promega, Madison, WI, USA). Cells were prepared for cell cycle evaluation using a Cell Cycle Test kit (BD Biosciences, San Jose, CA, USA). Apoptosis induction was determined by annexin V/propidium iodide (PI) (Bender Medsystems, Vienna, Austria), mitochondrial outer membrane permeability using a Mitochondrial Membrane Potential (MMP) Assay Kit (Cayman Chemical, Ann Arbor, MI, USA), and caspase‐3 activation with a fluorometric assay (MBL, Woburn, MA, USA). A cell cycle test, annexin V/PI assay, MMP Assay, and caspase‐3 assay were carried out by Flow Cytometry (FCM) using a FACS Diva software (BD Biosciences).
DNA microarray analysis
Total RNA was extracted using a PureLink RNA micro kit (Life Technologies, Carlsbad, CA, USA). RNA integrity was assessed using a 2100 Bioanalyzer (Agilent, Palo Alto, CA, USA). Double‐stranded cDNA and biotinylated cRNA were synthesized using a BioArray RNA labeling kit (Enzo, Farmingdale, NY, USA). Labeled RNA was then fragmented and hybridized to SurePrint G3 Human Gene Expression 8x60K version 2.0 (Agilent). The arrays were scanned using The SureScan Microarray Scanner G2600D (Agilent) and analyzed with Agilent Feature Extraction Software Version 11.5.1.1(Agilent).
Mutation analysis of p53
Reverse transcription–PCR was carried out to amplify the sequence targeting the ORF of p53 (GenBank Accession number NM_000546) as described previously.22
Quantitative real‐time RT‐PCR
The mRNA levels for Bim, nucleotide‐binding domain and leucine‐rich repeat containing gene (NLR) family, pyrin domain containing 3 (NLRP3), p21WAF1/CIP1, and porphobilinogen deaminase were measured using a LightCycler480 PCR System (Roche Diagnostics, Basel, Switzerland) as described previously.23 Primers and probes used in this study were summarized in Table S2. To normalize the results for variability in concentration and integrity of RNA and cDNA, the PBGD gene was used as an internal control for each sample.
PathScan stress and apoptosis signaling antibody array analysis
The PathScan Stress and Apoptosis Signaling Antibody Array Kit (Cell Signaling Technology, Beverly, MA, USA) allows for simultaneous detection of 19 different signaling molecules. Whole‐cell lysates were prepared and incubated on the slides overnight, followed by a biotinylated detection antibody cocktail. Streptavidin‐conjugated HRP and LumiGLO Reagent, containing in the kit, were then used to visualize by chemiluminescence. Slide images were captured with an image analyzer LAS3000 (Fujifilm, Tokyo, Japan) and spot signals were quantified (Multigauge version 3.0; Fujifilm).
Western blotting and antibodies
Western blot analysis was carried out as previously described.24 Analyses were undertaken using antibodies to p53 (DO‐1), acetylated histone‐H3 and ‐H4 (Merck, Darmstadt, Germany), caspase‐1, cleaved caspase‐1, Bim, BAX, Bcl‐2, Bcl‐xL, p21, IKBα, Iκ B kinase (IKK)α, IKKβ, IKKγ, and NLRP3 (Cell Signaling Technology), and β‐actin (Sigma, St. Louis, MO, USA).
Transfection and siRNA experiments
Transfection was performed with a Neon Transfection System MPK5000S (Invitrogen, Carlsbad, CA, USA). The transfection programs for KOB and LMY1 (No. 24) were run in such a manner that cell viability and transfection efficiency would be compatible (data not shown). Twelve hours after transfection, cells were treated with or without HBI‐8000 and processed for experiments. Two different siRNAs were prepared against each target as follows: Bim, Silencer Select Validated siRNA s195011 (#1) and Silencer Select siRNA s195012 (#2); NLRP3, Silencer Select siRNA s41555 (#1), s41556 (#2). As a control siRNA, Silencer negative control #1 (Applied Biosystems, Foster City, CA) was used.
Statistical analysis
Student's t‐test with Welch's correction was used to calculate statistical significance. **P‐values < 0.01 and *P‐values < 0.05 were regarded as significant.
Results
HBI‐8000 inhibits cell viability in ATL‐derived cell lines
To investigate the activity of HBI‐8000 in ATL cells, two model systems were used, both based on cells derived from ATL patients. First, the activity of HBI‐8000 was evaluated using seven ATL cell lines and the non‐ATL T‐cell line Jurkat, which is known as a good responder against HDACi.8 All cell lines tested showed decreased cell viability after 72 h of HBI‐8000 treatment (Fig. 1a). ST1 was relatively unresponsive and showed only modest inhibition. The IC50 values of ATL cells showing a response ranged from 0.7 to 1.7 μM (mean, 1.16 μM). These results indicated that sensitivities of ATL cell lines to HBI‐8000 are not inferior compared to Jurkat cells. HTLV‐1 infection, IL2‐dependence, or p53 status did not have an effect on the response to HBI‐8000.
Figure 1.

Apoptosis in adult T‐cell leukemia/lymphoma (ATL)‐derived cell lines induced by HBI‐8000. (a) ATL‐derived cell lines and Jurkat cells (2–5 × 105/mL) were treated with either the vehicle or indicated concentrations of HBI‐8000 for 72 h, then cell viability was evaluated using MTS assay. All experiments were carried out in triplicate and the results are expressed as the mean ± SD. (b–d) ATL‐derived cell lines and Jurkat cells (2–5 × 105/mL) were treated with either the vehicle or 4 μM HBI‐8000 for 48 h, and collected. (b) Annexin‐V/propidium iodide staining was carried out and the percentage of annexin‐V‐positive cells was evaluated. Experiments were carried out in triplicate and the results are expressed as the mean ± SD. (c) Cell cycle analysis. The percentage of cells in the sub‐G1, G1, S, and G2/M phase was evaluated. Increased numbers of cells in the G1 phase and those with G1 arrest were observed among KK1 and SO4 cells treated with HBI‐8000. (d) Evaluation of loss of mitochondrial membrane potential. Cells were incubated with JC‐1 dye and analyzed using FCM to determine the percentage with low JC‐1 red fluorescence. Except for KK1 cells, most of the cell lines showed decreased mitochondrial membrane potential (ΔψM). (e) Activated (cleaved) caspase‐3 was evaluated using an Apopcyto caspase‐3 colorimetric assay kit. Values for fold induction of signaling (HBI‐8000‐treated cells/non‐treated cells) are indicated.
Apoptosis induction by HBI‐8000 in ATL cells associated with annexin‐V binding, loss of mitochondrial membrane potential, and caspase‐3 activation
Apoptosis induction was measured using four commonly used assays: annexin‐V binding (Fig. 1b), accumulation of cells in the sub‐G1 cell cycle analysis peak (Fig. 1c), the loss of MMP (Fig. 1d), and activation of caspase‐3 (Fig. 1e). The data obtained from the four different methods used to detect cell death were consistent, and revealed that a significant level of apoptosis was induced in the KOB, LMY1, LMY2, LMWT5, and Jurkat lines, whereas the KK1, SO4, and ST1 lines showed only modest apoptosis. Consistent with previous findings,25 HBI‐8000 also induced G1 cell cycle arrest in KK1, SO4, and ST1 cells (Fig. 1c).
HBI‐8000 induces apoptosis in primary ATL cells
The effect of HBI‐8000 was also assessed using cells freshly obtained from ATL patients (Table S1). The cells were treated with various concentrations of HBI‐8000 for 72 h, after which cell viability was evaluated using an MTS assay. For the first screening step, we treated seven ATL samples with 1.25–100 μM HBI‐8000 (Table S1). For more detailed observations, we used 20 μM as the maximum dose and carried out MTS assays with six ATL samples, which included four new ATL samples, as well as samples 1 and 7 from the screening experiment (Fig. 2a, Table S1). Primary ATL cells, including two relapsed cases and two unfavorable chronic type cases, responded to HBI‐8000 in a dose‐dependent manner (mean IC50, 4.35 μM). HBI‐8000 showed cytotoxicity toward normal CD4 lymphocytes to some extent, whereas the ATL samples showed inhibited cell viability at significantly lower concentrations (Fig. S1). Importantly, ATL1 and ATL10, derived from patients who relapsed after both combination chemotherapy and mogamulizumab therapy, showed sensitivity to HBI‐8000 that was similar to other primary ATL cells. Thus, our results suggest that HBI‐8000 may have activity in patients who have developed resistance to standard therapeutic methods. Finally, we investigated the cell death pattern using an annexin‐V/PI assay with ATL samples (Fig. 2b). The ATL samples tested consisted of more than 50% annexin‐V‐positive cells, indicating that HBI‐8000 induced apoptotic cell death in primary ATL cells. In contrast, that of normal CD4 lymphocytes showed <30%.
Figure 2.
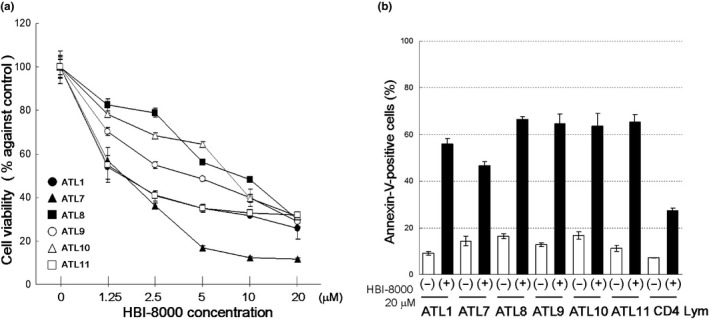
HBI‐8000 inhibits cell viability and induces apoptosis in primary adult T‐cell leukemia/lymphoma (ATL) cells. Cells (1 × 106/mL) were treated with either vehicle or indicated concentrations of HBI‐8000. (a) After 72 h, cell viability was evaluated using MTS assay. All experiments were carried out in triplicate and the results are expressed as the mean ± SD. (b) After 48 h, annexin‐V/propidium iodide staining was carried out and the percentage of annexin‐V‐positive cells was evaluated. Experiments were carried out in triplicate and the results are expressed as the mean ± SD.
HBI‐8000 caused acetylation of histones and activated tumor suppressor genes in ATL cells
It is well established that hyperacetylation of histones following HDAC inhibition can increase the gene expression of various tumor suppressors, such as p53 and p21. We confirmed that HBI‐8000 was effective for increasing the acetylation of histones H3/H4 in ATL cell lines and primary ATL cells (Fig. 3a,b), and then sought to determine the effect of HBI‐8000 on protein expressions of p53 and p21 by Western blot analysis. Previous results suggested that p53 status does not determine the response to HBI‐8000; however, cells with wild‐type p53 (KOB, LMY1) showed increased expression of p53 and p21. KK1 cells with mutated p53 showed accumulated p53 proteins and were not changed by HBI‐8000 treatment, but they nevertheless showed increased p21 expression (Fig. 3c). In primary ATL cells, ATL9 and 10 have wild‐type p53 (Table S1) and showed activation of p53. Reminiscent of the cell line KK1, ATL7 may possess mutant p53 in that it too showed accumulated p53 proteins. Surprisingly, none of the primary ATL samples showed increased p21 expression following HBI‐8000 treatment (Fig. 3d), yet they do not require p21 activation for HBI‐8000‐induced apoptosis.25 Only three cases (ATL1, 5, and 8), but we further investigated the expression of p21 mRNA after HBI‐8000 treatment. These cases have mutant p53, however, ATL1 showed upregulation of p21 mRNA. Thus, some ATL cells may have the ability to upregulate p21 mRNA or protein by HBI‐8000 but did not correlate with sensitivities to HBI‐8000. Our results also suggested that ATL cells with wild‐type p53 may be able to reactivate p53 with the addition of HBI‐8000. Interestingly, normal CD4 lymphocytes are harmed to some extent (Fig. S1) and the histones were acetylated (Fig. 3b) but their tumor suppressor genes were not activated by HBI‐8000 (Fig. 3d).
Figure 3.
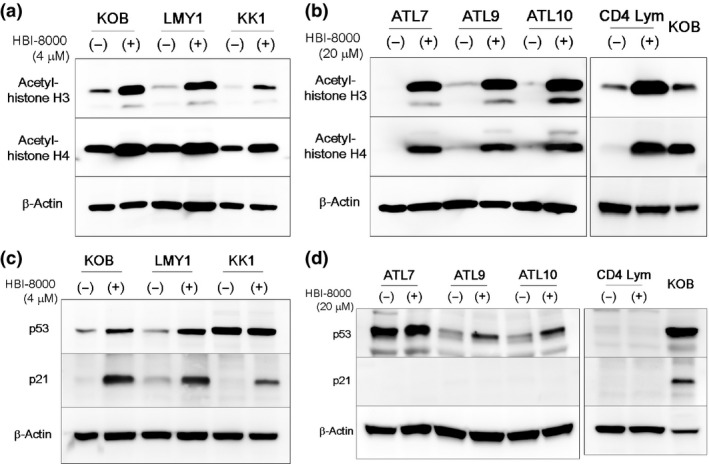
Analysis of acetylated histones, tumor suppressor genes, and antibody array in adult T‐cell leukemia/lymphoma (ATL) cell lines and primary ATL cells. (a–d) ATL cell lines (2–5 × 105/mL), primary ATL cells, and normal CD4 lymphocytes (1 × 106/mL) were treated with either the vehicle or the indicated concentrations of HBI‐8000 for 48 h, then collected. Whole‐cell lysates were prepared and Western blotting was carried out. In the membrane for CD4 lymphocytes, KOB cells treated with HBI‐8000 were used as positive control.
DNA microarray and antibody array analysis
To address the vital mechanisms of HBI‐8000‐induced ATL cell death, we used DNA microarray and protein expression array analyses. For the DNA microarray analysis, we used KOB and LMY1, which showed apoptotic changes with HBI‐8000 treatment, and compared gene expression profiles between treated and untreated cells. Genes with known functions related to apoptosis, cell cycle, cell death, or cell growth are arranged in order of change in the expression (log2 ratio) in either direction (Table 1). Among commonly observed changes, the upregulation of Chemokine (C‐C Motif) Ligands (CCL2 and CCL4) Interleukin (IL) 9, IL13, and as well as molecules related to cell‐growth signaling such as the JAK/signal transducer and activator of transcription pathway (IL9, IL13, and signal transducer and activator of transcription 4), and inflammasome‐related molecules (IL1β, tumor necrosis factor, γ‐interferon, and NLRP3) were notable. In contrast, apoptosis facilitating molecules such as FAS (CD95) and breast cancer 1, early onset were downregulated. These results seem to be in contradiction to the observed HBI‐8000‐induced ATL cell death. A possible explanation is that small numbers of apoptosis facilitators are activated by HBI‐8000 that can overcome anti‐apoptotic or cell‐growth signals. Considering that KOB and LMY1 undergo apoptosis by way of the intrinsic pathway (Fig. 1d), upregulation of BCL2L11 (Bim) is consistent with the observation.
Table 1.
Microarray analysis of KOB and LMY1 adult T‐cell leukemia/lymphoma cells
| Symbol | Gene | KOB | LMY1 |
|---|---|---|---|
| Upregulated genes | |||
| IL1B | Homo sapiens interleukin 1, β (IL1B), mRNA [NM_000576] | 11.0 | 2.5 |
| CCL4 | Homo sapiens chemokine (C‐C motif) ligand 4 (CCL4), transcript variant 1, mRNA [NM_002984] | 7.8 | 3.7 |
| IL9 | Homo sapiens interleukin 9 (IL9) | 7.5 | 6.0 |
| SERPINB2 | Homo sapiens serpin peptidase inhibitor, clade B (ovalbumin), member 2 (SERPINB2) | 7.3 | 4.6 |
| CCL2 | Homo sapiens chemokine (C‐C motif) ligand 2 (CCL2) | 6.3 | 5.3 |
| BCL2L11 | Homo sapiens BCL2‐like 11 (apoptosis facilitator) (BCL2L11) | 5.3 | 3.2 |
| CTLA4 | Homo sapiens cytotoxic T‐lymphocyte‐associated protein 4 (CTLA4) | 5.2 | 1.6 |
| IL13 | Homo sapiens interleukin 13 (IL13) | 5.1 | 4.6 |
| IL3RA | Homo sapiens interleukin 3 receptor, α (low affinity) (IL3RA) | 4.8 | 2.5 |
| GZMA | Homo sapiens granzyme A (granzyme 1, cytotoxic T‐lymphocyte‐associated serine esterase 3) (GZMA) | 4.7 | 3.4 |
| ID1 | Homo sapiens inhibitor of DNA binding 1, dominant negative helix‐loop‐helix protein (ID1) | 4.6 | 1.7 |
| NLRP3 | Homo sapiens NLR family, pyrin domain containing 3 (NLRP3) | 4.4 | 2.4 |
| IER3 | Homo sapiens immediate early response 3 (IER3) | 4.4 | 7.2 |
| BCL2A1 | Homo sapiens BCL2‐related protein A1 (BCL2A1) | 4.0 | 1.8 |
| IFNG | Homo sapiens interferon, gamma (IFNG) | 3.9 | 2.8 |
| TNF | Homo sapiens tumor necrosis factor (TNF) | 3.8 | 1.6 |
| SNAI1 | Homo sapiens snail homolog 1 (Drosophila) (SNAI1) | 3.6 | 2.5 |
| STAT4 | Homo sapiens signal transducer and activator of transcription 4 (STAT4) | 3.5 | 2.2 |
| DHRS2 | Homo sapiens dehydrogenase/reductase (SDR family) member 2 (DHRS2) | 3.0 | 2.6 |
| LGMN | Homo sapiens legumain (LGMN) | 2.7 | 4.6 |
| IL24 | Homo sapiens interleukin 24 (IL24) | 2.5 | 6.1 |
| Downregulated genes | |||
| IL4 | Homo sapiens interleukin 4 (IL4) | −3.7 | −1.2 |
| FAS | Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS) | −3.0 | −1.3 |
| TSC22D3 | Homo sapiens TSC22 domain family, member 3 (TSC22D3) | −2.4 | −2.1 |
| CHAC1 | Homo sapiens ChaC, cation transport regulator homolog 1 (E. coli) (CHAC1) | −2.3 | −2.1 |
| JMY | Homo sapiens junction mediating and regulatory protein, p53 cofactor (JMY) | −2.3 | −1.4 |
| SERPINH1 | Homo sapiens serpin peptidase inhibitor, clade H (HSP47), member 1, (SERPINH1) | −2.2 | −1.2 |
| HDAC9 | Homo sapiens histone deacetylase 9 (HDAC9) | −2.1 | −1.1 |
| PPP1R15A | Homo sapiens protein phosphatase 1, regulatory subunit 15A (PPP1R15A) | −2.1 | −1.1 |
| RNF7 | Homo sapiens ring finger protein 7 (RNF7) | −2.0 | −1.9 |
| HDAC7 | Homo sapiens histone deacetylase 7 (HDAC7) | −1.9 | −1.1 |
| IRAK4 | Homo sapiens interleukin‐1 receptor‐associated kinase 4 (IRAK4) | −1.9 | −1.1 |
| INPP5D | Homo sapiens inositol polyphosphate‐5‐phosphatase, 145 kDa (INPP5D) | −1.7 | −1.9 |
| IL9R | Homo sapiens interleukin 9 receptor (IL9R) | −1.6 | −1.7 |
| ADA | Homo sapiens adenosine deaminase (ADA) | −1.6 | −1.5 |
| BNIP3 | Homo sapiens BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) | −1.5 | −1.5 |
| FHL2 | Homo sapiens four and a half LIM domains 2 (FHL2) | −1.3 | −2.0 |
| CENPH | Homo sapiens centromere protein H (CENPH) | −1.2 | −1.5 |
| RAD51 | Homo sapiens RAD51 homolog (S. cerevisiae) (RAD51) | −1.2 | −1.7 |
| BRCA1 | Homo sapiens breast cancer 1, early onset (BRCA1) | −1.1 | −1.9 |
| MUC1 | Homo sapiens mucin 1, cell surface associated (MUC1) | −1.1 | −1.9 |
| CCNG2 | Homo sapiens cyclin G2 (CCNG2) | −1.0 | −2.1 |
Cells (2–5 × 105/mL) were treated with either vehicle or 4 μM HBI‐8000 for 16 h, then DNA microarray analysis was carried out. Upregulated genes and downregulated genes in KOB cells are arranged in descending and ascending orders of fold change (log2 ratio), respectively. Among them, we selected those with known functions related to apoptosis, cell cycle, cell death, and cell proliferation.
The findings from the protein expression array assays showed that cleavage of caspase‐3 and ‐7, and poly(ADP‐ribose) polymerase26 are common phenomena in cell lines and primary ATL samples (Table S3). These results support previous findings showing apoptosis induced by HBI‐8000 in ATL cells. Among other molecules, an apparent upregulation of IκBα was a common phenomenon in ATL cells except KK1.27 Western blotting revealed that there were tendencies for increased expression of IkBα and decreased expression of IKKs in both the cell lines and primary ATL cells (Fig. S2). These results indicate that HBI‐8000 contributes to ATL cell death partly through nuclear factor‐κB inhibition.
Analysis of intrinsic apoptotic pathway focusing on activation of Bim
Bim is a pro‐apoptotic protein that induces apoptosis by binding to and antagonizing anti‐apoptotic members of the Bcl‐2 family.28 To investigate the behavior of Bim, we undertook quantitative PCR for Bim and Western blot analysis for key molecules interacting with Bim (Fig. 4). Quantitative PCR confirmed upregulation of Bim by HBI‐8000 at the mRNA level. Importantly, this upregulation was commonly observed in KOB and LMY1 cells, as well as the other cells, but not in normal CD4 lymphocytes. In accordance with the PCR results, the protein expression of Bim was remarkably upregulated in KOB, LMY1, and all of the three primary ATL cells examined. Downregulation of Bcl‐2 and Bcl‐xL was apparent, whereas Bax showed no change in the KOB and LMY1 cell lines. Among the primary ATL cells, ATL9 and 10 showed downregulation of Bcl‐xL, and ATL7 and 10 showed moderate upregulation of Bax. These results support the previously noted speculation that small numbers of apoptosis facilitators are activated by HBI‐8000, which are able to overcome anti‐apoptotic or cell‐growth signals. Western blotting for normal CD4 lymphocytes was undertaken to compare with that of ATL samples and they did not alter expressions of Bax, Bim, Bcl‐2, or Bcl‐xL by HBI‐8000 (Fig. 4d).
Figure 4.
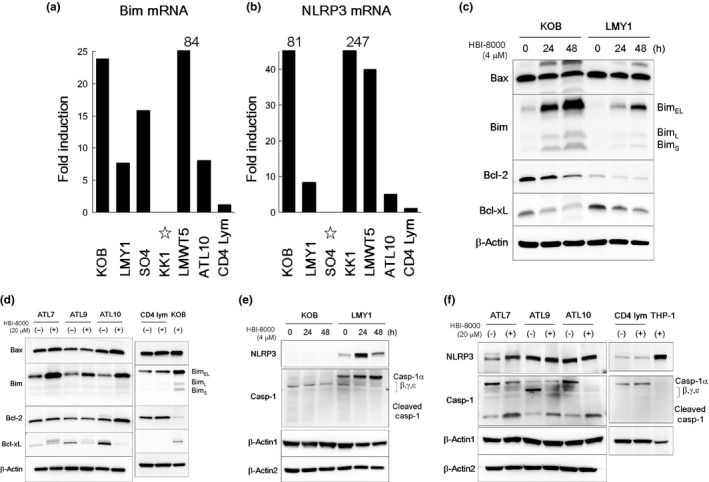
Analysis of intrinsic apoptotic and NLRP3 pathways in HBI‐8000‐induced cell death. (a, b) Adult T‐cell leukemia/lymphoma (ATL) cell lines (2–5 × 105/mL), primary ATL cells, and normal CD4 lymphocytes (1 × 106/mL) were treated with 4 or 20 μM HBI‐8000 for 24 h, and collected. Quantitative RT‐PCR assays for Bim, NLRP3, and porphobilinogen deaminase were carried out. Values for fold induction (HBI‐8000 treated/untreated cells) after calculation of the relative ratios as compared to porphobilinogen deaminase are indicated. Quantitative data for Bim in HBI‐8000‐treated KK1 cells and NLRP3 in HBI‐8000‐treated SO4 cells showed increases. However, the levels in non‐treated cells were lower than the detectable limit (star). Similarly, quantitative data for Bim in non‐treated LMWT5 cells, and NLRP3 in non‐treated KOB and KK1 cells showed quite low values. (c–f) Two ATL cell lines (2–5 × 105/mL), three types of primary ATL cells, and normal CD4 lymphocytes (1 × 106/mL) were treated for 48 and 24–48 h, respectively, with either the vehicle or the indicated concentrations of HBI‐8000. After collecting cells, Western blot analysis was carried out. In the membrane for CD4 lymphocytes, KOB cells treated with HBI‐8000 or THP‐1 cells were used as positive control. The antibody for procaspase‐1 used in this study is known to detect caspase‐1 isoforms (β, γ, δ) as well as caspase‐1α. Casp, caspase.
Analysis of activation of NLRP3 inflammasome signals
The inflammasome pathway is responsible for activation of inflammatory processes and induces cell pyroptosis, a process of programmed cell death distinct from apoptosis.29 NLRP3 is an inflammasome‐forming nucleotide‐binding oligomerization domain‐like receptor that activates procaspase‐1, leading to maturation of IL1β. Like other caspases, procaspase‐1 is activated in a proteolytic manner (cleaved caspase‐1).30 As our microarray analysis results strongly suggested the activation of NLRP3 inflammasome‐related molecules, we carried out further quantitative PCR assays for NLRP3, and Western blotting for NLRP3 and caspase‐1 (Fig. 4b,e,f). In accordance with the results of microarray analysis, quantitative PCR confirmed upregulation of NLRP3 by HBI‐8000 at the mRNA level in the ATL cell lines and primary ATL cells examined. Meanwhile, the level of mRNA and protein in normal CD4 lymphocytes did not upregulate NLRP3 and caspase‐1 in response to HBI‐8000. Western blot analysis showed that KOB cells did not express NLRP3 or procaspase‐1α, but rather showed decreased expression of procaspase‐1 isoforms. In contrast, LMY1 showed apparent upregulation of NLRP3 and procaspase‐1α, while the expression of caspase‐1 isoforms was decreased. Among the primary ATL cells, ATL7 and 10 showed decreased expression of procaspase‐1α and increased expression of cleaved caspase‐1, and ATL9 showed decreased expression of procaspase‐1 isoforms and increased expression of cleaved caspase‐1. Therefore, depending on the cell types, there was a tendency for NLRP3 and caspase‐1 to be activated by HBI‐8000.
Bim plays critical role in initiating HBI‐8000‐induced apoptosis
Cell viability assays revealed that targeting Bim (siRNA#1 and #2) significantly repressed HBI‐8000‐induced cell death in KOB and LMY1 cells (Fig. 5a). The proportion of intact cells in the si‐control group was 40–60%, whereas that in the si‐Bim group increased to approximately 80% after treatment with HBI‐8000. Similar inhibitory effects by si‐Bim were observed in annexin‐V/PI assay findings (Fig. 5b). Representative results of Western blotting using siRNA#1 were shown on Figure 5(c). In cells where NLRP3 induction by HBI‐8000 was detectable (LMY1), Western blotting revealed that si‐Bim suppressed the activation of Bim, but not that of NLRP3. These results indicate that Bim plays a critical role in initiating HBI‐8000‐induced apoptosis in KOB and LMY1 cells.
Figure 5.
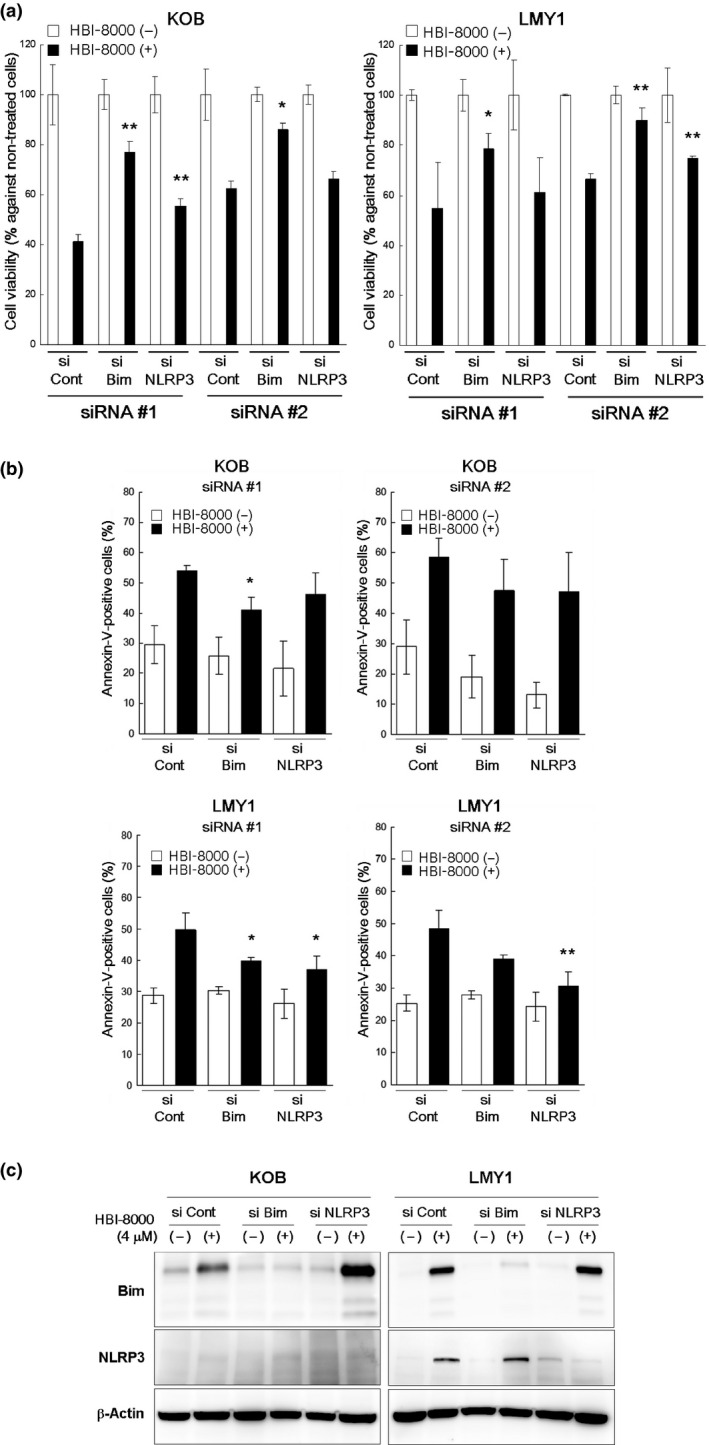
Knockdown experiments using siRNA of Bim and NLRP3 in HBI‐8000‐induced cell death. At 12 h after transfection, cells (1–2 × 105/mL) were incubated for 24 h with either the vehicle or 1–2 μM HBI‐8000. (a) Cell viability relative to untreated cells in each experiment was evaluated using MTS assay. (b) The percentage of annexin‐V‐positive cells was evaluated by FCM. Results in 5 (a, b) are expressed as the mean ± SD for three independent experiments and were analyzed using Student's t‐test with Welch's correction (*P < 0.05, **P < 0.01) as compared with HBI8000‐treated si‐Control cells. (c) After transfection using si‐Control, siRNA#1, or #2, cells (1–2 × 105/mL) were incubated for 24 h with either vehicle or 1–2 μM HBI‐8000. Cells were harvested and Western blot analysis was carried out. Representative results using siRNA#1 are shown.
Possible contribution of NLRP3 inflammasome in HBI‐8000‐induced cell death
We also carried out siRNA experiments targeting NLRP3. Cell viability assays revealed that siRNAs against NLRP3 significantly repressed HBI‐8000‐induced cell death in KOB and LMY1 cells (Fig. 5a). Similar inhibitory effects by si‐NLRP3 were observed in annexin‐V/PI assay findings (Fig. 5b). Significant inhibition of apoptosis was observed in LMY1 cells by use of siRNAs against NLRP3 and a similar tendency was seen in KOB cells, although it was not statistically significant. Western blot analysis revealed that si‐NLRP3 suppressed the activation of NLRP3 but not that of Bim in LMY1 cells (Fig. 5c). Together, these results suggest that activation of NLRP3 is important for HBI‐8000‐induced cell death of LMY1 cells.
Discussion
Accumulated evidence supports the notion that HDACi have therapeutic value for ATL.9, 10, 31 However, none of the approved HDACi therapies have been studied in regard to clinical efficacy for ATL. The China Food and Drug Administration recently granted approval for use of HBI‐8000, the world's first orally available HDACi for treatment of relapsed or refractory PTCL.18 Consistent with its activity for PTCL, HBI‐8000 induced cell cycle arrest and apoptosis in ATL‐derived cell lines and, perhaps more importantly, induced apoptosis of treatment‐naive or relapsed patient‐derived ATL cells. A phase I study of HBI‐8000 in non‐Hodgkin's lymphoma including ATL patients is underway in Japan, with phase II studies for ATL/PTCL in the planning stage.
The pro‐apoptotic molecule Bim has recently attracted increasing attention as a target for tumor therapy, with imatinib, gefitinib, bortezomib, and the Bim protein itself spotlighted as current and future Bim‐targeting therapeutic agents.32, 33 Bolden et al. investigated the ability of HDACi to affect the growth and survival of tumor cells whilst leaving normal cells relatively unharmed, and found that Bim is a key molecule in vorinostat‐induced apoptosis of cancer cells.34 It has been shown that HTLV‐1 Tax has a pro‐survival role in infected T cells by enhancing expression of the Bcl‐2 family of anti‐apoptotic proteins. Very recently, several reports pointed out the importance of the role of Bim in HTLV‐1 infected cells. Mühleisen et al. reported that HTLV‐1 Tax downregulates Bim expression by enhancing hypoxia‐inducible factor‐1α protein expression, while siRNA knockdown of the hypoxia‐inducible factor‐1α increased the expression level of Bim, and death receptor induced apoptosis in HTLV‐1‐infected leukemic T‐cell lines.35 Furthermore, Tanaka‐Nakanishi et al. found that an HTLV‐1 bZIP factor impairs transcription of Bim by disrupting FoxO3a function and acts to promote viability of HTLV‐1‐infected T cells by blocking their apoptosis.36 In addition, our previous studies indicated that death receptor‐mediated apoptotic signals in ATL cells are strongly inhibited at the mitochondrial level through the intrinsic apoptotic pathway.21 In the present study, we found that HBI‐8000 is a novel Bim activator and, as such, may be a promising agent for treatment of ATL.
Inflammasomes are protein complexes assembled following recognition of infection or cell damage signals, and play important roles in apoptotic and pyroptotic cell death.29
The NLR proteins show a variety of functions during innate immune response, among which NLRP3 is the most well known. It has been shown that deregulation of NLRP3 is associated with cancer pathogenesis, and viral infections may activate the NLRP3 inflammasome.37, 38, 39, 40 Previous reports have shown the role of the NLRP3 inflammasome in antiviral immunity towards influenza virus in vivo,41 and a very recent study indicated that the NLRP3 inflammasome pathway caused quiescent CD4 T‐cell death in HIV‐infected hosts by caspase‐1‐mediated pyroptosis.42 Therefore, it is not surprising that HTLV‐1‐infected CD4 T cells harbor an NLRP3‐induced cell death pathway. However, there is no useful method for detection of pyroptosis, such as the annexin‐V/PI assay for apoptosis, and distinguishing apoptotic cells from pyroptosis is difficult.43, 44
Our purpose in the present study did not include clarifying the mechanism of pyroptosis. Nevertheless, our microarray analysis results strongly suggest activation of NLRP3 inflammasome signaling. We focused on NLRP3 and caspase‐1, the initiator and executioner, respectively, of this pathway, and noted a clear tendency for some types of ATL cells to activate NLRP3 and caspase‐1 following treatment with HBI‐8000. Furthermore, siRNA experiments revealed that activation of NLRP3 partly contributed to HBI‐8000‐induced ATL cell death.
The importance of our results regarding NLRP3 activation may be explained from two aspects. First, there is no known previous report showing that antitumor drugs such as HDACi are responsible for inflammasome‐mediated cell death. Second, our results suggest that the antiviral response by NLRP3 inflammasome is reactivated by HBI‐8000 in ATL cells. This indicates the possibility that HBI‐8000 may be effective as an antiviral agent for patients who are HTLV‐1 carriers as well as an antitumor agent for patients with ATL.
Disclosure Statement
Hiroo Hasegawa received research funding from HUYA Bioscience International. Reid P. Bissonnette is an employee of HUYA Bioscience International. Mireille Gillings has a leadership position with HUYA Bioscience International. The other authors have no conflict of interest.
Supporting information
Fig. S1. Comparison of effects of HBI‐8000 on primary adult T‐cell leukemia/lymphoma cells with normal CD4 lymphocytes.
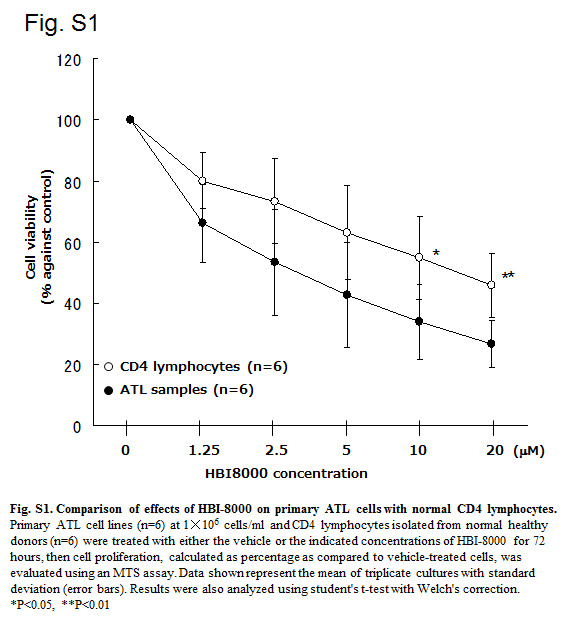{kind=link}
Fig. S2. Western blotting of IkBα and IKKs for adult T‐cell leukemia/lymphoma cell lines and primary cells.
{kind=link}
Table S1. Characteristics of chronic and acute adult T‐cell leukemia/lymphoma patients in this study.
Table S2. Primers and probes for quantitative real‐time RT‐PCR.
Table S3. Summary of the results of PathScan Stress and Apoptosis Signaling Antibody Array analysis for adult T‐cell leukemia/lymphoma cell lines and primary cells.
Acknowledgments
We wish to thank Miss Ai Ueda for cell culture and sample preparation. This study was supported in part by a Grant‐in‐aid for Scientific Research (23590641) from the Japan Society for the Promotion of Science, Health Labour Sciences Research Grant (Grant No. H23‐sinkou‐ippan‐016).
Cancer Sci 107 (2016) 1124–1133
Funding Information
Japan Society for the Promotion of Science (23590641); Ministry of Health, Labour and Welfare (Grant No. H23‐sinkou‐ippan‐016).
References
- 1. Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T‐cell leukaemia‐lymphoma. A report from the Lymphoma Study Group (1984–87). Br J Haematol 1991; 79: 428–37. [DOI] [PubMed] [Google Scholar]
- 2. Tsukasaki K, Tobinai K. Biology and treatment of HTLV‐1 associated T‐cell lymphomas. Best Pract Res Clin Haematol. 2013; 26: 3–14. [DOI] [PubMed] [Google Scholar]
- 3. Ishitsuka K, Tamura K. Human T‐cell leukaemia virus type I and adult T‐cell leukaemia‐lymphoma. Lancet Oncol. 2014; 15: e517–26. [DOI] [PubMed] [Google Scholar]
- 4. Yoshie O, Fujisawa R, Nakayama T et al Frequent expression of CCR4 in adult T‐cell leukemia and human T‐cell leukemia virus type 1‐transformed T cells. Blood 2002; 99: 1505–11. [DOI] [PubMed] [Google Scholar]
- 5. Ishida T, Utsunomiya A, Iida S et al Clinical significance of CCR4 expression in adult T‐cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res 2003; 9: 3625–34. [PubMed] [Google Scholar]
- 6. Ishida T, Joh T, Uike N et al Defucosylated anti‐CCR4 monoclonal antibody (KW‐0761) for relapsed adult T‐cell leukemia‐lymphoma: a multicenter phase II study. J Clin Oncol 2012; 30: 837–42. [DOI] [PubMed] [Google Scholar]
- 7. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014; 13: 673–91. [DOI] [PubMed] [Google Scholar]
- 8. Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS‐275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res 2003; 63: 3637–45. [PubMed] [Google Scholar]
- 9. Hasegawa H, Yamada Y, Tsukasaki K et al LBH589, a deacetylase inhibitor, induces apoptosis in adult T‐cell leukemia/lymphoma cells via activation of a novel RAIDD‐caspase‐2 pathway. Leukemia 2011; 25: 575–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen J, Zhang M, Ju W, Waldmann TA. Effective treatment of a murine model of adult T‐cell leukemia using depsipeptide and its combination with unmodified daclizumab directed toward CD25. Blood 2009; 113: 1287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pan D‐S, Yang Q‐J, Fu X et al Discovery of an orally active subtype‐selective HDAC inhibitor, chidamide, as an epigenetic modulator for cancer treatment. MedChemComm. 2014; 5: 1789–96. [Google Scholar]
- 12. Ning ZQ, Li ZB, Newman MJ et al Chidamide (CS055/HBI‐8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell‐mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol 2012; 69: 901–9. [DOI] [PubMed] [Google Scholar]
- 13. Gong K, Xie J, Yi H, Li W. CS055 (Chidamide/HBI‐8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS‐dependent apoptosis and differentiation in human leukaemia cells. Biochem J. 2012; 443: 735–46. [DOI] [PubMed] [Google Scholar]
- 14. Yao Y, Zhou J, Wang L et al Increased PRAME‐specific CTL killing of acute myeloid leukemia cells by either a novel histone deacetylase inhibitor chidamide alone or combined treatment with decitabine. PLoS ONE 2013; 8: e70522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang H, Guo Y, Fu M et al Antitumor activity of Chidamide in hepatocellular carcinoma cell lines. Mol Med Rep. 2012; 5: 1503–8. [DOI] [PubMed] [Google Scholar]
- 16. Liu L, Chen B, Qin S et al A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochem Biophys Res Commun 2010; 392: 190–5. [DOI] [PubMed] [Google Scholar]
- 17. Zhou Y, Pan DS, Shan S et al Non‐toxic dose chidamide synergistically enhances platinum‐induced DNA damage responses and apoptosis in Non‐Small‐Cell lung cancer cells. Biomed Pharmacother 2014; 68: 483–91. [DOI] [PubMed] [Google Scholar]
- 18. Shi Y, Dong M, Hong X et al Results from a multicenter, open‐label, pivotal phase II study of chidamide in relapsed or refractory peripheral T‐cell lymphoma. Ann Oncol 2015; 26: 1766–71. [DOI] [PubMed] [Google Scholar]
- 19. Kamihira S, Sugahara K, Tsuruda K et al Proviral status of HTLV‐1 integrated into the host genomic DNA of adult T‐cell leukemia cells. Clin Lab Haematol 2005; 27: 235–41. [DOI] [PubMed] [Google Scholar]
- 20. Maeda T, Yamada Y, Moriuchi R et al Fas gene mutation in the progression of adult T cell leukemia. J Exp Med 1999; 189: 1063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hasegawa H, Yamada Y, Harasawa H et al Sensitivity of adult T‐cell leukaemia lymphoma cells to tumour necrosis factor‐related apoptosis‐inducing ligand. Br J Haematol 2005; 128: 253–65. [DOI] [PubMed] [Google Scholar]
- 22. Hasegawa H, Yamada Y, Iha H et al Activation of p53 by Nutlin‐3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T‐cell leukemia cells. Leukemia 2009; 23: 2090–101. [DOI] [PubMed] [Google Scholar]
- 23. Sasaki D, Imaizumi Y, Hasegawa H et al Overexpression of enhancer of zeste homolog 2 with trimethylation of lysine 27 on histone H3 in adult T‐cell leukemia/lymphoma as a target for epigenetic therapy. Haematologica 2011; 96: 712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taniguchi H, Hasegawa H, Sasaki D et al Heat shock protein 90 inhibitor NVP‐AUY922 exerts potent activity against adult T‐cell leukemia‐lymphoma cells. Cancer Sci 2014; 105: 1601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wong JC, Guo L, Peng Z et al Application of p21 and klf2 reporter gene assays to identify selective histone deacetylase inhibitors for cancer therapy. Bioorg Med Chem Lett 2011; 21: 110–6. [DOI] [PubMed] [Google Scholar]
- 26. Ashkenazi A, Salvesen G. Regulated cell death: signaling and mechanisms. Annu Rev Cell Dev Biol 2014; 30: 337–56. [DOI] [PubMed] [Google Scholar]
- 27. Karin M, Ben‐Neriah Y. Phosphorylation meets ubiquitination: the control of NF‐[kappa]B activity. Annu Rev Immunol 2000; 18: 621–63. [DOI] [PubMed] [Google Scholar]
- 28. Bouillet P, Purton JF, Godfrey DI et al BH3‐only Bcl‐2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 2002; 415: 922–6. [DOI] [PubMed] [Google Scholar]
- 29. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol 2013; 13: 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Mol Cell 2002; 10: 417–26. [DOI] [PubMed] [Google Scholar]
- 31. Nishioka C, Ikezoe T, Yang J et al Histone deacetylase inhibitors induce growth arrest and apoptosis of HTLV‐1‐infected T‐cells via blockade of signaling by nuclear factor kappaB. Leuk Res 2008; 32: 287–96. [DOI] [PubMed] [Google Scholar]
- 32. Matsui H, Asou H, Inaba T. Cytokines direct the regulation of Bim mRNA stability by heat‐shock cognate protein 70. Mol Cell 2007; 25: 99–112. [DOI] [PubMed] [Google Scholar]
- 33. Akiyama T, Dass CR, Choong PF. Bim‐targeted cancer therapy: a link between drug action and underlying molecular changes. Mol Cancer Ther 2009; 8: 3173–80. [DOI] [PubMed] [Google Scholar]
- 34. Bolden JE, Shi W, Jankowski K et al HDAC inhibitors induce tumor‐cell‐selective pro‐apoptotic transcriptional responses. Cell Death Dis 2013; 4: e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Muhleisen A, Giaisi M, Kohler R, Krammer PH, Li‐Weber M. Tax contributes apoptosis resistance to HTLV‐1‐infected T cells via suppression of Bid and Bim expression. Cell Death Dis 2014; 5: e1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tanaka‐Nakanishi A, Yasunaga J, Takai K, Matsuoka M. HTLV‐1 bZIP factor suppresses apoptosis by attenuating the function of FoxO3a and altering its localization. Cancer Res 2014; 74: 188–200. [DOI] [PubMed] [Google Scholar]
- 37. Wei Q, Mu K, Li T et al Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest 2014; 94: 52–62. [DOI] [PubMed] [Google Scholar]
- 38. Gram AM, Frenkel J, Ressing ME. Inflammasomes and viruses: cellular defence versus viral offence. J Gen Virol 2012; 93: 2063–75. [DOI] [PubMed] [Google Scholar]
- 39. Dowling JK, O'Neill LA. Biochemical regulation of the inflammasome. Crit Rev Biochem Mol Biol 2012; 47: 424–43. [DOI] [PubMed] [Google Scholar]
- 40. Haneklaus M, O'Neill LA, Coll RC. Modulatory mechanisms controlling the NLRP3 inflammasome in inflammation: recent developments. Curr Opin Immunol 2013; 25: 40–5. [DOI] [PubMed] [Google Scholar]
- 41. Allen IC, Scull MA, Moore CB et al The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009; 30: 556–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Doitsh G, Galloway NL, Geng X et al Cell death by pyroptosis drives CD4 T‐cell depletion in HIV‐1 infection. Nature 2014; 505: 509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rayamajhi M, Zhang Y, Miao EA. Detection of pyroptosis by measuring released lactate dehydrogenase activity. Methods Mol Biol 2013; 1040: 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sagulenko V, Thygesen SJ, Sester DP et al AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ 2013; 20: 1149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Comparison of effects of HBI‐8000 on primary adult T‐cell leukemia/lymphoma cells with normal CD4 lymphocytes.
Fig. S2. Western blotting of IkBα and IKKs for adult T‐cell leukemia/lymphoma cell lines and primary cells.
Table S1. Characteristics of chronic and acute adult T‐cell leukemia/lymphoma patients in this study.
Table S2. Primers and probes for quantitative real‐time RT‐PCR.
Table S3. Summary of the results of PathScan Stress and Apoptosis Signaling Antibody Array analysis for adult T‐cell leukemia/lymphoma cell lines and primary cells.