

Abstract
Crizotinib is a standard treatment for advanced ALK‐positive non‐small‐cell lung cancer (NSCLC). We undertook this study to investigate the pharmacokinetics of crizotinib and clinical and pharmacogenomic factors that may increase the risk of adverse events (AEs). We defined clinically significant AEs as grade 4 hematological toxicity, grade ≥3 non‐hematological toxicity, and any grade of interstitial lung disease. Eight subjects with ALK‐positive NSCLC scheduled to receive crizotinib 250 mg twice daily were studied. Six patients were female and two were male, and most of the patients had low body weight with a median body weight of 46.8 kg (range, 42.4–61.0 kg). All patients developed AEs, five developing six clinically significant AEs. Six patients required dose reduction. In pharmacokinetic analysis, blood samples were obtained on days 1 and 15. The mean area under the plasma concentration–time curve from 0–12 h (AUC 0–12) on day 15 was significantly increased in patients with clinically significant AEs (n = 5) compared with those without (n = 3) (P = 0.04). Genetic polymorphisms of ABCB1 were analyzed. One patient with the ABCB1 1236TT‐2677TT‐3435TT genotype was an outlier, with an AUC 0–12 and peak concentrations on day 15 of 2.84× and 2.61× the mean, respectively, compared with those with other genotypes. Our results suggest that some Japanese NSCLC patients treated with crizotinib developed clinically significant toxicities that were related to altered pharmacokinetics parameters due to genotype and body weight factors.
Keywords: Crizotinib, EML4–ALK fusion protein, non‐small‐cell lung cancer, pharmacogenomics, pharmacokinetics
Identifying the molecular alterations that underlie the pathogenesis of lung cancer may allow dramatic progress in clinical outcomes. The echinoderm microtubule‐associated protein‐like 4 (EML‐4)–anaplastic lymphoma kinase (ALK) fusion gene, first described by Soda et al.1 is one of the driver oncogenes in non‐small‐cell lung cancer (NSCLC). It is known that approximately 5% of lung adenocarcinoma patients are ALK fusion gene‐positive (ALK‐positive).1, 2 Crizotinib (Pfizer, Mission, KS, USA), a multitarget tyrosine kinase inhibitor, showed favorable clinical activity in ALK‐positive NSCLC patients3, 4, 5, 6 and was approved for ALK‐positive NSCLC in 2011 in the USA and in March 2012 in Japan.
In phase 1 pharmacokinetic (PK) analyses of crizotinib carried out in the USA and Korea, the mean area under the plasma concentration–time curve (AUC) from 0 to 24 h and peak concentration (C max) (% coefficient variance [CV]) were 589 ng h/mL (34%; n = 4) and 91.4 ng/mL (34%; n = 9) for a single dose of 250 mg and 3641 ng h/mL (37%; n = 4) and 368 ng/mL (30%; n = 6) for multiple doses, respectively. Mean half‐life of crizotinib at the terminal phase was 43–51 h, giving an estimated steady state within 15 days.4 Further exploratory population PK analysis was carried out to identify factors affecting the AUC of crizotinib at steady state, including body weight, sex, age, and race. The C max, trough concentration (C trough), and AUC values were higher in Asian patients (n = 13), including Japanese, than in non‐Asian patients (n = 11) (C max, 506 vs 322 ng/mL, ratio 1.57 [90% confidence interval (CI), 1.16−2.13]; AUC, 4696 vs 3137 ng/h/mL, ratio 1.50 [90% CI, 1.10−2.04]). However, when adjusted for body weight or body surface area, these differences were not statistically significant, suggesting that ethnic/racial differences in the PK of crizotinib were unlikely.
Crizotinib is reported to be primarily metabolized by cytochrome P450 3A4 (CYP3A4) and to be a substrate of P‐glycoprotein.7 Expression of P‐glycoprotein has been reported to be influenced by ATP‐binding cassette sub‐family B member 1 (ABCB1) single nucleoside polymorphisms (SNPs). Previous studies have revealed a significant association between ABCB1 SNPs and the functionality of P‐glycoprotein.8, 9, 10, 11, 12 There are known ethnic differences in ABCB1 SNPs. It is also known that the PK and pharmacodynamics (PD) of some anticancer agents such as erlotinib differ in patients of different ethnicities.13, 14 These factors may lead to differences in intracellular versus extracellular concentrations of crizotinib.
The most common adverse events (AEs) in these crizotinib studies have been vision abnormalities (most frequently visual impairment, photophobia, or blurred vision), diarrhea, nausea, vomiting, constipation, elevated transaminase levels, edema, upper respiratory infection, dysgeusia, and dizziness.5 Grade 3 or higher AEs including neutropenia, elevated transaminase levels, fatigue, interstitial lung disease, pneumonia, and electrocardiogram QTc prolongation occasionally occurred.
Here, we investigated possible clinical and pharmacogenomic factors in the PK, PD, or pharmacogenomics (PGx) of crizotinib in Japanese patients with ALK‐positive NSCLC.
Patients and Methods
Patient selection criteria
Inclusion criteria were advanced ALK‐positive NSCLC scheduled for treatment with crizotinib therapy, age ≥20 years, and adequate organ function (serum total bilirubin ≤2.0 mg/dL, aspartate aminotransferase ≤150 IU/L, alanine aminotransferase ≤150 IU/L, serum creatinine ≤2.0 mg/dL, and SpO2 ≥90%). Exclusion criteria included: concomitant treatment with other anticancer agents, radiotherapy, or surgery; the inability to swallow tablets; gastrointestinal disorders that could affect the ingestion or absorption of crizotinib, such as watery diarrhea, intestinal paresis, ileus, or a history of gastrectomy or intestinal resection; intake of drugs or food that could act as potent cytochrome P450 3A4 or P‐glycoprotein inhibitors or inducers; and women of childbearing age unless using effective contraception.
Study design and outcome
The study was carried out under an open‐label observational design to evaluate the PK/PD/PGx of crizotinib in Japanese patients with ALK‐positive NSCLC. The primary objective was to characterize PK parameters of crizotinib in these patients. Secondary objectives were to investigate the relationship between PK parameters and PD, including toxicities and anticancer effects, and to explore any genetic factors, including ABCB1 SNPs.
This study (UMIN000009867) was designed and carried out at the National Cancer Center Hospital (Tokyo, Japan). The protocol was approved by the review board of the National Cancer Center Hospital on January 31, 2013, and the study was carried out in accordance with the ethical principles stated in the Declaration of Helsinki. All patients provided written informed consent.
Treatment and assessments
Patients received the standard crizotinib dose of 250 mg twice daily. Patients continued to receive therapy until disease progression, clinical deterioration, or intolerable AEs that did not improve with dose adjustment.
The ALK fusion gene was confirmed by real‐time RT‐PCR, immunohistochemistry staining with D5F3 antibody (Cell Signaling Technology, Danvers, MA, USA) or FISH. The ALK FISH testing was carried out using a Vysis ALK break‐apart probe set (Abbott Molecular, Abbott Park, IL, USA). The specimen was considered positive if more than 15% of scored tumor cells had split ALK 5′ and 3′ probe signals or had isolated 3′ signals.
Blood samples for PK analysis were collected immediately before and 0.5, 1, 2, 4, 6, 8, and 12 h after administration of crizotinib on days 1 and 15. Serial electrocardiography (ECG) was carried out in triplicate immediately before and 4 and 12 h after treatment with crizotinib on days 1 and 15 to assess for QTc prolongation.
Adverse events were evaluated using the Common Terminology Criteria for Adverse Events version 4.0. Objective tumor response according to the Response Evaluation Criteria in Solid Tumors (RECIST version 1.1) was evaluated in all eligible patients before treatment, 8 weeks after the start of treatment, and then every 3–4 months.15
Pharmacokinetic analysis
After blood was collected in EDTA‐containing tubes, plasma was separated within 30 min by centrifugation at 1500g for 10 min at 4°C and stored at –80°C until analysis. Plasma concentrations of crizotinib were measured by liquid chromatography–tandem mass spectrometry. Chromatographic separation of crizotinib and the internal standard, erlotinib‐d6, was carried out using an XBridge C18 HPLC column (2.1 × 5.0 mm, 3.5 μm) (Waters Corp., Milford, MA, USA) maintained at 40°C. Mobile phases A and B consisted of 0.1% formic acid aqueous solution and acetonitrile containing 0.1% formic acid, respectively. Separation was carried out using a gradation elution (from B of 15–90% over 5 min, B of 99% for 2.5 min, B of 15% for 3 min) at a flow rate of 0.3 mL/min with Nexera X2 ultra‐HPLC (Shimadzu Co., Kyoto, Japan). Quantitation was undertaken by selected reaction monitoring on a QTRAP5500 mass spectrometer (AB SCIEX, Framingham, MA, USA) with electrospray ionization in the positive mode. The selected reaction monitoring transitions were m/z 451.1–261.1 for crizotinib and m/z 400.2–278.0 for erlotinib‐d6. All data were acquired and analyzed using Analyst version 1.6.1 software (AB SCIEX). All sample analyses were carried out according to the internal assay quality guidelines. The lower limit of quantification was 5 ng/mL.
Pharmacokinetic variables of crizotinib and its metabolites were determined using the Phoenix WinNonlin PK program (Pharsight, Mountain View, CA, USA). The C max and time to maximum concentration were recorded directly from the data. The AUC with extrapolation to 12 h (AUC0–12h) was calculated by the trapezoidal rule. The linear trapezoidal rule was used for successively increasing concentration values, and the logarithmic trapezoidal rule for decreasing concentration values.
Genotyping
Genotyping assays were carried out for eligible patients who had sufficient DNA in their sample. DNA was extracted and the concentration was fixed at 10 ng/μL. Genotyping was undertaken using the i‐densy genetic testing platform (ARKRAY, Kyoto, Japan) and the QP‐system quenching probe system (J‐Bio 21, Tokyo, Japan) based on the principles of mutant detection. To detect the various genotypes, we used QProbe (Nippon Steel Kankyo Engineering, Tokyo, Japan).
We analyzed the SNPs ABCB1 1236C>T, 2677G>T/A, and 3435C>T (rs1128503, rs2032582, and rs1045642, respectively). The primers that we used were: ABCB1‐1236F, 5′‐cctgtgtctgtgaatYgccttgaag‐3′ (Y represents C or T); ABCB1‐1236R, 5′‐gtctgcccactctgcaccttc‐3′; ABCB1‐2677F, 5′‐aaatgttgtctggacaagcactg‐3′; ABCB1‐2677R, 5′‐aattaatcaatcatatttagtttgactcac‐3′; ABCB1‐3435F, 5′‐actgcagcattgctgagaac‐3′; and ABCB1‐3435R, 5′‐cagagaggctgccacatgctc‐3′. The probes that we used were: ABCB1‐1236, 5′‐ttcaggttcagacccttc‐TAMRA‐3′; ABCB1‐2677, 5′‐PACIFIC BLUE‐cccagaaccttctagttc‐3′; and ABCB1‐3435, 5′‐ ctgccctcacaatctcttc‐BODIPY FL‐3′.
Statistical analysis
Data are expressed as the geometric mean ± SD. The statistical significance of PK parameters was analyzed using a Wilcoxon signed‐rank test, with P < 0.05 considered significant. Progression‐free survival was defined as the time from the first day of crizotinib therapy to detection of the earliest signs of disease progression or death from any cause. Overall survival (OS) was defined as the time from the first day of crizotinib therapy to the last day on which the patient was confirmed to be alive or dead from any cause. Median survival was calculated using the Kaplan–Meier method. All statistical analyses were carried out with spss Statistics version 23.0 (SPSS, Chicago, IL, USA).
Results
Patient group
From March 2013 to June 2014, a total of eight patients were enrolled (Table 1, Fig. S1). All patients had adenocarcinoma. Six patients were female and two were male, with a median age of 59 years (range, 46–72 years). The median body weight was 46.8 kg (range, 42.4–61.0 kg), and the median body surface area was 1.47 m2 (range, 1.30–1.70 m2).
Table 1.
Characteristics of Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism (n = 8)
| No. of patients | ||
|---|---|---|
| Sex | Male/female | 2/6 |
| Age, years | Median (range) | 59 (46–72) |
| ECOG PS | 0/1/2/3 | 2/4/1/1 |
| Histologic type | Adenocarcinoma | 8 |
| Clinical stage | IV/recurrence | 7/1 |
| Brain metastasis | Yes/no | 5/3 |
| Height, cm | Median (range) | 158.2 (143.5–168.6) |
| Weight, kg | Median (range) | 46.8 (42.4–61) |
| BSA, m2 | Median (range) | 1.47 (1.30–1.70) |
| Smoking status | Never/ex‐smoker | 5/3 |
| Pack‐years | Median (range) | 30 (1–40) |
| T‐Bil, mg/dL | Median (range) | 0.6 (0.3–1.6) |
| AST, IU/L | Median (range) | 19 (13–30) |
| ALT, IU/L | Median (range) | 15 (7–38) |
| Cr, mg/dL | Median (range) | 0.59 (0.38–0.67) |
| CCr, mL/min | Median (range) | 120.4 (65.0–131.4) |
| Prior chemotherapy | Yes/no | 4/4 |
| Prior ALK‐TKI | Yes/no | 1/7 |
| Prior radiotherapy | Yes/no | 6/2 |
| Prior surgery | Yes/no | 1/7 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; BSA, body surface area; Cr, creatinine; CCr, creatinine clearance by the Cockcroft–Gault method; ECOG‐PS, Eastern Cooperative Oncology Group performance status; T‐Bil, total bilirubin; TKI, tyrosine kinase inhibitor.
Treatment efficacy and toxicity
Seven patients were positive for ALK with FISH and one was negative, but positive on immunohistochemistry. Five patients had a partial response, one had stable disease, and two had progressive disease as their best response. The overall response rate by RECIST was 62.5% (95% CI, 24.5–91.5) (Fig. S2). Of two patients with progressive disease, one developed a brain metastasis on day 60, and after treatment with stereotactic irradiation, she continued crizotinib treatment beyond progression. Another patient, who had a history of previous crizotinib treatment, developed liver metastasis on day 47. Seven patients eventually discontinued crizotinib treatment due to progressive disease, and one due to an AE. Six patients required dose reduction due to AEs. Median PFS was 9.8 (95% CI, 2.3–17.3) months and median OS was not applicable (Figs S3,S4).
Common hematological and non‐hematological AEs observed during crizotinib treatment are summarized in Table 2. All patients experienced at least one AE of any grade of toxicity, and five patients experienced clinically significant AEs: grade 3 or 4 alanine aminotransferase increased (n = 2), grade 3 aspartate aminotransferase increased (n = 1), grade 3 esophagitis (n = 1), grade 3 QTc prolongation (n = 1), and grade 3 interstitial nephritis (n = 1). We were able to manage these AEs with supportive care and reduced doses in seven patients.
Table 2.
Adverse events of any cause in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism treated with crizotinib (n = 8)
| Adverse event | Any grade, n | Grade 3, n | Grade 4, n |
|---|---|---|---|
| Diarrhea | 8 | 0 | 0 |
| ALT increased | 7 | 1 | 1 |
| AST increased | 7 | 1 | 0 |
| Visual disorder | 5 | 0 | 0 |
| Anorexia | 4 | 0 | 0 |
| Nausea | 4 | 0 | 0 |
| Vomiting | 4 | 0 | 0 |
| Constipation | 4 | 0 | 0 |
| Fatigue | 3 | 0 | 0 |
| Dysgeusia | 3 | 0 | 0 |
| QTc prolongation | 2 | 1 | 0 |
| Abdominal pain | 2 | 0 | 0 |
| Dizziness | 2 | 0 | 0 |
| Esophagitis | 1 | 1 | 0 |
| Interstitial nephritis | 1 | 1 | 0 |
| Skin eruption | 1 | 0 | 0 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase.
A female patient developed severe esophagitis with dysphagia and odynophagia 4 weeks after starting crizotinib. Gastrointestinal endoscopy revealed diffuse grade 3 esophagitis and the drug was discontinued. When her symptoms resolved 3 weeks after discontinuation, she was restarted at a reduced dose of 200 mg twice daily, but the esophagitis returned.
Another patient developed grade 3 QTc prolongation. Grade 2 nausea and anorexia developed shortly after starting crizotinib and the ECG 4 h after crizotinib was given on day 15 showed a QTc interval of 507 ms with an RR interval of 158 ms. After discontinuation of crizotinib for 5 days, her ECG normalized. She was restarted at a reduced dose of 200 mg twice daily until disease progression.
Pharmacokinetic analysis related to toxicity
The PK parameters of crizotinib on days 1 and 15 are presented in Table 3 and Figure S5. The geometric mean of AUC0–12 and C max measured in the Japanese patients in this study were generally similar to those in Asian patients obtained in the previous phase 1 study (Table 4). The geometric mean of AUC0–12 on day 15 was significantly increased in patients with clinically significant AEs (n = 5) compared to those without (n = 3) (Table 3, Fig. 1a). There was also a significant increase in the geometric mean of C max and C trough on day 15 in the patients with significant AEs.
Table 3.
Pharmacokinetic parameters of crizotinib in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism
| All patients n = 8 | Significant AE n = 5 | No significant AE n = 3 | P‐value | |
|---|---|---|---|---|
| AUC0–12, ng h/mL | ||||
| Day 1 | 872 ± 379 | 1040 ± 264 | 650 ± 519 | 0.57 |
| Day 15 | 5410 ± 3220 | 6540 ± 3660 | 3940 ± 277 | 0.04 |
| Ratio | 6.20 ± 3.55 | 6.28 ± 3.75 | 6.06 ± 4.01 | |
| C max, ng/mL | ||||
| Day 1 | 129 ± 54.0 | 156 ± 49.1 | 94.6 ± 44.4 | 0.14 |
| Day 15 | 525 ± 279 | 631 ± 312 | 387 ± 28.7 | 0.04 |
| C trough on day 15 | 422 ± 302 | 512 ± 354 | 305 ± 39.3 | 0.04 |
| Half‐life, min | ||||
| Day 1 | 447 ± 522 | 528 ± 645 | 338 ± 117 | 0.25 |
| Day 15 | 2010 ± 3780 | 1840 ± 1830 | 2310 ± 6190 | 1.00 |
| T max, min | ||||
| Day 1 | 304 ± 86.8 | 323 ± 97.8 | 275 ± 67.0 | 0.57 |
| Day 15 | 268 ± 198 | 245 ± 246 | 310 ± 68.9 | 0.79 |
AE, adverse event; AUC0–12, area under the plasma concentration–time curve from 0 to 12 h; C max, peak concentration; C trough, trough concentration; T max, time to maximum concentration.
Table 4.
Pharmacokinetic parameters (mean [coefficient of variation %]) of crizotinib on day 15 in this study of Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism compared with subjects in a global phase I study
| This trial | Global phase I trial | ||
|---|---|---|---|
| Japanese | Non‐Asian | Asian (8 Korean, 5 Japanese) | |
| n = 8 | n = 11 | n = 13 | |
| AUC0–12, ng h/mL | 5410 (60) | 3137 (55) | 4696 (11) |
| C max, ng/mL | 525 (53) | 322 (67) | 506 (23) |
AUC0–12, area under the plasma concentration–time curve from 0 to 12 h; C max, peak concentration.
Figure 1.
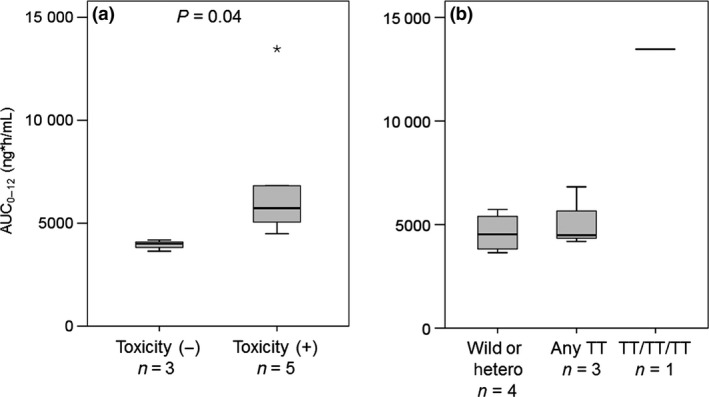
Mean area under the plasma concentration–time curve from 0–12 h (AUC 0–12) of crizotinib in Japanese patients with non‐small‐cell lung cancer with ABCB1 polymorphism. (a) Comparison of those with clinically significant adverse events with those without. (b) Comparison of AUC 0–12 of crizotinib by ABCB1 genotyping.
Genotype related to PK and toxicity
Figure 1(b) and Table 5 depict the genotype findings. For the ABCB1 1236C>T SNPs, we identified one CC, three CT, and four TT genotypes among all eight patients. One patient showed all TT alleles (TT‐TT‐TT, homozygous variant), and three patients had at least one TT allele. Regarding the relationship between ABCB1 genotype and PKs, one patient (#7) with all‐TT ABCB1 alleles was a pronounced outlier in AUC0–12 and C max on day 15 (13 467 ng h/mL and 1216 μg/mL, respectively). This was the same patient who developed grade 3 QTc prolongation.
Table 5.
Polymorphisms in ABCB1 in Japanese non‐small‐cell lung cancer patients (n = 8) treated with crizotinib
| Age, years | Sex | BW, kg | Detection method for ALK | Shrinkage rate,‡ % | PFS, days | Dose reduction | Reason for discontinuation | Clinically significant AE | ABCB1 polymorphism | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IHC | FISH,† % | 3435 | 2677 | 1236 | |||||||||
| #1 | 50 | Female | 53.0 | + | 40 | 87 | 60 | Yes | PD | Gr3 esophagitis | CC | GG | CC |
| #2 | 56 | Female | 42.8 | + | 70 | 57 | 302 | Yes | Ongoing | Gr3 ALT increased | CC | GG | CT |
| #3 | 72 | Male | 49.7 | + | 72 | 76 | 295 | No | PD | None | CC | GT or AT | TT |
| #4§ | 46 | Female | 45.0 | + | 31 | 66 | 47 | No | PD | None | CC | GG | CT |
| #5 | 48 | Male | 61.0 | Untested | 33 | 79 | 374 | Yes | PD | Gr3 AST increased and Gr4 ALT increased | CT | GT or AT | TT |
| #6 | 66 | Female | 42.4 | + | 4 | 43 | 323 | Yes | AE | Gr3 interstitial nephritis | CC | GT or AT | TT |
| #7 | 69 | Female | 43.3 | + | 29 | 67 | 138 | Yes | PD | Gr3 QTc prolongation | TT | TT | TT |
| #8 | 62 | Female | 48.6 | + | 70 | 36 | 301 | Yes | Ongoing | None | CC | GG | CT |
†Rate of positive cells in FISH (%). ‡Percent change at maximum reduction from baseline according to Response Evaluation Criteria in Solid Tumors version 1.1. §This patient had a previous history of crizotinib treatment. AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BW, body weight; Gr, grade; IHC, immunohistochemistry; PD, progressive disease; PFS, progression‐free survival.
Discussion
In this evaluation of the PKs of crizotinib in Japanese patients with ALK‐positive NSCLC, we found that patients appeared to develop toxicities of any grade, especially clinically significant toxicities, more frequently than in previous trials. Nevertheless, all AEs could be managed with appropriate supportive care and dose reduction, and only one patient required discontinuing of treatment due to an AE. These results suggest that the toxicity and PKs of crizotinib are related to the effects of body weight and ABCB1 SNPs.
The dose escalation phase I trial of 1001/NCT00585195 determined that the maximum tolerated dose of crizotinib was 250 mg twice daily.4, 16 Several later clinical studies reported the efficacy and safety of crizotinib in patients with malignant solid tumors, including advanced ALK‐positive NSCLC. Crizotinib is currently the standard of care for patients with advanced ALK‐positive NSCLC and is under development for ROS1‐rearranged NSCLC and MET mutated or amplified NSCLC.17, 18, 19, 20 Nevertheless, only a few PK studies of crizotinib have been reported, and most data were extracted primarily from documents submitted for drug approval.21, 22, 23 The phase I documents reported that that peak plasma concentration of crizotinib after a single oral dose of 50–300 mg was reached at approximately 4 h, followed by a multiexponential decline with a terminal half‐life of 43–51 h. After multiple doses, the steady state was reached within 15 days with a mean AUC of 3880 ng h/mL (CV 36%) and a mean C max of 411 ng/mL (CV 44%). Steady‐state C trough levels with a twice daily dose of 250 mg were stable with median C trough ranging from 242 to 319 ng/mL.23
In this study, Japanese patients appeared to develop toxicities of all grades more frequently than patients in the previous trials. The PK parameters at steady state, with a mean AUC of 5410 ng h/mL and a mean C max of 525 ng/mL, also appear to be higher than patients in the previous trials (Table 3). Moreover, the geometric mean of AUC0–12 on day 15 was significantly increased in the five patients with clinically significant AEs compared to the three without (6540 ± 3660 vs 3940 ± 277 ng h/mL respectively; P = 0.04). These results suggest a correlation between the toxicity and PK of crizotinib (Table 3). Potential reasons for the difference in toxicity and PK might include body weight, sex, race, and PGx. The previous population PK study compared parameters in 13 Asian (including eight Korean and five Japanese) and 11 non‐Asian (including white, black, and other) patients.24 Although mean values for crizotinib C max and AUC in Asian patients were 1.57× and 1.50× those in non‐Asian patients, respectively, these differences were lost on correction for body weight, and the population PK study accordingly concluded that the PK differences between Asian and non‐Asian subjects were due to body weight rather than race. In our results, the differences in crizotinib C max and AUC were lost on correction for body weight or body surface area (Table S1). However, the safety and efficacy profile suggested that no dose adjustment was required for Asian patients. The Japanese patients in that study also had mean C max and AUC values of 1.63× and 1.72× those in the non‐Asian patients, respectively. Due to lack of data access, including individual PK data and body weight in the non‐Asian patients, we could not statistically analyze the difference between our Japanese subjects and the previous non‐Asian subjects. The differences in toxicity and PK in the present study may be due to the very low weights (42.4–61.0 kg) of all of our subjects.
Crizotinib is reported to be a substrate of P‐glycoprotein.7 Expression of P‐glycoprotein has been shown to be influenced by ABCB1 SNPs. Previous studies have revealed a significant association between ABCB1 SNPs and the functionality of P‐glycoprotein.8, 9, 10, 11, 12 In particular, ABCB1 expression is largely influenced by three SNPs in the ABCB1 gene at positions 1236C>T, 2677G>T, and 3435C>T.12, 25, 26 These three SNPs occur frequently and are strongly linked, creating a common haplotype. Indeed, an increasing number of variant alleles, such as 1236TT‐2677TT‐3435TT, are known to be associated with lower expression.27 Interestingly, one patient with all‐TT ABCB1 alleles was a distinct outlier in AUC0–12 and C max on day 15 (2.84× and 2.61×, respectively) compared with patients with other genotypes. This patient also had grade 3 QTc prolongation. Because we could not interpret this outlier as a consequence of this subject's low weight, we suggest that PK parameters may be increased in patients with all‐TT ABCB1 alleles, and that these patients may be at risk of severe toxicity.
Several limitations of the study warrant mention. First, the sample size was very small because of the rarity of ALK‐positive NSCLC patients. Second, our results have not been validated in different patients. However, we consider that our speculations are valuable because of significant AEs to crizotinib in one patient with all‐TT ABCB1 alleles. Finally, because we could not access the individual PK data and body weight in non‐Asian patients in a previous study, we did not statistically analyze the difference between our Japanese subjects and non‐Asian subjects.
In conclusion, we evaluated the PK of crizotinib in Japanese patients with ALK‐positive NSCLC. Japanese patients in this study appeared to develop toxicities of any grade more frequently than patients in previous trials, especially clinically significant toxicities. Nevertheless, these could be managed with appropriate supportive care and dose reduction, and only one patient discontinued treatment due to an AE. These results suggest that the toxicity and PK of crizotinib are related to the effects of body weight and ABCB1 SNPs.
Disclosure Statement
None of the authors have a direct conflict of interest regarding this study but disclose a potential conflict of interest outside the submitted work, in the form of research grants: Y.F. from AstraZeneca, Eli Lilly, and Chugai; A.H. from AstraZeneca, Chugai, and Shimadzu; H.H. from Taiho, Merk Serono, Novartis, Astellas, and MSD; S.K. from AstraZeneca and ONO; Y.G. from Pfizer, Eli Lilly, and Abbie GK; H.N. from Merck Serono, Pfizer, Taiho, Eisai, Chugai, Eli Lilly, Novartis, Daiichi Sankyo, GlaxoSmithKline, Yakult, Quintiles, Astellas, AstraZeneca, Boehringer Ingelheim, and ONO; N.Y. from Chugai, Eli Lilly, Taiho, Eisai, Quintiles, Astellas, BMS, Novartis, Daiichi‐Sankyo, Pfizer, and Boehringer Ingelheim and has received honoraria from AstraZeneca, Pfizer, Eli Lilly, and Chugai; Y.O. from AstraZeneca, Chugai, Eli Lilly, ONO, BMS, Kyorin, Dainippon‐ Sumitomo, Pfizer, Taiho, Novartis, and Merck Serono and has received honoraria from AstraZeneca, Chugai, Lilly, ONO, BMS, Daiichi‐Sankyo, Nipponkayaku, Boehringer Ingelheim, Bayer, Pfizer, MSD, Taiho, Clovis, and Sanofi. The other authors have no conflict of interest.
Supporting information
Fig. S1. CONSORT diagram of this study.
Fig. S2. Tumor responses to crizotinib from baseline in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism.
Fig. S3. Progression‐free survival in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism treated with crizotinib.
Fig. S4. Overall survival in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism treated with crizotinib.
Fig. S5. Mean plasma concentration–time curves of crizotinib on days 1 (■) and 15 (○) (mean ± standard deviation) in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism.
Table S1. Pharmacokinetic parameters of crizotinib in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism adjusted by (A) body weight and (B) body surface area.
Acknowledgments
This work was supported by the grant from the Japanese Society of Clinical Pharmacology and Therapeutics and in part by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (KAKENHI 21590167). We greatly appreciate the patients who participated in this study and their families, and the assistance of the staff of the Department of Thoracic Oncology, National Cancer Center Hospital.
Cancer Sci 107 (2016) 1117–1123
Funding Information
Japanese Society of Clinical Pharmacology and Therapeutics; Japan Society for the Promotion of Science.
Trial registration ID: UMIN000009867; date of registration: January 1, 2013.
References
- 1. Soda M, Choi YL, Enomoto M et al Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature 2007; 448: 561–6. [DOI] [PubMed] [Google Scholar]
- 2. Kris MG, Johnson BE, Berry LD et al Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014; 311: 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shaw AT, Yeap BY, Solomon BJ et al Effect of crizotinib on overall survival in patients with advanced non‐small‐cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 2011; 12: 1004–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camidge DR, Bang YJ, Kwak EL et al Activity and safety of crizotinib in patients with ALK‐positive non‐small‐cell lung cancer: updated results from a phase 1 study. Lancet Oncol 2012; 13: 1011–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shaw AT, Kim DW, Nakagawa K et al Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med 2013; 368: 2385–94. [DOI] [PubMed] [Google Scholar]
- 6. Benjamin JS, Tony Mok, Dong?Wan Kim et al First‐Line Crizotinib versus Chemotherapy in ALK‐Positive Lung Cancer. N Engl J Med 2014; 371: 2167–2177. [DOI] [PubMed] [Google Scholar]
- 7. Johnson TR, Tan W, Goulet L et al Metabolism, excretion and pharmacokinetics of [C]crizotinib following oral administration to healthy subjects. Xenobiotica 2015; 45: 45–59. [DOI] [PubMed] [Google Scholar]
- 8. Hoffmeyer S, Burk O, von Richter O et al Functional polymorphisms of the human multidrug‐resistance gene: multiple sequence variations and correlation of one allele with P‐glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA 2000; 97: 3473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sakaeda T, Nakamura T, Okumura K. Pharmacogenetics of MDR1 and its impact on the pharmacokinetics and pharmacodynamics of drugs. Pharmacogenomics 2003; 4: 397–410. [DOI] [PubMed] [Google Scholar]
- 10. Marzolini C, Paus E, Buclin T, Kim RB. Polymorphisms in human MDR1 (P‐glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther 2004; 75: 13–33. [DOI] [PubMed] [Google Scholar]
- 11. Aarnoudse AL, van Schaik RH, Dieleman J et al MDR1 gene polymorphisms are associated with neuropsychiatric adverse effects of mefloquine. Clin Pharmacol Ther 2006; 80: 367–74. [DOI] [PubMed] [Google Scholar]
- 12. Fung KL, Gottesman MM. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim Biophys Acta 2009; 1794: 860–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamada A, Sasaki J, Saeki S et al Association of ABCB1 polymorphisms with erlotinib pharmacokinetics and toxicity in Japanese patients with non‐small‐cell lung cancer. Pharmacogenomics 2012; 13: 615–24. [DOI] [PubMed] [Google Scholar]
- 14. Rudin CM, Liu W, Desai A et al Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J Clin Oncol 2008; 26: 1119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Therasse P, Arbuck SG, Eisenhauer EA et al New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92: 205–16. [DOI] [PubMed] [Google Scholar]
- 16. Kwak EL, Bang YJ, Camidge DR et al Anaplastic lymphoma kinase inhibition in non‐small‐cell lung cancer. N Engl J Med 2010; 363: 1693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dietrich MF, Yan SX, Schiller JH. Response to crizotinib/erlotinib combination in a patient with a primary EGFR‐mutant adenocarcinoma and a primary c‐met‐amplified adenocarcinoma of the lung. J Thorac Oncol 2015; 10: e23–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paik PK, Drilon A, Fan PD et al Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 2015; 5: 842–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shaw AT, Ou SH, Bang YJ et al Crizotinib in ROS1‐rearranged non‐small‐cell lung cancer. N Engl J Med 2014; 371: 1363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamazaki S, Vicini P, Shen Z et al Pharmacokinetic/pharmacodynamic modeling of crizotinib for anaplastic lymphoma kinase inhibition and antitumor efficacy in human tumor xenograft mouse models. J Pharmacol Exp Ther 2012; 340: 549–57. [DOI] [PubMed] [Google Scholar]
- 21. Document of the United States Application for approval of crizotinib. Food and Drug Administration, 2011. [Google Scholar]
- 22. Document of the European Medicines Agency for approval of crizotinib. European Medicines Agency, 2012. [Google Scholar]
- 23. Hamilton G, Rath B, Burghuber O. Pharmacokinetics of crizotinib in NSCLC patients. Expert Opin Drug Metab Toxicol 2015; 11: 835–42. [DOI] [PubMed] [Google Scholar]
- 24. Ou SHI, Salgia R, Clark J. Comparison of crizotinib (PF‐02341066) pharmacokinetics between Asian and non‐Asian patients with advanced malignancies. J Thorac Oncol 2010; 5(suppl 5): S382. [Google Scholar]
- 25. Kroetz DL, Pauli‐Magnus C, Hodges LM et al Sequence diversity and haplotype structure in the human ABCB1 (MDR1, multidrug resistance transporter) gene. Pharmacogenetics 2003; 13: 481–94. [DOI] [PubMed] [Google Scholar]
- 26. Hodges LM, Markova SM, Chinn LW et al Very important pharmacogene summary: ABCB1 (MDR1, P‐glycoprotein). Pharmacogenet Genomics 2011; 21: 152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kimchi‐Sarfaty C, Marple AH, Shinar S et al Ethnicity‐related polymorphisms and haplotypes in the human ABCB1 gene. Pharmacogenomics 2007; 8: 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CONSORT diagram of this study.
Fig. S2. Tumor responses to crizotinib from baseline in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism.
Fig. S3. Progression‐free survival in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism treated with crizotinib.
Fig. S4. Overall survival in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism treated with crizotinib.
Fig. S5. Mean plasma concentration–time curves of crizotinib on days 1 (■) and 15 (○) (mean ± standard deviation) in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism.
Table S1. Pharmacokinetic parameters of crizotinib in Japanese non‐small‐cell lung cancer patients with ABCB1 polymorphism adjusted by (A) body weight and (B) body surface area.