

Abstract
The discovery of induced pluripotent stem cells (iPSC) and their application to patient-specific disease models offers new opportunities for studying the pathophysiology of neurological disorders. However, current methods for culturing iPSC-derived neuronal cells result in clustering of neurons, which precludes the analysis of individual neurons and defined neuronal networks. To address this challenge, we developed cultures of human neurons on micropatterned surfaces that promote neuronal survival over extended periods of time. This approach facilitates studies of neuronal development, cellular trafficking, and related mechanisms that require assessment of individual neurons and specific network connections. Importantly, micropatterns support the long-term stability of cultured neurons, which enables time-dependent analysis of cellular processes in living neurons. The approach described in this paper will allow mechanistic studies of human neurons, both in terms of normal neuronal development and function, as well as time-dependent pathological processes, and will provide a platform for testing of new therapeutics in neuropsychiatric disorders.
Introduction
Human induced pluripotent stem cells (iPSCs) share many properties with embryonic stem cells (ESCs), including self-renewal, pluripotency and a theoretically unlimited supply. Several reasons have made iPSCs derived from patients the system of choice, including the fact that the use of human ESCs presents significant ethical concerns due to the use of human embryos. One of the most promising advantages of iPSCs is their use in creating patient-specific disease models derived from an individual's own fibroblasts [1-2]. Such models offer the possibility of gaining a deeper understanding of disease mechanisms, but can also be useful for drug screening and personalized medicine [3-4]. Over the last ten years, numerous studies have utilized iPSC-derived neurons to explore mechanisms of neurodegenerative disorders such as Parkinson's disease (PD), Alzheimer's disease (AD) and Amyotrophic lateral sclerosis (ALS) [5]. However, these approaches are still limited, particularly for cultures that model neurological or psychiatric disorders and require resolution of individual neurons or defined networks, both of which are challenging because cells tend to form dense clusters during the differentiation process. Several approaches are in development to address these limitations, including the use of physical barriers that guide or compartmentalize neurons [6-8], chemically patterned substrates [9-10], and microfluidic devices that offer control over the positions and local environment surrounding the cells [11-12]. These methods, however, are not always straightforward to implement and have largely been limited to the patterning of primary neurons for only a few weeks [13-15]. For example, Zhang and coworkers reported three week cultures of cortical cells on polymeric substrates modified with a aminopropyl siloxane [16] and West and coworkers reported month-long cultures of primary neurons on glass slides patterned with poly(ethylene glycol) [17]. Microcontact printing of self-assembled monolayers of alkanethiolates on gold has also been used to pattern adherent cells [18]. The patterned monolayers have the benefits that they are straightforward to prepare and they maintain the patterned cells from one to four weeks [19-21]. In this paper we demonstrate that microcontact printing can be used to prepare patterned monolayers that maintain the positions of human iPSC-derived neuronal cells for periods in excess of 100 days, and we demonstrate how these cultures enable mechanistic studies of neuronal models.
Results
Design of micropatterned substrates for long-term neuronal culture
We patterned gold-coated glass slides with microcontact printing to generate surfaces presenting large arrays of identically-shaped cell-adhesive islands. We used a polydimethylsiloxane (PDMS) elastomer stamp that was inked with an ethanolic solution of octadecanethiol, dried, and then brought into contact with the gold surface (Figure 1A). We then backfilled the unstamped area of the substrate by immersing it in an ethanolic solution of a tri(ethylene glycol)-terminated alkanethiol, rendering the surrounding areas resistant to protein adsorption, and in turn preventing non-specific cell adhesion. After incubation of the substrate in ethanol for sterilization, we coated the protein-adhesive regions with buffered laminin solution for 1 hour at 37°C in order to render those regions permissive to cell attachment and spreading. We fabricated surfaces presenting one of three different shapes – flower, line and star – for use in immunocytochemistry and live cell imaging studies (Figure 1B).
Figure 1. Micropatterned substrate fabrication.
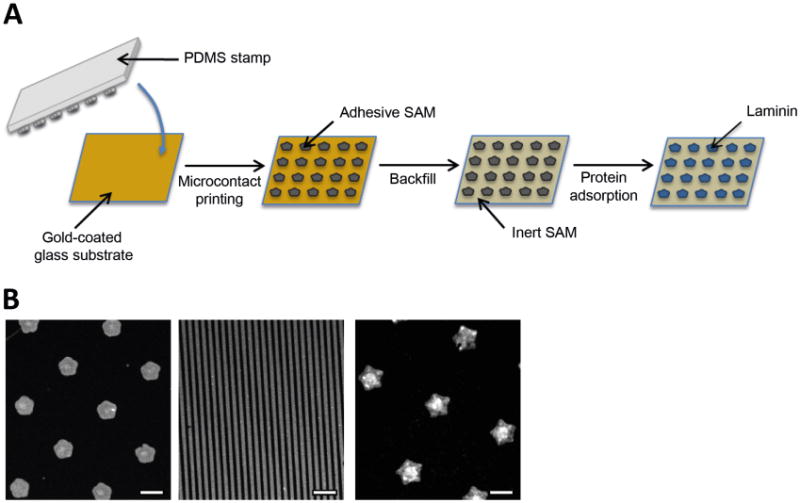
(a) Scheme showing the microcontact printing of gold-coated substrates. A polydimethylsiloxane (PDMS) stamp is used to pattern octadecanethiol onto a gold-coated glass substrate, rendering the desired areas protein-adhesive. Backfilling the substrate with an ethylene glycol-terminated molecule renders the surrounding areas resistant to protein deposition (and therefore resistant to cell adhesion). Coating the backfilled substrate with laminin makes the stamped “islands” cell-adhesive. (b) Immunofluorescent images of different micropatterned substrates showing surfaces presenting flower (1250μm2), line (10μm diameter) and star (2500μm2) patterns. Scale bar, 50μm.
Precise control of neuronal culture on micropatterned substrates
We generated midbrain dopaminergic neurons obtained from healthy human skin fibroblasts via reprogramming of iPSCs [22-23]. iPSCs were characterized previously for the expression of pluripotency markers, differentiation capacity through embryoid body formation, and normal genomic structure by karyotype analysis [23]. Differentiation of iPSCs into midbrain dopaminergic neurons was achieved by a previously published protocol [24]. Under conventional neuronal culture conditions 200,000 to 300,000 iPSC-derived neurons are plated onto regular unpatterned PDL/laminin-coated coverslips on day 25 to day 30 of differentiation. These conventional culture conditions result in uneven growth and neurite density, while plating cells onto micropatterned surfaces drives neuronal cultures towards a defined growth pattern allowing studies of individual neurons and neuronal networks (Figure 2A). Long-term culturing of human iPSC-derived neurons has not been possible so far due to neuronal clustering of cells during conventional culture conditions (Figure 2B). For micropatterned studies, substrates were either placed into one well of a 24-well plate or pre-designed live cell imaging substrates were used, coated with laminin and 150,000 to 300,000 neurons were seeded on day 25 to day 30 of differentiation. Once neurons attached to the surface, they separated into individual neuronal processes. We determined the specific size of single cell-adhesive areas and the distance from those areas to each other (for each flower-, star- and line-shaped patterning environment) to ensure that neurons evenly distributed onto the substrate. Under these optimized conditions, neurites were able to cross the cell-repellent regions to form connections with nearby neurites and cell bodies resulting in neuronal networks 20h after plating neurons onto a substrate with a flower-shaped pattern (Figure 2C, left image). At day 40 of the differentiation protocol, micropatterned neurons stained positive for the neuronal marker β-III-tubulin (Figure 2C, right image) as well as pre- and postsynaptic markers (data not shown). During early development iPSC-derived dopaminergic neurons initiate the expression of midbrain and dopaminergic markers, such as LMX1A, FOXA2 and tyrosine hydroxylase (TH). These markers are commonly used to assess the efficiency of neuronal differentiation. We compared neurons plated on micropatterned substrates with those grown on conventional unpatterned PDL/laminin-coated glass coverslips on day 70 of differentiation and were able to confirm that similar numbers of neurons plated on either systems stained positive for TH expression (Figure 2D). These results indicate that micropatterned culture conditions did not alter the maturation process in comparison with conventionally used unpatterned PDL/laminin-coated glass coverslip surfaces for iPSC-derived neuronal cultures.
Figure 2. Advantage of micropatterning technology over conventional protocol for iPSC-derived neuronal culturing.
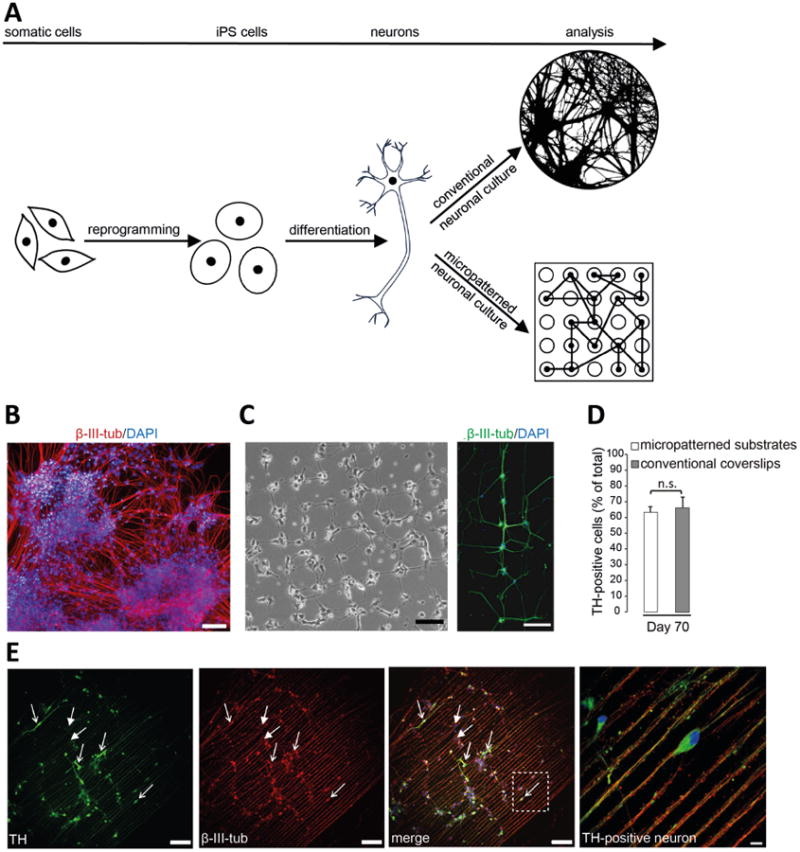
(a) Schematic of the differentiation of midbrain dopaminergic neurons from iPSCs, which have been reprogrammed from adult somatic cells. Culturing iPSC-derived neurons on conventional glass coverslips usually results in a less defined growth of neurons and neuronal processes, while culturing neurons on micropatterns enables the analysis of individual neurons and neuronal networks under controlled conditions. (b) Immunofluorescence image of iPSC-derived neurons grown on unpatterned PDL/laminin-coated coverslips following the conventional protocol. Cells were labeled with the neuronal marker β-III-tubulin (red) and DAPI (blue). Scale bar, 100μm. (c) Left: Bright field image taken 20h after plating iPSC-derived neurons onto a substrate with a flower-shaped pattern. Scale bar, 100μm. Right: Immunofluorescence image of iPSC-derived neurons on a substrate representing a flower-shaped pattern. Neurons were labeled with the neuronal marker β-III-tubulin (green) and DAPI (blue). Scale bar, 100μm. (d) Quantification of TH-positive neurons as percent of total number of cells cultured on micropatterned substrates or on conventional unpatterned PDL/laminin-coated coverslips at day 70 of differentiation; n.s.: not significant. (e) Immunofluorescence images of iPSC-derived neurons grown on a substrate with a line-shaped micropattern at day 70 of differentiation. Cells were labeled with the dopaminergic marker TH (green), the neuronal marker β -III-tubulin (red) and DAPI (blue). Examples of TH-positive (arrows with open arrowhead) and TH-negative (arrows with closed arrowhead) neurons are indicated. Scale bar, 100μm. Enlarged view of the boxed region shows a TH-positive neuron. Scale bar, 10μm.
Neurodegenerative diseases selectively target subpopulations of neurons, leading to the progressive failure of specific neuronal systems. The basis of such selective neuronal vulnerability has largely remained elusive. The factors that determine selective vulnerability in neurodegeneration are poorly understood conditions in vitro and in vivo. As a consequence, investigating these factors requires experimental approaches for monitoring the disease process in defined subtypes of neurons. The micropatterned substrates are well-suited for analyzing individual neurons within a culture as they separate cells based on their patterned spatial arrangement. We cultured iPSC-derived neurons on substrates with a line-shaped micropattern and immunolabeled them with an antibody against TH at day 70 of differentiation (Figure 2E). We were able to easily distinguish between TH-positive (arrows with open arrowhead) as well as TH-negative (arrows with closed arrowhead) neurons appearing in each of the parallel lines. Such experimental conditions allow examination of disease-associated processes in the more vulnerable dopaminergic neurons in comparison to the less vulnerable non-dopaminergic neurons. We next demonstrate that functional studies such as axonal transport of mitochondria and neurite outgrowth can be performed in these cultures. These methods will advance the use of iPSCs as patient-specific disease models, in particular in age-related disorders, by studying the timeline of pathological mechanisms in specific neuronal subtypes.
Long-term studies of mitochondrial dynamics under defined conditions
Mitochondrial dynamics are essential for maintaining organelle stability and function, and it is known that mitochondrial dysfunction is implicated in complex age-related neurodegenerative disorders including AD, PD and ALS [25-26]. The morphology of the mitochondrial network is in a constant state of flux, thereby retaining optimal populations of mitochondria. The transition between elongated reticular networks and punctate structures is dependent on two antagonistic processes, i.e. fission and fusion, both of which are vital for neuronal survival as they directly affect organelle number, shape and location [27]. In iPSC-derived neurons, grown on conventional unpatterned PDL/laminin-coated coverslips, the precise tracing of these events within individual neurons has been difficult to achieve, because neurite density varies dramatically due to uneven neuron growth (compare Figure 2B). Neurons grown on micropatterned surfaces extend their processes in a defined and uniform manner, which is dictated by the pattern design. This allows the precise evaluation of mitochondrial dynamics in individual neuronal processes in live neurons so that mitochondrial fission (Figure 3A, upper panel) or fusion (Figure 3A, lower panel) can be easily followed over time. To assess mitochondrial morphology in individual neuronal processes, we plated cells onto substrates patterned with lines 10μm wide and examined mitochondrial parameters after immunostaining with the mitochondrial marker TOM20 and the neuronal marker β-III-tubulin (Figure 3B). Conversion of the TOM20 signal into a binary image facilitated the visualization of mitochondrial structures for subsequent analysis with ImageJ software. The analysis of mitochondrial parameters in individual neuronal processes of patterned neurons at different time points revealed a change in mitochondrial morphology over time (Figure 3C). While mitochondria appeared punctated at day 40 of differentiation, they were more elongated and interconnected at day 100. We quantitated the length of individual mitochondria (also known as aspect ratio) as well as their interconnectivity (also known as form factor) using ImageJ analysis on the basis of the binarized TOM20 signal as described previously [28]. The results showed a time-dependent increase of mitochondrial length as well as interconnectivity when comparing 40 and 100 days old neurons (Figure 3C).
Figure 3. Analysis of mitochondrial dynamics in long-term neuronal cultures.
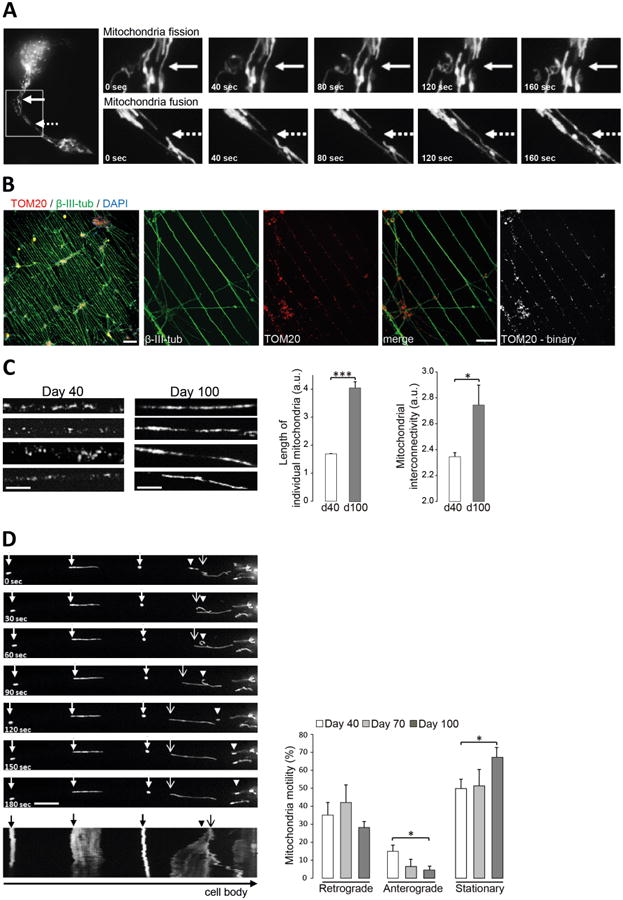
(a) Image series showing mitochondrial trafficking as fusion (solid arrow; upper panel) and fission (dashed arrow, lower panel) events in one neuronal process over 160 seconds. iPSC-derived neurons were plated onto a live cell imaging substrate representing a flower-shaped micropattern. Mitochondria were visualized by transduction of mito-GFP BacMam 2.0. (b) Immunofluorescence images of iPSC-derived neurons grown on a substrate with line-shaped pattern shown in low magnification (10×; Scale bar, 100μm) or higher magnification (40×; Scale bar, 20μm). Cells were labeled with neuronal marker β-III-tubulin (green), mitochondrial marker TOM20 (red) and DAPI (blue). TOM20 signal was converted into a binary image for analysis of mitochondrial structures. (c) Mitochondrial length and interconnectivity in individual neuronal processes of iPSC-derived neurons grown on substrates with line-shaped pattern was analyzed after 40 (d40) and 100 days (d100) of differentiation using ImageJ software. *p<0.05; ***p<0.001. Scale bar, 5μm. (d) Image series showing retrograde moving (arrowhead), anterograde moving (arrow with open arrowhead) or stationary (arrow with closed arrowhead) mitochondria over 180 seconds. Corresponding kymograph is shown at the bottom of the image series. Scale bar, 10μm. Mitochondrial trafficking was followed after 40, 70 and 100 days of differentiation and retrograde moving, anterograde moving (≥ 10μm/3 minutes in either direction) and stationary mitochondria were analyzed using ImageJ software. The percentage motility of mitochondria along the neuronal process was calculated as a percentage of the total number of mitochondria imaged per process. *p<0.05.
Axonal transport of mitochondria using micropatterned long-term culture of human neurons
Mitochondria not only show dynamic movement within the mitochondrial network, but also show motility throughout neurons [29]. The trafficking of mitochondria to areas of higher energy requirements, such as synapses, where mitochondrial densities fluctuate, further highlights the importance of efficient mitochondrial dynamics in neurons. Specific trafficking machineries enable mitochondrial transport to synaptic areas of increased energy demand or to the cell body for degradation [30]. Mitochondria move along microtubules using motor and adaptor proteins. Anterograde movement (towards the axon terminal) is dependent on kinesin motors, while retrograde movement utilizes dynein motors [31]. Impaired mitochondrial transport has been shown previously to be implicated in numerous age-related neurodegenerative disorders [32]. A number of model systems have been established to study mitochondrial movement in neurons [33-35]. We used an insect cell virus (baculovirus) coupled with a mammalian promotor to label cellular organelles. We transduced iPSC-derived neurons 16 hours prior to imaging with the BacMam CellLight® Mitochondria-GFP construct, which utilizes the leader sequence of E1 alpha-pyruvate dehydrogenase (3.1 kDa) to label mitochondria. By adjusting the relative number of viral particles, it was possible to efficiently label the mitochondria in mature iPSC-derived neurons and carry out continuous observation of mitochondrial movement with no cytotoxicity. We followed mitochondrial trafficking in individual neurons at day 40, day 70 and day 100 of differentiation and analyzed retrograde (towards the cell body) and anterograde (away from the cell body) mitochondrial movements (where a movement required at least a 10μm displacement in one direction) as well as stationary mitochondria as percentage of mitochondrial motility (Figure 3D). The corresponding kymograph (Figure 3D, bottom) illustrates mitochondrial movement (x axis) over time (y axis), in which the stationary mitochondria are displayed as straight lines and the moving mitochondria as diagonal lines in either direction. Overall, individual mitochondria exhibited a variety of dynamic behaviors as shown in Supplemental Movie 1. For example, some mitochondria were largley static while others moved at high speed, some stopped abruptly before continuing to move, or reversed direction entirely. Overall, the micropatterned neurons cultured for 100 days showed a decrease in mitochondrial motility towards the anterograde direction in comparison with neurons at day 40, while the percentage of stationary mitochondria was greater in neurons at day 100 compared to day 40. Approximately one-half of the mitochondria were stationary in neurites at day 40 and day 70, whereas 70% were stationary at day 100. These results demonstrate the feasibility of quantitating mitochondrial dynamics in human neurons cultured on micropatterned surfaces over long periods. The transport of organelles, e.g. mitochondria, is particularly important in neurons, because their functions are needed throughout the axon. Defects in axonal transport are thus of special interest to degenerative diseases of the nervous system.
Organization and dynamic network formation of neurons on micropatterned substrates
Neurons are highly polarized cells with two molecularly and functionally distinct domains that emerge from the cell body: a single thin, long axon that transmits signals, and multiple shorter dendrites that are specialized to receive signals. The ability of neurons to polarize is crucial for synaptic transmission, and knowledge of the mechanisms that govern neuronal polarization is fundamental to our understanding of normal neural development, plasticity as well as neuropsychiatric diseases. Mouse primary and human iPSC-derived neurons from genetic models of neurodegenerative diseases have been used to study disease-associated changes of neuronal organization or network formation, primarily focusing on neurite outgrowth phenotypes [36-37]. Further, high throughput screening studies for human neurons have been developed to assess phenotypic outcomes including neurite outgrowth and branching [38]. However, it remains very challenging to reliably examine these processes in iPSC-derived neurons under conventional culture conditions due to aforementioned clustering of cells. To study outgrowth as an early process in the formation of neuronal networks, we imaged iPSC-derived neurons 50 hours after plating onto a substrate patterned with an array of flower-shaped features (Supplemental Movie 2). As shown in Figure 4A, neurons cultured on patterned substrates allowed for easy tracking of neurite outgrowth processes from one cell to another over time.
Figure 4. Dynamic network formation of iPSC-derived neurons.
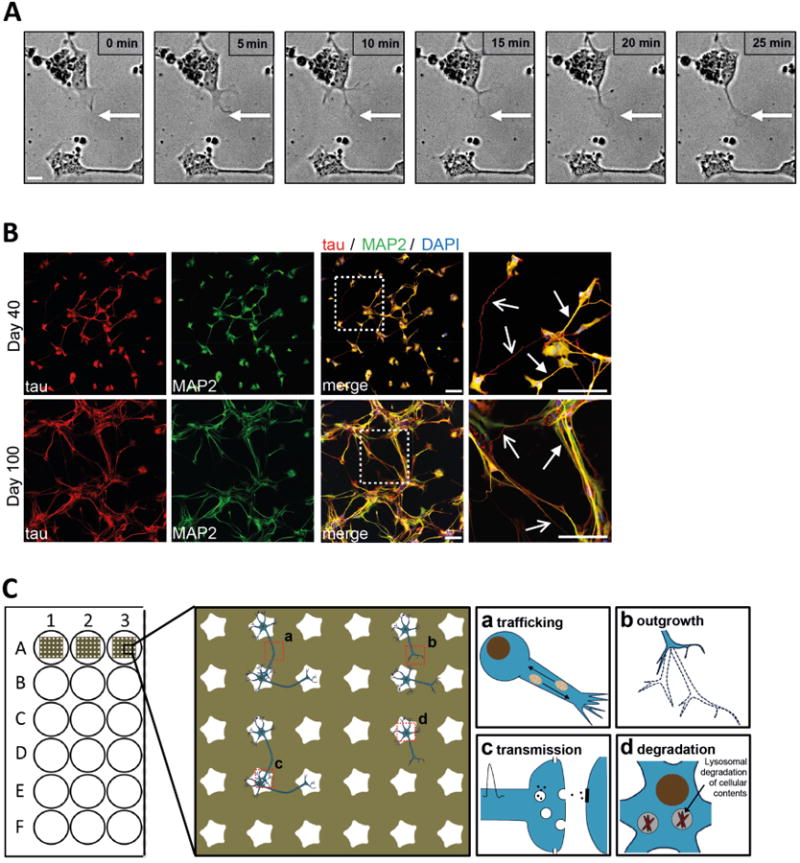
(a) Bright field images showing neurite outgrowth towards a nearby cell body over 25 minutes. iPSC-derived neurons were seeded onto a live cell imaging substrate with a flower-shaped micropattern on day 30 of differentiation and bright field images were taken 50 hours after seeding. Scale bar, 10μm. (b) Immunofluorescence images of iPSC-derived neurons grown on a substrate with a star-shaped micropattern. Cells were fixed and labeled with the axonal marker tau (red), the dendritic marker MAP2 (red) and DAPI (blue) at day 40 (top panel) or day 100 (bottom panel) of differentiation. Enlarged views of the boxed regions show tau-positive axons (arrows with open arrowheads) and MAP2-positive dendrites (arrows with closed arrowheads). Scale bar, 100μm. (c) Schematic of the potential use of micropatterned substrates for studies of cell dynamics, such as mitochondrial dynamics (a), neuronal outgrowth (b), synaptic transmission (c) and cellular degradation pathways, e.g. lysosomal degradation of aggregated proteins or cellular contents (d).
The process of network formation depends on the development of neuronal polarity with a specified axon as well as several dendrites and requires precise regulation of microtubule and actin dynamics. Microtubule-associated proteins (MAP) control the dynamic properties of microtubules, which differ in their complement of MAPs. For example, MAP2 is found mostly in dendrites and tau is found mainly in axons. Micropatterned substrates provide a suitable environment to study neuronal polarity because they allow for the distinction of axons and dendrites on the surface due to the separation of cells on cell-adhesive areas. Neurons were plated on day 30 of differentiation onto star-shaped micropatterned substrates and either immunostained 10 days later (day 40 of differentiation) or 70 days later (day 100 of differentiation). Using antibodies against tau (to label axons; Figure 4B, arrow with open arrowhead), and MAP2 (to label dendrites; Figure 4B, arrow with closed arrowhead), we could visualize the formation of individual connections to neighboring cell bodies. Culturing neurons until day 100 of differentiation on star-shaped micropatterned substrates resulted in a progressively more organized and denser neuronal network, further demonstrating the utility of micropatterns in studies of neuronal maturation and network development.
Discussion
The use of iPSCs for modeling neurological disorders addresses a critical need in medicine, yet technical limitations in neuronal cultures have made the analysis of individual neurons and neuronal networks very challenging. The clustering of human neurons during the differentiation process makes it difficult to image single cell bodies and to track the fates of individual neurons over time. The challenge, in particular, of culturing human neurons in a micropatterned environment is in providing conditions that ensure survival of single cells on a cell-adhesive site as well as to maintain the defined pattern during long-term neuronal culture. In this work, we demonstrate that the use of patterned monolayers overcomes limitations such as neuronal clustering and enables the long-term culture of human neurons under well-defined conditions. We give examples of the use of these patterned cultures in the quantitative tracking of mitochondrial movement, evaluation of mitochondrial morphology, analysis of neurite outgrowth and neuronal polarity in specific subtypes of neurons. These applications have not been possible with the use of conventional iPSC culture formats, and serve to illustrate the significance of the work presented in this paper.
Earlier reports that cultured human stem cell-derived neuronal cells on patterned surfaces were directed towards short-term applications - one study reported culture of neural progenitor cells on patterned surfaces for only 2 days [39] and a second study investigated the adherence and differentiation capacity of human cord blood-derived neural stem cells for 7 days [40]. Other examples cultured primary neurons on micropatterned substrates for several days to a few weeks [13, 15, 17]. It is not clear why the patterned monolayers are effective at maintaining the patterned neurons for periods of months in culture. Previous examples of patterned cell cultures on monolayers of alkanethiolates on gold have found that the cells typically remain in their initial positions for one to four weeks [19-21]. In those cases, it is likely that the protein resistant oligo(ethylene glycol) groups were oxidatively damaged, compromising the protein-resistant property. We did not alter those patterned substrates to achieve the long-term culture of neurons reported in this work; rather, the stability must reflect either a less oxidative (or otherwise reactive) environment near the cells or a lack of migratory phenotype in these cells. In any event, enabling the culture of human iPSC-derived neurons for more than 100 days, our method enables studies of disease-relevant mechanisms and phenotypes in individual neurons and networks over periods of several months. In particular, we monitored changes in neuronal development, mitochondrial morphology as well as trafficking in living neurons.
Neurons are postmitotic and highly energy-dependent and therefore particularly vulnerable to alterations in cellular bioenergetics and increased stress that may occur as a direct or indirect result of mitochondrial dysfunction. These cellular processes are also implicated in age-related neurodegenerative disorders, such as AD, PD and ALS, indicating a common pathological thread [25-26]. Integral changes in mitochondria have long been thought to contribute to the pathological events during aging, notably in relation to complex I deficiency of the electron chain and the accumulation of mitochondrial DNA (reviewed in [25, 41]). But so far, mitochondrial morphology as well as trafficking within individual neurons has been challenging to study because neuronal processes must be adherent, stable and accessible for high-resolution imaging.
Using our optimized micropatterned substrates we found that alterations of mitochondrial parameters change over time in normal healthy dopaminergic neurons; e.g. an increase in mitochondrial length and interconnectivity as well as a reduction in anterograde transport towards more stationary mitochondria from day 40 to day 100 of differentiation. It is well established that adaptations in mitochondrial dynamics are required during development and aging to maintain mitochondrial function, but the precise nature of these changes has not been studied in human neurons [42]. Fusion and fission events are mediated through different specialized proteins (reviewed in [43]). While fission events have been shown to be preceded by a sustained fall in mitochondrial membrane potential [44], fusion mixes mitochondrial contents and is known to benefit mtDNA stability and allows the spreading of metabolites, enzymes, and mitochondrial gene products throughout the mitochondrial compartment and also serves to repair abnormal mitochondrial phenotypes [45]. The extensive fusion of mitochondria observed at later time points in our studies might indicate a compensatory response to mitochondrial damage. Hyperfused mitochondria may be resistant to apoptosis [46] and escape autophagosomal degradation [47].[48]. Fission, conversely, generates fragmented mitochondria that are necessary for transportation purposes as well as selecting damaged or aged mitochondria for autophagy. Changes in both of these processes have been shown to contribute to neurodegenerative and other disorders [26]. Further, mitochondrial trafficking is important in neurons since active transport of mitochondria supplies local energy needs [49]. Several studies have shown that defects in the microtubule-based machinery can cause or contribute to a number of human neurodegenerative diseases [32, 42, 50-51].
Over the past decades, many studies have explored the cellular and molecular mechanisms that underlie neuronal polarization primarily using mouse primary neurons. Also, iPSC-derived neurons from genetic models of neurodegenerative diseases have been used to study disease-associated changes of neuronal organization or network formation, primarily focusing on neurite outgrowth phenotypes [36-37]. Further, high throughput screening studies for human neurons have been developed to assess phenotypic readouts such as neurite outgrowth and branching and suggested its use for compound screening as well as drug development [38]. However, it has been very challenging to reliably examine these processes in iPSC-derived human neurons under conventional culture conditions due to aforementioned clustering of cells. There have been attempts to culture non-iPSC-derived cells in microfluidic platforms to establish a more organized environment for studies of neurite outgrowth, but those platforms are designed to guide the neurites towards a predetermined direction. In addition, this approach has not been utilized in iPSC-derived neurons [52]. We found that neurons plated on star-shaped micropatterned substrates initially formed individual connection to neighboring cell bodies either by the axon or the dendrites and were able to form a more dense and organized network over time. Micropatterned substrates provide an excellent environment to study the development of neuronal polarity in human neurons, because they allow for the distinction of axons and dendrites on the surface due to their separation on cell-adhesive areas.
Using micropatterned surfaces for the study of progressive phenotypes in individual neurons and neuronal networks fills a gap in the currently available techniques and will be important in defining mechanisms in neurodegenerative disorders. Our approach can be combined with various other imaging and molecular biology techniques, including the immunocytochemistry or live cell imaging applications, but will also be useful to screen for accumulation of synaptic or perinuclear components in individual neurons and examine cellular kinetics will live-cell imaging, electrophysiology or high-resolution single-molecule tracking (Figure 4C).
The discovery of iPSCs has transformed our ability to study complex tissue phenotypes in the laboratory and has exciting implications for personalized medicine. Yet, culturing iPSC-derived cell lines can be very challenging. This has been the case with neuronal cells and our report of a method that can pattern adult human neurons for periods of several months represents a critical advance in harnessing the potential of iPSC-derived neurons. We believe studying biological processes such as mitochondrial transport and dynamic network formation in these neuronal cultures will be important for understanding disease-relevant mechanisms of clinically significant human brain pathologies such as PD, AD and ALS.
Experimental section
Micropatterned substrate fabrication
Arrayed surfaces were fabricated using microcontact printing, as described previously [18]. In brief, we started with gold substrates (220 A on top of a 40 A Ti adhesion layer), deposited with an electron beam vapor deposition system (Thermionics, CA). Substrates were rinsed with 100% ethanol, and dried with a stream of nitrogen. Polydimethylsiloxane stamps were inked with octadecanethiol (10mM, Sigma, #74731) in ethanol, dried with a stream of nitrogen, and brought into contact with the clean gold for 1 minute. The printed gold was then cleaned with 100% ethanol, dried with nitrogen and backfilled in triethylene glycol mono-11-mercaptoundecyl ether (10 mM, Sigma, #673110) in ethanol overnight at 4°C. Surfaces were then rinsed well in 100% ethanol and dried with a stream of nitrogen for storage until use.
Preparation of micropatterned substrates for neuronal cell culture
The preparation of micropatterned substrates for neuronal cell culture took place in an aseptic environment and all reagents used were sterile. To sterilize micropatterned substrates, they were rinsed with 100% ethanol and placed in a petri dish containing enough 100% ethanol to fully submerge them for 15 minutes. Substrates were removed from the petri dish using a sterilized fine forceps and completely air dried before placing them into a 24-well plate (about 15 minutes). For live cell imaging micropatterned substrates, which were glued to drilled dishes (see below), ethanol rinsing and drying was done in the same manner. When completely dried, substrates were coated with laminin (5ug/ml, Roche, # 11243217001) in PBS and incubated at 37°C in a humidified incubator for 1 hour. Before seeding cells onto substrates, laminin was aspirated off and substrates were washed once with PBS. For live cell imaging studies, an 18mm whole was drilled into 35mm plastic dishes. Micropatterned substrates were glued onto a drilled dish using Sylgard (Electron Microscopy Sciences, #21236) adhesive, which was cured briefly at 80°C on a temperature-controlled hotplate.
iPSC culture and neural differentiation
The iPSC line was generated from skin fibroblasts of a healthy individual and was reprogrammed through retroviral expression of OCT4, SOX2, cMYC, KLF4 as described previously [22]. The line was characterized for expression of pluripotency markers (OCT4, Tra-1-60, SSEA4, Nanog), genomic integrity through G-banding karyotype analysis and teratoma analysis [22-23, 53]. iPSCs were cultured and maintained as described previously [22-23]. Differentiation of iPSCs into midbrain dopaminergic neurons was done according to published protocols [24]. To better control consistency of neuralization, cells were first passaged en bloc at day 10 to 15 by mechanical dissociation of thickened cell layer into 2mm2 blocks followed by plating onto PDL/laminin coated 10cm dishes. At day 25 to 30 of differentiation, neural blocks were passed by accutase treatment onto laminin coated micropatterned substrates (for details see above) and incubated until analysis. Neuralization growth factors were withdrawn at day 40 and neurons were maintained in Neurobasal media (Life Technologies, #21103-049) containing Neurocult SM1 supplement (Stemcell technologies, #05711). The medium was replaced every 3 days.
Immunocytochemistry
Neurons were fixed for 20 minutes in 4% paraformaldehyde in PBS, washed twice with PBS and permeabilized with 0.3% Triton X-100 in PBS for 10 minutes. Cells were blocked in 1-2% BSA, 5% normal goat serum in 0.3% Triton X-100/PBS for 1 hour at room temperature. Neurons were incubated with the following primary antibodies: anti β -III-tubulin (Covance # MMS-435P, 1:1000 or Covance # MRB-435P, 1:1000), TOM20 (BD Bioscience # 612278, 1:300), Tyrosine Hydroxylase (Merck-millipore # 657012, 1:1000), tau (DAKO # A002401-2, 1:300), MAP2 (Sigma # M4403, 1:300). Primary antibodies were incubated overnight, washed in 0.3% Triton X-100/PBS and then incubated with Alexa-conjugated anti-rabbit or anti-mouse antibodies at 1:500 for 1 hour at room temperature. Micropatterned substrates were mounted in DAPI Fluoromount (Southern Biotech).
Microscopy
Immunostainings were visualized on an inverted Leica confocal microscope (Leica TCS SPE-II with DMI4000 RYBC Leica TCS SPE confocal unit RYBV) equipped with 10×/0.30 CS, 40×/1.15 Oil CS and 63×/1.30 Oil CS objectives and appropriate filter sets (excitation: 405nm/25 mW, 488nm/10 mW, 561nm/20 mW). Phase-contrast images were taken using an inverted EVOS XL Imagin6g System (# AME3300) equipped with long working distance phase contrast objectives and LED illumination. Images were acquired with a digital 3 MP CMOS color camera.
Live cell imaging
Time lapse studies were performed using a live cell imaging Zeiss Axiovert Fluorescence microscope equipped with a Hamamatsu Orca-ER camera and a self-contained incubation chamber providing 37°C and 5% CO2. Frames were taken every 2 seconds for a total period of 3 minutes for mitochondrial trafficking studies as well as for imaging mitochondrial fusion and fission events using a 63× oil objective (Plan-Apochromat, 1.4 NA). For visualization of mitochondria, cells were transduced with CellLight® Mito-GFP, BacMam 2.0 (Invitrogen) 16 hours prior to imaging. The staining media was replaced with fresh neuronal media before imaging. Time-lapse imaging of neurons for neuronal outgrowth studies was carried out using a 20× phase-contrast objective. Frames were taken every 2 minutes for a period of 25 minutes.
Image analysis
ImageJ version 1.43 software (NIH, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/) was used to analyze parameters of mitochondrial morphology and motility as described previously [28, 54]. In detail, the length of single mitochondria (aspect ratio), mitochondrial network formation (form factor) and mitochondrial trafficking parameters (including the number of moving mitochondria and their direction) were evaluated. Fluorescence microscopy images were optimized by adjusting the contrast and subsequently binarized by conversion to 8 bit images. After reduction of unspecific noise of the fluorescence signal, a threshold was applied to the images to define mitochondrial structures. The threshold was kept equal within one experiment. Analysis of the aspect ratio as well as the form factor included the use of the ‘analyze particle’ function in ImageJ program to preclude from inclusion of unspecific background. At least 3 fields of view were used per coverslip, and several thousand mitochondrial structures were analyzed per condition (= neurons at day 40 or day 100). For quantification, all samples within one experiment were stained simultaneously and imaged with identical settings. For analysis of mitochondrial trafficking studies, kymographs were created using the ‘Multiple Kymograph’ plugin in ImageJ software from time-lapse images. From these kymographs the movement of mitochondria was classified as retrograde (moved ≥10μm in the retrograde direction), anterograde (moved ≥10μm in the anterograde direction), or stationary (moved < 10μm during the duration of the 3 minutes video). The percent motility of mitochondria along the neural process (retrograde, anterograde, stationary) was calculated as a percentage of the total number of mitochondria imaged per neuronal process.
Statistical analysis
Student's t-test (two-sided) was used for analysis of TH-positive cells as percent of total (Figure 2D), mitochondria length (aspect ratio) as well as mitochondrial interconnectivity (form factor) (Figure 3C). One-way ANOVA with Dunnet's post-hoc test was used for quantifications of percent of mitochondrial motility (Figure 3D). p values less than 0.05 were considered significant. Statistical calculations were performed with GraphPad Prism Software, Version 5.0 (http://www.graphpad.com/scientific-software/prism/). All data shown is representative of experiments from at least 3 separate differentiated culture sets.
Supplementary Material
Acknowledgments
This work was supported by NIH (R01NS076054 to DK). LFB was supported by a fellowship within the postdoc program of the German Academic Exchange Service (DAAD).
Contributor Information
Lena F. Burbulla, The Ken and Ruth Davee Department of Neurology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA
Kristin G. Beaumont, Departments of Biomedical Engineering, Chemistry and Cell and Molecular Biology, Northwestern University, Evanston, IL 60208, USA
Milan Mrksich, Email: milan.mrksich@northwestern.edu, Departments of Biomedical Engineering, Chemistry and Cell and Molecular Biology, Northwestern University, Evanston, IL 60208, USA.
Dimitri Krainc, Email: krainc@northwestern.edu, The Ken and Ruth Davee Department of Neurology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
References
- 1.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Cell. 2008;134:877. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Cell. 2007;131:861. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Grskovic M, Javaherian A, Strulovici B, Daley GQ. Nature reviews Drug discovery. 2011;10:915. doi: 10.1038/nrd3577. [DOI] [PubMed] [Google Scholar]
- 4.Anson BD, Kolaja KL, Kamp TJ. Clin Pharmacol Ther. 2011;89:754. doi: 10.1038/clpt.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sterneckert JL, Reinhardt P, Scholer HR. Nature reviews Genetics. 2014;15:625. doi: 10.1038/nrg3764. [DOI] [PubMed] [Google Scholar]
- 6.Shi P, Nedelec S, Wichterle H, Kam LC. Lab on a chip. 2010;10:1005. doi: 10.1039/b922143c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. Nature methods. 2005;2:599. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi P, Scott MA, Ghosh B, Wan D, Wissner-Gross Z, Mazitschek R, Haggarty SJ, Yanik MF. Nature communications. 2011;2:510. doi: 10.1038/ncomms1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleinfeld D, Kahler KH, Hockberger PE. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1988;8:4098. doi: 10.1523/JNEUROSCI.08-11-04098.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi P, Shen K, Kam LC. Developmental neurobiology. 2007;67:1765. doi: 10.1002/dneu.20553. [DOI] [PubMed] [Google Scholar]
- 11.Amadio S, De Ninno A, Montilli C, Businaro L, Gerardino A, Volonte C. BMC neuroscience. 2013;14:121. doi: 10.1186/1471-2202-14-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunello CA, Jokinen V, Sakha P, Terazono H, Nomura F, Kaneko T, Lauri SE, Franssila S, Rivera C, Yasuda K, Huttunen HJ. Journal of nanobiotechnology. 2013;11:11. doi: 10.1186/1477-3155-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogt AK, Wrobel G, Meyer W, Knoll W, Offenhausser A. Biomaterials. 2005;26:2549. doi: 10.1016/j.biomaterials.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 14.Kelly S, Regan EM, Uney JB, Dick AD, Mc Geehan JP, Bristol Biochip G, Mayer EJ, Claeyssens F. Biomaterials. 2008;29:2573. doi: 10.1016/j.biomaterials.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Czondor K, Garcia M, Argento A, Constals A, Breillat C, Tessier B, Thoumine O. Nature communications. 2013;4:2252. doi: 10.1038/ncomms3252. [DOI] [PubMed] [Google Scholar]
- 16.Kuddannaya S, Bao J, Zhang Y. ACS applied materials & interfaces. 2015;7:25529. doi: 10.1021/acsami.5b09032. [DOI] [PubMed] [Google Scholar]
- 17.Hardelauf H, Waide S, Sisnaiske J, Jacob P, Hausherr V, Schobel N, Janasek D, van Thriel C, West J. The Analyst. 2014;139:3256. doi: 10.1039/c4an00608a. [DOI] [PubMed] [Google Scholar]
- 18.Mrksich M, Dike LE, Tien J, Ingber DE, Whitesides GM. Experimental cell research. 1997;235:305. doi: 10.1006/excr.1997.3668. [DOI] [PubMed] [Google Scholar]
- 19.Kane RS, Takayama S, Ostuni E, Ingber DE, Whitesides GM. Biomaterials. 1999;20:2363. doi: 10.1016/s0142-9612(99)00165-9. [DOI] [PubMed] [Google Scholar]
- 20.Kilian KA, Bugarija B, Lahn BT, Mrksich M. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4872. doi: 10.1073/pnas.0903269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thery M, Racine V, Pepin A, Piel M, Chen Y, Sibarita JB, Bornens M. Nature cell biology. 2005;7:947. doi: 10.1038/ncb1307. [DOI] [PubMed] [Google Scholar]
- 22.Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:5970. doi: 10.1523/JNEUROSCI.4441-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Cell. 2011;146:37. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L. Nature. 2011;480:547. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin MT, Beal MF. Nature. 2006;443:787. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 26.Westermann B. Nature reviews Molecular cell biology. 2010;11:872. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 27.Chan DC. Annual review of genetics. 2012;46:265. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 28.Burbulla LF, Fitzgerald JC, Stegen K, Westermeier J, Thost AK, Kato H, Mokranjac D, Sauerwald J, Martins LM, Woitalla D, Rapaport D, Riess O, Proikas-Cezanne T, Rasse TM, Kruger R. Cell death & disease. 2014;5:e1180. doi: 10.1038/cddis.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saxton WM, Hollenbeck PJ. Journal of cell science. 2012;125:2095. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai Q, Zakaria HM, Simone A, Sheng ZH. Current biology : CB. 2012;22:545. doi: 10.1016/j.cub.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacAskill AF, Kittler JT. Trends in cell biology. 2010;20:102. doi: 10.1016/j.tcb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Perlson E, Maday S, Fu MM, Moughamian AJ, Holzbaur EL. Trends in neurosciences. 2010;33:335. doi: 10.1016/j.tins.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller KE, Sheetz MP. The Journal of cell biology. 2006;173:373. doi: 10.1083/jcb.200510097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris RL, Hollenbeck PJ. The Journal of cell biology. 1995;131:1315. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waters J, Smith SJ. Pflugers Archiv : European journal of physiology. 2003;447:363. doi: 10.1007/s00424-003-1182-0. [DOI] [PubMed] [Google Scholar]
- 36.Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, Zhang SC, Abbeduto L, Bhattacharyya A. Stem cells and development. 2014;23:1777. doi: 10.1089/scd.2014.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, Hoing S, Hargus G, Heck SA, Dhingra A, Wu G, Muller S, Brockmann K, Kluba T, Maisel M, Kruger R, Berg D, Tsytsyura Y, Thiel CS, Psathaki OE, Klingauf J, Kuhlmann T, Klewin M, Muller H, Gasser T, Scholer HR, Sterneckert J. Cell stem cell. 2013;12:354. doi: 10.1016/j.stem.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 38.Sirenko O, Hesley J, Rusyn I, Cromwell EF. Assay and drug development technologies. 2014;12:536. doi: 10.1089/adt.2014.592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, Beaumont KG, Kim HJ, Topol A, Ladran I, Abdelrahim M, Matikainen-Ankney B, Chao SH, Mrksich M, Rakic P, Fang G, Zhang B, Yates JR, 3rd, Gage FH. Molecular psychiatry. 2015;20:361. doi: 10.1038/mp.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buzanska L, Ruiz A, Zychowicz M, Rauscher H, Ceriotti L, Rossi F, Colpo P, Domanska-Janik K, Coecke S. Acta neurobiologiae experimentalis. 2009;69:24. doi: 10.55782/ane-2009-1726. [DOI] [PubMed] [Google Scholar]
- 41.Papa S, De Rasmo D. Trends in molecular medicine. 2013;19:61. doi: 10.1016/j.molmed.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 42.Chen H, Chan DC. Human molecular genetics. 2009;18:R169. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan DC. Annual review of cell and developmental biology. 2006;22:79. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 44.Twig G, Graf SA, Wikstrom JD, Mohamed H, Haigh SE, Elorza A, Deutsch M, Zurgil N, Reynolds N, Shirihai OS. American journal of physiology Cell physiology. 2006;291:C176. doi: 10.1152/ajpcell.00348.2005. [DOI] [PubMed] [Google Scholar]
- 45.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Cell. 2010;141:280. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. Developmental cell. 2001;1:515. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 47.Rambold AS, Kostelecky B, Lippincott-Schwartz J. Autophagy. 2011;7:1568. doi: 10.4161/auto.7.12.17992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakada K, Inoue K, Hayashi J. Biochemical and biophysical research communications. 2001;288:743. doi: 10.1006/bbrc.2001.5838. [DOI] [PubMed] [Google Scholar]
- 49.Hollenbeck PJ, Saxton WM. Journal of cell science. 2005;118:5411. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chevalier-Larsen E, Holzbaur EL. Biochimica et biophysica acta. 2006;1762:1094. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 51.De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Annual review of neuroscience. 2008;31:151. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- 52.Dinh ND, Chiang YY, Hardelauf H, Baumann J, Jackson E, Waide S, Sisnaiske J, Frimat JP, van Thriel C, Janasek D, Peyrin JM, West J. Lab on a chip. 2013;13:1402. doi: 10.1039/c3lc41224e. [DOI] [PubMed] [Google Scholar]
- 53.Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, Hargus G, Deleidi M, Lawson T, Bogetofte H, Perez-Torres E, Clark L, Moskowitz C, Mazzulli J, Chen L, Volpicelli-Daley L, Romero N, Jiang H, Uitti RJ, Huang Z, Opala G, Scarffe LA, Dawson VL, Klein C, Feng J, Ross OA, Trojanowski JQ, Lee VM, Marder K, Surmeier DJ, Wszolek ZK, Przedborski S, Krainc D, Dawson TM, Isacson O. Science translational medicine. 2012;4:141ra90. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burbulla LF, Schelling C, Kato H, Rapaport D, Woitalla D, Schiesling C, Schulte C, Sharma M, Illig T, Bauer P, Jung S, Nordheim A, Schols L, Riess O, Kruger R. Human molecular genetics. 2010;19:4437. doi: 10.1093/hmg/ddq370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.