

Abstract
Background
The purpose of this study was to evaluate the safety and effectiveness of iloperidone for the prevention of relapse in schizophrenia.
Methods
Study subjects were adults with schizophrenia who started on oral open-label iloperidone titrated to an initial target dose of 12 mg/day (6 mg twice daily) and then stabilized on a flexible-dose iloperidone regimen (range 8–24 mg/day) for up to 24 weeks. Subjects meeting stabilization criteria then entered the relapse-prevention phase and were randomized 1:1 in a double-blind fashion to continue with iloperidone or placebo withdrawal for up to 26 weeks or until meeting relapse or other withdrawal criteria.
Results
A total of 303 subjects were randomized to the relapse-prevention phase; 153 continued to receive iloperidone, and 150 were withdrawn to placebo. The modal total daily dose for iloperidone in all phases of the study was 12 mg/day. The pre-defined unblinded interim analysis upon reaching 68 relapse events confirmed the hypothesis that iloperidone (n = 97) was more effective than placebo (n = 96) in preventing relapse events, and the trial was stopped early. The estimated relapse rates were 63.4 % (Kaplan–Meier [KM] estimate) for placebo compared with 20.4 % (KM estimate) for those continuing to receive iloperidone (log rank test: p < 0.0001). The mean time to relapse was 71 days for placebo and 139 days for iloperidone (hazard ratio 4.7; 95 % confidence interval 2.7–8.3; p < 0.0001). The safety profile observed in previous short-term studies was also reaffirmed in this maintenance treatment setting. The most common treatment-emergent adverse events (TEAEs) in the stabilization phase were dizziness (11.6 %), somnolence (8.3 %), and dry mouth (6.8 %). Rates of reported extrapyramidal disorder or akathisia during stabilization were 2.5 and 3.7 %, respectively.
Conclusions
Flexible dosing of iloperidone for maintenance-phase therapy, with a modal dose of 12 mg/day was effective in preventing relapse in subjects previously stabilized on iloperidone. The adverse event profile for iloperidone was consistent with other studies, and the low extrapyramidal symptom and akathisia burden during stabilization was sustained during the course of the study.
ClinicalTrials.gov identifier: NCT01291511.
Electronic supplementary material
The online version of this article (doi:10.1007/s40263-016-0345-4) contains supplementary material, which is available to authorized users.
Key Points
| Iloperidone is an effective antipsychotic for the prevention of relapse during maintenance treatment of schizophrenia in patients previously stabilized on iloperidone. |
| The approved dose range of iloperidone for acute treatment is 12–24 mg/day given twice a day; in this relapse-prevention trial, most subjects were treated successfully with a dose between 12 and 16 mg/day. |
| The adverse event profile for iloperidone in this study was consistent with those in the fixed-dose acute iloperidone studies, showing very low propensity to cause extrapyramidal symptoms or akathisia. |
Introduction
Iloperidone is a second-generation “atypical” antipsychotic currently available as an oral formulation. Its primary mechanism of action is combined D2/5HT2A antagonism, with greater affinity for the 5HT2A receptor than for the D2 receptor [1]. Whereas iloperidone shares a high affinity for both D2 and 5HT2A receptors, it has a unique receptor-binding profile that includes very strong affinity for the noradrenergic alpha 1 (NEα1) receptor [2]. The tolerability profile of iloperidone is most notable for a very low propensity to cause either antipsychotic-induced akathisia or antipsychotic-induced extrapyramidal symptoms (EPS). The low propensity for akathisia or EPS is seen across the entire therapeutic dose range, and the need for anticholinergics or other anti-EPS or anti-akathisia therapies is relatively infrequent [3–7].
Iloperidone first received regulatory approval for the treatment of adults with schizophrenia in May 2009 in the USA, with subsequent approvals in Israel, Argentina, and Mexico. The US FDA approval was based on two pivotal short-term (4- and 6-week) acute trials [3, 6, 8]. In addition, three 52-week maintenance studies have been conducted using a non-inferiority design to compare outcomes in individuals who were randomized and successfully stabilized on either iloperidone or haloperidol [7]. The pooled results demonstrated that the 52-week relapse rate for iloperidone at a mean daily dose of 12.5 mg was comparable to that for haloperidol 12.5 mg (52-week relapse estimate by Kaplan–Meier [KM] method was 43.5 % relapse for iloperidone vs. 41.2 % relapse for haloperidol). From a statistical standpoint, iloperidone was non-inferior to haloperidol within the margin stated in the study design [7]. The study reported here (REPRIEVE [RElapse PRevention, Iloperidone EVidence Evaluation]) was a double-blind placebo-controlled randomized withdrawal study with the primary objective of demonstrating that continuation of iloperidone has statistically and clinically significant relapse-prevention benefits during maintenance phase therapy.
Methods
Overview of Design
Upon entering the study, subjects began taking iloperidone in an open-label crossover design using a standard up-titration schedule to discontinue any prior antipsychotic while simultaneously reaching the therapeutic target dose of iloperidone 12 mg on a twice-daily regimen. Response to iloperidone was then established by flexible-dose open-label iloperidone treatment for at least 12 weeks. Following stabilization, subjects entered the double-blind relapse-prevention phase and were randomized 1:1 to either continue on the same flexible-dose regimen of iloperidone or to stop iloperidone in favor of a matched placebo. Subjects were followed for up to 26 weeks and were withdrawn upon showing signs of relapse or upon conclusion of the study, based on efficacy results following a predefined interim analysis (IA). The primary endpoint was a comparison of the time until first relapse event between groups. Secondary objectives included assessing the long-term safety and tolerability of the flexible-dose iloperidone regimen as well as the course of symptom response.
Patient Population
This was an international multicenter trial conducted at 66 study sites between 7 February 2011 and 21 March 2014: USA – 27 sites (289 total subjects); India – 15 sites (118 total subjects), and Ukraine – 24 sites (228 total subjects). Study subjects included men and women (aged 18–65 years, inclusive) with a Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV) diagnosis of schizophrenia for at least 1 year, currently treated as outpatients with a need for ongoing psychiatric treatment, and who had a clinical reason for changing their current antipsychotic medication. The objective of many other key enrollment criteria was to establish a population known to be vulnerable to relapse and have a history of reasonable therapeutic response to first-line antipsychotics. Therefore, subjects needed to have had a history of at least one acute exacerbation of schizophrenia that was successfully treated. In addition, subjects with a history of treatment-resistant schizophrenia based on a recent history of poor response to two or more antipsychotic trials were excluded, as were those with a high level of symptoms as defined by a Positive and Negative Syndrome Scale (PANSS) total score (PANSST) greater than 100. Other key exclusion criteria included having a primary diagnosis other than schizophrenia, being on iloperidone at the time of the screening visit, being medically unstable, having a history of serious suicidal ideation within the prior 6 months or suicidal behavior within the last 2 years, or having a positive history of significant cardiovascular illness (e.g., myocardial infarction, cardiac arrhythmia) or baseline QTcF ≥ 450 ms. Included subjects had a body mass index >17 and <40 kg/m2.
The study protocol and all amendments were reviewed by either the Independent Ethics Committee or Institutional Review Board for each center. The study was conducted according to the ethical principles of the Declaration of Helsinki. Informed consent was obtained from each subject in writing before any study-specific procedures were performed.
Study Phases
The study design consisted of three phases after baseline screening: (1) an initial “cross-titration phase” with the goal of changing from the previous antipsychotic to iloperidone monotherapy, whereby the subject’s current antipsychotic was discontinued; (2) a “stabilization phase” with the goal of establishing a clinically appropriate iloperidone dose and meeting stabilization criteria for the ensuing relapse-prevention phase; and (3) a double-blind “relapse-prevention phase,” where subjects were randomized to stay on or withdraw from iloperidone and followed for up to 26 weeks until either showing signs of relapse or reaching the final end-of-study (EOS) visit. These phases are shown in the CONSORT (Consolidated Standards Of Reporting Trials) flow diagram (Fig. 1) and described in greater detail in the following sections. For the analysis, data from the initial cross-titration and stabilization phases were combined. Of note is that the Independent Data Monitoring Committee (iDMC) reviewed the results of the IA and determined that the study had successfully met its efficacy endpoint. Once the iDMC’s decision was announced, new patient enrollment stopped, and subjects who had not completed the double-blind relapse-prevention phase were discontinued after completing their next (and final) study visit.
Fig. 1.
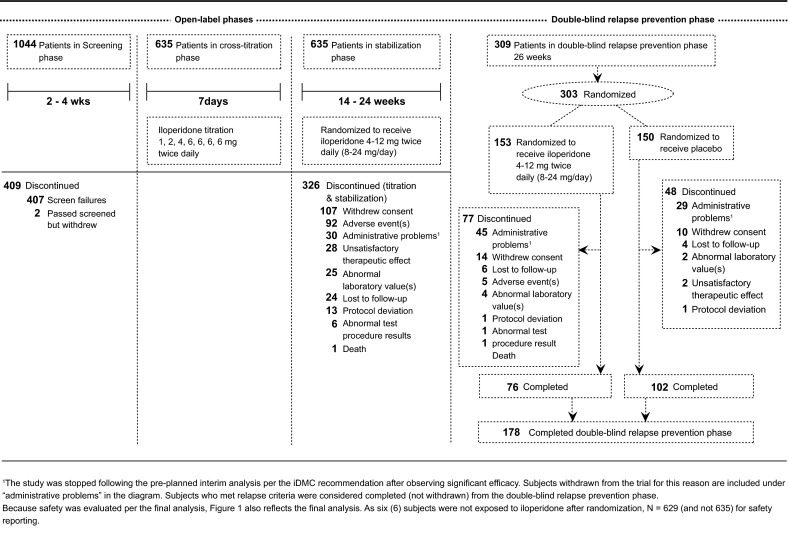
CONSORT (Consolidated Standards of Reporting Trials) flow diagram of study design
Initial Cross-Titration Phase
Consenting subjects meeting inclusion/exclusion criteria began an initial crossover to, or were started on, open-label iloperidone tablets taken twice a day to reach a target dose of 12 mg/day by day 4. The initial up-titration regimen was fixed, whereby the doses were increased from 2 → 4 → 8 → 12 mg/day. The dosage was then maintained at 12 mg/day for the following 3 days. The prior antipsychotic had to be completely discontinued by day 4 of the initial iloperidone up-titration period, with the specific down-titration schedule determined based on investigator judgment.
All concomitant illnesses were treated in accordance with prevailing medical practice. Subjects who were prescribed other medications with known central nervous system effects (e.g., anti-parkinsonian medications, beta-blockers, anxiolytics, or sedative/hypnotics) for at least 6 weeks prior to entering the study were maintained at these stable doses during the study. Per the protocol, zolpidem was permitted throughout the study as a rescue medication for insomnia; lorazepam was permitted as a rescue medication for agitation, severe restlessness, or insomnia; and anticholinergics were permitted for the treatment of EPS.
Stabilization Phase
After reaching the target dose of 12 mg/day at the end of the first week, subjects were evaluated weekly for the first 2 weeks and then every other week until meeting stabilization or exclusion criteria. This phase lasted 14 weeks unless the subject needed more time to meet randomization criteria; thus, the stabilization phase could be extended for a maximum of 24 weeks. During this time, the investigator could adjust the iloperidone dose up or down in increments of 4 mg/day based on clinical judgment, within a permitted range of a total daily dose of 8–24 mg/day. Stability was defined as reaching and remaining at a prespecified symptom criteria threshold (e.g., Clinical Global Impression of Severity [CGI-S] ≤4, five individual PANSS positive symptoms [delusions, conceptual disorganization, hallucinatory behavior, suspiciousness/persecution, hostility] and uncooperative item scores ≤4, as well as a PANSST ≤70) and not requiring any new acute treatment service intervention. Subjects needed to remain clinically stable for at least 12 weeks and not require any dose adjustments for at least 4 weeks before being eligible for the relapse-prevention phase described next.
Relapse-Prevention Phase
Subjects who met full stabilization criteria then entered the relapse-prevention phase and were randomized on a 1:1 ratio (administered via an interactive voice response system [IVRS]). Study drug and placebo were provided by the sponsor with identical packaging, labeling, administration schedule, appearance, taste, and odor. Subjects, investigators, individuals performing the assessments, and data analysts remained blind to the identity of the treatment from the time of randomization until database lock.
Subjects assigned to iloperidone remained on the same dose as had been determined for stabilization. Any subsequent dose increase of double-blind study medication for symptom control would automatically meet relapse criteria, whereas dose reductions for tolerability issues were not considered to be relapse criteria.
Subjects were evaluated weekly after randomization for the first 4 weeks, every other week between weeks 5 and 14, and then every 4 weeks for up to a maximum of 26 weeks. Each study visit after randomization included an assessment to see whether the subject met relapse criteria, and subjects and caregivers were asked to notify study staff immediately in the event of any signs of impending relapse that began between study visits. Subjects who met relapse criteria were considered to have met the study endpoint criteria, received an EOS assessment, and then received standard of care treatment for schizophrenia. Other reasons for early discontinuation included subject withdrawal and sponsor-initiated termination after the IA decision to end the study early (see Sect. 2.4).
Efficacy Assessments
The primary efficacy endpoint was time to relapse or impending relapse, defined as any of the following: hospitalization due to worsening of schizophrenia; increase (worsening) of the PANSST of ≥30 % from randomization, PANSST score confirmed at a second visit conducted within 1–7 days; clinically significant emergent or worsening suicidal, homicidal, or aggressive behavior; a CGI-Improvement (CGI-I) score of 6 (much worse) or 7 (very much worse) after randomization; a dose increase in study medication or a need for additional open-label antipsychotic treatment.
A pre-specified IA was to be performed by the iDMC after 60 events (relapse or impending relapse) had occurred. If significant efficacy was observed (at 0.001 α level), the study was considered to have met the research endpoint. At that point, further enrollment of new subjects and follow-up of enrolled subjects would be halted.
Secondary efficacy variables included the change from randomization to endpoint for PANSST and subscale (positive, negative, and general psychopathology) scores, 18-item Brief Psychiatric Rating Scale (BPRS), CGI-I, CGI-S and the Sheehan Disability Scale (SDS).
Safety Assessments
Safety assessments were conducted throughout all phases of the study and included collection of adverse events (AEs), clinical laboratory evaluations, 12-lead electrocardiograms (ECGs), body weight, vital signs, Columbia Suicide Severity Rating Scale (C-SSRS), and movement disorder rating scales (Barnes Akathisia Rating Scale [BARS], Abnormal Involuntary Movement Scale [AIMS], and Simpson Angus Scale [SAS]).
Statistical Analyses
Analyses Performed
Sample size calculation was based on the primary endpoint of time to relapse using a 2-sided log rank test at the 0.048 level of significance, and a true hazard ratio (HR) of 1.8 (i.e., the hazard of the placebo group meeting relapse criteria relative to the hazard of the iloperidone group meeting relapse criteria).
The log-rank test was used to evaluate the primary outcome of time to relapse or impending relapse between the iloperidone and placebo groups. An independent biostatistics team conducted a predefined unblinded IA utilizing a group sequential testing procedure with an O’Brien–Fleming stopping boundary after 68 observed events (relapse or impending relapse). Once those event numbers were reached, additional time was needed for the statistical team and the iDMC to prepare, analyze, and communicate their decision to end the study. The final dataset had more subjects with more events than the dataset used for the IA. Studies that are halted because of an IA review may present results in two ways: the data available for the IA review and a final analysis, including the entire data from all study visits and observations. Because the study was stopped early as planned in the protocol as a result of significance examined at a level of α = 0.001 in the IA, results from the IA are considered to be the primary results, and the final results (examined at an α = 0.048) including the data between the IA and the actual ending are considered supportive. Plots of KM product limit estimates were used to summarize the survival distributions. In addition, a Cox regression model was used to provide an estimated HR and corresponding 95 % confidence interval (CI).
Change from baseline at randomization for the secondary efficacy variables such as PANSS total and subscale scores, 18-item BPRS, SDS, CGI-I, and CGI-S was analyzed using an analysis of covariance (ANCOVA) model with treatment and site as main effects and the score at randomization as a covariate. The treatment-by-site interaction and treatment by baseline interaction were tested and analyzed only in an ad hoc exploratory fashion. No adjustment for multiple comparisons was made.
Efficacy in the control of symptoms analysis was based on all randomized subjects who received at least one dose of blinded study medication and from whom at least one efficacy measurement was obtained while on study medication. The safety population was defined for both the combined initial cross-titration and stabilization phases and the relapse-prevention phase. For each phase, the safety population included all subjects who received at least one dose of study medication during the corresponding phase. Descriptive statistics by study phase for each of the safety assessments were calculated. Conclusions on the secondary endpoints and safety data were based on the final analysis results.
Results
Subject Disposition
The final analysis dataset consisted of 635 subjects entering the stabilization phase, with 303 (48 %) entering the relapse-prevention phase, including 153 subjects randomized to continue with iloperidone and 150 to switch to placebo. The IA data consisted of 587 subjects entering the stabilization phase, with 195 (33 %) continuing on in the relapse-prevention phase. Of those 195 subjects, 99 were randomized to continue with iloperidone and 96 discontinued iloperidone but continued taking matched placebo (Fig. 1).
Demographic data are presented for the final analysis (Table 1). Distribution of subjects by age, sex, race, and baseline PANSS scores was balanced in the iloperidone and placebo treatment groups. At the time of the first screening visit, the majority (90 %) of study participants reported taking antipsychotic medications for the treatment of schizophrenia. Data for the IA are available in the Electronic Supplementary Material.
Table 1.
Demographic summary by treatment group: final analysis, enrolled/randomized subjects
| Characteristics | Cross-titration/stabilization phase | Relapse-prevention phase | ||
|---|---|---|---|---|
| Total (N = 635) |
Iloperidone (N = 153) |
Placebo (N = 150) |
Total (N = 303) |
|
| Age (years) | 39.6 ± 11.3 | 38.4 ± 11.3 | 38.2 ± 11.1 | 38.3 ± 11.2 |
| Male sex | 400 (63.0) | 96 (62.7) | 82 (54.7) | 178 (58.7) |
| BMI (kg/m2) | 26.8 ± 5.6 | 26.5 ± 5.5 | 25.6 ± 5.5 | 26.1 ± 5.5 |
| Race | ||||
| Caucasian | 280 (44.1) | 74 (48.4) | 77 (51.3) | 151 (49.8) |
| Black | 201 (31.7) | 31 (20.3) | 28 (18.7) | 59 (19.5) |
| Asian | 123 (19.4) | 38 (24.8) | 40 (26.7) | 78 (25.7) |
| Pacific Islander | 2 (0.3) | 1 (0.7) | 0 | 1 (0.3) |
| Other | 29 (4.6) | 9 (5.9) | 5 (3.3) | 14 (4.6) |
| Age at diagnosis (years) | 26.0 ± 8.7 | 25.4 ± 8.7 | 26.5 ± 8.8 | 25.9 ± 8.8 |
| PANSST at baselinea | 76.8 ± 11.6 | 55.6 ± 10.8 | 55.2 ± 10.1 | 55.3 ± 10.4 |
| CGI-S, last study visit, N b,c | – | 112 | 105 | 217 |
| CGI-S | – | 3.0 ± 0.9 | 3.6 ± 1.1 | – |
Data are presented as mean ± standard deviation or n (%) unless otherwise indicated
BMI body mass index, DBRP double-blind RP, CGI-S Clinical Global Impression of Severity, PANSS T Positive and Negative Syndrome Scale Total Score, RP relapse prevention
aBaseline at the beginning of the respective phase
bPercentages are based on the total number of subjects with available data in each treatment group
cRP completion visit includes observations from last visit during DBRP period for subjects who completed the study and RP completion visit that could not be remapped to a previous scheduled visit for subjects who discontinued the DBRP phase
Dosing
The modal total daily dose for iloperidone during the combined initial cross-titration and stabilization phase and the subsequent relapse-prevention phase based on the final analysis was 12 mg in each. The average total daily dose for iloperidone during the combined initial cross-titration and stabilization phase and subsequent relapse-prevention phase was 13.6 ± 4.5 mg (Fig. 2) and 15.0 ± 4.9 mg, respectively.
Fig. 2.

Average iloperidone total daily dose during the initial cross-titration and stabilization phase
Primary Efficacy
Relapse Prevention
The IA was completed after a total of 68 relapse events. As mentioned, because results met the study hypothesis that continuation on iloperidone is more effective for relapse prevention than placebo, the iDMC recommended the study be stopped.
The IA showed that continued iloperidone treatment was more effective than placebo in relapse prevention (log rank test: p < 0.0001), with estimated 26-week relapse rates of 63.4 % (KM estimate; 95 % CI 52.7–74.1) for the placebo group compared with 20.4 % (KM estimate; 95 % CI 12.9–31.4) for those continuing with iloperidone. The mean time to relapse based on KM estimates was 71 days for placebo and 139 days for iloperidone, with a Cox regression HR estimate of 4.7 (95 % CI 2.7–8.3) favoring iloperidone (p < 0.0001) (Fig. 3a; Table 2).
Fig. 3.

Kaplan–Meier plots of time to relapse or impending relapse. a Interim analysis population (N = 195). Interim analysis mean time to relapse: placebo 71 days, iloperidone 139 days; interim analysis Cox regression hazard ratio estimate: 4.7 (95 % confidence interval 2.7–8.3; p < 0.0001, log-rank test). b Final analysis population. Final analysis mean time to relapse: placebo 95 days; iloperidone 140 days; final analysis Cox regression hazard ratio estimate: 5.2 (95 % confidence interval 3.2–8.4; p < 0.0001, log-rank test). Survival estimates are displayed with 95 % Hall–Wellner confidence bands in color shading. The primary efficacy endpoint—time to relapse or impending relapse—is defined as the time from the first dose of double-blind study medication to the assessment at which the first time relapse or impending relapse is identified. For subjects not relapsing, the time to relapse is censored to the last double-blind relapse-prevention period study visit, including the end of study visit, early termination visit, or unscheduled visit for the double-blind relapse-prevention period. Two subjects lost to follow-up after randomization were excluded from the analysis population
Table 2.
Secondary efficacy variables: cross-titration/stabilization and relapse-prevention phases: final analysis
| Visit | Cross-titration/stabilization (N = 629) |
Relapse prevention | |
|---|---|---|---|
| Iloperidone (N = 151) |
Placebo (N = 150) |
||
| PANSS total score, change from baseline to last visit | |||
| N | 607 | 150 | 149 |
| Baseline mean (SD) | 76.8 (11.54) | 55.6 (10.76) | 55.2 (10.08) |
| Change mean (SE) | −14.0 (14.05) | 1.1 (1.12)* (Adj) | 12.4 (1.15) (Adj) |
| CGI-S, last visit | |||
| N | 609 | 112 | 105 |
| Median | 3.0 | 3.0 | 4.0 |
| SDS total score, change from baseline to last visit | |||
| N | 586 | 142 | 128 |
| Baseline mean (SD) | 15.6 (7.42) | 8.8 (6.47) | 8.0 (6.31) |
| Change mean (SE) | −4.6 (7.62) | −0.2 (0.54)** (Adj) | 1.8 (0.61) (Adj) |
OC data from the safety set are presented for the PANSS in the cross-titration/stabilization phase; LOCF data are presented for the PANSS and SDS for the relapse-prevention phase. The CGI-S reflects OC data, as this measure does not include a baseline assessment. Baseline refers to the respective study phases shown at the top of each column that displays data. Cross-titration/stabilization baseline is defined as the last available assessment prior to the start of medication in the cross-titration phase. Relapse prevention baseline is defined as the last available assessment prior to the first dose of double-blind study medication. Change from cross-titration/stabilization baseline is calculated as post value minus cross-titration/stabilization baseline value. A negative change indicates improvement. Change from relapse prevention baseline is calculated as post value minus relapse prevention baseline value. A negative change indicates improvement. P values are based on an ANCOVA model with treatment and site as main effects and relapse prevention baseline as a covariate. Adjusted change is the least squared mean change obtained from the ANCOVA model
Adj adjusted, ANCOVA analysis of covariance, CGI-S Clinical Global Impression of Severity, LOCF last observation carried forward, OC observed cases, PANSS Positive and Negative Syndrome Scale, SD standard deviation, SDS Sheehan Disability Scale, SE standard error
* p < 0.0001, ** p = 0.0062, log-rank test
The supportive final analysis confirmed the IA results favoring iloperidone; the time to relapse or impending relapse was significantly longer by the log-rank test (p < 0.0001) for the iloperidone treatment group than for the placebo treatment group (140 and 95 days, respectively) (Fig. 3b). The relapse rate was lower in the iloperidone treatment group than in the placebo treatment group (17.9 vs. 64.0 %, respectively; p < 0.0001). The demonstrated HR estimate was 5.2 (95 % CI 3.2–8.4).
The IA of time to relapse after 68 events was supported by the results of the final analysis, which included the entire efficacy data collected in the study, after a total of 104 events (Fig. 3b).
Symptoms and Severity of Illness Results
The secondary efficacy results for course of symptoms (PANSST) and overall severity of illness (CGI-S) used the final analysis dataset and are presented in Table 2. Evaluation of the PANSST demonstrated a significantly smaller increase (least squared mean change in the ANCOVA model) from the relapse-prevention baseline to the last study visit in the iloperidone treatment group (1.1) compared with the placebo treatment group (12.4; p < 0.0001). Similar results were observed based on the same parameters for the 18-item BPRS (iloperidone group 4.2; placebo group 12.1; p < 0.0001).
The median CGI-S score was 3.0 (i.e., mildly ill) for the iloperidone treatment group and 4.0 (i.e., moderately ill) for the placebo treatment group at the last study visit (final analysis). No statistically significant differences occurred between the iloperidone and placebo groups on the CGI-S.
The median SDS total score at the last study visit was 8.0 for the iloperidone treatment group and 9.0 for placebo-treated subjects. The least squared mean change from relapse-prevention baseline to the last study visit was significantly lower in the iloperidone treatment group (–0.2) than in the placebo treatment group (1.8; p = 0.0062, last observation carried forward) (Table 2).
Safety
Adverse Events Overview
Cross-titration and stabilization phase Of the 629 subjects who had some exposure to open-label iloperidone during these combined phases, 433 (69 %) experienced a treatment-emergent AE (TEAE) (Table 3), of which 345 TEAEs were suspected to be related to study drug. The most common TEAEs (≥5 %) suspected to be related to study drug were dizziness (10.7 %), somnolence (8.1 %), and dry mouth (6.7 %). A total of 21 subjects (3.3 %) treated with iloperidone experienced a serious AE (SAE). The SAE was considered to be possibly attributed to study drug for 33 % of these subjects. The most common SAE was schizophrenia (1.4 %). No other SAEs occurred in more than one subject. Two subjects in the study died while receiving iloperidone (one in each phase). The death during the titration and stabilization phase was attributed to alcohol poisoning and was considered unrelated to study medication.
Table 3.
Incidence of treatment-emergent adverse events (5 % incidence in any group): cross-titration and stabilization phase
| Adverse event | Total (N = 629) |
|---|---|
| Subjects with at least one TEAE | 433 (68.8) |
| Possibly drug-related TEAE | 345 (54.8) |
| Subjects with one or more serious TEAE | 21 (3.3) |
| Schizophrenia | 15 (2.4) |
| Insomnia | 35 (5.6) |
| Dizziness | 73 (11.6) |
| Headache | 40 (6.4) |
| Weight increased | 34 (5.4) |
| Nausea | 32 (5.1) |
| Somnolence | 52 (8.3) |
| Dry mouth | 43 (6.8) |
Data are presented as n (%)
TEAE treatment-emergent adverse event
Relapse-prevention phase A total of 60 of the 151 subjects who continued on double-blind iloperidone and 54 of the 150 who went on to placebo (36.0 %) experienced a TEAE (Table 4). During the relapse-prevention phase, 23.3 % of all subjects had at least one TEAE that was suspected to be related to study drug; the percentage of subjects with at least one TEAE suspected to be related to study drug was similar between treatment groups. No TEAEs suspected to be related to study drug had an incidence ≥5 % in the iloperidone treatment group. Six subjects (4.0 %) treated with iloperidone versus four (2.7 %) treated with placebo experienced an SAE. As in the previous phase, the most common SAE was schizophrenia: 1.3 % in iloperidone-treated subjects and 1.3 % in placebo-treated subjects. No other SAEs occurred in more than one subject. The second case of death in the study occurred in the relapse-prevention phase and involved a 47-year-old female whose cause of death was initially labeled as “unknown”; consequently, the investigator assigned a potentially causal relationship to study drug. The subject’s final cause of death was reported as “probable sudden cardiac arrhythmia/natural”. Of note, a routine ECG recorded the day before her death was rated as unchanged from baseline, with a QTcF of 410 ms.
Table 4.
Incidence of treatment-emergent adverse events: relapse-prevention phase
| Adverse event | Iloperidone (N = 151) |
Placebo (N = 150) |
Total (N = 301) |
|---|---|---|---|
| Subjects with at least one TEAE | 60 (39.7) | 54 (36.0) | 114 (37.9) |
| Possibly drug-related TEAE | 37 (24.5) | 33 (22.0) | 70 (23.3) |
| Subjects with one or more serious TEAE | 6 (4.0) | 4 (2.7) | 10 (3.3) |
| Schizophrenia | 3 (2.0) | 15 (10.0) | 18 (6.0) |
| Insomnia | 3 (2.0) | 7 (4.7) | 10 (3.3) |
| Dizziness | 4 (2.6) | 4 (2.7) | 8 (2.7) |
| Headache | 3 (2.0) | 5 (3.3) | 8 (2.7) |
| Weight increased | 2 (1.3) | 4 (2.7) | 6 (2.0) |
| Nausea | 2 (1.3) | 0 | 2 (0.7) |
| Somnolence | 2 (1.3) | 0 | 2 (0.7) |
| Dry mouth | 0 | 0 | 0 |
Data are presented as n (%)
TEAE treatment-emergent adverse event
Akathisia and Extrapyramidal Disorder
Cross-titration and stabilization phase Akathisia at baseline prior to starting iloperidone and defined by a BARS score ≥1 was present in 7 % of subjects; however, by the end of the stabilization phase it was present in only 3 % of subjects receiving open-label iloperidone. A total of 23 subjects (3.7 %) reported akathisia as a TEAE at one point during this phase while receiving open-label iloperidone.
A total of 16 subjects (2.5 %) reported extrapyramidal disorder as a TEAE. Subjects’ mean SAS total score, reflecting neuroleptic-induced parkinsonism was 0.8 at initial baseline prior to starting the iloperidone crossover. After stabilization on iloperidone, the mean SAS score was 0.5. No clinically meaningful changes in dyskinesia scores as measured by the AIMS were observed (range 0.4–0.6).
Relapse-prevention phase One subject (1 %) treated with iloperidone versus 0 treated with placebo reported akathisia as a TEAE during the relapse-prevention phase. The rate of akathisia in the placebo group (8 %) was greater than reported in the iloperidone treatment group (5 %) at completion, according to the BARS (score ≥1).
Two subjects (1.3 %) treated with iloperidone versus 0 treated with placebo reported extrapyramidal disorder as a TEAE. Mean SAS total scores were 0.3 and 0.4 for iloperidone and placebo subjects, respectively, at completion. No clinically meaningful changes in the AIMS scores were observed (range 0.2–0.4). Medications for EPS included trihexyphenidyl (iloperidone 4.6 %; placebo 4.0 %), benzatropine (0.7 % in both treatment groups), and diphenhydramine (iloperidone 0.7 %; placebo 2.0 %).
Clinical Laboratory Parameters
Two clinically notable differences in hematology or chemistry parameters occurred: 4.5 and 2.6 % of subjects treated with iloperidone had elevations in blood urea nitrogen levels and low-density lipoprotein (LDL), respectively, versus 0 in subjects treated with placebo.
Weight
Cross-titration and stabilization phase Mean change from baseline in weight at the end of the cross-titration and stabilization phase was 0.7 kg in subjects treated with iloperidone; 13.6 % of subjects treated with iloperidone experienced a ≥7 % increase in weight from baseline at the end of this phase, and 3.4 % experienced a ≥7 % decrease in weight over the same period. Increased weight reported as an AE occurred in 5.4 % of subjects during the phase.
Relapse-prevention phase Mean change from baseline in weight from the initial study baseline to the end of the study was 0.02 kg in subjects treated with iloperidone versus 0.73 kg in subjects treated with placebo, whereas 25.2 % of subjects treated with iloperidone versus 20 % of subjects treated with placebo experienced a ≥7 % increase in weight from initial baseline at the end of the study. In total, 12.6 % of iloperidone subjects and 10.7 % of placebo subjects experienced a decrease in weight of ≥7 % from initial baseline measurements. Increased weight reported as an AE occurred in 1.3 % of iloperidone subjects and 2.7 % of placebo subjects in this phase. No notable between-group differences in waist circumference were observed at the end of the study.
Cardiovascular Parameters
Cross-titration and stabilization phase Mean change from baseline in QTcF for subjects treated with iloperidone was 6.4 ms. One subject had a QTcF > 500 ms (baseline QTcF: 412.7 ms), and six subjects (1.0 %) had an increase from baseline in QTcF ≥ 60 ms, the protocol-specified criterion for discontinuation.
Relapse-prevention phase The mean change from baseline in QTcF for subjects treated with iloperidone was 4.9 versus 1.0 ms in subjects treated with placebo. No subjects in this phase had an increase from baseline in QTcF ≥ 60 ms or a QTcF > 500 ms.
Discussion
Consistent with results of earlier studies comparing iloperidone with haloperidol, iloperidone therapy is effective for maintenance-phase treatment of schizophrenia [7]. Continuing on iloperidone therapy was statistically and clinically effective in preventing relapse. Subjects stabilized on iloperidone who were subsequently randomized to placebo withdrawal were three times more likely to relapse than individuals continuing to receive the medication. Furthermore, iloperidone showed statistical and clinical benefits in delaying the time until relapse relative to placebo among the subgroup of subjects who did eventually relapse. On the basis of the observed early efficacy at the preplanned IA, the iDMC recommended an early stop for the trial. The final analysis confirmed the efficacy demonstrated at the interim.
Results of the analyses for psychiatric symptoms and illness severity support the robust relapse-prevention findings. Subjects randomized to iloperidone during the relapse-prevention phase continued to maintain their symptom response achieved in the stabilization phase. Additionally, the adjusted mean CGI-S scores indicated that subjects in the iloperidone treatment group were mildly ill, whereas subjects in the placebo treatment group were moderately ill.
The average daily dose for iloperidone-treated subjects across study phases in this trial was 12–16 mg, with a modal dose for both the initial cross-titration/stabilization and relapse-prevention phases of 12 mg/day. Given that investigators were permitted to treat patients by optimizing iloperidone dose during stabilization with a dose range between 12 and 24 mg, it seems that investigators judged the 12- to 16-mg dose range as optimal for most subjects. The alternative hypothesis of dose-related AEs presenting a tolerability ceiling of 16 mg was also considered, but it seems unlikely given that the majority of those subjects receiving a dose >16 mg/day did not down-titrate iloperidone during the trial. Data from an earlier pharmacokinetic/pharmacodynamic modeling study of the dose–response characteristics of iloperidone suggests there is, in fact, a therapeutic response association with plasma concentrations between 5 and 8 ng/ml without any further efficacy at higher plasma levels [9]. The iloperidone dosage range that typically results in this plasma concentration is 12–16 mg/day.
Further, the relative absence of dose escalation in this report of the REPRIEVE study suggests that, within the parameters of the study population, 12–16 mg is a reasonable initial target dose for schizophrenia outpatients who are starting on iloperidone. The caveat is that treatment-resistant patients were excluded such that the clinical dosing inference cannot be generalized to choosing a target dose for relatively treatment-resistant patients.
The safety and tolerability profile characterized by the 3210 subjects exposed to iloperidone across nine studies in the clinical trials program is reinforced by the current study and is consistent with those in other iloperidone trials [10–12]. The most common AE was dizziness, occurring in 11.6 % of the subjects during the initial cross-titration and stabilization phase. As expected, the incidence of dizziness was much lower during the relapse-prevention phase (2.6 %), presumably due to accommodation of α1 antagonism. A recently reported switch study using iloperidone also demonstrated that the AEs associated with α1 antagonism were transient in nature [10, 11]. Reported somnolence was also less frequent during the relapse-prevention phase relative to the stabilization period. Rates of schizophrenia (worsening or exacerbation) as a TEAE were constant in subjects receiving iloperidone across the trial. As expected, for subjects randomized to placebo, these rates increased during the relapse-prevention phase.
No new safety concerns with long-term use of iloperidone appeared in the analysis. Any observed laboratory abnormalities were expected based on those reported in previous clinical studies. Although increases in the QTc interval were more common in the iloperidone treatment group than in the placebo treatment group, this is not surprising given that iloperidone is known to have some propensity to lengthen the interval [1]. In the initial cross-titration and stabilization phase, the mean change from baseline in QTcF for subjects treated with iloperidone was 6.4 ms; in the relapse-prevention phase, the mean change from baseline in QTcF for subjects treated with iloperidone was 4.9 versus 1.0 ms in subjects treated with placebo. The change in QTcF from baseline was slightly lower than that observed in previous iloperidone studies [5, 7]. A possible explanation for this finding may be the exclusion of subjects with baseline QTc values ≥450 ms.
Results of the REPRIEVE trial support the minimal impact of iloperidone on rates of common neurological AEs associated with antipsychotic treatment (akathisia, EPS), and its modest association with sedation and changes in body weight and metabolic variables demonstrated in previous research [1, 3, 6, 7, 10, 11]. The prevalence rate for akathisia associated with iloperidone use is likely very low; iloperidone appears to be associated with some of the lowest rates of occurrence among currently available atypical antipsychotic agents [13]. Akathisia causes profound suffering in patients and, in extreme cases, is associated with suicide [14]. Akathisia is also associated with poor response to acute treatment, as well as treatment withdrawal [15, 16]. Furthermore, akathisia is notoriously difficult to diagnose and often is missed in clinical practice [17, 18]. While the molecular basis for the low EPS and akathisia profile is unknown, one hypothesis is that the very high α1 antagonism in the CNS reverses the adverse effects of striatal D2 receptor blockade [19].
Another feature of the iloperidone safety profile is relatively modest effects associated with AEs not related to movement disorders. Iloperidone is unique in that other antipsychotic options with very low rates of EPS or akathisia tend to be more likely to cause sedation and dyslipidemia. For the subjects receiving iloperidone in the current trial, rates of sedation were 3.7 % in the cross-titration and stabilization phase and decreased to 1.3 % for iloperidone versus 0.7 % for placebo during the double-blind phase. Total and LDL cholesterol levels and triglycerides were reduced or unchanged in those receiving iloperidone across all phases of the study. In the relapse-prevention phase, more subjects with baseline elevated total cholesterol values shifted to normal values while taking iloperidone relative to placebo. Similarly, the longer-term iloperidone treatment reported here had no medically relevant impact on glucose levels.
The mean change in weight from the start of open-label therapy to the last study visit was 0.02 kg for randomized subjects continuing on iloperidone and 0.73 kg for those randomized to placebo, with most change occurring during cross-titration and stabilization. Increased weight reported as an AE occurred in 5.4 % of subjects receiving iloperidone during cross-titration and stabilization and occurred less frequently for iloperidone subjects than for placebo subjects during the relapse-prevention phase (1.3 and 2.7 %, respectively). During the double-blind phase, mean weight decreased slightly for subjects in both treatment groups. Looking across all phases of the study, it appears that starting iloperidone is associated with modest weight gain during stabilization, but the overall trajectory of weight changes seems to plateau by the time the subjects enter the relapse-prevention phase of the study.
Iloperidone would be helpful in several clinical circumstances. For many patients with persistent symptoms, iloperidone represents an efficacious medication to which they have not yet been exposed. Because akathisia, EPS, sedation, and metabolic derangements remain very significant problems, even in the era of atypical antipsychotics, iloperidone may be selected on the basis of its overall favorable tolerability profile. Furthermore, when a need exists for switching medications, either by down-titrating a current drug or by directly changing it, iloperidone may be used flexibly, with either cross-titration or immediate switch strategies. The results of the present study support these observations. The choice of medication in these situations would need to be balanced with a potentially inconvenient twice-daily dosing regimen, need for titration to avoid orthostatic hypotension, and considerations around QT interval prolongation.
Limitations
The primary limitation of the study design is that the length of the placebo-controlled phase of the study lasted for only 6 months. However, ethical considerations around the use of placebo in this patient population necessarily informed the study design. While the flexible-dosing regimen in this study allowed for a more real-world experience, it also prevented the assessment of dose–response.
Conclusions
The REPRIEVE study demonstrated that flexible dosing of iloperidone for maintenance therapy (with a median dose range of 12–16 mg/day) was safe and effective in preventing relapse or impending relapse in subjects previously stabilized on iloperidone. The analysis of safety indicated no new safety signals with respect to the use of iloperidone.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors thank the subjects and investigators who participated in this study.
Compliance with Ethical Standards
Funding
This research was supported by Novartis Pharmaceuticals Corporation and Vanda Pharmaceuticals, Inc. Editorial assistance, supported by Vanda, was provided by Aesculapius Consulting, Inc. Although the sponsor was involved in the design, collection, analysis, interpretation, and fact checking of information, the content of this manuscript, the ultimate interpretation, and the decision to submit it for publication in CNS Drugs was made by the authors independently. The authors had full control of the content of this manuscript. Open access was funded by Vanda.
Conflict of interest
Peter Weiden has been a consultant for Alkermes, Delpor, Forum, Johnson & Johnson (Janssen Pharmaceuticals), Lundbeck, Otsuka, Novartis, Sunovion, Teva, and Vanda. He has also received research support from AbbVie, Alkermes, Forum, Forest Laboratories, Genentech, Johnson & Johnson (Janssen Pharmaceuticals), Otsuka, Neurocrine, Novartis, Reckitt Benckiser Pharmaceuticals, and Takeda. Dr. Weiden has been a member of speaker’s bureaus for Forum, Jazz, Johnson & Johnson (Janssen Pharmaceuticals), Lundbeck, Otsuka, Novartis, and Sunovion, and he is also a stockholder with Delpor. Raymond Manning is an investigator at the California Neuropsychopharmacology Clinical Research Institute, Los Angeles facility, and received reimbursement from Novartis for travel to the investigator meeting. Curt Wolfgang was an employee of Vanda Pharmaceuticals Inc. at the time of this study and owns stock related to employment. Dr. Wolfgang is currently an employee of GlycoMimetics, Inc. J. Michael Ryan is an employee of Novartis Pharmaceuticals Corporation and owns stock related to that employment. Linda Mancione and Saeed Ahmed are employees of Novartis Pharmaceuticals Corporation. Guangyang Han is an employee of Novartis Pharmaceuticals Corporation and owns stock and has stock options related to employment. Mallery Mayo is an employee of Vanda Pharmaceuticals, Inc., has received stock options related to that employment, and formerly worked for an agency contracted to Novartis (2013).
Ethical standards
The study protocol and all amendments were reviewed by either the Independent Ethics Committee or Institutional Review Board for each center. The study was conducted according to the ethical principles of the 1964 Declaration of Helsinki. Informed consent was obtained from each subject in writing before any study-specific procedures were performed.
Author contributions
Study concept and design (Wolfgang, Weiden, Ahmed); acquisition, analysis, or interpretation of data (all authors); drafting of the manuscript (Weiden, Mayo); critical revision of the manuscript for important intellectual content (all authors); statistical analysis (Han).
References
- 1.Fanapt (iloperidone) [prescribing information]. Washington, DC: Vanda Pharmaceuticals Inc.; 2014.
- 2.Kalkman HO, Subramanian N, Hoyer D. Extended radioligand binding profile of iloperidone: a broad spectrum dopamine/serotonin/norepinephrine receptor antagonist for the management of psychotic disorders. Neuropsychopharmacology. 2001;25:904–914. doi: 10.1016/S0893-133X(01)00285-8. [DOI] [PubMed] [Google Scholar]
- 3.Cutler AJ, Kalali AH, Mattingly GW, Kunovac J, Meng X. Long term safety and tolerability of iloperidone: results from a 25-week, open-label extension trial. CNS Spectr. 2013;18:43–54. doi: 10.1017/S1092852912000764. [DOI] [PubMed] [Google Scholar]
- 4.Weiden PJ. Iloperidone for the treatment of schizophrenia: an updated clinical review. Clin Schizophr Relat Psychoses. 2012;6:34–44. doi: 10.3371/CSRP.6.1.5. [DOI] [PubMed] [Google Scholar]
- 5.Cutler AJ, Kalali AH, Weiden PJ, Hamilton J, Wolfgang CD. Four-week, double-blind, placebo- and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 Suppl 1):S20–S28. doi: 10.1097/JCP.0b013e318169d4ce. [DOI] [PubMed] [Google Scholar]
- 6.Weiden PJ, Cutler AJ, Polymeropoulos MH, Wolfgang CD. Safety profile of iloperidone: a pooled analysis of 6-week acute-phase pivotal trials. J Clin Psychopharmacol. 2008;28(2 Suppl 1):S12–S19. doi: 10.1097/JCP.0b013e3181694f5a. [DOI] [PubMed] [Google Scholar]
- 7.Kane JM, Lauriello J, Laska E, Di Marino M, Wolfgang CD. Long-term efficacy and safety of iloperidone: results from 3 clinical trials for the treatment of schizophrenia. J Clin Psychopharmacol. 2008;28(2 Suppl 1):S29–S35. doi: 10.1097/JCP.0b013e318169cca7. [DOI] [PubMed] [Google Scholar]
- 8.Potkin SG, Litman RE, Torres R, Wolfgang CD. Efficacy of iloperidone in the treatment of schizophrenia: initial phase 3 studies. J Clin Psychopharmacol. 2008;28(2 Suppl 1):S4–S11. doi: 10.1097/JCP.0b013e3181692787. [DOI] [PubMed] [Google Scholar]
- 9.Baroldi P, Wolfgang CD, Fisher D. A pharmacokinetic (PK)-pharmacodynamic (PD) relationship exists for efficacy of iloperidone, a novel investigational atypical antipsychotic agent. Paper presented at: American Psychiatric Association Annual Meeting, May 19–24, 2007, San Diego, CA.
- 10.Weiden PJ, Citrome L, Alva G, Brams M, Glick ID, Jackson R, et al. A trial evaluating gradual- or immediate-switch strategies from risperidone, olanzapine, or aripiprazole to iloperidone in patients with schizophrenia. Schizophr Res. 2014;153:160–168. doi: 10.1016/j.schres.2013.11.042. [DOI] [PubMed] [Google Scholar]
- 11.Citrome L, Weiden PJ, Alva G, Glick ID, Jackson R, Mattingly G, et al. Switching to iloperidone: an omnibus of clinically relevant observations from a 12-week, open-label, randomized clinical trial in 500 persons with schizophrenia. Clin Schizophr Relat Psychoses. 2015;8:183–195. doi: 10.3371/CSRP.CIWE.103114. [DOI] [PubMed] [Google Scholar]
- 12.Potkin SG, Preskorn S, Hochfeld M, Meng X. A thorough QTc study of 3 doses of iloperidone including metabolic inhibition via CYP2D6 and/or CYP3A4 and a comparison to quetiapine and ziprasidone. J Clin Psychopharmacol. 2013;33:3–10. doi: 10.1097/JCP.0b013e31827c0314. [DOI] [PubMed] [Google Scholar]
- 13.Lohr JB, Eidt CA, Alfaraj AA, Soliman MA. The clinical challenges of akathisia. CNS Spectr. 2015;20:4–14. doi: 10.1017/S1092852915000838. [DOI] [PubMed] [Google Scholar]
- 14.Shear MK, Frances A, Weiden P. Suicide associated with akathisia and depot fluphenazine treatment. J Clin Psychopharmacol. 1983;3:235–236. doi: 10.1097/00004714-198308000-00006. [DOI] [PubMed] [Google Scholar]
- 15.Levinson DF, Simpson GM, Singh H, et al. Fluphenazine dose, clinical response, and extrapyramidal symptoms during acute treatment. Arch Gen Psychiatry. 1990;47:761–768. doi: 10.1001/archpsyc.1990.01810200069010. [DOI] [PubMed] [Google Scholar]
- 16.Van Putten T, May PR, Marder SR. Response to antipsychotic medication: the doctor’s and the consumer’s view. Am J Psychiatry. 1984;141:16–19. doi: 10.1176/ajp.141.1.16. [DOI] [PubMed] [Google Scholar]
- 17.Hirose S. The causes of underdiagnosing akathisia. Schiz Bull. 2003;29:547–558. doi: 10.1093/oxfordjournals.schbul.a007027. [DOI] [PubMed] [Google Scholar]
- 18.Weiden PJ, Mann JJ, Haas G, et al. Clinical nonrecognition of neuroleptic-induced movement disorders: a cautionary study. Am J Psychiatry. 1987;144:1148–1153. doi: 10.1176/ajp.144.9.1148. [DOI] [PubMed] [Google Scholar]
- 19.Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18:285–288. doi: 10.1017/S1092852913000850. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.