

Abstract
Most patients with non‐small cell lung cancer (NSCLC) harboring common epidermal growth factor receptor (EGFR) mutations, such as deletions in exon 19 or the L858R mutation in exon 21, respond dramatically to EGFR tyrosine kinase inhibitors (EGFR‐TKI), and their sensitivities to various EGFR‐TKI have been well characterized. Our previous article showed the in vitro sensitivities of EGFR exon 18 mutations to EGFR‐TKI, but little information regarding the sensitivities of other uncommon EGFR mutations is available. First, stable transfectant Ba/F3 cell lines harboring EGFR L858R (Ba/F3‐L858R), L861Q (Ba/F3‐L861Q) or S768I (Ba/F3‐S768I) mutations were created and their drug sensitivities to various EGFR‐TKI were examined. Both the Ba/F3‐L861Q and Ba/F3‐S768I cell lines were less sensitive to erlotinib, compared with the Ba/F3‐L858R cell line, but their sensitivities to afatinib were similar to that of the Ba/F3‐L858R cell line. The Ba/F3‐L861Q cell line was similarly sensitive and the Ba/F3‐S768I cell line was less sensitive to osimertinib, compared with the Ba/F3‐L858R cell line. The results of western blot analyses were consistent with these sensitivities. Next, similar experiments were also performed using the KYSE270 (L861Q) and KYSE 450 (S768I) cell lines, and their results were compatible with those of the transfectant Ba/F3 cell lines. Our findings suggest that NSCLC harboring the EGFR L861Q mutation might be sensitive to afatinib or osimertinib and that NSCLC harboring the EGFR S768I mutation might be sensitive to afatinib. Overall, afatinib might be the optimal EGFR‐TKI against these uncommon EGFR mutations.
Keywords: Afatinib, epidermal growth factor receptor‐tyrosine kinase inhibitors, L861Q, S768I, uncommon epidermal growth factor receptor mutation
Activating mutations in the epidermal growth factor receptor (EGFR) gene occur in 40% of non‐small cell lung cancer (NSCLC) patients among Asians1 and in 20% of those among Caucasians.2, 3 The most common EGFR mutations are in‐frame exon 19 deletions and the exon 21 L858R point mutation, which constitute approximately 90% of all EGFR mutations.4 Most patients with NSCLC harboring exon 19 deletions or the L858R mutation respond dramatically to the first generation (1G) reversible EGFR tyrosine kinase inhibitors (EGFR‐TKI) gefitinib and erlotinib5, 6, 7, 8, 9 and to the second generation (2G) irreversible EGFR‐TKI afatinib.10, 11 Recently, third generation (3G) EGFR‐TKI, which are mutant‐selective and irreversible inhibitors, have been developed.12, 13, 14, 15, 16 3G EGFR‐TKI are reportedly effective for patients with NSCLC harboring the EGFR T790M mutation, which is the most common mechanism of acquired resistance to EGFR‐TKI.13, 14, 15, 16 To date, both the experimental and clinical efficacy of various types of EGFR‐TKI for common EGFR mutations have been reported, while less information about the sensitivities of uncommon EGFR mutations is available. The uncommon EGFR mutations, which include exon 18 mutations, S768I in exon 20, L861Q in exon 21 or insertions in exon 20, account for approximately 10% of EGFR mutations in NSCLC.17 Several studies, although very small, have shown that 1G EGFR‐TKI are less effective in patients with NSCLC harboring such uncommon EGFR mutations, compared with patients harboring common EGFR mutations.18, 19, 20, 21, 22, 23, 24, 25, 26 In contrast, our previous study, in which the sensitivities to various EGFR‐TKI for exon 18 mutations were investigated in vitro, showed the efficacy of afatinib or neratinib against exon 18 mutations.27 Indeed, afatinib was reported to be clinically effective for patients with NSCLC harboring uncommon EGFR mutations, including exon 18 mutations in a recent post‐hoc analysis.28 Although this study also indicated the efficacy of afatinib in patients with NSCLC harboring EGFR L861Q or S768I mutations, it remains unclear what EGFR‐TKI is optimal for patients with NSCLC harboring these uncommon EGFR mutations. In this study, we focused on the EGFR L861Q and S768I mutations and investigated the in vitro sensitivities of cells carrying these mutations to various EGFR‐TKI.
Materials and Methods
Structure of epidermal growth factor receptor
The crystal structure of EGFR in this study was modified based on the crystal structure (ID, 4G5J) deposited in the Protein Data Bank (PDB), which showed the structure of wild‐type EGFR in complex with afatinib.29 The modified picture was drawn using the PyMOL Molecular Graphics System (Version 1.7.4) (Schrodinger, New York, NY, USA), as previously described.27
Cell culture and reagents
The Ba/F3 cell line was maintained in IL‐3 additive RPMI1640 medium (Sigma‐Aldrich, St. Louis, MO, USA) with 10% FBS (Sigma‐Aldrich). Conditioned medium from WEHI‐3 cells was used as a source of IL‐3, as previously described.27 The KYSE270 and KYSE450 cell lines (human esophageal cancer cell lines) were maintained in a 1:1 mixture of RPMI1640 and F12 (Nissui Pharmaceutical, Tokyo, Japan) with 2% FBS according to a previously reported method.30, 31, 32 According to the Catalogue of Somatic Mutations in Cancer (COSMIC) database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), these cell lines carry uncommon EGFR mutations (KYSE270, L861Q and KYSE450, S768I, respectively). All the cell lines were maintained in a 5% CO2‐humidified atmosphere at 37°C. Erlotinib, afatinib and osimertinib were purchased from Selleck Chemicals (Houston, TX, USA).
Plasmid construction, viral production and stable transfectants
We constructed retrovirus vectors expressing enhanced green fluorescent protein (EGFP) and EGFR L858R, L861Q and S768I. The methods used in this section have been previously described.33 The retroviral vector pBABE, carrying the full‐length cDNA of wild‐type EGFR, was purchased from Addgene (Cambridge, MA, USA). A pBABE construct encoding EGFR L858R, L861Q or S768I was then generated using the Prime STAR Mutagenesis Basal Kit (TaKaRa, Shiga, Japan). The primers used for introducing each mutant EGFR were as follows: L858R‐F, GCGGGCCAAACTGCTGGGTGC; L858R‐R, AGCAGTTTGGCCCGCCCAAAAATCTGTGATCTTG; L861Q‐F, GCCAAACAGCTGGGTGCGGAAGAGAA; L861Q‐R, ACCCAGCTGTTTGGCCAGCCCAAAATC; S768I‐F, ATGGCCATCGTGGACAACCCCCACGT and S768I‐R, GTCCACGATGGCCATCACGTAGGCTTC. All the mutations were confirmed by sequencing. The stable viral transfectant Ba/F3 cell lines were designated as Ba/F3‐EGFP, Ba/F3‐L858R, Ba/F3‐L861Q and Ba/F3‐S768I, respectively.
Western blot analysis
A western blot analysis was performed as described previously.34 Rabbit antibodies specific for EGFR, phospho‐EGFR, caspase‐3, cleaved‐caspase‐3 and β‐actin were obtained from Cell Signaling (Beverly, MA, USA). To evaluate the influence of reagents on phosphorylation and apoptosis, the cells were stimulated for 3 and 24 h, respectively.
IL‐3 independent cell growth assay
A total of 1 × 103 Ba/F3 cells were plated in each well of a 96‐well plate and grown in RPMI medium with 10% FBS in the absence of IL‐3 for 72 h. The cell growth was examined using a 3‐(4, 5‐di‐methylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT; Sigma‐Aldrich) assay, as described previously.35
In vitro growth inhibition assay
The growth‐inhibitory effects of various EGFR‐TKI were examined using an MTT assay, as described previously.34 When the transfectant Ba/F3 cell lines were used, the cells were cultured without IL‐3. The experiment was performed in triplicate.
Statistical analysis
The results of experiments were presented as the mean values ± SD or the mean of independent triplicate experiments and were analyzed using the Student t‐test. The statistical analyses were two‐tailed and were performed using Microsoft Excel (Microsoft, Redmond, WA, USA). A P‐value less than 0.05 was considered statistically significant.
Results
EGFR L861Q or S768I mutations have an oncogenic activity similar to that of the EGFR L858R mutation
The crystal structure of EGFR was drawn using the PyMOL Molecular Graphics System based on crystal structure information from PDB ID 4G5J. The structure of EGFR is shown in Figure 1a. Codon 768 is located in the alpha‐C helix, and both codons 858 and 861 are located in the activation loop. To assess the effects of EGFR‐TKI on these EGFR mutations properly, Ba/F3 cell lines harboring each EGFR mutation (L858R, L861Q, or S768I) were created using a retroviral method. EGFR overexpression was confirmed by a western blot analysis (Fig. 1b), and an IL‐3 independent cell growth assay was performed. The Ba/F3 cell line is well known to be dependent on IL‐3 but to be rendered IL‐3 independent by induction with activating mutations of tyrosine kinase oncogenes.36 Although the Ba/F3 and Ba/F3‐EGFP cell lines could not grow in the absence of IL‐3, all the Ba/F3 cell lines harboring each EGFR mutation (L858R, L861Q or S768I) were able to grow IL‐3 independently (Fig. 1c). These results indicate that both EGFR L861Q and S768I are activating oncogenic mutations similar to the common mutation EGFR L858R.
Figure 1.
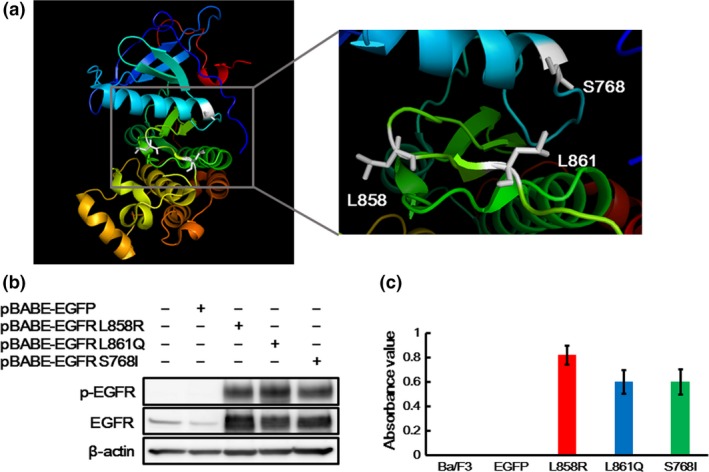
Crystal structure and oncogenic activities of EGFR L861Q and S768I mutations. (a) Structure of epidermal growth factor receptor (EGFR). The figures were drawn using the PyMOL Molecular Graphics System based on the crystal structure information from PDB ID 4G5J. Codon 768 is located in the alpha‐C helix, and both codons 858 and 861 are located in the activation loop. (b) Expressions of EGFR in the transfectant Ba/F3 cell lines. EGFR overexpression was confirmed by a western blot analysis. EGFR was strongly expressed in the Ba/F3 cell lines harboring each EGFR mutation. The phosphorylation level of EGFR was also elevated in the Ba/F3‐L861Q and Ba/F3‐S768I cell lines, similar to that in the Ba/F3‐L858R cell line. β‐actin was used as an internal control. p‐EGFR, phospho‐EGFR. (c) IL‐3 independent cell growth assay. A cell growth assay without IL‐3 was performed using an MTT assay. The Ba/F3 and Ba/F3‐EGFP cell lines could not grow in the absence of IL‐3, while all the Ba/F3 cell lines harboring each EGFR mutation could grow IL‐3 independently. Columns, mean of independent triplicate experiments; error bars, SD.
Different sensitivities to various epidermal growth factor receptor‐tyrosine kinase inhibitors in each transfectant Ba/F3 cell line
The Ba/F3 cell line is often used as a model system for assessing the effects of kinase oncogenes and their sensitivities to inhibitors.36 Therefore, to investigate the sensitivities to various EGFR‐TKI, growth inhibition assays in each transfectant Ba/F3 cell line were performed using an MTT assay. To evaluate the difference in sensitivities to various EGFR‐TKI, erlotinib, afatinib and osimertinib (1G, 2G and 3G EGFR‐TKI, respectively) were used, because these EGFR‐TKI are widely used in clinical settings. The 50% inhibitory concentration (IC50) values of all the EGFR‐TKI examined in this study are summarized in Table 1. The Ba/F3‐L861Q and Ba/F3‐S768I cell lines were less sensitive to erlotinib, compared with the Ba/F3‐L858R cell line. In contrast, their sensitivities to afatinib were similar to that of the Ba/F3‐L858R cell line. The Ba/F3‐L861Q cell line was similarly sensitive and the Ba/F3‐S768I cell line was less sensitive to osimertinib, compared with the Ba/F3‐L858R. (Fig. 2a).
Table 1.
IC50 values of various EGFR‐TKIs in the transfectant Ba/F3 cell lines
| EGFR‐TKI (nM) | EGFR mutation | ||
|---|---|---|---|
| L858R | L861Q | S768I | |
| Erlotinib | 4.5 | 92 | 146 |
| Afatinib | 0.2 | 0.5 | 0.7 |
| Osimertinib | 2.5 | 9 | 49 |
IC50, 50% inhibitory concentration; EGFR, epidermal growth factor receptor; TKI, tyrosine kinase inhibitor.
Figure 2.
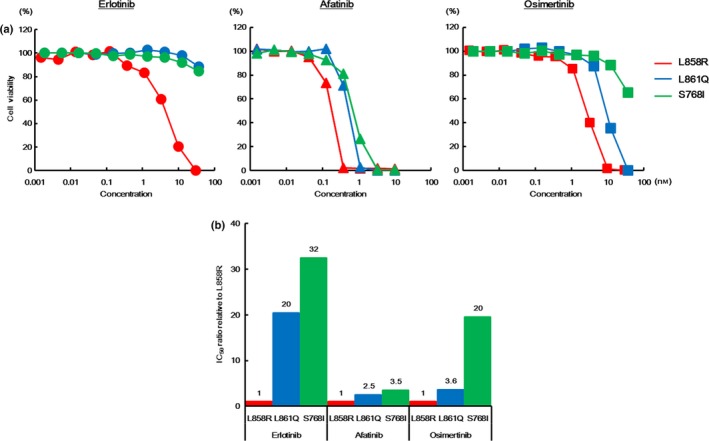
Sensitivities to various epidermal growth factor receptor‐tyrosine kinase inhibitors (EGFR‐TKI) in the transfectant Ba/F3 cell lines (L858R, L861Q and S768I). (a) Growth inhibitory curves of various EGFR‐TKI in the transfectant Ba/F3 cell lines. The cells were exposed to each concentration of various EGFR‐TKI for 72 h, and the growth inhibitory effects were evaluated using an MTT assay. The Ba/F3‐L861Q and Ba/F3‐S768I cell lines were less sensitive to erlotinib, compared with the Ba/F3‐L858R cell line. In contrast, the sensitivities of the Ba/F3‐L861Q and Ba/F3‐S768I cell lines to afatinib were similar to that of the Ba/F3‐L858R cell line. The Ba/F3‐L861Q cell line was similarly sensitive and the Ba/F3‐S768I cell line was less sensitive to osimertinib, compared with the Ba/F3‐L858R cell line. Lines, mean of independent triplicate experiments. (b) IC50 ratios of various EGFR‐TKI in the transfecant Ba/F3 cell lines. To evaluate the difference in drug sensitivities to various EGFR‐TKI in each EGFR mutation, the IC50 ratios relative to the Ba/F3‐L858R cell line were calculated. The IC50 values of erlotinib in the Ba/F3‐L861Q and the Ba/F3‐S768I cell lines were both much higher than that in the Ba/F3‐L858R cell line (IC50 ratio, 20‐fold and 32‐fold, respectively). In contrast, there was no such difference in the IC50 values of afatinib among these cell lines. The IC50 ratios of afatinib in the Ba/F3‐L861Q and the Ba/F3‐S768I cell lines were 2.5‐fold and 3.5‐fold, respectively. The IC50 value of osimertinib in the Ba/F3‐L861Q cell line was not relatively high, compared with that in the Ba/F3‐L858R cell line (3.6‐fold), whereas that in the Ba/F3‐S768I cell line was much higher than that in the Ba/F3‐L858R cell line (20‐fold).
Next, we estimated the IC50 ratios relative to the Ba/F3‐L858R cell line to evaluate the difference in drug sensitivities to various EGFR‐TKI in each EGFR mutation distinctly. As shown in Figure 2b, the IC50 values of erlotinib in both the Ba/F3‐L861Q and Ba/F3‐S768I cell lines were much greater than that of the Ba/F3‐L858R cell line (IC50 ratio, 20‐fold and 32‐fold, respectively). In contrast, there was no such difference in the IC50 values of afatinib among the Ba/F3‐L858R, Ba/F3‐L861Q and Ba/F3‐S768I cell lines. The IC50 ratios of afatinib in the Ba/F3‐L861Q and Ba/F3‐S768I cell lines relative to the Ba/F3‐L858R cell line were less than 5‐fold (2.5‐fold and 3.5‐fold, respectively) (Fig. 2b). The IC50 value of osimertinib in the Ba/F3‐L861Q cell line was not relatively high, compared with that of the Ba/F3‐L858R cell line (IC50 ratio, 3.6‐fold), whereas the IC50 value of osimertinib in the Ba/F3‐S768I cell line was much higher than that in the Ba/F3‐L858R cell line (IC50 ratio, 20‐fold) (Fig. 2b). These results suggest that NSCLC harboring the EGFR L861Q mutation might be sensitive to afatinib or osimertinib, and that NSCLC harboring the EGFR S768I mutation might be sensitive to afatinib.
Differences in epidermal growth factor receptor inhibitory effects of various epidermal growth factor receptor‐tyrosine kinase inhibitors among EGFR L858R, L861Q, and S768I mutations
Next, to estimate the EGFR inhibitory effects of these EGFR‐TKI for each EGFR mutation, western blot analyses were performed. Afatinib inhibited the phosphorylation of EGFR to almost the same degree in all of the transfectant Ba/F3 cell lines, whereas erlotinib inhibited the phosphorylation of EGFR to a lesser degree in both the Ba/F3‐L861Q and Ba/F3‐S768I cell lines than in the Ba/F3‐L858R cell line (Fig. 3). In particular, the phosphorylation of EGFR persisted in the Ba/F3‐L861Q and Ba/F3‐S768I cell lines even in the presence of 100 nM of erlotinib (Fig. 3). Osimertinib inhibited the phosphorylation of EGFR to almost the same degree in the Ba/F3‐L861Q cell line but to a lesser degree in the Ba/F3‐S768I cell line, compared with that in the Ba/F3‐L858R cell line (Fig. 3). The phosphorylation of EGFR also strongly persisted in the Ba/F3‐S768I cell line in the presence of 100 nM of osimertinib (Fig. 3). These results are consistent with those of the growth inhibitory assays, indicating that the difference in sensitivities to various EGFR‐TKI is caused by the difference in the EGFR inhibitory effects of various EGFR‐TKI on each EGFR mutation.
Figure 3.
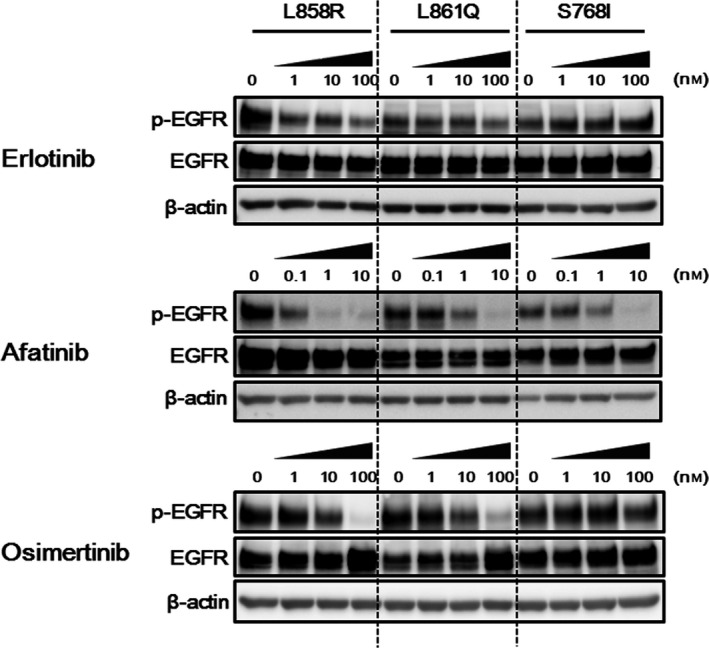
Difference in epidermal growth factor receptor (EGFR) inhibitory effects of various EGFR‐TKI among the transfectant Ba/F3 cell lines (L858R, L861Q and S768I). Three hours after the cells were treated with the indicated concentrations of drugs, the samples were collected. Afatinib inhibited the phosphorylation of EGFR to almost the same degree in all the transfectant Ba/F3 cell lines, whereas erlotinib inhibited the phosphorylation of EGFR to a lesser degree in both the Ba/F3‐L861Q and Ba/F3‐S768I cell lines, compared with the Ba/F3‐L858R cell line. Osimertinib inhibited the phosphorylation of EGFR to almost the same degree in the Ba/F3‐L861Q cell line and to a lesser degree in the Ba/F3‐S768I cell line, compared with that in the Ba/F3‐L858R cell line. β‐actin was used as an internal control. p‐EGFR, phospho‐EGFR.
KYSE270 and KYSE450 cell lines exhibit a tendency similar to that of the transfectant Ba/F3 cell lines
Next, we investigated the effects of various EGFR‐TKI in EGFR‐mutated cancer cell lines to comfirm the experimental results that were obtained using the Ba/F3 cell lines. No NSCLC cell line harboring uncommon EGFR mutations, such as EGFR L861Q or S768I mutations, could be found. Instead, the eshopageal cancer cell lines KYSE270 (L861Q) and KYSE450 (S768I) were used to evaluate the sensitivities to various EGFR‐TKI, the EGFR signal and apoptosis. Both the KYSE270 and KYSE450 cell lines were most sensitive to afatinib and least sensitive to erlotinib (Fig. 4a). Both cell lines were intermediately sensitive to osimertinib. Among them, osimertinib was as effective against the KYSE270 cell line as afatinib but was much less effective than afatinib against the KYSE450 cell line (Fig. 4a). The IC50 values of erlotinib, afatinib and osimertinib in the KYSE 270 cell line were 0.7, 0.004 and 0.03 μM, respectively, and those of erlotinib, afatinib and osimertinib in the KYSE 450 cell line were 1.1, 0.004 and 0.4 μM, respectively (Fig. 4b).
Figure 4.
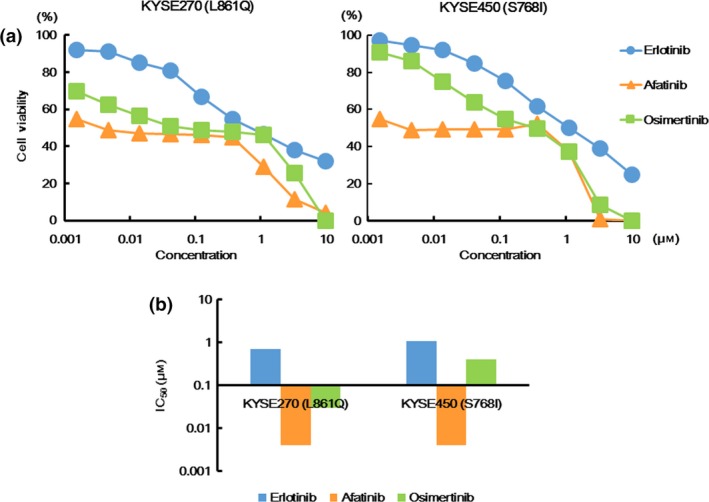
Sensitivities to various EGFR‐TKI in the KYSE270 and KYSE450 cell lines. (a) Growth inhibitory curves of various EGFR‐TKI in the KYSE270 and KYSE450 cell lines. The cells were exposed to each concentration of various EGFR‐TKI for 72 h, and the growth inhibitory effects were evaluated using an MTT assay. Both the KYSE270 (L861Q) and KYSE450 (S768I) cell lines were most sensitive to afatinib and least sensitive to erlotinib. Osimertinib was as effective for the KYSE270 cell line as afatinib, but was much less effective for the KYSE450 cell line than afatinib. Lines, mean of independent triplicate experiments. (b) IC50 values of various EGFR‐TKI in the KYSE270 and KYSE450 cell lines. The IC50 values of erlotinib, afatinib and osimertinib in the KYSE 270 cell line (L861Q) were 0.7, 0.004 and 0.03 μM, respectively, while those of erlotinib, afatinib and osimertinib in the KYSE 450 cell line (S768I) were 1.1, 0.004 and 0.4 μM, respectively.
In both cell lines, afatinib markedly inhibited the phosphorylation of EGFR, compared with erlotinib (Fig. 5a). Osimertinib intermediately inhibited the phosphorylation. In particular, in the KYSE450 cell line, its EGFR inhibitory effect was much weaker than that of afatinib (Fig. 5a). In the KYSE270 cell line, afatinib and osimertinib elevated the expression level of cleaved caspase‐3, whereas only afatinib elevated this level in the KYSE450 cell line (Fig. 5b). Based on the results of western blot analyses, the difference in the EGFR inhibitory effects and the induction of apoptosis reflected the different sensitivities to various EGFR‐TKI. These results were compatible with those of the transfectant Ba/F3 cell lines.
Figure 5.
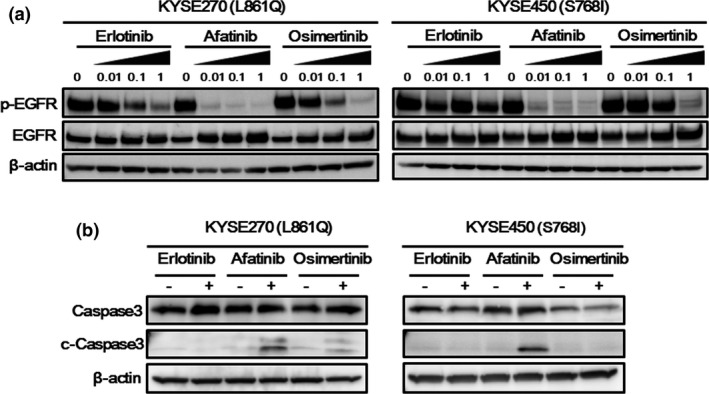
Western blotting in the KYSE270 and KYSE450 cell lines. (a) Phosphorylation of epidermal growth factor receptor (EGFR). Three hours after the cells were treated with the indicated drug concentrations, the samples were collected. In both the KYSE270 (L861Q) and the KYSE450 (S768I) cell lines, afatinib markedly inhibited the phosphorylation of EGFR, compared with erlotinib. Osimertinib intermediately inhibited the phosphorylation. In the KYSE450 cell line, especially, the EGFR inhibitory effect of osimertinib was much weaker than that of afatinib. β‐actin was used as an internal control. p‐EGFR, phospho‐EGFR. (b) Expression of apoptosis‐related molecules. Twenty‐four hours after the cells were exposed to each drug (0.1 μM), the samples were collected. In the KYSE270 cell line (L861Q), afatinib and osimertinib induced an expression of cleaved caspase‐3, whereas only afatinib induced the expression of cleaved caspase‐3, in the KYSE450 cell line (S768I). β‐actin was used as an internal control. c‐Caspase3, cleaved caspase‐3.
Discussion
Lung cancer is the leading cause of cancer‐related mortality worldwide, accounting for 20% of all cancer‐related deaths.37, 38 NSCLC accounts for approximately 85% of all lung cancers, and the EGFR mutation frequency in patients with NSCLC is relatively high (Asia, 47%; North America, 22%; and Europe, 15%).38 Therefore, NSCLC harboring uncommon EGFR mutations should not be overlooked, although uncommon EGFR mutations account for only 10% of all EGFR mutations in NSCLC.17 In the present study, the uncommon EGFR mutations L861Q and S768I were focused on, and the in vitro differences in sensitivities to various EGFR‐TKI and the efficacy of afatinib for both EGFR L861Q and S768I mutations were revealed for the first time.
Most studies examining uncommon EGFR mutations are small retrospective studies or case reports, and clinical data is limited because this population is “uncommon” (G719X, 5%; S768I, 1%; L861Q, 3%; exon 20 insertions, 3%).17 Although several articles have reported patients with NSCLC harboring uncommon mutation who responded to 1G EGFR‐TKI, getitinib or erlotinib,39, 40 many studies have shown that these uncommon EGFR mutations were less sensitive to 1G EGFR‐TKI (objective response rate, less than 50%), compared with common EGFR mutations.18, 19, 20, 21, 22, 23, 24, 25, 26 Preclinical studies showing a lower efficacy of 1G EGFR‐TKI for such uncommon mutations supports these clinical data.27, 41, 42, 43 Our previous study has shown that EGFR exon 18 mutations are less sensitive to 1G EGFR‐TKI, compared with common mutations,27 and a similar tendency for EGFR L861Q or S768I mutations was observed in the present study. These findings are consistent with previous clinical and in vitro studies.18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 37, 38, 39 In contrast, a combined post‐hoc analysis revealed a favorable progression‐free survival and treatment response for afatinib in patients with NSCLC harboring uncommon EGFR mutations, including G719X, L861Q and S768I mutations (objective response rate, 77.1%; disease control rate, 84.2%; progression free survival, 10.7 months).28 Our previous in vitro study has demonstrated the efficacy of afatinib or neratinib for the exon 18 mutation, including G719X.27 Furthermore, in our present study, the EGFR L861Q or S768I mutation was considered to be sensitive to afatinib. Thus, these results were consistent with the favorable outcome of afatinib‐treated patients with these uncommon EGFR mutations.28 In contrast, like 1G EGFR‐TKI, afatinib might be less effective for most exon 20 insertions in preclinical and clinical studies.24, 25, 28, 42, 43 Few reports regarding the effectiveness of 3G EGFR‐TKI against patients with NSCLC harboring uncommon EGFR mutations are available. Recent preclinical studies revealed that osimertinib might be partially effective for exon 20 insertions.42, 43 Our previous in vitro study showed a lower efficacy of 3G EGFR‐TKI, including osimertinib, for EGFR exon 18 mutations,27 and the present in vitro study indicated that osimertinib might be effective against the EGFR L861Q mutation, but is likely to be less effective against the EGFR S768I mutation. Considering these findings including our studies, 18, 19, 20, 21, 22, 23, 27, 28, 41 afatinib might be optimal for patients with NSCLC harboring the EGFR uncommon mutations including exon 18 mutations, L861Q and S768I in clinical settings.
We also found that the different EGFR inhibitory effects of EGFR‐TKI reflected the different sensitivities to various EGFR‐TKI. Several studies, in which affinities between kinases and various EGFR‐TKI were investigated, indicate that the different EGFR inhibitory effects of EGFR‐TKI might be associated with the different structural‐based interactions.25, 44 However, a similar study for 3G EGFR‐TKI has not yet been reported. Both 2G and 3G EGFR‐TKI are irreversible small‐molecule inhibitors that covalently bind to EGFR kinase by targeting the cysteine‐797 residue, located in the ATP binding pocket, and 3G EGFR‐TKI are considered to be more specific for EGFR sensitizing mutations than 2G EGFR‐TKI.12, 13, 14 Nevertheless, in our studies, 3G EGFR‐TKI seemed to be less effective against the uncommon EGFR mutations. Although the detailed mechanism remains unclear, we speculated that this difference in the response of cell lines harboring uncommon EGFR mutations to afatinib and osimertinib might be caused by different structural‐based interactions based on the present findings. Further investigations, including structural analyses, are needed to clarify our hypothesis.
This study had several limitations. First, neither the binding affinities between tyrosine kinases and EGFR‐TKI nor the protein structures could be analyzed. Instead, we determined the crystal structure of EGFR using the PDB database. Second, we did not use NSCLC cell lines, but rather artificially transfected Ba/F3 cell lines and esophageal cancer cell lines, because we could not find NSCLC cell lines harboring uncommon EGFR mutations such as the EGFR L861Q or S768I mutations. However, the Ba/F3 cell line has often been used in many studies investigating EGFR mutations,24, 27, 36, 41, 42 and trends similar to the results obtained using transfectant Ba/F3 cell lines were confirmed in the KYSE cell lines, even though they are not NSCLC cell lines. Third, we could not analyze clinical data or samples. The number of patients with NSCLC harboring such uncommon mutations is too small to show such data. Large‐scale clinical trials should be performed but might be difficult in such small patient subgroups. Therefore, studies, like ours, investigating the in vitro sensitivities to various EGFR‐TKI might be valuable.
In conclusion, based on our present in vitro findings, NSCLC harboring the EGFR L861Q mutation might be sensitive to afatinib or osimertinib, and NSCLC harboring the EGFR S768I mutation might be sensitive to afatinib. Our previous study also indicated that afatinib or neratinib might be effective against NSCLC harboring EGFR exon 18 mutations. Taken together, these findings suggest that afatinib might be optimal for NSCLC patients harboring the uncommon EGFR mutations including exon 18 mutations, L861Q and S768I in clinical settings. Our in vitro study may help clinicians to select an appropriate EGFR‐TKI in such subsets of NSCLC patients. To confirm these findings, further clinical and basic research is needed.
Disclosure Statement
Y. Togashi has received a lecture fee from Boehringer‐Ingelheim, T. Mitsudomi has received lecture fees from Astra‐Zeneca, Boehringer‐Ingelheim, Chugai and Pfizer and research funding from Astra‐Zeneca, Boehringer‐Ingelheim, Chugai and Pfizer. K. Nishio has received lecture fees from Chugai, Daiichi Sankyo and Sumitomo Bakelite. The other authors do not have any potential conflicts of interest to report.
Acknowledgments
We thank Ms Tomoko Kitayama and Ms Ayaka Kurumatani for their technical assistance.
Cancer Sci 107 (2016) 1134–1140
Funding Information
This study was supported in part by a Grant‐in‐Aid for Research Activity start‐up (Y. Togashi; 15H06754). Japan Society for the Promotion of Science.
References
- 1. Yatabe Y, Kerr KM, Utomo A et al EGFR mutation testing practices within the Asia Pacific region: results of a multicenter diagnostic survey. J Thorac Oncol 2015; 10: 438–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosell R, Moran T, Queralt C et al Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009; 361: 958–67. [DOI] [PubMed] [Google Scholar]
- 3. Kris MG, Johnson BE, Berry LD et al Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014; 311: 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci 2007; 98: 1817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitsudomi T, Morita S, Yatabe Y et al Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010; 11: 121–8. [DOI] [PubMed] [Google Scholar]
- 6. Zhou C, Wu YL, Chen G et al Erlotinib versus chemotherapy as first‐line treatment for patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (OPTIMAL, CTONG‐0802): a multicentre, open‐label, randomised, phase 3 study. Lancet Oncol 2011; 12: 735–42. [DOI] [PubMed] [Google Scholar]
- 7. Fukuoka M, Wu YL, Thongprasert S et al Biomarker analyses and final overall survival results from a phase III, randomized, open‐label, first‐line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non‐small‐cell lung cancer in Asia (IPASS). J Clin Oncol 2011; 29: 2866–74. [DOI] [PubMed] [Google Scholar]
- 8. Rosell R, Carcereny E, Gervais R et al Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol 2012; 13: 239–46. [DOI] [PubMed] [Google Scholar]
- 9. Inoue A, Kobayashi K, Maemondo M et al Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin–paclitaxel for chemo‐naive non‐small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann Oncol 2013; 24: 54–9. [DOI] [PubMed] [Google Scholar]
- 10. Sequist LV, Yang JC, Yamamoto N et al Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013; 31: 3327–34. [DOI] [PubMed] [Google Scholar]
- 11. Wu YL, Zhou C, Hu CP et al Afatinib versus cisplatin plus gemcitabine for first‐line treatment of Asian patients with advanced non‐small‐cell lung cancer harbouring EGFR mutations (LUX‐Lung 6): an open‐label, randomised phase 3 trial. Lancet Oncol 2014; 15: 213–22. [DOI] [PubMed] [Google Scholar]
- 12. Zhou W, Ercan D, Chen L et al Novel mutant‐selective EGFR kinase inhibitors against EGFR T790M. Nature 2009; 462: 1070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walter AO, Sjin RT, Haringsma HJ et al Discovery of a mutant‐selective covalent inhibitor of EGFR that overcomes T790M‐mediated resistance in NSCLC. Cancer Discov 2013; 3: 1404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cross DA, Ashton SE, Ghiorghiu S et al AZD9291, an irreversible EGFR TKI, overcomes T790M‐mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014; 4: 1046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Janne PA, Yang JC, Kim DW et al AZD9291 in EGFR inhibitor‐resistant non‐small‐cell lung cancer. N Engl J Med 2015; 372: 1689–99. [DOI] [PubMed] [Google Scholar]
- 16. Sequist LV, Soria JC, Goldman JW et al Rociletinib in EGFR‐mutated non‐small‐cell lung cancer. N Engl J Med 2015; 372: 1700–9. [DOI] [PubMed] [Google Scholar]
- 17. Roengvoraphoj M, Tsongalis GJ, Dragnev KH et al Epidermal growth factor receptor tyrosine kinase inhibitors as initial therapy for non‐small cell lung cancer: focus on epidermal growth factor receptor mutation testing and mutation‐positive patients. Cancer Treat Rev 2013; 39: 839–50. [DOI] [PubMed] [Google Scholar]
- 18. Wu JY, Wu SG, Yang CH et al Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res 2008; 14: 4877–82. [DOI] [PubMed] [Google Scholar]
- 19. Arcila ME, Nafa K, Chaft JE et al EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther 2013; 12: 220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beau‐Faller M, Prim N, Ruppert AM et al Rare EGFR exon 18 and exon 20 mutations in non‐small‐cell lung cancer on 10 117 patients: a multicentre observational study by the French ERMETIC‐IFCT network. Ann Oncol 2014; 25: 126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Watanabe S, Minegishi Y, Yoshizawa H et al Effectiveness of gefitinib against non‐small‐cell lung cancer with the uncommon EGFR mutations G719X and L861Q. J Thorac Oncol 2014; 9: 189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Otsuka T, Mori M, Yano Y et al Effectiveness of tyrosine kinase inhibitors in Japanese patients with non‐small cell lung cancer harboring minor epidermal growth factor receptor mutations: results from a multicenter retrospective study (HANSHIN Oncology Group 0212). Anticancer Res 2015; 35: 3885–91. [PubMed] [Google Scholar]
- 23. Chiu CH, Yang CT, Shih JY et al Epidermal growth factor receptor tyrosine kinase inhibitor treatment response in advanced lung adenocarcinomas with G719X/L861Q/S768I mutations. J Thorac Oncol 2015; 10: 793–9. [DOI] [PubMed] [Google Scholar]
- 24. Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non‐small‐cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012; 13: e23–31. [DOI] [PubMed] [Google Scholar]
- 25. Yasuda H, Park E, Yun CH et al Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 2013; 5: 216ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Naidoo J, Sima CS, Rodriguez K et al Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: clinical outcomes and response to erlotinib. Cancer 2015; 121: 3212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kobayashi Y, Togashi Y, Yatabe Y et al EGFR exon 18 mutations in lung cancer: molecular predictors of augmented sensitivity to afatinib or neratinib as compared with first‐ or third‐generation TKIs. Clin Cancer Res 2015; 21: 5305–13. [DOI] [PubMed] [Google Scholar]
- 28. Yang JC, Sequist LV, Geater SL et al Clinical activity of afatinib in patients with advanced non‐small‐cell lung cancer harbouring uncommon EGFR mutations: a combined post‐hoc analysis of LUX‐Lung 2, LUX‐Lung 3, and LUX‐Lung 6. Lancet Oncol 2015; 16: 830–8. [DOI] [PubMed] [Google Scholar]
- 29. Solca F, Dahl G, Zoephel A et al Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther 2012; 343: 342–50. [DOI] [PubMed] [Google Scholar]
- 30. Shimada Y, Imamura M, Wagata T et al Characterization of 21 newly established esophageal cancer cell lines. Cancer 1992; 69: 277–84. [DOI] [PubMed] [Google Scholar]
- 31. Tanaka H, Shibagaki I, Shimada Y et al Characterization of p53 gene mutations in esophageal squamous cell carcinoma cell lines: increased frequency and different spectrum of mutations from primary tumors. Int J Cancer 1996; 65: 372–6. [DOI] [PubMed] [Google Scholar]
- 32. Tanaka H, Shimada Y, Imamura M et al Multiple types of aberrations in the p16 (INK4a) and the p15(INK4b) genes in 30 esophageal squamous‐cell‐carcinoma cell lines. Int J Cancer 1997; 70: 437–42. [DOI] [PubMed] [Google Scholar]
- 33. Maegawa M, Arao T, Yokote H et al Epidermal growth factor receptor lacking C‐terminal autophosphorylation sites retains signal transduction and high sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci 2009; 100: 552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Togashi Y, Hayashi H, Terashima M et al Inhibition of beta‐Catenin enhances the anticancer effect of irreversible EGFR‐TKI in EGFR‐mutated non‐small‐cell lung cancer with a T790M mutation. J Thorac Oncol 2015; 10: 93–101. [DOI] [PubMed] [Google Scholar]
- 35. Togashi Y, Mizuuchi H, Kobayashi Y et al An activating ALK gene mutation in ALK IHC‐positive/FISH‐negative nonsmall‐cell lung cancer. Ann Oncol 2015; 26: 1800–1. [DOI] [PubMed] [Google Scholar]
- 36. Jiang J, Greulich H, Janne PA et al Epidermal growth factor‐independent transformation of Ba/F3 cells with cancer‐derived epidermal growth factor receptor mutants induces gefitinib‐sensitive cell cycle progression. Cancer Res 2005; 65: 8968–74. [DOI] [PubMed] [Google Scholar]
- 37. DeSantis CE, Lin CC, Mariotto AB et al Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin 2014; 64: 252–71. [DOI] [PubMed] [Google Scholar]
- 38. Midha A, Dearden S, McCormack R. EGFR mutation incidence in non‐small‐cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII). Am J Cancer Res 2015; 5: 2892–911. [PMC free article] [PubMed] [Google Scholar]
- 39. Hellmann MD, Reva B, Yu H et al Clinical and in vivo evidence that EGFR S768I mutant lung adenocarcinomas are sensitive to erlotinib. J Thorac Oncol 2014; 9: e73–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klughammer B, Brugger W, Cappuzzo F et al Examining treatment outcomes with erlotinib in patients with advanced non‐small cell lung cancer whose tumors harbor uncommon EGFR nutations. J Thorac Oncol 2016; 11: 545–55. [DOI] [PubMed] [Google Scholar]
- 41. Kancha RK, von Bubnoff N, Peschel C et al Functional analysis of epidermal growth factor receptor (EGFR) mutations and potential implications for EGFR targeted therapy. Clin Cancer Res 2009; 15: 460–7. [DOI] [PubMed] [Google Scholar]
- 42. Hirano T, Yasuda H, Tani T et al In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non‐small‐cell lung cancer. Oncotarget 2015; 6: 38789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang M, Xu X, Cai J et al NSCLC harboring EGFR exon‐20 insertions after the regulatory C‐helix of kinase domain responds poorly to known EGFR inhibitors. Int J Cancer 2016; 139: 171–6. [DOI] [PubMed] [Google Scholar]
- 44. Davis MI, Hunt JP, Herrgard S et al Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 2011; 29: 1046–51. [DOI] [PubMed] [Google Scholar]