

Abstract
Reactive oxygen species (ROS) are an important endogenous source of DNA damage and oxidative stress in all cell types. Deficiency in tuberin resulted in increased oxidative DNA damage in renal cells. In this study, the role of tuberin in the regulating of ROS and NADPH oxidases was investigated. Formation of ROS and activity of NADPH oxidases were significantly higher in mouse embryonic fibroblasts and in primary culture of rat renal proximal tubular epithelial tuberin‐deficient cells compared to wild‐type cells. In addition, expression of NADPH oxidase (Nox)1, Nox2, and Nox4 (Nox isoforms) was higher in mouse embryonic fibroblasts and renal proximal tubular epithelial tuberin‐deficient cells compared to wild‐type cells. Furthermore, activity levels of NADPH oxidases and protein expression of all Nox isoforms were higher in the renal cortex of rat deficient in tuberin. However, treatment of tuberin‐deficient cells with rapamycin showed significant decrease in protein expression of all Nox. Significant increase in protein kinase C βII expression was detected in tuberin‐deficient cells, whereas inhibition of protein kinase C βII by bisindolylmaleimide I resulted in decreased protein expression of all Nox isoforms. In addition, treatment of mice deficient in tuberin with rapamycin resulted in significant decrease in all Nox protein expression. Moreover, protein and mRNA expression of all Nox were highly expressed in tumor kidney tissue of patients with tuberous sclerosis complex compared to control kidney tissue of normal subjects. These data provide the first evidence that tuberin plays a novel role in regulating ROS generation, NADPH oxidase activity, and Nox expression that may potentially be involved in development of kidney tumor in patients with tuberous sclerosis complex.
Keywords: Kidney, renal cells, reactive oxygen species, tumor suppressor gene, tumorigenesis
Reactive oxygen species (ROS) originate from different sources, including the mitochondrial electron transport chain, xanthine oxidase, myeloperoxidase, NADPH oxidases (Nox enzymes), and lipoxygenase.1, 2, 3 Organisms also possess enzymatic systems that physiologically generate ROS. The catalytic core of the phagocyte NADPH oxidase is the membrane‐integrated flavocytochrome b 558, comprising the two subunits p22phox and gp91phox, the latter of which contains a complete electron‐transferring apparatus (from NADPH to molecular oxygen) with binding sites for heme, flavin adenine dinucleotide (FAD), and NADPH.4, 5 NADPH oxidase enzymes and lipoxygenase are responsible for the production of ROS in response to hormones, growth factors, and cytokines.6, 7, 8 Among these oxygen metabolites are superoxide anions (O2 −), hydrogen peroxide (H2O2), and hydroxyl radicals (∙OH).9 These species are not all equally reactive with their prospective targets. Many of them have very short half‐lives, leading to little relevance in terms of signaling. The NADPH oxidases of the Nox family have been shown to generate ROS in certain cancer cells.10 These oxidases are proposed to play a role in a variety of events such as signaling for cell growth or cell death, oxygen sensing, and inflammatory processes.11
Reactive oxygen species product has deleterious effects on DNA, lipids, and proteins. Several studies have highlighted the importance of ROS as a second messenger in numerous cellular processes, including cell proliferation, gene expression, adhesion, differentiation, senescence, and apoptosis.12, 13 In addition, the superoxide‐generating oxidase Mox1, homologous to gp91phox, is highly expressed in colon and to a lesser extent in prostate, uterus, and vascular smooth muscle.14 NADPH oxidase 4 was found to be highly expressed in kidney,15 suggesting the significant role of Nox in ROS generation. Protein kinase C (PKC) is also known to induce cellular ROS and PKC‐dependent activation of the reduced form of NADPH oxidase has been shown to be responsible, in part, for increased oxidative stress.16
Renal tumors found in patients with tuberous sclerosis complex (TSC) are mainly benign angiomyolipoma and are rarely renal cell carcinoma.17 Tuberin encodes by tuberous sclerosis complex 2 (TSC2) and its deficiency resulted in accumulation of significant levels of oxidative DNA damage, in the form of 8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine, in kidney tumor of Eker rat.18 The precise mechanism by which tuberin regulates ROS generation and Nox expression in renal cells is not known. These data showed a novel role of tuberin as a key molecule in the regulation of ROS generation, superoxide‐producing NADPH oxidases, Nox isoforms in renal cells, in kidney from rodents and in tumor kidney of TSC patients.
Materials and Methods
Cell culture
Mouse embryonic fibroblast (MEF) cells
Tsc2 −/−, Tsc2 +/−, and Tsc2 +/+ mouse embryonic fibroblast (MEF) cells were generously provided by Dr. D. J. Kwiatkowski (Harvard Medical School, Boston, MA, USA). The cells were tested and authenticated by Dr. Kwiatkowski's laboratory. Cells were grown in DMEM supplemented with 10% FBS and serum‐deprived overnight. All cell lines were grown at 37°C in a humidified atmosphere of 5% CO2.
Renal primary proximal tubular epithelial cells
Fresh renal primary proximal tubular epithelial (RPTE) cells were isolated from kidney cortex of wild‐type and Tsc2 +/− rats at age of 4 months and cultured as previously described.19 The cells were tested and characterized for mutation in TSC2 by genotyping as previously described.20
Measurement of intracellular ROS production
The peroxide‐sensitive fluorescent probe 2′,7′‐dichlorodihydrofluorescein diacetate (DCF‐DA; Molecular Probes, Carlsbad, CA, USA) was used to assess the generation of intracellular ROS as described previously.21 Cells were grown in 6‐well plates and serum‐deprived overnight. Cells were washed with Hanks' balanced salt solution without phenol red and then incubated for 30 min in the dark at 37°C with the same solution containing the peroxide‐sensitive fluorophore DCF‐DA (Molecular Probes) at 5 μmol/L. The DCF‐DA fluorescence was detected at excitation and emission wavelengths of 488 and 520 nm, respectively, as measured with a multiwall fluorescence plate reader (Wallac 1420 Victor2; PerkinElmer Life Sciences, Waltham, MA).
Nicotinamide adenine dinucleotide phosphate oxidase assay
The NADPH oxidase activity was measured by the lucigenin‐enhanced chemiluminescence method using a microplate reader counter as described previously.22 Photon emission expressed as relative light units (RLU) was measured every 30 s for 5 min in a luminometer. A buffer blank was subtracted from each reading before calculation of the data. Superoxide production was expressed as the rate of RLU/min/mg protein. Protein concentration was determined with the Bradford reagent23 using BSA as a standard.
Treatment with mammalian target of rapamycin and PKC inhibitor
The MEF cells were grown to 80–90% confluency in 60‐mm Petri dishes and serum‐deprived overnight. Cells were then treated with different concentrations of rapamycin (0, 20, 40, 60, or 100 nM) or bisindolylmaleimide I (BMI; PKC inhibitor) (0, 2.5, or 5 μM) for 24 h. Rapamycin and BMI were purchased from Cayman Chemical (Ann Arbor, MI, USA). Cells were lysed in a lysis buffer as described previously.24 Cell lysates were used for Western blot analysis.
Protein extraction and immunoblot analysis
Protein concentration of the cell lysates was determined with the Bradford reagent23 using BSA as a standard. Western blot analysis was carried out as previously described.25 Tuberin, p‐p70S6K, and p70S6K antibodies were purchased from Cell Signaling Technology (Danvers, MA). GAPDH, PKCβII, and Nox4 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Nox1 was purchased from EMD Millipore (Billerica, MA, USA) and Nox2 from Abcam (Cambridge, MA, USA). Mouse β‐actin antibody was purchased from Oncogene Research Products (La Jolla, California). Rapamycin was purchased from Calbiochem (Billerica, MA, USA). Proteins were visualized by ECL solution. Expression of each protein was quantified by densitometry using NIH Image 1.62 software (Imagej.NIH.gov).
mRNA analysis by RT‐PCR
RNA was extracted from kidney tissue or MEF cells using the RNeasy Mini kit (Qiagen, Valencia, CA, USA). RNA was quantitated by spectrophotometry at 260 nm, and its integrity tested by formaldehyde/agarose gel electrophoresis. Primer sequences of Nox1, Nox2, and Nox4 as well as GAPDH were used as described by Li et al.26 The amplification for each Nox and GADPH was carried out using a Mastercycler Thermal Cycler (Eppendorf, Hauppauge, NY) programmed for 25 cycles. The amplified PCR products were separated on 2% agarose gels. The PCR products were analyzed on ethidium bromide stained gel. The yield was integrated for each sample on an image analyzer and the ratio of each isoform of Nox to GAPDH then calculated.
Animals
Rats
Male wild‐type (TSC2 +/+) and Eker mutant (TSC2 +/−) rats were purchased from a breeding colony maintained in‐house at the University of Texas M.D. Anderson Cancer Center (Smithville, TX, USA). The animals were allowed food and water ad libitum during the experiments. Animals were killed at 4 months for nephrectomy. Kidneys were quickly removed and snap frozen in liquid nitrogen for biochemical analysis.
Mice
Two‐month‐old male TSC2‐deficient (TSC2 +/−) mice were purchased from The Jackson Laboratory (Bar Harbor, MN, USA). The animals were allowed food and water ad libitum prior to and during the experiments. At age of 3 months, mice were divided into two groups of four mice each. Group 1 mice (controls) were injected with an equal amount of DMSO. Group 2 mice were injected i.p. with 2 mg/kg body weight rapamycin in DMSO 5 days/week for 4 weeks. Injections were carried out under isoflurane inhalation anesthesia (Abbott, Abbott Park, IL, USA). Animals were euthanized and the kidneys were removed rapidly for dissection and biochemical analysis. The Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio (San Antonio, TX, USA) approved these animal studies.
Human tissues
Human kidney tissues from de‐identified cases from TSC patients with renal angiomyolipoma and unrelated healthy people were obtained from the Tissue Bank for Development Disorders (University of Maryland, Baltimore, MD, USA) and San Antonio Cancer Institute Core (San Antonio, TX, USA). The study was approved by the Institutional Review Board of The University of Texas Health Science Center at San Antonio.
Statistics
Data are presented as mean ± standard error. Statistical differences were determined using anova followed by Student Dunnett's (Treatment. vs. Control) test using one trial analysis. P‐values <0.01 and 0.05 were considered statistically significant.
Results
Tuberin regulates ROS, NADPH‐dependent oxidase activity, and Nox expression in RPTE cells
Production of ROS in primary proximal tubular epithelial cells isolated from kidney of wild‐type and mutant Eker rats were evaluated. Intracellular ROS production was measured using the peroxide‐sensitive fluorescent probe DCF‐DA. The data in Figure 1(a) show that deficiency in tuberin resulted in more than 3‐fold increase in ROS formation in TSC2 +/− cells compared to wild‐type cells. In addition, NADPH‐dependent ROS generation measured by lucigenin‐enhanced chemiluminescence was significantly increased in TSC2 +/− cells compared to wild‐type cells (Fig. 1b). Moreover, cells expressing low levels of tuberin protein showed higher protein expression of Nox1, Nox2, and Nox4 that associated with increase in mammalian target of rapamycin (mTOR) activity (measured by protein expression of P‐p70S6K) compared to wild‐type cells (Fig. 1c). These data indicate that loss of one allele of TSC2 is sufficient to regulate ROS generation and increase the downstream expression of Nox protein.
Figure 1.
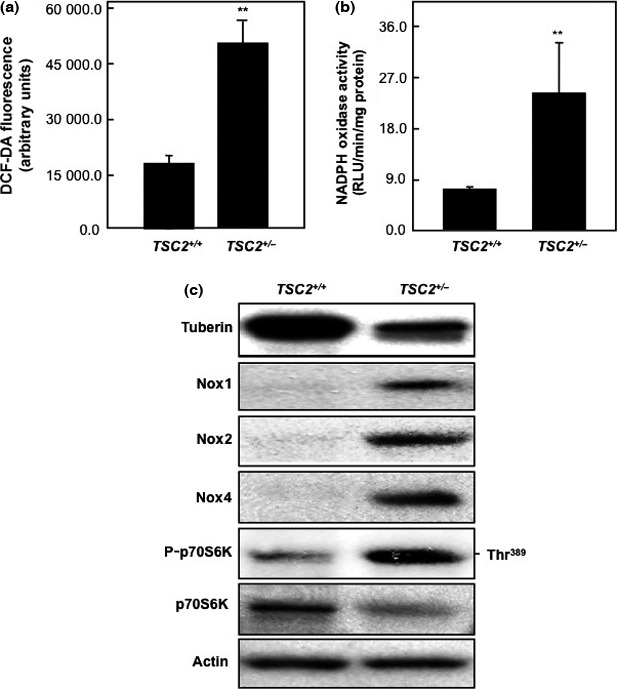
Tuberin deficiency increases reactive oxygen species (ROS) formation and NADPH‐dependent oxidase activity in renal primary proximal tubular epithelial (RPTE) cells isolated from wild‐type and TSC2 +/− rats. (a) Intracellular ROS production was measured using the peroxide‐sensitive fluorescent probe 2′,7′‐dichlorodihydrofluorescein diacetate (DCF‐DA) in RPTE cells. Deficiency in tuberin significantly increased ROS formation in TSC2 +/− cells compared to wild‐type cells. The data were quantitated, and the results are expressed as the means ± SE. (b) NADPH oxidase activity measured by lucigenin‐enhanced chemiluminescence in RPTE cell homogenates. Significant increases in NADPH oxidase activity were detected in TSC2 +/− cells compared to wild‐type cells. Production of NADPH oxidase activity is expressed as relative light units (RLU)/mg protein/min and normalized as a percentage of the control. **P < 0.01, significant difference from wild‐type cells. (c) Deficiency in tuberin results in upregulation of NADPH oxidase (Nox) expression in primary culture of RPTE cells. Cell lysates were prepared and protein extracts were loaded onto 7% SDS–polyacrylamide gels and transferred to a PVDF membrane. The membrane was incubated with anti‐tuberin or anti‐Nox1, anti‐Nox2, anti‐Nox4, anti‐P‐p70S6K, and anti‐p70S6K followed by specific HRP‐conjugated secondary antibodies. Actin was used as a loading control.
Tuberin deficiency resulted in increased NADPH oxidase activity and Nox protein expression in kidney cortex of Eker rat
To determine if tuberin deficiency in kidney of Eker rat is associated with upregulation of NADPH oxidase and Nox expression, kidney cortex homogenates from wild‐type and normal Eker rat were analyzed by Western blot. Tuberin deficiency is associated with significant increase in NADPH oxidase activity compared to kidney tissue from wild‐type rats (Fig. 2a). Tuberin expression in kidney from Eker rats is significantly reduced compared to wild‐type rats (Fig. 2b). In addition, kidney tissues from Eker rats showed significant increase in Nox1, Nox2, and Nox4 protein expression that associated with increase in mTOR activity (measured by protein expression of P‐p70S6K) compared to kidney tissue from wild‐type rats (Fig. 2b,c). These data demonstrate that tuberin is an important regulator of most of Nox isoforms.
Figure 2.
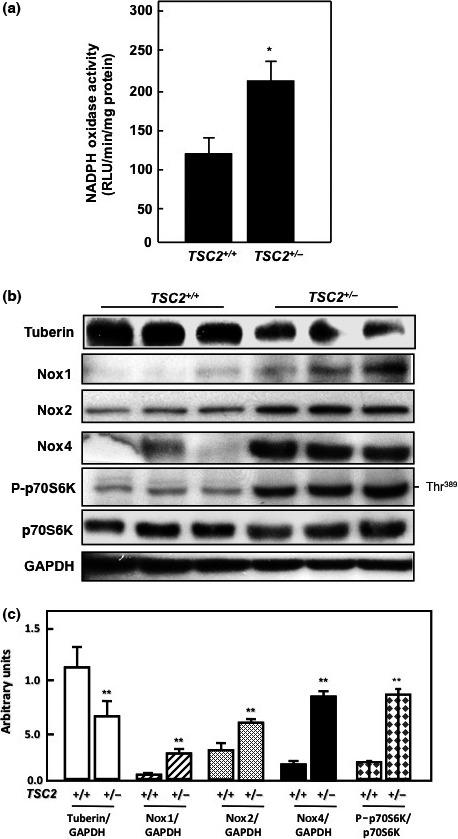
Deficiency in tuberin increases NADPH‐dependent oxidase generation in kidney cortex of TSC2 +/− Eker rat. (a) NADPH‐dependent superoxide generation was measured in control and tuberous sclerosis complex (TSC) kidney cortex. Values are the means ± SE of three independent experiments. *P < 0.01, significant difference from wild‐type rats. RLU, relative light units. (b) Deficiency in tuberin is associated with increased NADPH oxidase (Nox)1, Nox2, and Nox4 expression in kidney tissue of Eker rat. GAPDH were used as a loading control. (c) Histograms represent means ± SE. **P < 0.01, significant difference from wild‐type rats.
Loss of tuberin increases ROS formation, NADPH‐dependent oxidase activity, and Nox expression in MEF cells
To determine whether complete genetic deficiency of tuberin is similarly associated with increase ROS generation and NADPH oxidase activity, cell lysates of MEF expressing different copy numbers of TSC2 (TSC2 +/+, TSC2 +/−, and TSC2 −/−) were analyzed. Loss of tuberin in null cells or deficiency of tuberin in heterozygous cells was associated with significant increase in intracellular ROS production compared to wild‐type MEF cells (Fig. 3a). In addition, loss of tuberin in MEF cells showed 6‐fold increase in NADPH oxidase activity, whereas deficiency in tuberin showed approximately 2.5‐fold increase compared to wild‐type cells, suggesting the important role of tuberin in regulating NADPH oxidase activity in cells (Fig. 3b). Moreover, loss of tuberin in TSC2 −/− cells resulted in significant increase in Nox1, Nox2, and Nox4 protein (Fig. 3c) and mRNA expression (Fig. 3d) compared to wild‐type cells. Furthermore, TSC2 +/− cells showed a significant increase only in Nox2 and Nox4 protein and mRNA expression compared to wild‐type cells (Fig. 3c,d). These data suggest that tuberin is involved in the regulation of most Nox isoforms and indicate the important role of tuberin as a key molecule in protecting cells from accumulation of ROS and Nox generation.
Figure 3.
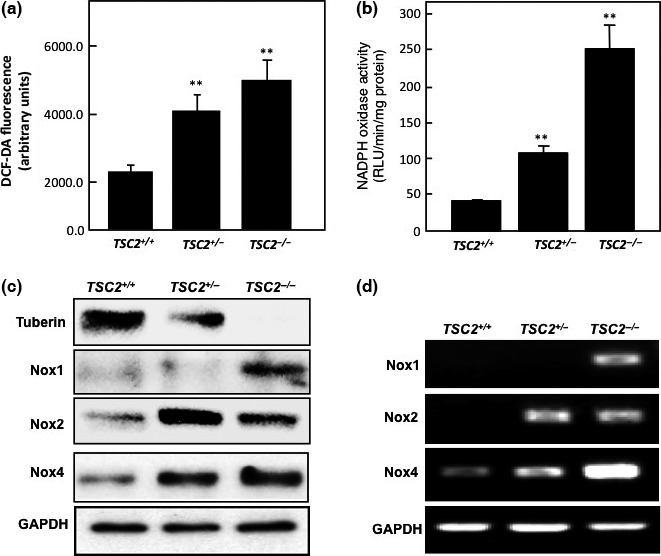
Deficiency in tuberin increases reactive oxygen species (ROS) formation and NADPH‐dependent oxidase activity in mouse embryonic fibroblast (MEF) cells. (a) 2′,7′‐Dichlorodihydrofluorescein diacetate (DCF‐DA) fluorescence in MEF cells. Intracellular ROS production was measured using the peroxide‐sensitive fluorescent probe DCF‐DA. The data were quantitated and the results are expressed as the means ± SE. (b) NADPH‐dependent ROS generation measured by lucigenin‐enhanced chemiluminescence in MEF cell homogenates. Production was expressed as relative light units (RLU)/mg protein/min and normalized as a percentage of the control. **P < 0.01, significant difference from wild‐type cells. (c, d) Deficiency in tuberin results in upregulation of NADPH oxidase (Nox) expression in MEF cells. Western blot analysis (c) and RT‐PCR analysis (d) showed that loss of tuberin in TSC2 −/− cells resulted in significant increase in protein and mRNA of Nox1, Nox2, and Nox4. TSC2 +/− cells showed a significant increase in protein and mRNA of Nox2 and Nox4 compared to wild‐type cells. GAPDH was used as loading control. PCR products were analyzed on an ethidium bromide stained gel.
Introduced tuberin into tuberin‐null cells decreases ROS generation, NADPH oxidase activity, and Nox expression
To confirm the role of tuberin as a key molecule in regulation of oxidative stress in cells, tuberin‐null cells were infected with Ad‐TSC2. The MEF TSC2 −/− cells were used to measure ROS generation using the peroxide‐sensitive fluorescent probe DCF‐DA. Data in Figure 4(a) show that introducing tuberin into null MEF cells resulted in significant decrease in ROS generation, close to the levels in wild‐type cells. In addition, introducing tuberin into null cells resulted in significant decrease in NADPH oxidase to 50% less than in wild‐type cells (Fig. 4b). Moreover, introducing tuberin to null cells (confirmed by Western blot analysis) resulted in decrease Nox2 and Nox4 protein expression to the levels expressed in wild‐type cells (Fig. 4c). These data confirm that tuberin is an upstream target of Nox.
Figure 4.
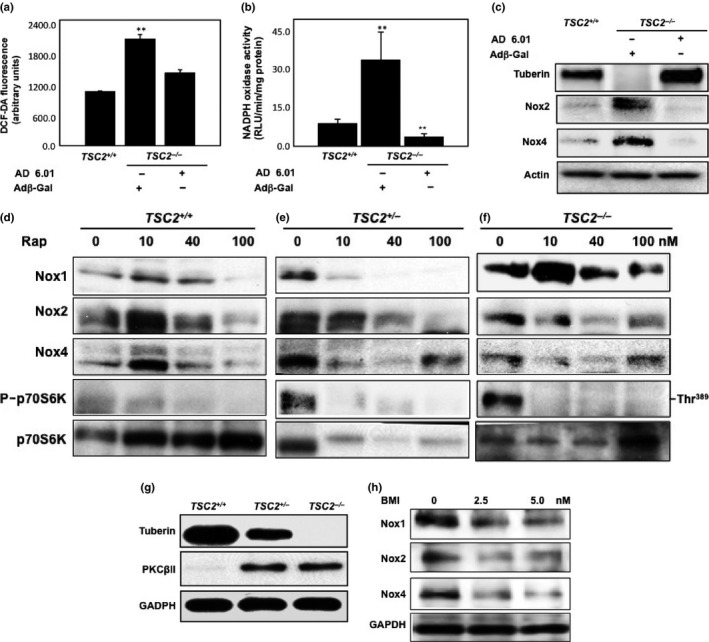
Introduction of TSC2 cDNA into tuberin‐deficient cells restored the wild‐type pattern of reactive oxygen species (ROS), decreased NADPH oxidase (Nox) activity, and abolished Nox isoform expression. Tuberin‐null cells were grown on a 6‐well plate and infected with an adenovirus expressing tuberin (Ad‐TSC2). An otherwise identical adenovirus expressing β‐Gal (Adβ‐Gal) was used as a control. (a) Intracellular ROS production was measured using the peroxide‐sensitive fluorescent probe 2′,7′‐dichlorodihydrofluorescein diacetate (DCF‐DA) in wild‐type and infected null‐tuberin cells with β‐Gal or Ad‐TSC2 (AD 6.01). The data were quantitated and the results are expressed as the means ± SE. (b) NADPH‐dependent ROS generation measured by lucigenin‐enhanced chemiluminescence in wild‐type and infected null‐tuberin cells with β‐Gal or Ad‐TSC2 (AD 6.01) homogenates. Production is expressed as relative light units (RLU)/mg protein/min and normalized as a percentage of the control. **P < 0.01, significant difference from wild‐type cells. (c) Introduction of TSC2 cDNA into tuberin‐deficient cells abolished Nox protein expression. Cell lysates of wild‐type and infected null‐tuberin cells with β‐Gal or Ad‐TSC2 (AD 6.01) were prepared. Western blot was carried out and the membrane was incubated with antibody specific for tuberin, Nox2, and Nox4, followed by HRP‐conjugated secondary antibody. The proteins were visualized with ECL. Actin expression served as a loading control. (d–f) Rapamycin blocks activation of mammalian target of rapamycin (mTOR) activation to decrease expression of Nox subunits in mouse embryonic fibroblast (MEF) cells. MEF cells were treated with different concentrations of rapamycin (0–100 nM) for 24 h. MEF cell lysates of TSC2 +/+, TSC2 +/−, and TSC2 −/− were prepared and protein extracts and analyzed by Western blot. Data showed a slight decrease in Nox1 in TSC2 +/− cells treated with 100 nM rapamycin. Rapamycin treatment at concentrations 40–100 nM showed a decrease in protein expression of Nox2. In addition, the higher concentration of rapamycin (40 nM) showed a significant decrease in Nox4 in TSC2 +/− and TSC2 −/− cells. (g, h) Tuberin‐deficient cells expressed higher protein kinase C (PKC) protein; treating the cells with PKC inhibitor significantly decreased Nox protein expression. (g) Significant increase in PKCβII expression was detected in TSC2 −/− and TSC2 +/− cells compared to TSC2 +/+ cells by Western blot analysis. (h) TSC2 −/− cells showed higher expression of all Nox isoforms were treated with 2.5 and 5.0 nm of bisindolylmaleimide I (BMI; PKC inhibitor) for 24 h before harvesting for Western blot analysis. Data showed significant decrease in the protein expression of Nox1, Nox2, and Nox4 compared to non‐treated cells, suggesting that PKC is an upstream target of Nox. GAPDH was used as loading control.
Inhibition of mTOR decreases Nox expression in MEF cells
We explored the role of mTOR in the regulation of Nox protein expression in all three MEF cell types. To investigate whether rapamycin can decrease Nox protein expression, MEF cells were treated with rapamycin. A significant decrease in Nox1 protein expression was evident in wild‐type and TSC2 +/− cells treated with 10–100 nM rapamycin (Fig. 4d–f), whereas slight decrease in Nox1 was shown in TSC2 −/− cells treated with 100 nM rapamycin compared to non‐treated cells. In addition, rapamycin treatment at higher concentrations 40–100 nM showed a decrease in protein expression of Nox2 in all three cells compared to non‐treated cells (Fig. 4d–f). In contrast, treatment with 100 nM rapamycin significantly decreased Nox4 expression in wild‐type cells, whereas 40 nM rapamycin significantly decreased Nox4 expression in TSC2 +/− and TSC2 −/− cells. Note that TSC2 −/− cells showed higher levels of mTOR activity compared to TSC2 +/− cells and wild‐type cells, but the activity was completely blocked by rapamycin treatment.
Tuberin deficiency increases PKC expression, while inhibition of PKC decreases Nox expression in MEF cells
We explored the mechanism by which PKC is involved in the regulation of Nox in MEF cells. To investigate whether tuberin is associated with decreased PKC expression and regulates Nox expression, MEF wild‐type (TSC2 +/+), hetero (TSC2 +/−), and null (TSC2 −/−) cell lysates were subjected to Western blot analysis. Both TSC2 +/− and TSC2 −/− cells showed high expression of PKCβII compared to its non‐detectable expression in wild‐type cells (Fig. 4g). Cells were treated with different concentrations of PKC inhibitor BMI (0, 2.5, and 5 μM) to test whether inhibition of PKC can decrease Nox expression in tuberin‐null cells. Cells treated with BMI showed significant decrease in Nox1, Nox2, and Nox4 protein expression (Fig. 4h). These data suggest that tuberin blocks the protein expression of PKC and controls expression of all Nox isoforms.
Inhibition of mTOR decreases Nox expression in kidney of tuberin‐deficient mice
To confirm the in vitro data that rapamycin blocks mTOR and downregulates Nox expression in tuberin‐deficient cells, TSC2 +/− mice were treated with rapamycin (2 mg/kg body weight), or with vehicle (DMSO) for 4 weeks. Homogenates of kidney cortex were examined for Nox1, Nox2, Nox4, and P‐p70S6K activity protein expression by Western blot analysis. Kidney tissues from mice treated with rapamycin showed decrease in expression of all Nox proteins in kidney compared to non‐treated mice (Fig. 5). Inhibition of mTOR activity (measured by p70S6K phosphorylation) by rapamycin is associated with decrease in expression of all Nox proteins. Collectively, these data obtained in vitro and in vivo strongly suggest that tuberin/mTOR is an upstream target of Nox.
Figure 5.
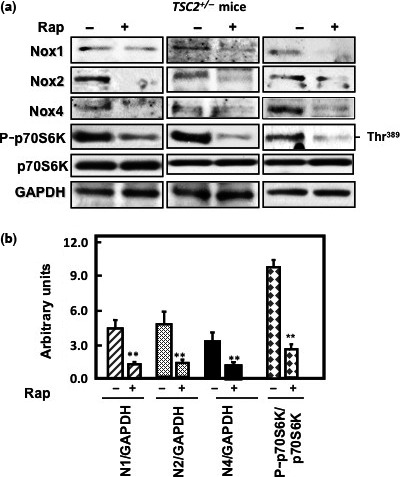
Rapamycin blocks mammalian target of rapamycin (mTOR) activation‐decreased NADPH oxidase (Nox) subunit expression in kidney tissue of TSC2 +/− mice. (a) Western blot analysis was carried out using kidney cortex homogenate of mice treated or not treated with rapamycin (Rap). Mice treated with rapamycin showed a significant decrease in phosphorylation of p70S6K at Thr389. Inactivation of mTOR resulted in decrease the protein expression of Nox in rapamycin‐treated mice. GAPDH was used as a loading control. (b) Histograms represent means ± SE. **P < 0.01, significant difference from non‐treated mice.
Tuberin deficiency resulted in increased protein expression and mRNA of Nox in kidney tumor of TSC patients
To determine the relevance of our findings in different cell lines and two animal models of TSC2 +/− to humans, tissue homogenates of control kidney and kidney tumors from patients with TSC were analyzed. Normal kidney tissues from control group showed higher protein expression of tuberin compared to kidney tumors from TSC patients. The decrease in tuberin expression in kidney tumor was associated with significant increase in Nox1, Nox2, and Nox4 protein expression (Fig. 6a,b) as well as mRNA expression (Fig. 6c,d). In addition, decrease in tuberin expression was associated with significant increase in mTOR activity in tumor tissue compared to control tissue (Fig. 6a,b). These data confirmed that tuberin is the key molecule in regulating oxidative stress and Nox expression in renal cells.
Figure 6.
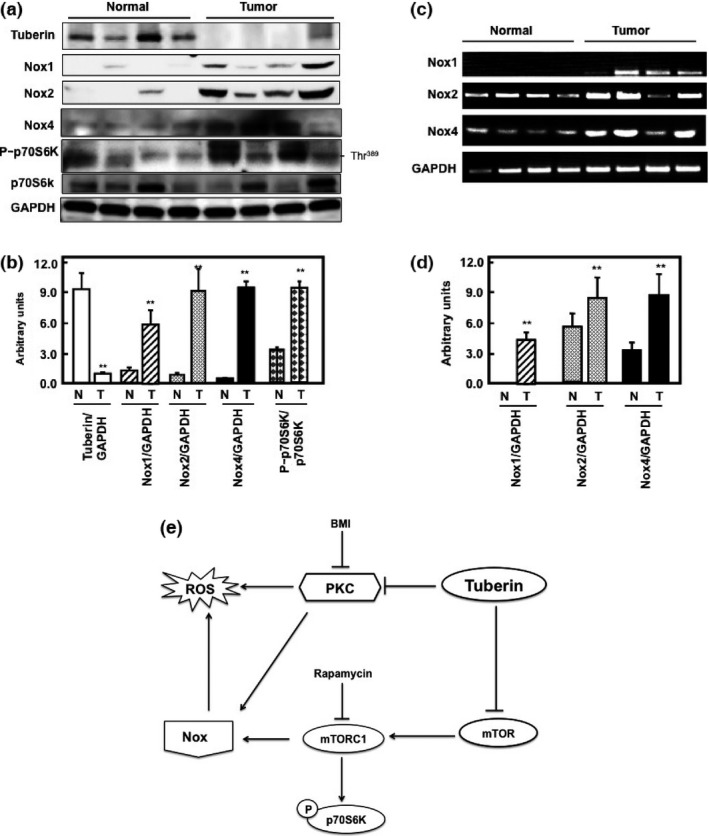
Tuberin deficiency increases expression of NADPH oxidase (Nox) subunits in tumor tissue of patients with tuberous sclerosis. (a) Western blot analysis showed that tuberin deficiency increases Nox1, Nox2, and Nox4 protein expression in tumor tissues. Deficiency in tuberin is associated with higher mammalian target of rapamycin (mTOR) activity (measured by phosphorylation of p70S6K at Thr389) in tumor tissue of patients compared to control kidney samples. (c) RT‐PCR was carried out in RNA isolated from normal and tumor tissue using specific human Nox PCR primers (Nox1, Nox2, and Nox4) or GAPDH as loading control. PCR products were analyzed on an ethidium bromide stained gel. Tuberin deficiency increases mRNA expression of Nox subunits in tumor tissues compared to normal control tissues. (b, d) Histograms represent means ± SE (n = 4). **P < 0.01, significant difference from normal subjects. GAPDH was used as loading control. (e) Proposed model of the regulation of reactive oxygen species (ROS) and Nox by tuberin. Tuberin blocks protein kinase C (PKC) and mTOR activities to decrease ROS generation and Nox expression, indicating that tuberin is the key molecule that controls ROS generation and Nox expression in renal cells. Inhibition of PKC by bisindolylmaleimide I (BMI) and mTOR by rapamycin results in decreased ROS generation and Nox expression.
Discussion
This study identifies tuberin as a key molecule in regulating ROS generation, NADPH oxidase activity, and Nox expression. Several approaches were used to conclusively show that tuberin regulates ROS generation and Nox expression. First, measurement of ROS levels, NADPH oxidase activity, and Nox expression in primary culture of rat renal proximal tubular epithelial cells showed that deficiency in tuberin resulted in higher levels of ROS and NADPH oxidase activity as well as higher Nox protein expression compared to wild‐type cells. Second, kidney cortex of Eker rats showed higher activity of NADPH and Nox protein expression, associated with increased mTOR activity, compared to kidney cortex from wild‐type rats. Third, MEF cells genetically deficient in tuberin showed higher levels of ROS and NADPH oxidase activity, and expressed higher protein and mRNA levels of all Nox isoforms, compared to wild‐type cells. Fourth, upregulation of tuberin by introduction of TSC2 cDNA into tuberin‐deficient cells resulted in significant decreases in ROS generation, NADPH oxidase activity, and protein expression of Nox isoforms. Moreover, to investigate the mechanism by which the tuberin/mTOR pathway regulates Nox expression, MEF cells treated with rapamycin showed a significant decrease in Nox protein expression, associated with decreased mTOR activity. In addition, inhibition of PKC activity by BMI inhibitor resulted in significant decrease in expression of all Nox, indicating the role of tuberin in regulating PKC to decrease ROS and Nox expression. Furthermore, rapamycin treatment of mice deficient in tuberin resulted in significant decreases in protein expression in all Nox isoforms. Lower levels of tuberin expression were associated with increases in protein and mRNA expression in all Nox in tumor tissue from TSC patients compared to control kidney tissue from healthy subjects. Taken together, these data suggest the strong role of tuberin in regulating oxidative stress and ROS generation in renal cells.
A major source of ROS is a family of NADPH oxidases that includes Nox1, Nox2, and Nox4. Nox4 is highly expressed in kidney compare to other Nox isoforms. Nox4 a major candidate for the origin of kidney.27, 28, 29, 30 Reactive oxygen species are chemically reactive products that damage several biomolecules including DNA, protein, and lipids. The data of renal primary tubular cells showed that cells deficient in tuberin accumulate excessive amounts of ROS and NADPH oxidase activity compared to cells isolated from wild‐type rats. In addition, tuberin‐deficient cells clearly expressed higher protein levels of Nox, Nox2, and Nox4, associated with increased mTOR activity, compared to wild‐type cells. These data suggest that tuberin blocks generation of ROS through inhibiting the activity of PKC and mTOR. Moreover, higher levels of Nox protein and mRNA expression in tubular cells suggest its function as an oxygen sensor for ROS production.
Acute tubular necrosis secondary to ischemic renal failure is a serious clinical problem. Nox‐generated ROS are implicated in the pathogenesis of a variety of kidney‐related diseases such as renal hypertension, diabetic nephropathy, and atherosclerosis.31 Our data of kidney cortex from tuberin‐deficient rats showed higher activity of NADPH oxidase and higher protein expression of Nox1, Nox2, and Nox4 compared to kidney cortex from wild‐type rats. These data confirm the specificity of tubular cells in expressing most Nox isoforms, as more than 80% of tubular cells are present in the cortex region of the kidney. In addition, tubular cells are the cells of origin of renal cell carcinoma and most other tumors in kidney. Therefore, these data suggest that higher expression of Nox and excessive levels of ROS may cause genetic mutations that are involved in kidney tumorigenesis.
Reactive oxygen species are known to be important mediators of many pathophysiological processes in renal diseases, probably by causing cell death through necrosis or apoptosis.32 MEF cells that are genetically null in tuberin show significant accumulation of ROS levels as well as higher activity of NADPH oxidase activity compared to heterozygous cells and wild‐type cells. In addition, higher expression of protein and mRNA of Nox1, Nox2, and Nox4 were detected in null‐tuberin cells compared to heterozygous cells and wild‐type cells, suggesting that tuberin strongly contributes to the regulation of ROS, NADPH oxidase activity, and increased Nox expression. These data were further confirmed when cDNA carrying tuberin was introduced into null cells and showed significant decrease in ROS generation, NADPH oxidase activity, and protein expression of Nox isoforms, indicating the novel role of tuberin as a key molecule in controlling ROS generation.
The serine–threonine kinase mTOR controls a variety of cellular functions and is implicated in a variety of cancers.33 Tuberin‐deficient cells as well as kidney cortex of TSC2 +/− mice express higher levels of mTOR activity, which was completely abolished by treating the cells and mice with rapamycin. Inhibition of mTOR activity by rapamycin in TSC2 +/− mice resulted in downregulation of Nox1, Nox2, and Nox4 protein expression. In addition, null‐tuberin cells treated with rapamycin showed significant decreases in protein expression of Nox2 and Nox4 but not in Nox1, suggesting an alternative mechanism of regulation of Nox1.
Protein kinase C isoforms are involved in a variety of cellular signaling pathways.34 These proteins have been shown to contain a unique structural feature that is susceptible to oxidative modification. Previous studies showed that PKCβII is required for TPA‐induced ROS production in myeloid leukemia cells.35 Another study showed that PKCβII mediated invasion that its plays, suggesting a major role in the promotion of colon carcinogenesis.36 In addition, a previous study showed that mutations in TSC2 activates Rac1 and increases ROS production.37 Another study showed that TSC2 responds to ROS at the peroxisome and identified that peroxisomes are signal organelles involved in mTOR regulation.38 Our tuberin‐null cells treated with PKC inhibitor showed significant decreases in protein expression of Nox1, Nox2, and Nox4 compared to non‐treated cells, suggesting the deficiency in tuberin resulted in increased PKC activity and led to accumulation of Nox protein. These data suggest that inhibition of PKC activity in tuberin‐deficient cells is an important step to prevent accumulation of ROS and oxidative stress.
Several studies have shown increased cell proliferation is due to the overproduction of ROS, whereas inhibition of NADPH oxidases was associated with decreased proliferation of cancer cells. Different organs with cancer show higher levels of Nox expression, including Nox4 in melanoma, Nox5 in prostate cancer, and Nox1 in glioblastoma and colon cancer.39, 40, 41, 42, 43, 44, 45 Other studies also showed that protein and mRNA of Nox1 are overexpressed beginning at the adenoma (precancerous) stage,46 consistent with our data in tumor tissue of TSC patients. Overexpression of Nox1, Nox2, and Nox4 protein and mRNA was correlated with deficiency in tuberin expression and elevated mTOR activity in tumor tissues. Although Nox overexpression seems to be a feature of many cancer cells, altered expression of many genes is a frequent feature of cancer.
In summary, these data comprise the first report of a novel role of tuberin in regulating ROS generation and Nox expression. These data confirmed the important role of tuberin as a key molecule in protecting cells from oxidative stress and from accumulating ROS. In addition, the data showed a novel mechanism by which the tuberin/PKC/mTOR pathway regulates ROS and Nox protein in several cell lines and animal models as well as human kidney tumor from TSC patients (Fig. 6e). These data shed light on the molecular mechanisms by which tuberin regulates Nox‐derived ROS in kidney tumorigenesis. Further studies to investigate the link between Nox isoforms and tumor cell phenotype is required to justify the consideration of Nox enzymes as drug targets for certain cancer treatments.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported in part by grants from the American Heart Association and the Merit Review Award from South Texas Veterans Healthcare System (to S.L.H.). This work is dedicated to the soul of Dr. Abboud.
Cancer Sci 107 (2016) 1092–1100
Funding Information
American Heart Association; South Texas Veterans Healthcare System.
References
- 1. Cave AC, Brewer AC, Narayanapanicker A et al NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal 2006; 8: 691–728. [DOI] [PubMed] [Google Scholar]
- 2. Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001; 269: 131–40. [DOI] [PubMed] [Google Scholar]
- 3. Deng S, Kruger A, Kleschyov AL, Kalinowski L, Daiber A, Wojnowski L. Gp91phox‐containing NAD(P)H oxidase increases superoxide formation by doxorubicin and NADPH. Free Radic Biol Med 2007; 42: 466–73. [DOI] [PubMed] [Google Scholar]
- 4. Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 2006; 290: 2–10. [DOI] [PubMed] [Google Scholar]
- 5. Lo W, Bravo T, Jadhav V, Titova E, Zhang JH, Tang J. NADPH oxidase inhibition improves neurological outcomes in surgically‐induced brain injury. Neurosci Lett 2007; 414: 228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vernon PJ, Tang D. Autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid Redox Signal 2013; 18: 677–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Altenhöfer S et al The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci 2012; 69: 2327–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murdoch CE, Alom‐Ruiz SP, Wang M et al Role of endothelial Nox2 NADPH oxidase in angiotensin II‐induced hypertension and vasomotor dysfunction. Basic Res Cardiol 2011; 106: 527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med 2009; 47: 1239–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hsieh CH, Shyu WC, Chiang CY, Kuo JW, Shen WC, Liu RS. NADPH oxidase subunit 4‐mediated reactive oxygen species contribute to cycling hypoxia‐promoted tumor progression in glioblastoma multiforme. PLoS ONE 2011; 6: e23945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pendyala S, Gorshkova IA, Usatyuk PV et al Role of Nox4 and Nox2 in hyperoxia‐induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal 2009; 11: 747–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Serrander L, Cartier L, Bedard K et al NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 2007; 406: 105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seyer‐Hansen K, Hansen J, Gundersen HJ. Renal hypertrophy in experimental diabetes. A morphometric study. Diabetologia 1980; 18: 501–5. [DOI] [PubMed] [Google Scholar]
- 14. Sharma K et al Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 2008; 118: 1645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Susztak K, Ramachandrarao S, Qiu G et al Glucose‐induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006; 55: 225–33. [PubMed] [Google Scholar]
- 16. Lee HB, Yu MR, Song JS, Ha H. Reactive oxygen species amplify protein kinase C signaling in high glucose‐induced fibronectin expression by human peritoneal mesothelial cells. Kidney Int 2004; 65: 1170–9. [DOI] [PubMed] [Google Scholar]
- 17. Habib SL. Insight into mechanism of oxidative DNA damage in angiomyolipomas from TSC patients. Mol Cancer 2009; 8: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Habib SL, Phan MN, Patel SK, Li D, Monks TJ, Lau SS. Reduced constitutive 8‐oxoguanine‐DNA glycosylase expression and impaired induction following oxidative DNA damage in the tuberin deficient Eker rat. Carcinogenesis 2003; 24: 573–82. [DOI] [PubMed] [Google Scholar]
- 19. Habib SL, Bhandari BK, Sadek N, Abboud‐Werner SL, Abboud HE. Novel mechanism of regulation of the DNA repair enzyme OGG1 in Tuberin‐deficient cells. Carcinogenesis 2010; 31: 2022–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ. Tsc2(+/−) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest 1999; 104: 687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lebel CP et al Evaluation of the probe 2′,7′ ‐dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 1992; 5: 227–31. [DOI] [PubMed] [Google Scholar]
- 22. Lee HB, Ischiropoulos H, Bondy SC. Reactive oxygen species‐regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol 2003; 14(Suppl 3): S241–5. [DOI] [PubMed] [Google Scholar]
- 23. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 1976; 72: 248–54. [DOI] [PubMed] [Google Scholar]
- 24. Habib SL, Kasinath BS, Arya RR, Vexler S, Velagapudi C. Novel mechanism of reducing tumourigenesis: upregulation of the DNA repair enzyme OGG1 by rapamycin‐mediated AMPK activation and mTOR inhibition. Eur J Cancer 2010; 46: 2806–20. [DOI] [PubMed] [Google Scholar]
- 25. Liang S, Cuevas G, Tizani S et al Novel mechanism of regulation of fibrosis in kidney tumor with tuberous sclerosis. Mol Cancer 2013; 29: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H, Han W, Villar VA et al D1‐like receptors regulate NADPH oxidase activity and subunit expression in lipid raft microdomains of renal proximal tubule cells. Hypertension 2009; 53: 1054–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. You YH, Okada S, Ly S et al Role of Nox in diabetic kidney disease. Am J Physiol Renal Physiol 2013; 304: F840–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Babelova A, Avaniadi D, Jung O et al Role of Nox4 in murine models of kidney disease. Free Radic Biol Med 2012; 53: 842–53. [DOI] [PubMed] [Google Scholar]
- 29. Kuroda J, Nakagawa K, Yamasaki T et al The superoxide‐producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 2005; 10: 1139–51. [DOI] [PubMed] [Google Scholar]
- 30. Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovasc Res 2005; 65: 16–27. [DOI] [PubMed] [Google Scholar]
- 31. Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008; 57: 1446–54. [DOI] [PubMed] [Google Scholar]
- 32. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, aging. Cell 2005; 120: 483–95. [DOI] [PubMed] [Google Scholar]
- 33. Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature 2013; 497: 217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cosentino‐Gomes D, Rocco‐Machado N, Meyer‐Fernandes JR. Cell signaling through protein kinase C oxidation and activation. Int J Mol Sci 2012; 13: 10697–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Datta R, Yoshinaga K, Kaneki M, Pandey P, Kufe D. Phorbol ester‐induced generation of reactive oxygen species is protein kinase c beta‐dependent and required for SAPK activation. J Biol Chem 2000; 275: 41000–3. [DOI] [PubMed] [Google Scholar]
- 36. Zhang J, Anastasiadis PZ, Liu Y, Thompson EA, Fields AP. Protein kinase C (PKC) betaII induces cell invasion through a Ras/Mek‐, PKC iota/Rac 1‐dependent signaling pathway. J Biol Chem 2004; 279: 22118–23. [DOI] [PubMed] [Google Scholar]
- 37. Suzuki T, Das SK, Inoue H et al Tuberous sclerosis complex 2 loss‐of‐function mutation regulates reactive oxygen species production through Rac1 activation. Biochem Biophys Res Commun 2008; 368: 132–7. [DOI] [PubMed] [Google Scholar]
- 38. Zhang J, Kim J, Alexander A et al A tuberous sclerosis complex signaling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat Cell Biol 2013; 10: 1186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brar SS, Corbin Z, Kennedy TP et al NOX5 NAD(P)H oxidase regulates growth and apoptosis in DU 145 prostate cancer cells. Am J Physiol Cell Physiol 2003; 285: C353–69. [DOI] [PubMed] [Google Scholar]
- 40. Lim SD, Sun C, Lambeth JD et al Increased Nox1 and hydrogen peroxide in prostate cancer. Prostrate 2005; 62: 200–7. [DOI] [PubMed] [Google Scholar]
- 41. Kawahara T, Kohjima M, Kuwano Y et al Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am J Physiol Cell Physiol 2005; 288: C450–7. [DOI] [PubMed] [Google Scholar]
- 42. Fu X, Beer DG, Behar J, Wands J, Lambeth D, Cao W. cAMP‐response element‐binding protein mediates acid‐induced NADPH oxidase NOX5‐S expression in Barrett esophageal adenocarcinoma cells. J Biol Chem 2006; 281: 20368–82. [DOI] [PubMed] [Google Scholar]
- 43. Lambeth JD. From SOD to Phox to Nox: a personal account of the evolving views of reactive oxygen species. Recent Advances and Research Updates 2003; 4: 31–40. [Google Scholar]
- 44. Geiszt M, Lekstrom K, Brenner S et al NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91(phox) in the regulated production of superoxide by phagocytes. J Immunol. 2003; 171: 299–306. [DOI] [PubMed] [Google Scholar]
- 45. Szanto I, Rubbia‐Brandt L, Kiss P et al Expression of NOX1, a superoxide‐generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J Pathol. 2005; 207: 164–76. [DOI] [PubMed] [Google Scholar]
- 46. Fukuyama M, Rokutan K, Sano T, Miyake H, Shimada M, Tashiro S. Overexpression of a novel superoxide‐producing enzyme, NADPH oxidase 1, in adenoma and well‐differentiated adenocarcinoma of the human colon. Cancer Lett 2005; 221: 97–104. [DOI] [PubMed] [Google Scholar]