

Abstract
Recent investigations revealed genetic analysis provides important information in management of gliomas, and we previously reported grade II–III gliomas could be classified into clinically relevant subgroups based on the DNA copy number aberrations (CNAs). To develop more precise genetic subgrouping, we investigated the correlation between CNAs and mutational status of the gene encoding isocitrate dehydrogenase (IDH) of those tumors. We analyzed the IDH status and CNAs of 174 adult supratentorial gliomas of astrocytic or oligodendroglial origin by PCR‐based direct sequencing and comparative genomic hybridization, respectively. We analyzed the relationship between genetic subclassification and clinical features. We found the most frequent aberrations in IDH mutant tumors were the combined whole arm‐loss of 1p and 19q (1p/19q codeletion) followed by gain on chromosome arm 7q (+7q). The gain of whole chromosome 7 (+7) and loss of 10q (−10q) were detected in IDH wild‐type tumors. Kaplan–Meier estimates for progression‐free survival showed that the tumors with mutant IDH, −1p/19q, or +7q (in the absence of +7p) survived longer than tumors with wild‐type IDH, +7, or −10q. As tumors with +7 (IDH wild‐type) showed a more aggressive clinical nature, they are probably not a subtype that developed from the slowly progressive tumors with +7q (IDH mutant). Thus, tumors with a gain on chromosome 7 (mostly astrocytic) comprise multiple lineages, and such differences in their biological nature should be taken into consideration during their clinical management.
Keywords: Astrocytomas, chromosome 7, DNA copy number, genetic subgrouping, IDH
The histological diagnosis of gliomas is defined by the tumor cell type and their malignant features including mitosis, nuclear atypism, necrosis, and vascular proliferation.1, 2 However, the systems presently used to define these tumors have limitations because interpreting cellularity, anaplasia, or even cell type is not purely objective. Furthermore, the clinical outcome of the tumor varies widely within the same histologic grade, which is partly due to interobserver discrepancies in diagnosis.3, 4, 5, 6 Progress in the genetic analysis of gliomas has validated the genetic subgrouping of these tumors. Better understanding of glioma development may help identify groups that are at risk for early progression, and specific genetic aberrations may serve as reliable predictors of the clinical outcome.7, 8, 9 Since Cairncross et al.10 reported that allelic loss of chromosome 1p is a statistically significant marker of prognosis for anaplastic oligodendrogliomas, many studies on DNA copy number aberrations (CNAs) in gliomas have reported the utility of CNAs in tumor classification.11, 12, 13, 14, 15, 16, 17, 18, 19 The results from these studies indicated the following: 1p/19q codeletion (1p/19q codel) marks a subset of oligodendroglial tumors,12, 15 the gain of whole chromosome 7 is the most frequent aberration in high‐grade astrocytomas,13 the gliomas with losses on chromosomal arms 9p and 10q and gain on chromosome 7 show trends toward short survival,14 the gain on chromosomal arm 7q appears to be a key early event in a subgroup of astrocytomas,16 and +7q and 1p/19q codel are mutually exclusive in low‐grade gliomas.16 Many of the cytogenetic analyses were carried out for glioblastomas, but we have also previously shown that grade II–III adult supratentorial gliomas could be classified into clinically relevant subgroups based on the CNAs.20 The astrocytomas of these grades contained a wide variety of genetic subgroups in contrast to oligodendrogliomas and glioblastomas, which are characterized with 1p/19q codel and +7/−10q, respectively. Our data showed that tumors with +7 had a worse outcome than tumors with 7q, although it was not confirmed that the former could develop from the latter; that is, it is not clear that a stepwise DNA copy number gain on chromosome 7, beginning as partial gain of 7q, develops along with glioma progression. As astrocytomas are generally refractory to adjuvant therapy compared to oligodendrogliomas and the treatment strategy for astrocytomas should be established according to their biological/genetic nature, further characterization of these astrocytic tumors with gain of chromosome 7 is warranted. Application of information provided by genetic analysis to the treatment of cancers has been a major challenge in medical oncology as well as neuro‐oncology. If genetic subtypes predict outcome and therapeutic responses, these criteria could help establish rational and objective means for analyzing treatment protocols and a diagnostic system for predicting prognosis in grade II–III gliomas.
Recently, many studies focusing on genetic prognostic markers for gliomas have highlighted the clinical significance of the mutational status of the gene coding isocitrate dehydrogenase 1 (IDH1).21 Mutations in IDH1 are associated with improved prognosis in glioblastoma and are considered to develop during the early stages of tumorigenesis of astrocytoma and oligodendroglioma,22, 23, 24 which usually progress to a higher‐grade tumor. Although mutant IDH1 is not a predictive marker for radiation or chemotherapy, this information could help in deciding the adjuvant therapy for gliomas, and thus might serve as a critical factor for genetic subgrouping of the tumors. Therefore, we wished to determine how the IDH1 status fits in our CNA‐based genetic subgrouping of gliomas and investigated the IDH1 status in 174 adult supratentorial astrocytic and oligodendroglial gliomas. We evaluated the correlation of the IDH1 status and the genetic subgrouping in order to create an accurate tumor classification system.
Materials and Methods
Tissue samples and clinical characteristics
We reviewed the cases of neuroepithelial tumors (gliomas) surgically treated at Keio University Hospital (Tokyo, Japan) and Fujita Health University Hospital (Toyoake, Japan) between 1994 and 2012, and selected institutionally diagnosed astrocytomas or oligodendrogliomas including mixed gliomas (oligo‐astrocytomas). In general, the patients underwent maximum safe resection of the tumor, and adjuvant therapy according to existence of residual tumor and the tumor grade, that is, both chemotherapy and radiotherapy in most cases. When the tumor was diagnosed as grade II and gross totally resected, the patients were frequently observed without adjuvant therapy. Infratentorial gliomas, which have been reported to have genetic features distinct from supratentorial tumors,25 and pediatric tumors, which previously have been reported to have genetic features distinct from adult tumors,26 were also excluded from the study. After the review, the study cases were selected based on availability of tissue samples and were followed up for longer than 36 months, or until tumor progression, to collect the clinical information. One hundred and seventy‐four tissue samples were obtained through the Departments of Neurosurgery and Pathology at Keio University and at Fujita Health University. The tumor tissues had been fixed in 10% neutral buffered formalin and processed in paraffin for routine light microscopy. The samples were re‐evaluated by neuropathologists (M.A.), and were classified according to the 2007 edition of WHO criteria.1 We obtained clinical data from patient records. This study was approved by the local ethical review board of Keio University and Fujita Health University. Informed consent was obtained from the patient(s) and/or guardian(s).
DNA isolation and comparative genomic hybridization
DNA preparation from microdissected tissue using degenerate oligonucleotide‐primed PCR was carried out according to the method validated in a previous study.27 DNA aliquot extracted from formalin‐fixed and paraffin‐embedded tissue as mentioned above was subjected to degenerate oligonucleotide‐primed PCR. Metaphase spreads were prepared from phytohemaglutinin‐stimulated human peripheral blood lymphocytes from a normal healthy male. Comparative genomic hybridization was carried out according to the procedure described by Hirose et al.27
Polymerase chain reaction‐based sequencing of IDH1 gene
The IDH1 gene including codon 132 (exon 4) was amplified using forward 5′‐CGGTCTTCAGAGAAGCCATT‐3′ and reverse 5′‐CACATTATTGCCAACATGAC‐3′ primers. Amplification was undertaken in a total volume of 20 μL, consisting of 5–100 ng/mL DNA solution, Platinum Taq (Fisher Scientific, Pittsburgh, PA, USA) DNA polymerase (0.5 U), 10 mM dNTP Mix, 50 mM MgCl2, 10× PCR buffer, 0.25 mM primer (sense, antisense).22 The reaction mixture was subjected to 40 cycles of rapid PCR consisting of initial denaturation at 95°C for 3 min, followed by 95°C for 30 s, annealing at 56°C for 30 s, extension at 72°C for 40 s, and final extension at 72°C for 10 min. We identified PCR products exhibiting IDH1 fragments of 121 bp in length by electrophoresis. Subsequently, 0.5 μL PCR product was mixed with DMSO, BigDye Sequencing Buffer (Fisher Scientific, Pittsburgh, PA, USA) and the same primer as for PCR. After heating at 93°C for 3 min, 4 μL Ready Reaction Mix (Fisher Scientific, Pittsburgh, PA, USA) was added and re‐amplified in a total volume of 20 μL. The PCR protocol was set for 25 cycles of rapid PCR consisting of initial denaturation at 96°C for 1 min, followed by 96°C for 10 s, annealing at 50°C for 5 s, extension at 60°C for 4 min. According to the manufacturer's instructions, the re‐amplified PCR products were sequenced using the BigDye Terminator version 3.1 Cycle Sequencing Kit (Fisher Scientific, Pittsburgh, PA, USA) and analyzed on an ABI 3100 (Applied Biosystems, Pittsburgh, PA, USA). Based on the sequencing results, each case of IDH1 mutation was distributed either positive or negative.
Statistical analysis
We analyzed relationships of clinical variables and genetic subgroups based on chromosomal CNAs and IDH1 mutation. Progression‐free survival (PFS) and overall survival (OS) were the minimum time from surgery to disease progression confirmed by MRI and to death, respectively. Patients who were alive at last follow‐up or who had no documented time to progression at last follow‐up were regarded as censored. Kaplan–Meier analysis was used to estimate event‐free rates. The log–rank test (at a two‐sided 5% significance) was used to compare two groups. Cox regression multivariate analysis was used for determining independent prognostic factors.
Results
Genetic features of WHO grade II‐III gliomas
We analyzed both CNAs and IDH1/2 status in a total of 174 primary adult supratentorial glioma samples and found IDH1/2 mutations in 125 tumors and wild‐type IDH1/2 in 49 tumors. All mutations at codon 132 in IDH1 were found to belong to the CGT to CAT (R132H) type mutation. In cases with IDH1 wild type, IDH2 analysis (mutation at codon 172) was carried out, and only one case showed IDH2 mutation (see Table S1). Based on WHO criteria, we classified the mixed gliomas as oligodendroglial tumors.1 The clinical features and CNAs of the cases were partly shown in our previous study.20 These results are summarized in Table S1.
Correlation of IDH1/2 status with histology and genetic subgrouping is summarized in Table 1. As previously reported, mutations of IDH1/2 were frequently detected in oligodendrogliomas and low‐grade astrocytomas, although wild‐type IDH1/2 was detected in higher‐grade astrocytomas. Concerning genetic subgrouping, 1p/19q codeletion was closely correlated with IDH1/2 mutation with one exceptional case that had wild‐type IDH1 and mutant IDH2. In tumors with gain on chromosome 7, the most frequent aberration in IDH1/2 mutant tumors was the gain on 7q, whereas the most frequent aberration in IDH1/2 wild‐type tumors was the gain of 7 (gain of both 7q and 7p) and +7/−10q. These results suggest that, based on the IDH1/2 status, tumors with gain on chromosome 7 (mostly astrocytic tumors) could be classified into at least two lineages, although both of them share gain of 7q.
Table 1.
Correlation between histology and genetic aberrations in grade II–III astrocytomas
| Histology | IDH1/2 mutant | IDH1/2 wild type | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1p19q codeleted | 1p intact | 1p19q codeleted | 1p intact | ||||||||
| +7q | +7 | +7/−10q | 1p/7 intact | +7q | +7 | +7/−10q | 1p/7 intact | ||||
| Astrocytic tumor | |||||||||||
| Diffuse astrocytoma | 6 | 17 | 3 | 0 | 13 | 0 | 0 | 4 | 2 | 2 | 47 |
| Anaplastic astrocytoma | 5 | 5 | 5 | 0 | 4 | 0 | 0 | 8 | 15 | 11 | 53 |
| Oligodendroglial tumor | |||||||||||
| Oligodendroglioma | 19 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 21 |
| Anaplastic oligodendroglioma | 11 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 14 |
| Oligoastrocytoma | 12 | 3 | 2 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 24 |
| Anaplastic oligoastrocytoma | 7 | 0 | 2 | 0 | 1 | 0 | 0 | 2 | 0 | 3 | 15 |
| Total | 60 | 25 | 12 | 0 | 28 | 0 | 0 | 14 | 19 | 16 | 174 |
Correlation between genetic and clinical features of WHO grade II‐III gliomas
Kaplan–Meier curves for PFS and OS of the study cases using two representative CNAs, gain on chromosome 7 (regardless of limited gain of 7q or not; +Ch7) and 1p/19q codel, and IDH1/2 status are shown in Figure 1. The survival was longer in IDH1/2 mutant cases than in IDH1/2 wild‐type cases for both PFS (median: total IDH1/2 mutant tumors, 63 months; total IDH1/2 wild type, 12 months; P < 0.0001) and OS (median: total IDH1/2 mutant tumors, undefined; total IDH1/2 wild type, 28 months; P < 0.0001). Even without 1p/19q codel tumors, which are known to have better prognosis, IDH1/2 mutant cases showed longer survival than IDH1/2 wild‐type cases (PFS, median: IDH1/2 mutant, 51 months; IDH1/2 wild type, 12 months; P < 0.0001; OS, median: IDH1/2 mutant, undefined; IDH1/2 wild type, 28 months; P < 0.0001). Interestingly, the curve for IDH1/2 mutant +Ch7 tumors overlapped well with that for the IDH1/2 mutant and of Ch7 and 1p intact (i.e., tumors without classical astrocytic/oligodendroglial genetic markers), whereas the curve for IDH1/2 wild‐type +Ch7 tumors overlapped well with those of IDH1/2 wild type and Ch7/1p intact tumors. The survival curves for PFS of all the tumors with 1p/19q codel overlapped well with the curves for IDH1/2‐mutant +Ch7 and IDH1/2‐mutant Ch7/1p intact tumors, whereas OS of 1p/19q codel tumors was longer than that of the other IDH1/2‐mutant tumors, although the difference was not statistically significant (P = 0.07). Taken together, our data suggest that, in agreement with previous studies, IDH1/2 mutation is a critical factor with great impact on survival of patients with WHO grade II–III supratentorial gliomas, and that astrocytic tumors (infiltrative gliomas without 1p/19q codel) should be classified by IDH1/2 status but not by the region of the gain on chromosome 7 because +7q tumors and +7 tumors are clinically distinct, whereas IDH1‐mutant tumors without 1p/19q codel showed similar prognosis regardless of gain on chromosome 7.
Figure 1.
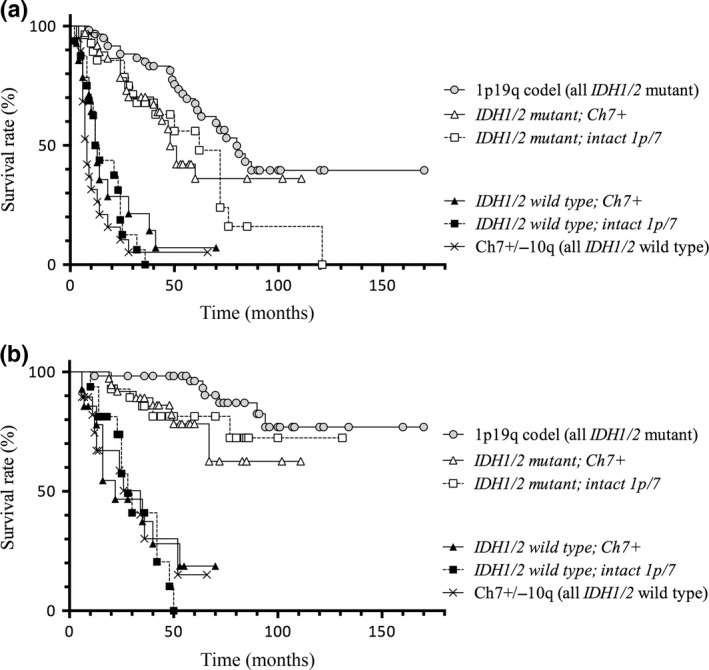
Kaplan–Meier curves for progression‐free survival (PFS) (a) and overall survival (OS) (b) of WHO grade II–III tumors defined by IDH1/2 status. Survival was longer in IDH1/2 mutant cases than in IDH1/2 wild type cases for both PFS (median: IDH1/2 mutant, 63 months; IDH1 wild type, 12 months; P < 0.0001) and OS (median: IDH1/2 mutant, undefined; IDH1/2 wild type, 28 months; P < 0.0001). Even without 1p/19q‐codeleted (codel) tumors, which are known to have better prognosis, IDH1/2 mutant cases showed significantly longer survival than IDH1/2 wild type cases (PFS, median: IDH1/2 mutant, 51 months; IDH1/2 wild type, 12 months; P < 0.0001; OS, median: IDH1/2 mutant, undefined; IDH1/2 wild type, 28 months; P < 0.0001).
Cox regression multivariate analysis that stratified for the effect of surgery or radiotherapy showed that surgery and IDH1/2 status were strong predictors for long PFS and OS (P < 0.001; Tables 2,3, respectively). In summary, the present study showed that +7q is the change only observed in IDH‐mutated gliomas, that IDH status had stronger impact than the manner of DNA copy number gain on chromosome 7, and that 10q loss (hallmark of glioblastoma) was not found in IDH mutant tumor.
Table 2.
Multivariate analysis of progression‐free survival in patients with grade II–III glioma
| Variable | Hazard ratio (95% CI) | P‐value |
|---|---|---|
| Age | 1.006 (0.991–1.021) | 0.446 |
| Gender | 0.801 (0.532–1.207) | 0.289 |
| KPS | 0.964 (0.939–0.989) | 0.005 |
| WHO grade II versus III | 0.808 (0.634–1.030) | 0.085 |
| MIB‐1 | 0.993 (0.976–1.009) | 0.389 |
| Tumor type (astrocytic vs oligodendroglial) | 1.065 (0.821–1.383) | 0.635 |
| Surgery (TR + STR) versus (PR + biopsy) | 0.379 (0.243–0.590) | <0.001 |
| Radiotherapy versus none | 0.692 (0.413–1.157) | 0.160 |
| Chemotherapy versus none | 1.174 (0.696–1.982) | 0.547 |
| IDH1/2 mutant versus wild type | 0.262 (0.133–0.518) | <0.001 |
CI, confidence interval; KPS,; MIB‐1, positive rate (%); PR, partial resection; STR, subtotal resection; TR, total resection.
Table 3.
Multivariate analysis of overall survival in patients with grade II–III glioma
| Variable | Hazard ratio (95% CI) | P‐value |
|---|---|---|
| Age | 1.022 (1.001–1.044) | 0.043 |
| Gender | 1.090 (0.585–2.034) | 0.786 |
| KPS | 0.948 (0.911–0.986) | 0.008 |
| WHO grade II versus III | 0.628 (0.414–0.955) | 0.030 |
| MIB‐1 | 0.989 (0.967–1.012) | 0.357 |
| Tumor type (astrocytic vs oligodendroglial) | 1.581 (1.039–2.405) | 0.033 |
| Surgery (TR + STR) versus (PR + biopsy) | 0.259 (0.123–0.547) | 0.001 |
| Radiotherapy versus none | 0.438 (0.289–0.848) | 0.012 |
| Chemotherapy versus none | 1.757 (0.701–4.402) | 0.229 |
| IDH1/2 mutant versus wild type | 0.218 (0.100–0.474) | <0.001 |
CI, confidence interval; KPS,; MIB‐1, positive rate (%); PR, partial resection; STR, subtotal resection; TR, total resection.
Discussion
Surgery is the first line of treatment for gliomas, and the extent of resection has a great impact on patient survival.28 However, in general, glioma is not cured by surgical resection, and therefore, adjuvant therapy must be considered as the next step of the treatment. The decision for adjuvant treatment is based on histological diagnosis; chemotherapy and radiotherapy are generally administered for high‐grade gliomas, whereas the treatment strategy for low‐grade tumors is still controversial. A major reason for this ambiguity is the variability in the clinical features of WHO grade II, and even grade III, tumors. This clinical heterogeneity makes it difficult to establish a treatment strategy for gliomas based on their biological nature, and many studies have recently attempted to develop a clinically relevant genetic subgrouping of these tumors.29, 30 We had previously reported that grade II–III gliomas are classified into clinically relevant subgroups according to their chromosomal DNA CNAs.20 This study reported that non‐ependymal supratentorial gliomas are basically classified into three categories: tumors with gain on chromosome 7, tumors with 1p/19q codel, and tumors with intact chromosomes 7 and 1p/19q. Tumors with gain of chromosome 7, mostly astrocytic tumors, and tumors with loss of 1p/19q, mostly oligodendroglial tumors, were then classified into subgroups on the basis of genetic profiles as follows: tumors with gain on chromosome arm 7q (+7q), tumors with gain of whole chromosome 7 (+7), tumors with gain of 7 and loss of 10q (+7/−10q), and tumors with 1p/19q codel. Of them, tumors with +7q and those with 1p/19q codel, that is, low‐grade tumors of two major lineages, showed long survival. However, in contrast to the tumors with 1p/19q codel, which usually show good response to genotoxic treatments, it is not clear that tumors with gain on chromosome 7 have common clinical feature(s) and whether the +7q tumor is really the early stage of +7 and +7/−10q tumors. Thus, it is ambiguous whether tumors with gain on chromosome 7 at various stages are included in the same lineage. If these tumors are composed of multiple lineages, the ideal treatment should also differ between them.
As the gain on 7q, as well as 1p/19q codel, was associated with longer survival, whereas +7 and +7/−10q were associated with shorter survival, it could be presumed that +7q would occur in the early stage of glioma progression, and that the tumor might develop malignancy along with gain on 7p to show gain on whole chromosome 7. However, we found that this hypothesis was not reasonable because IDH1/2 mutation, which is considered to occur at an early stage of tumorigenesis,21, 22, 23, 31 does not coexist with +7 when the tumors are of higher grade. Our data suggested that the tumors with a gain on chromosome 7 were divided into at least two lineages: tumors with gain of 7q (only) and mutant IDH1/2 and those with gain of whole chromosome 7 and wild‐type IDH1/2. Furthermore, from the clinical point of view, +7 tumors progress in a similar way to 1p/Ch7 intact and IDH1/2 wild‐type tumors. Thus, the present study led to a new hypothetical pathway of glioma development. In early stages, IDH1/2 mutation occurs in a subset of glioma progenitor cells, and these cells progress to 1p/19q codel tumors and others (including +7q). These types of tumors might progress to a higher grade; however, in contrast to 1p/19q codel tumors, which progress with a gain of +7, the majority of +7q tumors would progress without additional gain of 7p because our study for paired sets of primary and recurrent +7q tumors did not show an association between tumor progression and gain of 7p (data not shown). However, IDH1/2 wild‐type progenitor cells could progress to tumors with +7, +7/−10q or intact 1p/Ch7. Briefly, most tumors with +7q and those with 7 belong to distinct lineages developed from precursors characterized by different IDH1/2 status. In contrast to tumors with gain on chromosome 7, there is a lack of evidence to indicate that tumors with 1p/19q codel are composed of multiple lineages. In addition, we recently published a study showing IDH1/2‐mutated gliomas without 1p/19q codel were mostly characterized by TP53 mutation, but +7 tumors did not show this mutation.32 Thus, again, +7q tumors and +7 tumors are genetically distinct in terms of both IDH1/2 and TP53 status, and IDH1/2‐mutated tumors could be divided into two groups, 1p/19q‐codeleted TP53 wild‐type tumors and 1p/19q non‐deleted TP53‐mutant tumors. The +7q tumors belong to the latter group, and +7 tumors, mostly both IDH1/2 and TP53 wild type, are clearly distinct tumors in both clinical and genetic aspects (Fig. 2a). Thus, by collecting data on CNAs and IDH status together with detailed clinical information, we now present a hypothetical glioma progression model. In summary, different from our previous hypothetical glioma progression pathway, tumors with gain on chromosome 7 are classified into three groups: +7q (IDH1/2 mutant), +7 (IDH1/2 wild type), and +7/−10q (similar to primary glioblastoma, IDH1/2 wild type), whereas tumors with 1p/19q codel develop in a single pathway (Fig. 2b).
Figure 2.
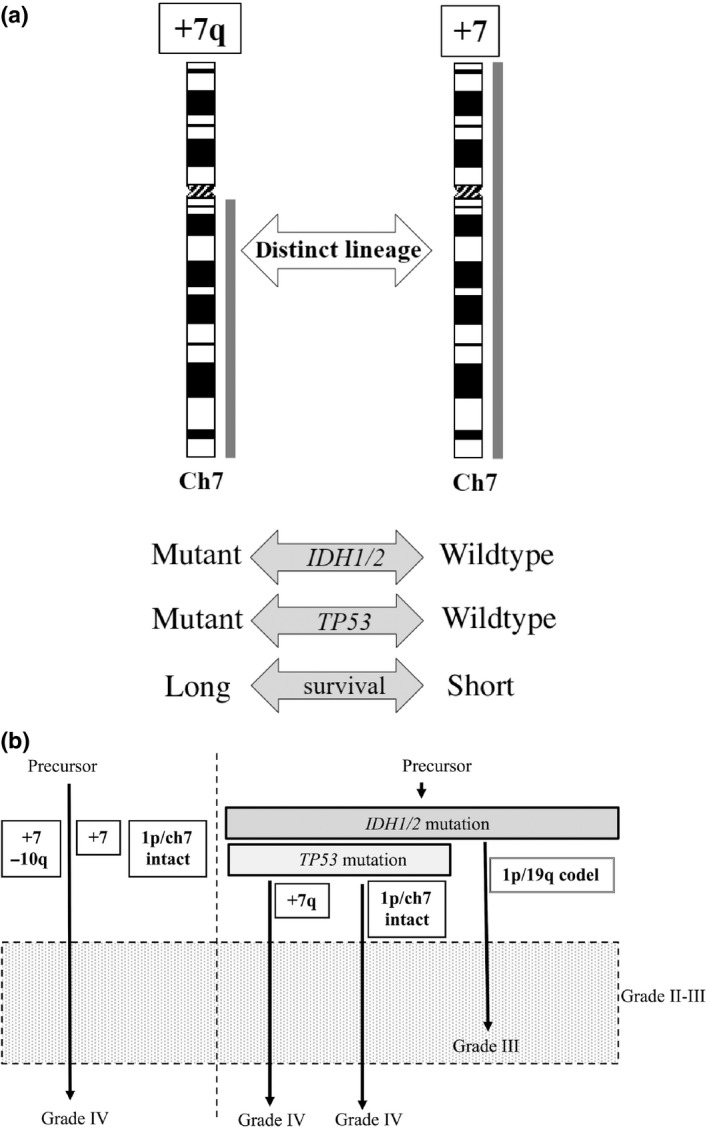
(a) Comparison between tumors with gain limited to chromosomal arm 7q (+7q) and those with gain of whole chromosome 7 (+7). Thick lines to the right of chromosome scheme represent region of relative copy number gain. The +7q tumors and +7 tumors are genetically distinct in terms of both IDH1/2 and TP53 status,32 and show clearly distinct clinical aspects. (b) Schematic illustration of hypothetical genetic pathways associated with glioma development. Tumors are divided on the basis of IDH1/2 status, and representative aberrations (i.e., the gain of 7q or codeletion [codel] of 1p/19q) follow IDH1/2 mutation as an early event in astrocytic or oligodendroglial tumor lineages, respectively. Gain of whole chromosome 7 occurs in IDH1/2 wild type tumors. Note that +7q tumor and +7 (whole) belong to two distinct lineages defined by the IDH1/2 and TP53 status.32
We noted early tumor progression in four cases with IDH1 mutation. As all of these patients underwent biopsy only during surgery, the extent of tumor resection might be important, thus it suggests that management of adjuvant therapy for grade II–III gliomas should be considered in accordance with both genetic (CNAs and IDH1/2) and clinical (extent of tumor resection) information. In some cases, histologically low‐grade gliomas might be treated as high‐grade gliomas. With the guidance of subdivided genetic classification of gliomas, we expect that precise treatments that are customized for each tumor lineage may be applied to adult supratentorial grade II–III astrocytic and oligodendroglial gliomas. Still a small proportion of +7 tumors might develop from +7q tumors with IDH1/2 mutation because the status of gain of chromosome 7 and IDH1/2 mutation were not completely matched, however, our data suggested that +7 tumors with mutant IDH1/2 should be managed in a similar manner to +7q tumors (Fig. 3). Recent studies reported that grade II–III gliomas classified by genetic features could be relatively easily analyzed.33, 34, 35 Together with those studies, the present data emphasizes the significance of genetic investigation in the clinical management of gliomas, and provides a basis for refined classification of grade II–III gliomas. The present study was retrospective and treatments were not uniform, and has limitations in investigating correlations between genetic subgroup and prognosis. Future studies with cases treated in a uniform manner would clearly validate the genetic classification of WHO grade II–III gliomas.
Figure 3.
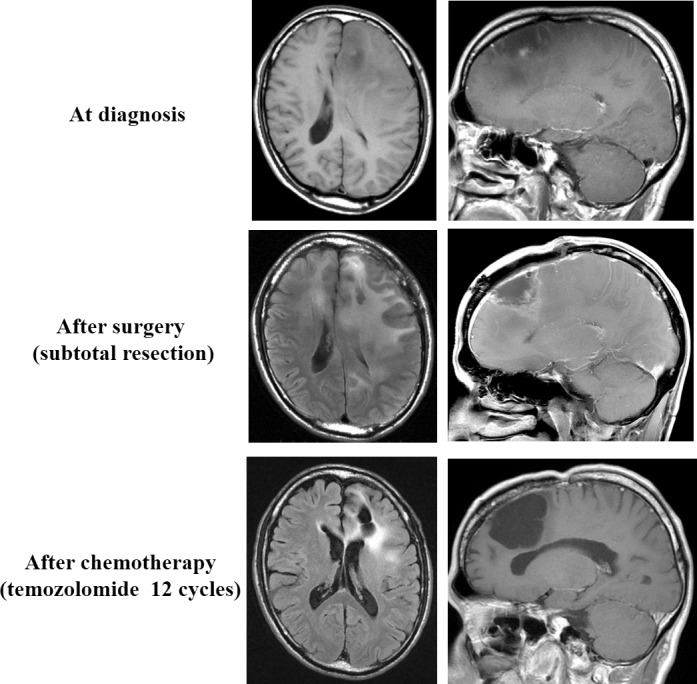
A representative case with IDH mutant tumor. Although the tumor had gain of whole chromosome 7, this patient has survived without tumor progression over 7 years. Extent of tumor resection appeared important especially in management of the IDH1/2 mutant tumor without 1p/19q codeletion.
Disclosure Statement
Yuichi Hirose received a research grant from Daiichi Sankyo Company. The other authors have no conflict of interest.
Supporting information
Table S1. Clinical and genetic features of study cases with grade II–III gliomas. †This case showed IDH2 mutation. Ast, astrocytic; F, female; M, male; NU, nitrosourea; Oligo, oligodendroglial; PAV, procarbazine + ACNU + vincristine; PR, partial resection; STR, subtotal resection; TMZ, temozolomide; TR, total resection.
Acknowledgments
We would like to thank Ms. Kiyomi Koide, Ms. Naoko Tsuzaki (Keio University), and Ms. Fujiko Sueishi (Fujita Health University) for their excellent technical assistance. This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT S1001034 and JSPS KAKENHI nos. 23592141, 23791617, and 24592178) and by grants from Fujita Health University (nos. 0748, 1048, and 1109).
Cancer Sci 107 (2016) 1159–1164
Funding Information
Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT S1001034 and JSPS KAKENHI nos. 23592141, 23791617, and 24592178); Fujita Health University (0748, 1048, and 1109).
References
- 1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. Lyon, France: IARC Press, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daumas‐Duport C. Histological grading of gliomas. Curr Opin Neurol Neurosurg 1992; 5: 924–31. [PubMed] [Google Scholar]
- 3. Vertosick FT Jr, Selker RG, Arena VC. Survival of patients with well‐differentiated astrocytomas diagnosed in the era of computed tomography. Neurosurgery 1991; 28: 496–501. [DOI] [PubMed] [Google Scholar]
- 4. Coons SW, Johnson PC, Scheithauer BW, Yates AJ, Pearl DK. Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer 2000; 79: 1381–93. [DOI] [PubMed] [Google Scholar]
- 5. Aldape K, Simmons ML, Davis RL, et al Discrepancies in diagnoses of neuroepithelial neoplasms: the San Francisco bay area adult glioma study. Cancer 2000; 88: 2342–9. [PubMed] [Google Scholar]
- 6. van den Bent MJ. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician's perspective. Acta Neuropathol 2010; 120: 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burton EC, Lamborn KR, Forsyth P, et al Aberrant p53, mdm2, and proliferation differ in glioblastomas from long‐term compared with typical survivors. Clin Cancer Res 2002; 8: 180–7. [PubMed] [Google Scholar]
- 8. Nutt CL, Mani DR, Betensky RA, et al Gene expression‐based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 2003; 63: 1602–7. [PubMed] [Google Scholar]
- 9. Gravendeel LA, Kouwenhoven MC, Gevaert O, et al Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res 2009; 69: 9065–72. [DOI] [PubMed] [Google Scholar]
- 10. Cairncross JG, Ueki K, Zlatescu MC, et al Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 1998; 90: 1473–9. [DOI] [PubMed] [Google Scholar]
- 11. Mohapatra G, Bollen AW, Kim DH, et al Genetic analysis of glioblastoma multiforme provides evidence for subgroup within the grade. Genes Chromosom Cancer 1998; 21: 195–206. [PubMed] [Google Scholar]
- 12. Bigner SH, Matthews MR, Rasheed BK, et al Molecular genetic aspects of oligodendrogliomas including analysis by comparative genomic hybridization. Am J Pathol 1999; 155: 375–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kunwar S, Mohapatra G, Bollen A, Lamborn KR, Prados M, Feuerstein BG. Genetic subgroups of anaplastic astrocytomas correlate with patient age and survival. Cancer Res 2001; 61: 7683–8. [PubMed] [Google Scholar]
- 14. Burton EC, Lamborn KR, Feuerstein BG, et al Genetic aberrations defined by comparative genomic hybridization distinguish long‐term from typical survivors of glioblastoma. Cancer Res 2002; 62: 6205–10. [PubMed] [Google Scholar]
- 15. Jeuken JW, Sprenger SH, Vermeer H, Kappelle AC, Boerman RH, Wesseling P. Chromosomal imbalances in primary oligodendroglial tumors and their recurrences: clues about malignant progression detected using comparative genomic hybridization. J Neurosurg 2002; 96: 559–64. [DOI] [PubMed] [Google Scholar]
- 16. Hirose Y, Aldape KD, Chang S, Lamborn K, Berger MS, Feuerstein BG. Grade II astrocytomas are subgrouped by chromosome aberrations. Cancer Genet Cytogenet 2003; 142: 1–7. [DOI] [PubMed] [Google Scholar]
- 17. Nigro JM, Misra A, Zhang L, et al Integrated array‐comparative genomic hybridization and expression array profiles identify clinically relevant molecular subtypes of glioblastoma. Cancer Res 2005; 65: 1678–86. [DOI] [PubMed] [Google Scholar]
- 18. Maher EA, Brennan C, Wen PY, et al Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res 2006; 66: 11502–13. [DOI] [PubMed] [Google Scholar]
- 19. Yin D, Ogawa S, Kawamata N, et al High‐resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol Cancer Res 2009; 7: 665–77. [DOI] [PubMed] [Google Scholar]
- 20. Hirose Y, Sasaki H, Miwa T, et al Whole genome analysis from microdissected tissue revealed adult supratentorial grade II–III gliomas are divided into clinically relevant subgroups by genetic profile. Neurosurgery 2011; 69: 376–90. [DOI] [PubMed] [Google Scholar]
- 21. Yan H, Williams Parsons D, Jin G, et al IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360: 765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 2009; 174: 1149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sonoda Y, Kumabe T, Nakamura T, et al Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Sci 2009; 100: 1996–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gorovets D, Kannan K, Shen R, et al IDH mutation and neuroglial developmental features define clinically distinct subclasses of lower grade diffuse astrocytic glioma. Clin Cancer Res 2012; 18: 2490–501. [DOI] [PubMed] [Google Scholar]
- 25. Miwa T, Hirose Y, Sasaki H, Ikeda E, Yoshida K, Kawase T. Genetic characterization of adult infratentorial gliomas. J Neurooncol 2009; 91: 251–5. [DOI] [PubMed] [Google Scholar]
- 26. Miwa T, Hirose Y, Sasaki H, Ezaki T, Yoshida K, Kawase T. Single‐copy gain of chromosome 1q is a negative prognostic marker in pediatric nonependymal, nonpilocytic gliomas. Neurosurgery 2011; 68: 206–12. [DOI] [PubMed] [Google Scholar]
- 27. Hirose Y, Aldape K, Takahashi M, Berger MS, Feuerstein BG. Tissue microdissection and degenerate oligonucleotide‐primed polymerase chain reaction (DOP‐PCR) is an effective method to analyze genetic aberrations in invasive tumors. J Mol Diag 2001; 3: 62–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanai N, Berger MS. Recent surgical management of gliomas. Adv Exp Med Biol 2012; 746: 12–25. [DOI] [PubMed] [Google Scholar]
- 29. Hartmann C, Hentschel B, Tatagiba M, et al Molecular markers in low‐grade gliomas: predictive or prognostic? Clin Cancer Res 2011; 17: 4588–99. [DOI] [PubMed] [Google Scholar]
- 30. Kim Y‐H, Nobusawa S, Mittelbronn M, et al Molecular classification of low‐grade diffuse gliomas. Am J Pathol 2010; 177: 2708–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lai A, Kharbanda S, Pope WB, et al Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol 2011; 29: 4482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nakae S, Sasaki H, Hayashi S, et al PCR‐based simple subgrouping is validated for classification of gliomas and defines negative prognostic copy number aberrations in IDH mutant gliomas. PLoS ONE 2015; 10: e0142750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eckel‐Passow JE, Lachance DH, Molinaro AM, et al Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 2015; 372: 2499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cancer Genome Atlas Research Network , Brat DJ, Verhaak RG, et al Comprehensive, integrative genomic analysis of diffuse lower‐grade gliomas. N Engl J Med 2015; 372: 2481–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Suzuki H, Aoki K, Chiba K, et al Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet 2015; 47: 458–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical and genetic features of study cases with grade II–III gliomas. †This case showed IDH2 mutation. Ast, astrocytic; F, female; M, male; NU, nitrosourea; Oligo, oligodendroglial; PAV, procarbazine + ACNU + vincristine; PR, partial resection; STR, subtotal resection; TMZ, temozolomide; TR, total resection.