

Abstract
First described in 1985, intermediate cell histiocytosis is a rare disorder of the cutaneous dendritic cell group with a varied clinical presentation and evolution. The pathologic substrate is constituted by the proliferation of indeterminate cells (ICs) that are immunophenotypically characterized by the positivity of CD1a, CD68, and faint/focal S100, plus the negativity for CD207 (langerin). The authors present the case of a healthy elderly woman who presented generalized dome-shaped reddish cutaneous nodules over her trunk, neck, face, and extremities over a period of 18 months. A laboratory and imaging work-up ruled out internal involvement. The skin biopsy was consistent with IC histiocytosis. The patient was treated with narrowband ultraviolet B phototherapy, which resulted in an excellent short-term outcome.
Keywords: Histiocytosis, Skin Diseases, Phototherapy
CASE REPORT
A 76-year-old Caucasian woman sought medical care complaining of the presence of scattered nodules all over her body surface. She referred the onset of the appearance of a few dispersed lesions on the neck 18 months before. Since then, new lesions had appeared in the inframammary region, abdomen, and back, which centrifugally spread to the lower and upper limbs, and finally to the face involving the nose and ears. The patient was reluctant to seek medical care until the lesions began to appear in exposed areas, especially on her face. During this period, she maintained in good health.
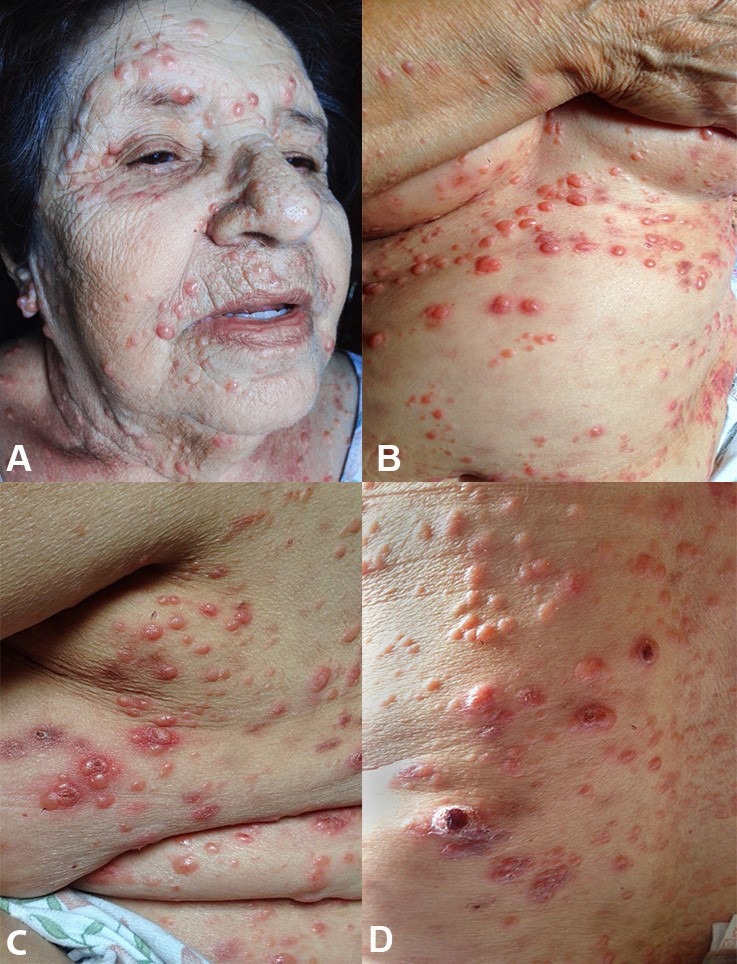
She had recently used a corticoid cream over her face with subjective improvement. She denied fever, weight loss, or any other complaint except for hopelessness caused by her appearance. Her medical history included the diagnosis of hypertension and the regular use of valsartan, levanlodipine, clopidogrel, and simvastatin; she also had a myocardial infarction 10 years ago, which had been treated with an angioplasty. The physical examination showed an apparently depressed patient, weighing 78 kg, with a height of 1.58 m, and normal vital signs. The skin examination revealed multiple reddish or brown dome-shaped, non-pruriginous, painless papules of varying sizes (1-5 mm) of firm consistency that eventually ulcerated (Figure 1). The remaining examination was normal.
Figure 1. Skin examination showing in A - disseminated nodular lesions over the face; B - over the trunk and the inframammary region; C - over the lateral face of the thorax; and D - over the back.
The laboratory work-up, which included a total blood cell count and erythrocyte sedimentation rate, electrolytes, renal function tests, calcium, hepatic enzymes, uric acid, thyroid function, protein electrophoresis, β2 microglobulin, and immunoglobulin dosage, was within the normal range. Serology for HIV1 and HIV2, hepatitis B, hepatitis C, syphilis, antinuclear antibody, and anti-DNA were negative. A bone radiological inventory ruled out any lesion. A thoracic tomography disclosed signs consistent with pulmonary emphysema. The positron emission tomography-computed tomography scans showed multiple hypermetabolic cutaneous lesions, but no other suspicious lesions were described. The magnetic resonance imaging showed no evidence of central nervous system disease. Therefore, she was deemed free of internal disease.
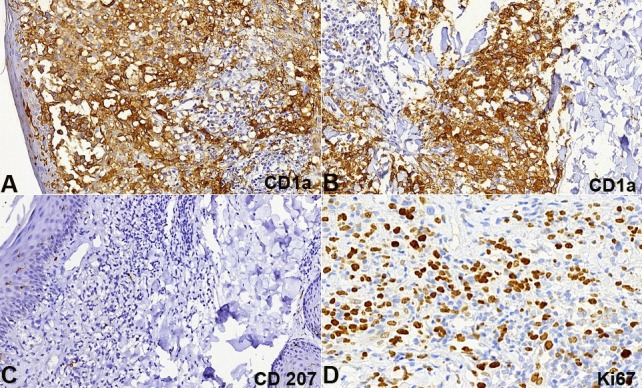
The patient was submitted to a skin biopsy, which revealed a dense superficial dermal infiltrate composed of histiocytoid cells with oval-shaped nuclei sometimes presenting longitudinal chromatin grooves. Sparse multinucleated cells and plasma cells were also present. Small lymphocytes surrounded groups of histiocytes. The epidermis showed spongiosis, lymphocytes exocytosis, and a focally ulcerated area (Figures 2A, B). Immunostains were focally positive for S100 (Figure 2C) and CD68 (Figure 2D); diffusely positive for CD1a (Figures 3A, B) and were negative for CD207 (langerin) (Figure 3C). The Ki67 labeling index was about 60% (Figure 3D). Based on these findings, the diagnosis was concluded as an indeterminate dendritic cell tumor; also called indeterminate cell (IC) histiocytosis. The BRAF V600 mutation was negative in the neoplastic cells (sequencing analysis of BRAF gene mutations technique).
Figure 2. Histology and Immunostains of the skin biopsy. A - Dense infiltrate in the dermis (H&E, 100X); B - Infiltrate composed by histiocytoid cells, lymphocytes, plasma cells and multinucleated cells (H&E, 400X); C - S100 partially positive in epidermal Langerhans cells and dermal infiltrate (anti-S100, 200X); D - CD68 positive in histiocytoid cells of the dermal infiltrate (anti-CD68, 200X).
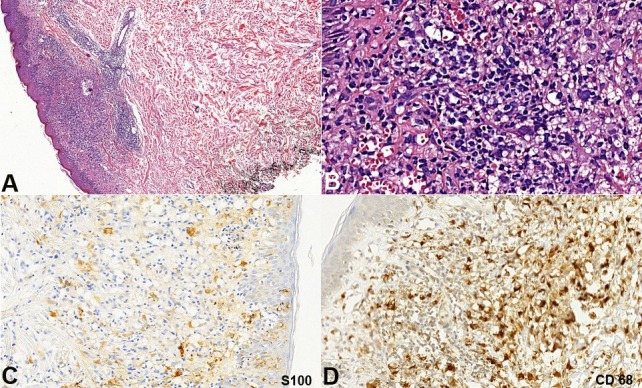
Figure 3. Photomicrography of the Immunostains of the skin biopsy in A and B - CD1a diffusely positive in dermal histiocytoid cells and epidermal Langerhans cells (anti-CD1a, 200X); C - CD207 (Langerin) negative in the demal infiltrate and positive in the epidermal Langerhans cells (anti-CD207, 200X); D - Ki67 positive in about 60% of the dermal cells infiltrate (anti-Ki67, 400X).
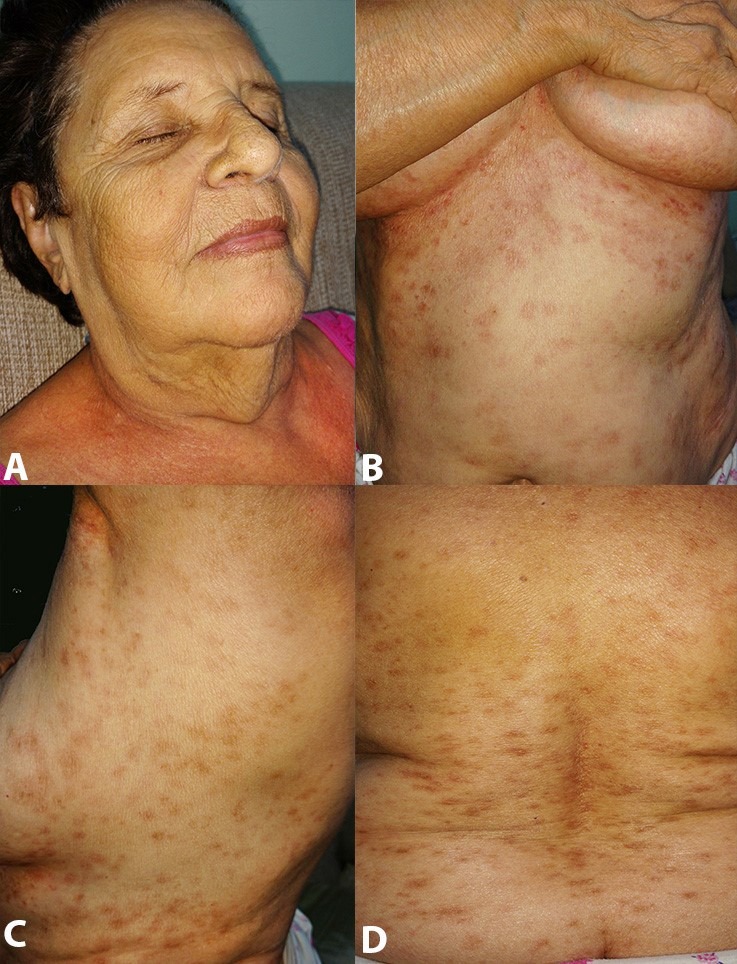
With the diagnosis of IC histiocytosis of exclusive cutaneous involvement (single multifocal system), corticosteroid (prednisone 0.5 mg/kg/day) was started but the patient’s blood pressure increased. Muscular pain and headache ensued and another treatment modality needed to be scheduled. Taking into account the patient’s intolerance to the intermediate steroids dose, age, and comorbidities, a reasonable option was local therapy, so the patient was treated with narrowband ultraviolet B (UVB) phototherapy three times a week for 2 months. The lesions started effacing after the first month of the phototherapy and completely subsided on the third month leaving local hyperpigmentation. The patient is now at the sixth month of follow-up and is completely symptomless (Figure 4); she did not report any adverse reactions.
Figure 4. Cutaneous examination after the sixth month of therapy. The face is free of lesions - A; and some erythematous scar-like lesions remain on the trunk - B, C and D.
DISCUSSION
Histiocytoses comprise a group of disorders characterized by the proliferation of monocytes, macrophages, and dendritic cells, which are not involved in a response to primary disease. The nomenclature of histiocytosis has changed substantially over the last half century, which is now based on the primary involved cell in the pathophysiology of the disease.
The bone marrow pluripotent stem cells, under the influence of (i) granulocyte-macrophage colony stimulating factor (GM-CSF); and (ii) tumor necrosis factor alpha (TNFα) differentiate into a particular group of specialized cells with the functions of antigen presentation and phagocytosis–the dendritic cells.1 These cells move into the blood stream and migrate to the dermal and epidermal layers of the skin. Within the tissue (skin) the dendritic cell precursors under the action of the transforming growth factor β1 (TGF-β1) develop the Birbeck granules and therefore will differentiate into the Langerhans cells. The other cells will not suffer the action of such a cytokine and will remain as two different populations of dendritic cells.2 Along with different subpopulations of lymphocytes, the epidermal Langerhans cells and other dermal antigen-presenting cells (ICs and dendritic cells) make up the major component of the skin’s immune system. The proliferation of such cells will categorize the histiocytoses in (i) Langerhans cell histiocytosis (LCH): (ii) non-Langerhans cell histiocytosis (NLCH); and (iii) IC histiocytosis (ICH).3 Although the latter has been proposed to be a variant of the NLCH,4 in 2008 the World Health Organization (WHO) incorporated ICH into the tumors of the hematopoietic and lymphoid tissue tumors.5
The precise origin of the IC is under debate. While some researchers believe that they are precursors of Langerhans cells, which, when en route to the epidermis, remained arrested into the dermis and did not acquire the Birbeck granules,6-8 other researchers believe they are “veiled dendritic cells” that migrate from the skin to the regional lymph nodes.9 Additionally, experimental evidence points toward the myeloid lineage for the bone-marrow-derived dendritic cells by presenting myeloid lineage markers, such as CD13 and CD33.10-13
Thus, ICs are dendritic elements of the skin (specifically of the dermis, but occasionally the epidermis as well), which express CD1a, CD68, and feeble S100. The absence of the Birbeck granules–and therefore the lack of the expression of CD 207 (langerin)–differentiates IC from Langerhans cells.
In 1985, Wood et al.8 first described the IC histiocytosis. Despite being much more common in adults,14 ICH has been reported in all ages and no gender predominance has been observed. Clinically, ICH is characterized by cutaneous (sparing mucosae) pinky to reddish, varying sized, non-itchy, painless papules or nodules that appear in otherwise healthy persons. A reactive form triggered by cutaneous diseases, such as scabies and pityriasis rosea, has been reported.15,16 Such lesions may be single, a discrete group of lesions, or multiple generalized papules spread over the trunk, face, and extremities, which may present spontaneous remission, stable disease, or remission and recurrence.4,9,16-18 The exclusive cutaneous presentation is the rule; however, bone and corneal involvement have been reported.19,20 The clinical differential diagnosis of ICH is represented by generalized eruptive histiocytosis, a non-Langerhans cell histiocytosis, but in this condition the histiocytes do not express CD1a and S100 protein. The ICH histopathological differential diagnosis is represented by Langerhans cell histiocytosis where the dendritic cells display cytoplasmic Birbeck granules at transmission electron microscopy exam and positivity to CD207 (langerin).
The rarity of this entity imposes certain difficulties in establishing any relationship with other malignancies. However, concomitant or not, cases of ICH have been reported in association with low-grade B-cell lymphoma18,21 and other hematologic malignancies years after their initial diagnosis.
The optimal treatment for ICH is not clear, but similarly to LCH, identifying the initial presentation and organ or system involvement is critical for choosing the appropriate modality of therapy (a systemic work-up prior to the initiation of treatment should be considered). Case reports describe a wide range of effective therapies that include UVB phototherapy, thalidomide, methotrexate, and surgical excision for solitary lesions.22,23
As far as we know, based on published data, this is the third case treated with narrowband UVB with very few side effects, good tolerability, and an excellent short-term result.24,25
Footnotes
Zerbini MCN, Sotto MN, Campos FPF, et al. Indeterminate cell histiocytosis successfully treated with phototherapy. Autopsy Case Rep [Internet]. 2016;6(2):33-38. http://dx.doi.org/10.4322/acr.2016.038
REFERENCES
- 1.Thomas R, Lipsky PE. Dendritic cells: origin and differentiation. Stem Cells. 1996;14(2):196-206. http://dx.doi.org/10.1002/stem.140196. PMid: [DOI] [PubMed] [Google Scholar]
- 2.Toebak MJ, Gibbs S, Bruynzeel P, Scheper J, Rustemeyer T. Dendritic cells: biology of the skin. Contact Dermat. 2009;60(1):2-20. http://dx.doi.org/10.1111/j.1600-0536.2008.01443.x. PMid: [DOI] [PubMed] [Google Scholar]
- 3.Bakry OA, Samaka RM, Kandil MA, Younes SF. Intermediate cell histiocytosis with naïve cells. Rare Tumors. 2013;5(1):e13. http://dx.doi.org/10.4081/rt.2013.e13. PMid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ventura F, Pereira T, Luz Duarte M. Intermediate cell histiocytosis in association with acute myeloid leukemia. Dermatol Res Pract 2010;2010:569345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weiss LM, Chan JKC, Fletcher CDM. Other rare dendritic cell tumors. In: Swerdlow SH, Campo E, Harris NL et al. editors. World Health Organization classification of tumors of the hematopoietic and lymphoid tissues. Lyon: WHO Press; 2008. chap. 14; p. 365. [Google Scholar]
- 6.Vener C, Soligo D, Berti E, et al. Indeterminate cell histiocytosis in association with later occurrence of acute myeloblastic leukaemia. Br J Dermatol. 2007;156(6):1357-61. http://dx.doi.org/10.1111/j.1365-2133.2007.07880.x. PMid: [DOI] [PubMed] [Google Scholar]
- 7.Kolde G, Brocker EB. Multiple skin tumors of indeterminate cells inadult. J Am Acad Dermatol. 1986;15(4 Pt 1):591-7. http://dx.doi.org/10.1016/S0190-9622(86)70209-0. PMid: [DOI] [PubMed] [Google Scholar]
- 8.Wood GS, Hu CH, Beckstead LH, Turner RR, Winkelmann RK. The intermediate cell proliferative disorder: report of a case manifesting as an unusual cutaneous histiocytosis. J Dermatol Surg Oncol. 1985;11(11):1111-9. http://dx.doi.org/10.1111/j.1524-4725.1985.tb01399.x. PMid: [DOI] [PubMed] [Google Scholar]
- 9.Berti E, Gianotti R, Alessi E. Unusual cutaneous histiocytosis expressing an intermediate immunophenotype between Langerhans’ cells and dermal macrophages. Arch Dermatol. 1988;124(8):1250-3. http://dx.doi.org/10.1001/archderm.1988.01670080062020. PMid: [PubMed] [Google Scholar]
- 10.Thomas R, Davis LS, Lipsky PE. Isolation and characterization of human peripheral blood dendritic cells. J Immunol. 1993;150(3):821-34. PMid: [PubMed] [Google Scholar]
- 11.Lenz A, Heine M, Schuler G, Romani N. Human and murine dermis contain dendritic cells: isolation by means of a novel method and phenotypical and functional characterization. J Clin Invest. 1993;92(6):2587-96. http://dx.doi.org/10.1172/JCI116873. PMid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Doherty U, Peng M, Gezelter S, et al. Human peripheral blood contains two dendritic cell sub-sets: one mature and one immature. Immunology. 1994;82(3):487-93. PMid: [PMC free article] [PubMed] [Google Scholar]
- 13.Peters JH, Gieseler R, Thiele B, Steinbach F. Dendritic cells: from ontogenetic orphans to myelomonocytic descendants. Immunol Today. 1996;17(6):273-8. http://dx.doi.org/10.1016/0167-5699(96)80544-5. PMid: [DOI] [PubMed] [Google Scholar]
- 14.Rodrígues-Jurado R, Vidaurri-de la Cruz H, Durán-Mckinster C, Ruíz-Maldonado R. Indeterminate cell histiocytosis. Clinical and pathological study in a pediatric patient. Arch Pathol Lab Med. 2003;127(6):748-51. PMid: [DOI] [PubMed] [Google Scholar]
- 15.Hashimoto K, Fujiwara K, Mehregan A. Current topics of immunohistochemistry as applied in skin tumors. J Dermatol. 1993;20(9):521-32. http://dx.doi.org/10.1111/j.1346-8138.1993.tb01333.x. PMid: [DOI] [PubMed] [Google Scholar]
- 16.Ratzinger G, Burgdorf WHC, Metze D, Zelger BG, Zelger B. Indeterminate cell histiocytosis: fact or fiction? J Cutan Pathol. 2005;32(8):552-60. http://dx.doi.org/10.1111/j.0303-6987.2005.00382.x. PMid: [DOI] [PubMed] [Google Scholar]
- 17.Miracco C, Raffaelli M, de Santi MM, Fimiani M, Tosi P. Solitary cutaneous reticulum cell tumor. Enzyme immunohistochemical and electron-microscopic analogies with IDRC sarcoma. Am J Dermatopathol. 1988;10(1):47-53. http://dx.doi.org/10.1097/00000372-198802000-00006. PMid: [PubMed] [Google Scholar]
- 18.Rezk S, Spagnolo D, Brynes R, Weiss L. Indeterminate cell tumor: a rare dendritic neoplasm. Am J Surg Pathol. 2008;32(12):1868-76. http://dx.doi.org/10.1097/PAS.0b013e31818593d6. PMid: [DOI] [PubMed] [Google Scholar]
- 19.Flores-Stadler EM, Gonzales-Crussi F, Greene M, Thangavelu M, Kletzel M, Chou PM. Indeterminate-cell histiocytosis: immunophenotypic and cytogenetic findings in an infant. Med Pediatr Oncol. 1999;32(4):250-4. http://dx.doi.org/10.1002/(SICI)1096-911X(199904)32:4<250::AID-MPO2>3.0.CO;2-#. PMid: [DOI] [PubMed] [Google Scholar]
- 20.Calatayud M, Güell JL, Gris O, Puig J, Arrondo E, Huguet P. Ocular involvement in a case of systemic indeterminate cell histiocytosis: a case report. Cornea. 2001;20(7):769-71. http://dx.doi.org/10.1097/00003226-200110000-00021. PMid: [DOI] [PubMed] [Google Scholar]
- 21.Bettington A, Lai J, Kennedy C. Indeterminate dendritic cell tumour presenting in a patient with follicular lymphoma. Pathology. 2011;43(4):372-5. http://dx.doi.org/10.1097/PAT.0b013e32834685b7. PMid: [DOI] [PubMed] [Google Scholar]
- 22.Toth B, Katona M, Harsing J, Szepesi A, Karpati S. Indeterminate cell histiocytosis in a pediatric patient: successful treatment with Thalidomide. Pathol Oncol Res. 2012;18(2):535-8. http://dx.doi.org/10.1007/s12253-011-9405-8. PMid: [DOI] [PubMed] [Google Scholar]
- 23.Fournier J, Ingraffea A, Pedvis-Leftick A. Successful treatment of indeterminate cell histiocytosis with low-dose methotrexate. J Dermatol. 2011;38(9):937-9. PMid: [DOI] [PubMed] [Google Scholar]
- 24.Bard S, Torchia D, Connelly E, Duarte A, Badiavas E, Schachner L. S100-negative indeterminate cell histiocytosis in an Africa American child responsive to narrow ultraviolet B. Pediatr Dermatol. 2011;28(5):524-7. http://dx.doi.org/10.1111/j.1525-1470.2011.01305.x. PMid: [DOI] [PubMed] [Google Scholar]
- 25.Ishibashi M, Ouchi T, Tanikawa A, Ishiko A. Indeterminate cell histiocytosis successfully treated with ultraviolet B phototherapy. Clin Exp Dermatol. 2008;33(3):301-4. http://dx.doi.org/10.1111/j.1365-2230.2007.02667.x. PMid: [DOI] [PubMed] [Google Scholar]